


 Marjorie Busby4
Marjorie Busby4

- 1 Department of Neurology, University of South Carolina School of Medicine, Columbia, SC, USA
- 2 The Neurogenetics Foundation, Cranbury, NJ, USA
- 3 Neuroscience Institute, Saint Francis Medical Ceter, Trenton, NJ, USA
- 4 University of North Carolina, Chapel Hill, NC, USA
Significance: Aortic arch (AA) atheroma and AA atheroma progression are independent risk factors for recurrent vascular events in stroke/transient ischemic attack (TIA) patients. Total homocysteine level (tHcy) is an independent risk marker for atherosclerosis including that found in AA. The purpose of this study was to prospectively test the association between AA atheroma progression and tHcy. Methods: This is a cohort study of 307 consecutive hospitalized stroke/TIA patients undergoing transesophageal echocardiogram (TEE) as a part of their clinical workup. Measurable AA atheroma was detected in 167 patients of whom 125 consented to a protocol-mandated follow-up TEE at 12 months. Patients had evaluation for vascular risk factors, dietary factors (folate, B12 and pyridoxine), and methylene tetrahydrofolate reductase (MTHFR) polymorphism. One hundred eighteen stroke/TIA patients had tHcy, acceptable paired AA images, and detailed plaque measurements. An increase by ≥1 grade of AA atheroma was defined as progression. Results: Of the 118 patients, 33 (28%) showed progression and 17 (14%) showed regression of their index arch lesion at 1 year. tHcy (≥14.0 μmol/l) was significantly associated with progression on both univariate (RR = 3.4, 95% CI 2.0–5.8) and multivariate analyses (adjusted RR = 3.6, 95% CI 2.2–4.6). The changes in AA plaque thickness (r2 = 0.11; p < 0.001) and AA plaque area (r2 = 0.08; p = 0.002) correlated with tHcy. tHcy was associated with change in plaque thickness over 12 months, independent of age, dietary factors, renal function and MTHFR polymorphism (Standardized β-coefficient 0.335, p = 0.02). Conclusions: Our results validate the association and a linear correlation between tHcy and progression of AA atheroma.
Introduction
Significant aortic arch (AA) atheroma is the second most prevalent cardioembolic risk factor for stroke after atrial fibrillation, and is present in 16–20% of all stroke and transient ischemic attack (TIA) patients (Sen et al., 2000; Cheitlin et al., 2003). While there is no definitive treatment (Tunick and Kronzon, 2000), AA atheroma is an independent risk factor for new and recurrent stroke (Amarenco et al., 1994; The French Study of Aortic Plaques in Stroke Group, 1996). Recently, we have shown that AA atheroma progression is an independent risk factor for recurrent vascular events in stroke/TIA patients (Sen et al., 2007). Detection and measurement of AA atheroma is a safe procedure in stroke/TIA patients using the well-established, validated, and cost-effective method of transesophageal echocardiogram (TEE) (Daniel et al., 1991; McNamara et al., 1997; Cheitlin et al., 2003). In a retrospective analysis of sequential TEE data, we have shown that AA atheroma has a high rate of progression (Sen et al., 2002); in 29% of stroke/TIA patients AA atheroma worsened ≥1 grade in 9 months compared to <10% that worsened annually in the carotid arteries (Olin et al., 1998; Liapis et al., 2000; Schminke et al., 2000). The study was limited by the fact that methods to control for the location of those measurements had not been developed and factors that may influence the association between homocysteine and atherosclerosis such as diet and genetic polymorphism were not assessed.
In order to respond to these issues, we designed a prospective 12-month study to analyze sequential changes in AA atheroma and to examine the role of stroke risk factors in promoting the progression of AA atheroma. Measurements of the rate of progression were obtained at two time points by using standardized criteria to quantitate plaque thickness and measure sectional areas. In order to ensure accurate sequential measurements at the same location, we established imaging landmarks in each segment.
Hyperhomocysteinemia is an independent risk factor for atherosclerotic vascular disease including cerebrovascular disease (Coull et al., 1990; Stampfer et al., 1992; Malinow et al., 1993; Spence et al., 1999), coronary artery disease (Stampfer et al., 1992), peripheral vascular disease (Malinow et al., 1989), and aortic plaque thickness (Konecky et al., 1997; Tribouilloy et al., 2000). Common factors known to influence homocysteine levels are age, intake of vitamin B12, folic acid levels, and genetic methylene tetrahydrofolate reductase (MTHFR) polymorphism (Kaul et al., 2006). Therefore, an additional objective of this study was to assess whether the association between homocysteine and AA atheroma progression is independent of these determinants of homocysteine level.
Materials and Methods
Three hundred and seven consecutive patients with stroke/TIA underwent TEE assessment within 1 month of symptom onset, as a part of their clinical evaluation to determine stroke etiology. All patients received a brain CT and/or MRI to confirm the diagnosis of stroke, had assessment of stroke risk factors, and were classified into stroke sub-types based on clinical assessment of etiology. Exclusion criteria were: absence of measurable aortic atheroma on the first TEE, age <18 years, intracerebral hemorrhage, subarachnoid hemorrhage, coma and serious medical conditions limiting life expectancy. Of these patients, 167 had evidence of aortic atheroma (measurable plaque ≥1 mm in ascending, arch or descending segment) and 125/167 eligible patients consented to a protocol-mandated follow-up TEE at 12 months. One hundred eighteen (79 strokes and 39 TIAs) had adequate paired aortic images allowing detailed measurements of the AA plaque. Qualifying patients underwent a fasting plasma homocysteine level measurement as part of their stroke risk assessment at the time of admission for the index cerebrovascular event prior to the initial TEE. Of these 118 patients, 116 provided blood samples for assessment of MTHFR polymorphism and 110 filled out a self-administered food frequency questionnaire (FFQ) just prior to the 12-month TEE. All patients enrolled in the study signed informed consent and the study was approved by the Biomedical Institutional Review Board of the University of North Carolina at Chapel Hill.
Tee Assessment of AA Atheroma
A comprehensive TEE with detailed imaging of the aorta was performed using a Hewlett-Packard 21364A omniplane probe. Details of the imaging procedure and quantification of AA atheroma have been previously described (Sen et al., 2000). Briefly, the proximal and mid-ascending aorta were imaged at a probe depth of approximately 30 cm with a multi-plane angle of 100–150° to view the vessel in the long axis. Examination of the descending thoracic aorta was accomplished by advancing the probe to the distal esophagus, imaging the aorta in cross-section (at 0°), and then slowly withdrawing the probe to image the more proximal segments. As the transducer reached the AA, the multi-plane angle was rotated to between 0° and 90° to acquire sequential short axis views.
Digital images were acquired of the diseased areas in each segment of the AA with annotation of the distance of the transducer from the incisors. Identical locations in the AA were evaluated on the 1-year examination, using the depth of the transducer, plaque morphology, and (when available) surrounding anatomical landmarks for guidance.
Two observers blinded to clinical data independently quantified atheroma size for each imaged segment. Plaque thickness was measured as the maximal thickness of the intimal and medial layers and graded as mild (<1 mm), moderate (1–3.9 mm), or severe (≥4 mm), using the criteria of Amarenco et al. (1994) AA atheroma progression was defined as an increase in maximal thickness of the plaque in the AA by ≥1 grade(s) and regression defined as a decrease in maximal thickness by ≥1 grade(s). There was good interobserver reliability between the two observers, in the assessment of aortic plaque progression and regression in the ascending (κ = 0.77), arch (κ = 0.85) and descending segment (κ = 0.86) of thoracic aorta. Excellent intraobserver reliabilities were noted for the first observer (κ = 0.93–1.00) and the second observer (κ = 0.91–0.94). Addition to grading of plaque thickness, cross-sectional plaque area was measured by tracing the outline of the atheroma as described by Khoury et al. (1998) and Ti et al. (2003).
Demographics, Risk Factors and Etiology of Stroke
Cerebrovascular risk factors were classified as follows: age was based on the age at the time of presentation for initial TEE and dichotomized around a mean age of 65 years; hypertension was based on a previous diagnosis or prescription of anti-hypertensive medication; diabetes was based on a previous diagnosis with or without treatment with an anti-diabetic agent; hypercholesterolemia was based on a previous diagnosis or prescription of cholesterol-lowering medications; a cigarette smoker was defined as smoking 1 cigarette daily. A TIA was defined as a brief episode of neurologic dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than 1 h, and without evidence of acute infarction on diffusion MRI scan (Albers et al., 2002). Patients suspected of complicated migraine, seizures or presenting with vague neurological symptoms were excluded. Stroke was defined as a persistent neurological deficit of sudden onset (non-convulsive) lasting more than 24 h with confirmation by infarction noted on diffusion weighted MRI or follow-up CT, when MRI was not feasible (example Pacemaker). Stroke etiology was classified using the Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria (Adams et al., 1993).
Homocysteine Measurement
Patients without renal failure (creatinine ≤2.5 mg/dl) were included for assessment of fasting plasma homocysteine level (tHcy). None were taking medications (example: Dilantin) known to elevate tHcy. Fasting blood samples were transported on ice and plasma separated and stored at 4°C until measurement of total tHcy using high performance liquid chromatography with fluorescence detection.
MTHFR Polymorphism
Following informed consent, blood samples were collected and genomic DNA was extracted (Puregene Systems, Gentra). The MTHFR polymorphism was genotyped as previously described by PCR using the following primers: 5′-TGAAGGAGAAGGTGTCTGCGGGA-3′ and 5′-AGGACGGTGCGGTGAGAGTG-3′ (Frosst et al., 1995). A 198 base pair (bp) product is amplified and following digestion with Hinf1 (New England Biolabs), the common “C” allele of the C677T polymorphism remains undigested (Frosst et al., 1995). However, digestion of the PCR product containing the “T” polymorphism results in the generation of two fragments (175 and 23 bp) which are resolved by gel electrophoresis (6% polyacrylamide gel). The allele frequency was obtained by direct gene counting.
Dietary Assessment Using Food Frequency Questionnaire
The Block FFQ, a validated questionnaire (Subar et al., 2001) was administered to all study patients. The 1998 version of the full-diet Block FFQ (Block98) was used to convert information on food and supplement consumption to average daily energy and nutrient intake using values from the USDA Nutrient Database for Standard. The Block 98 is an eight-page, scannable, quantitative instrument that includes 109 food and beverage items (including alcohol) and three multiple and nine single vitamin and mineral supplement items; We chose to examine intake of folic acid, vitamin B12, and pyridoxine in this analysis because of their importance in homocysteine metabolism.
Statistical Analysis
Continuous variables describing demographic characteristics and biochemical parameters were expressed as mean ± standard deviation. Univariate analysis using the Chi square test and Risk Ratio (RR) analysis investigated the association between each stroke risk factor and progression of AA atheroma. Subsequently, multiple logistic regression and adjusted Relative Risk (adj. RR) analysis explored the association between AA atheroma progression and all stroke risk factors using methods described by Zhang and Yu (1998). This allows assessment of whether stroke risk factors independently increased the chance of detecting progression of AA atheroma by TEE. Kolmogorov–Smirnov (K–S) test was used to assess normality of distribution of continuous variables. The variables that deviated from normal distribution were log-transformed prior to conducting tests applicable to normally distributed variables. Linear regression analysis was used to test the association between tHcy and interval change in plaque thickness as well as cross-sectional plaque area, measured on the sequential TEEs. Multiple linear regression analysis was used to test if the association between tHcy and AA atheroma progression was independent of determinants of tHcy including age, intake of vitamin B12, folic acid levels, and genetic MTHFR polymorphism. Statistical analysis was performed using SAS version 9.1.3 (Cary, NC, USA).
Results
The characteristics of the 118 patients who had a protocol-mandated follow-up TEE are described in Table 1. They had a mean age of 65 ± 12 years; 53% were males and 25% were black. The majority had a history of hypertension or elevated cholesterol, while smaller proportions had diabetes mellitus, history of recent tobacco use, or elevated homocysteine levels. On the baseline study, the largest lesion was mild in 8 (7%), moderate in 72 (61%) and severe in 38 (32%). Of these 118 patients, 33 (28%) showed progression and 16 (14%) had regression of their index lesion at 1 year. Among the 85 patients who did not progress, there were no significant differences between the sub-group of patients who regressed (N = 16) and those who did not (N = 69) in stroke risk factors including lipid profiles and fasting homocysteine levels. Hence this entire group (N = 85) is justifiably compared with the progression group (N = 33). Laboratory parameters including tHcy were assessed at similar time intervals (p = 0.91) from the index event among the progression group (2.8 ± 7.3 days) and the no-progression group (3.0 ± 7.2 days). Among the stroke risk factors, age ≥65 years, hypertension, diabetes, high cholesterol and smoking were not associated with atherosclerotic progression (Figure 1). Only tHcy (≥14.0 μmol/l) was significantly associated with progression on both univariable (RR = 3.4, 95% CI 2.0–5.8) and multivariable analysis (RR = 3.6, 95% CI 2.2–4.6, adjusted for the vascular risk factors). The change in plaque thickness over 12 months was higher (p < 0.0001) in patients with homocysteine level ≥14.0 mcm/l (0.90 ± 0.22 mm) than those with homocysteine level <14.0 mcm/l (0.08 ± 0.10 mm). The tHcy on baseline examination was higher (p = 0.001) in patients with progression of AA atheroma (14.6 ± 1.3 mcm/l) than in those without progression (9.7 ± 0.4 mcm/l). Although in a limited sample (N = 85), the convalescent tHcy (measured >3 months from index event) was higher in patients with progression of AA atheroma (N = 27, 13.9 ± 1.6 mcm/l) than in those without progression (N = 58, 11.7 ± 0.9 mcm/l), the difference was not significant (p = 0.2). There were no significant differences between the progression and non-progression groups in total cholesterol (189 ± 44 vs. 184 ± 39 mg/dl), LDL cholesterol (111 ± 44 vs. 105 ± 35 mg/dl), HDL cholesterol (51 ± 16 vs. 53 ± 17 mg/dl) and serum triglycerides (158 ± 130 vs. 149 ± 82 mg/dl).
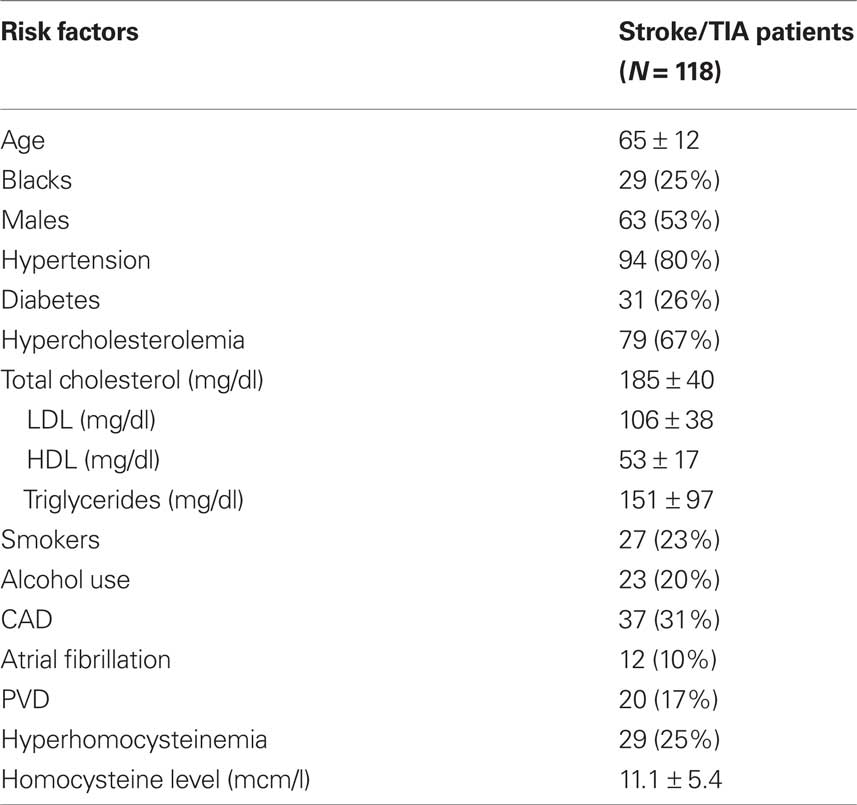
Table 1. Baseline clinical characteristics of stroke/TIA patients (N = 118, 79 strokes and 39 TIAs) assessed for progression of aortic arch atheroma.
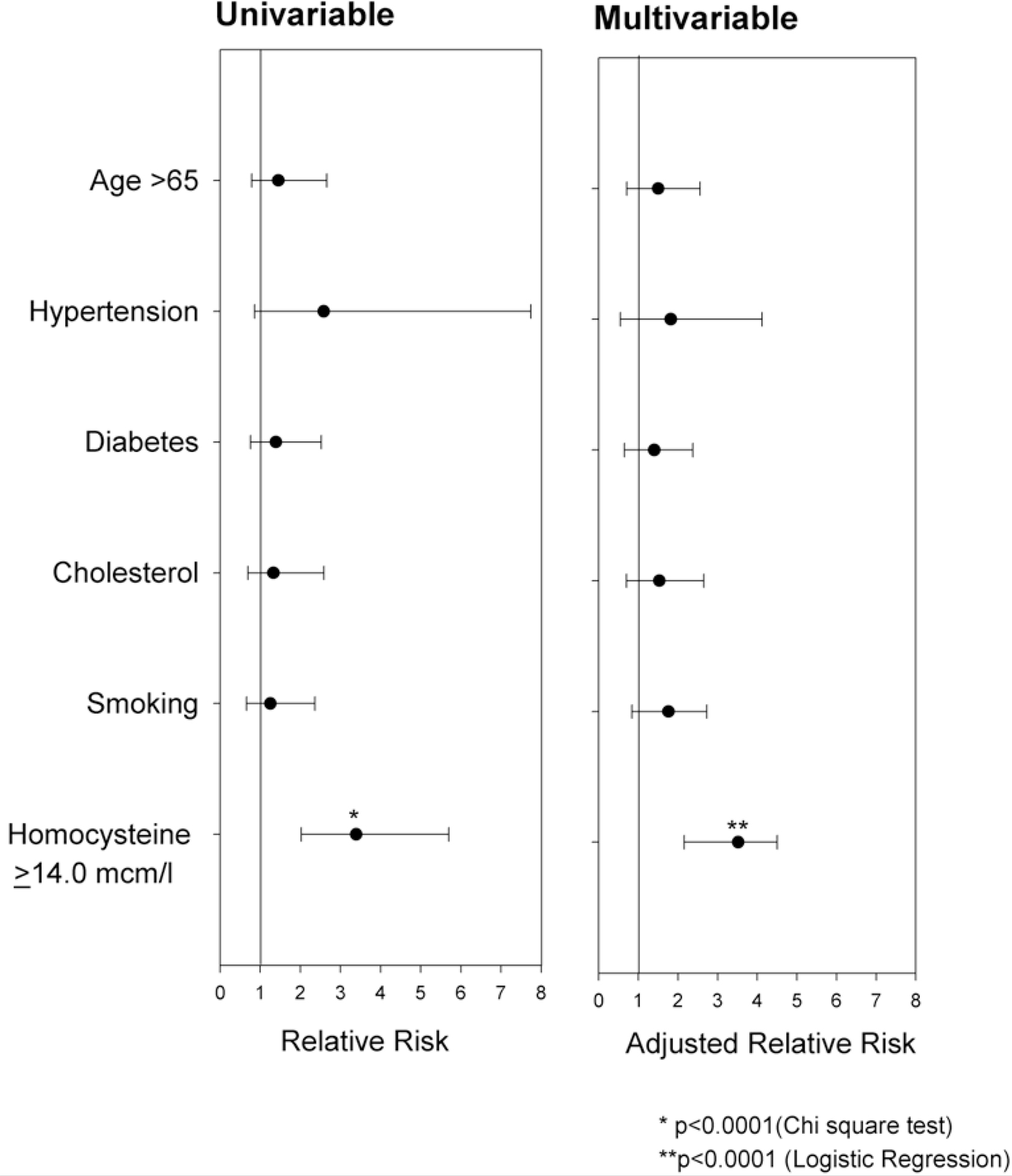
Figure 1. Results of uni- and multivariable analysis investigating the contribution of vascular risk factors to AA plaque progression (≥1 grade over a period of 12 months) in patients with stroke and TIA.
The frequency of the MTHFR 677T allele in the 116 subjects that participated in the study was 0.328. There were 55 MTHFR 677C/C homozygotes (45.2%), 46 677C/T heterozygotes (44.1%), and 15 677T/T homozygotes (10.7%). The observed MTHFR genotype frequencies did not deviate from those predicted by the Hardy-Weinberg equilibrium (p = 0.38, NS). ANOVA showed that MTHFR genotype was not a significant determinant of plasma homocysteine concentration (p = 0.43).
Of the 118 subjects, nutritional data was available in 110 subjects. Although the progression group had a higher mean energy intake (1546 kCal) compared with those who did not progress (mean energy intake = 1306 kCal), the difference did not attain statistical significance (p = 0.1). There was no significant difference between the progression and no-progression groups in the energy adjusted folate (295 vs. 287 mcg, p = 0.69), energy adjusted pyridoxine (1.48 vs. 1.42 mg, p = 0.65) and energy adjusted Vitamin B12 (3.22 vs. 3.77 μg, p = 0.30).
There was no significant correlation (r2 = 0.01, p = 0.31) between baseline AA plaque thickness on initial TEE and fasting plasma homocysteine level. Also, a significant correlation (r2 = 0.01, p = 0.29) was not noted between baseline AA plaque area and fasting plasma homocysteine level. However, a correlation (r2 = 0.11; p < 0.001) between baseline fasting homocysteine levels and the change (Δ) in AA plaque thickness was observed, as shown in Figure 2A. Likewise, there was a correlation (r2 = 0.08; p = 0.002) between baseline fasting homocysteine levels and the change (Δ) in AA plaque area, as shown in Figure 2B. Plasma tHcy was associated with change in plaque thickness over 12 months, independent of age, dietary factors, renal function and MTHFR polymorphism (Standardized β-coefficient 0.335, p = 0.02) as noted in Table 2.
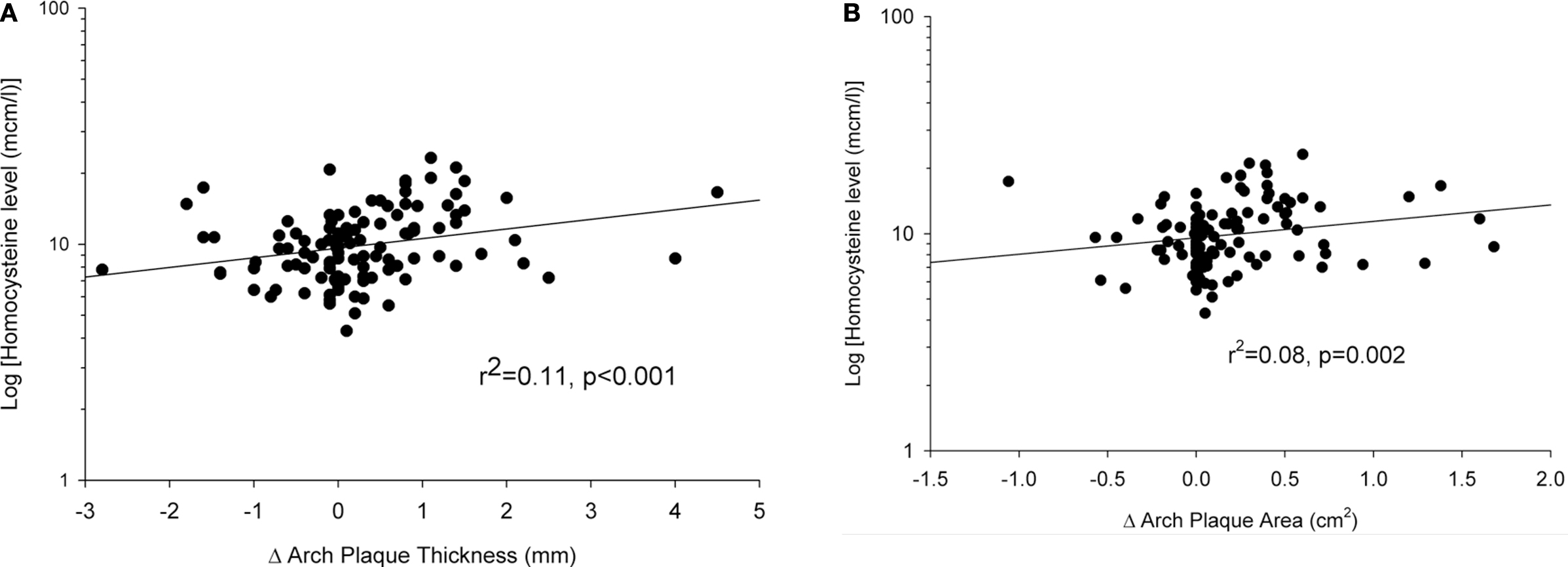
Figure 2. (A) A correlation (r2 = 0. 11, p < 0.001) was noted between the change (Δ) in AA plaque thickness over a 12-month interval and fasting plasma homocysteine level (log-transformed), depicted in a semi-log scatter plot. (B) A correlation (r2 = 0.08, p = 0.002) was noted between the change (Δ) in AA plaque area over a 12-month interval and fasting plasma homocysteine level (log-transformed), depicted in a semi-log scatter plot.
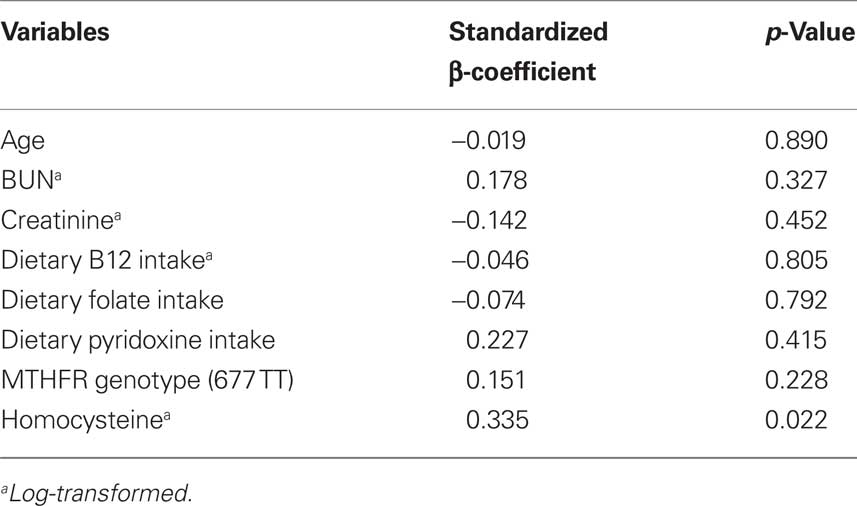
Table 2. Covariates which may influence tHcy and its association with the change in AA plaque thickness over 12 months (dependent variable), tested using multiple linear regression analysis.
Discussion
This 12-month study indicates that AA atheroma is a dynamic process: 28% of stroke/TIA patients showed plaque progression and 14% experienced regression. It confirms our prior finding that progression of AA atheroma is associated with hyperhomocysteinemia (≥14.0 μmol/l) and that these changes were not influenced by traditional vascular stroke risk factors. In that study, we reported a higher rate of progression (37%) and regression (22%); however, a composite of changes were measured in the ascending, arch, and descending thoracic aorta. When patients with changes only in the arch were selected, progression was noted in 29% and regression in 9%, similar to the present study. The present study has the advantage of a prospective design utilizing methodological details discussed earlier, aimed at improving the accuracy of measurement of aortic plaque.
Montgomery et al. prospectively re-evaluated 30 patients with moderate to severe aortic plaque noted on initial biplanar or multiplanar TEE (obtained as part of a workup for cardiac disease or an embolic event). Over an average of a 1-year period, progression was reported in 23% and regression in 10% (Montgomery et al., 1996). In a small group of 16 patients with familial hypercholesterolemia taking pravastatin, Pistavos et al. (1998) using monoplanar TEE noted a rate of progression of 19% and a rate of regression of 38% over 2 years. More recently Geraci and Weinberger (2000), using supraclavicular B-mode ultrasonography of the proximal AA in 89 patients evaluated for transient neurological symptoms or dizziness, noted a progression rate of 19% and a regression rate of 18% over an average of 7.7 months (range 3–18 months). Compared to these studies, we report a similar rate of progression (28%). Parenthetically, in the extracranial carotid arteries, atheroma appears to progress at a lesser rate over longer follow-up. Thus, in the internal carotid artery a progression rate of 15–19% has been reported over 1.5–3.0 years (Olin et al., 1998; Liapis et al., 2000; Schminke et al., 2000). These studies do not report regression of carotid atheroma. The risk factors associated with atheroma progression were not investigated in any of these reports. Our rate of regression of AA atheroma noted on TEE (14%) is similar to that reported in the initial TEE series (10%) (Montgomery et al., 1996), but is lower than that reported in patients on statins (Pistavos et al., 1998). Newer imaging modalities including MRI, Multi-detector CT (MDCT) and electron-beam CT (EBCT) have been shown to be promising approaches to measure AA progression (Rumberger, 2001; Takahashi and Stanford, 2005; Corti, 2006). Several studies testing the effect of statins and lipid lowering agents have used MRI to image changes in the thoracic aorta (Pistavos et al., 1998; Corti et al., 2005; Yonemura et al., 2005). Yet, none of these studies have reported the association of AA progression with tHcy.
Nevertheless, all prior studies utilizing transesophageal imaging to evaluate AA atheroma suffer from similar limitations. Use of a grading system may limit the ability to detect progression or regression of AA atheroma due to a “ceiling effect.” For example, an aortic plaque of 4.6 mm thickness progressing to a thickness of 7 or 8 mm may be graded as severe in both instances and hence may be incorrectly regarded as not to have progressed. Secondly, use of thickness as the only variable does not consider the effect of aortic remodeling that is associated with atherosclerosis. Age related loss of elasticity frequently results in distention and unfolding of the AA and accompanies aortic atherosclerosis (Shimojo et al., 1991). Hence, use of plaque area may be a better means of quantification compared to IMT in terms of capturing these modeling changes.
The current study has a few limitations that merit comment. First, generalizability may be limited by the requirement that individuals with stroke/TIA have evidence of ≥1 mm aortic atheroma on initial TEE and agree to a follow-up TEE, 12 months later. Second, as TEE is a semi-invasive test, measurements of AA atheroma were not performed at intermediate time points. Third, the lack of association between traditional risk factors and change in plaque thickness may indicate a lack of sufficient power to detect such relationships. Alternatively, treatment of hypertension, diabetes and hypercholesterolemia may have modified the impact of these risk factors. Finally, multivariable logistic regression models can generally handle 1 predictor for every 8–10 outcome events. With only 33 patients demonstrating progression of AA atheroma and inclusion of six covariates in the model, there may be a concern about over-fitting. This is unlikely as using fewer (≤4) covariates in a step-wise logistic regression model, yielded similar independent association between hyperhomocysteinemia and progression of AA atheroma.
We report that elevated homocysteine levels (≥14.0 μmol/l) are significantly associated with progression of AA atheroma independent of the selected determinants of tHcy. Though, admission tHcy is known to be elevated as an acute phase reactant after a stroke (Howard et al., 2000), this is an unlikely explanation for the association between tHcy and aortic plaque progression. Admission tHcy were performed at similar intervals from the index event in both the progression and the no-progression group. Further, initial stroke severity (measured by admission NIH stroke scale) did not influence the association between tHcy and plaque progression. Limited data on convalescent tHcy although concordant with admission tHcy in its association with plaque progression, did not reach statistical significance. It is possible that homocysteine may mediate endothelial dysfunction resulting in plaque progression (Coppola et al., 2000). Alternatively, hyperhomocysteinemia may produce a hypercoagulable state that may result in thrombus deposition on the atheromatous plaque (Coppola et al., 2000). Since hyperhomocysteinemia can be treated with vitamin therapy (Folic acid, B6 and B12), it can be speculated that such treatment may prevent the progression of AA atheroma. Indeed, a recent study has indicated that progression of carotid intima-media thickness may be arrested and regression promoted by treating hyperhomocysteinemia with folic acid 2.5 mg, pyridoxine 25 mg and cyanocobalamin 250 μg daily (Hackam et al., 2000). Three recent large, multicenter, double-blind, randomized studies have evaluated the impact of homocysteine lowering therapy for secondary prevention of stroke and MI in high-risk individuals. The VISP (Vitamin Intervention for Stroke Prevention) study followed patients for a 24-month period, did not include a placebo-arm and did not show a reduction in incidence of stroke or composite vascular events (Toole et al., 2004). The NORVIT (Norwegian Vitamin Trial), despite a longer follow-up, (median follow-up of 40 months) and a placebo-arm did not reveal a decrease in composite vascular events (Bønaa et al., 2006), but did show a non-significant 17% decrease in stroke. The Heart Outcomes Prevention Evaluation-2 (HOPE-2) trial with the strength of the longest follow-up of 60 months, the largest sample size, and a placebo-arm showed a significant 25% reduction in incidence of stroke (Lonn et al., 2006). In light of the results of our study as well as the carotid IMT study (Hackam et al., 2000), it appears that the effect of homocysteine on a surrogate outcome such as atherosclerosis progression is more immediate and easily detectable in a smaller sample size, compared to the recurrence of vascular events.
Our study supports an independent correlation between tHcy and atherosclerosis, and suggests that tHcy may serve as a mediator of aortic plaque progression. Thus, tHcy should be ascertained in stroke and TIA patients with AA atheroma. If elevated, progression may be amenable to vitamin therapy. The results neither establish a causal relationship between homocysteine and cardiovascular risk, nor if tHcy is a marker of existing disease burden. Further studies are needed to elucidate the role of tHcy in atherogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding for the study was provided by PHS grant 1K23NS02117 and in part by grants M01RR00046 and UL1RR025747 from the National Center of Research Resources.
References
Adams, H. P., Bendixen, B. H., Jap Kappelle, L., Biller, J., Love, B. B., Gordon, D. L., Marsh, E. E,, and The TOAST Investigators. (1993). Classification of subtypes of acute ischemic stroke: definitions for use in a multicenter clinical trial. Stroke 24, 35–41.
Albers, G. W., Caplan, L. R., Easton, J. D., Fayad, P. B., Mohr, J. P., Saver, J. L., and Sherman, D. G. (2002). TIA Working Group. Transient ischemic attack – proposal for a new definition. N. Engl. J. Med. 347, 1713–1716.
Amarenco, P., Cohen, A., Tzourio, C., Bertrand, B., Hommel, M., Besson, G., Chauvel, C., Touboul, P.-J., and Bousser, M. G. (1994). Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N. Engl. J. Med. 331, 1474–1479.
Bønaa, K. H., Njølstad, I., Ueland, P. M., Schirmer, H., Tverdal, A., Steigen, T., Wang, H., Nordrehaug, J. E., Arnesen, E., and Rasmussen, K. (2006). NORVIT trial investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N. Engl. J. Med. 354, 1578–1588.
Cheitlin, M. D. Armstrong, W. F., Aurigemma, G. P., Beller, G. A., Bierman, F. Z., Davis, J. L., Douglas, P. S., Faxon, D. P., Gillam, L. D., Kimball, T. R., Kussmaul, W. G., Pearlman, A. S., Philbrick, J. T., Rakowski, H., Thys, D. M., Antman, E. M., Smith, S. C. Jr, Alpert, J. S., Gregoratos, G., Anderson, J. L., Hiratzka, L. F., Hunt, S. A., Fuster, V., Jacobs, A. K., Gibbons, R. J., Russell, R. O., American College of Cardiology, American Heart Association, and American Society of Echocardiography. (2003). ACC/AHA/ASE Guideline update for clinical application of echocardiography by the American College of Cardiology and American Heart Association Inc.
Coppola, A., Davi, G., De Stefano, V., Mancini, F. P., Cerbone, A. M., and Di Minno, G. (2000). Homocysteine, coagulation, platelet function, and thrombosis. Semin. Thromb. Hemost. 26, 243–254.
Corti, R. (2006). Noninvasive imaging of atherosclerotic vessels by MRI for clinical assessment of the effectiveness of therapy. Pharmacol. Ther. 110, 57–70.
Corti, R., Fuster, V., Fayad, Z. A., Worthley, S. G., Helft, G., Chaplin, W. F., Muntwyler, J., Viles-Gonzalez, J. F., Weinberger, J., Smith, D. A., Mizsei, G., and Badimon, J. J. (2005). Effects of aggressive versus conventional lipid-lowering therapy by simvastatin on human atherosclerotic lesions: a prospective, randomized, double-blind trial with high-resolution magnetic resonance imaging. J. Am. Coll. Cardiol. 46, 106–112.
Coull, B. M., Malinow, M. R., Beamer, N., Sexton, G., Nordt, F., and de Garmo, P. (1990). Elevated plasma homocyst(e)ine concentration as a possible independent risk factor for stroke. Stroke 21, 572–576.
Daniel, W. G., Erbel, R., Kasper, W., Visser, C. A., Engberding, R., Sutherland, G. R., Grube, E., Hanrath, P., Maish, B., and Denning, K. (1991). Safety of transesophageal echocardiography: a multicenter survey of 10419 examinations. Circulation 83, 817–821.
Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., Boers, G. J., den Heijer, M., Kluijtmans, L. A., van den Heuvel, L. P., and Rozen, R. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 10, 111–113.
Geraci, A., and Weinberger, J. (2000). Natural history of aortic arch atherosclerotic plaque. Neurology 54, 749–751.
Hackam, D. G., Peterson, J. C., and Spence, J. D. (2000). What level of plasma homocyst(e)ine should be treated? Effect of vitamin therapy on progression of carotid atherosclerosis in patients with homocyst(e)ine levels above and below 14 micromol/L. Am. J. Hypertens. 13, 105–110.
Howard, V. J., Sides, E. G., Newman, G. C., Cohen, S. N., Howard, G., Malinow, M. R., Toole, J. F., and Stability of Plasma Homocyst(e)ine in Acute Stroke Patients (SHASP) Study Investigators. (2000). Changes in plasma homocyst(e)ine in the acute phase after stroke. Stroke 33, 473–478.
Kaul, S., Zadeh, A. A., and Shah, P. K. (2006). Homocysteine hypothesis for atherothrombotic cardiovascular disease: not validated. J. Am. Coll. Cardiol. 48, 914–923.
Khoury, Z., Rinat, C., Benhorin, J., Stern, S., and Keren, A. (1998). Quantification of atherosclerotic burden in the descending aorta by transesophageal echocardiography: inter- and intraobserver variability. Echocardiography 15, 59–66.
Konecky, N., Malinow, M. R., Tunick, P. A., Freedberg, R. S., Rosenzweig, B. P., Katz, E. S., Hess, D. L., Upson, B., Leung, B., Perez, J., and Kronzon, I. (1997). Correlation between plasma homocyst(e)ine and aortic atherosclerosis. Am. Heart J. 133, 534–540.
Liapis, C., Kakisis, J., Papavassiliou, V., Ntanou, A., Kontopoulou, S., Kaperonis, E., Koumakis, K., and Gogas, J. (2000). Internal carotid artery stenosis: rate of progression Eur. J. Vasc. Endovasc. Surg. 19, 111–117.
Lonn, E., Yusuf, S., Arnold, M. J., Sheridan, P., Pogue, J., Micks, M., McQueen, M. J., Probstfield, J., Fodor, G., Held, C., Genest, J. Jr, and Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. (2006). Heart outcomes prevention evaluation (HOPE) 2 investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 354, 1567–1577.
Malinow, M. R., Kang, S. S., Taylor, L. M., Wong, P. W. K., Coull, B., Inahara, T., Mukerjee, D., Sexton, G., and Upson, B. (1989). Prevalence of hyperhomocyst(e)inemia in patients with peripheral arterial occlusive disease. Circulation 79, 1180–1188.
Malinow, M. R., Nieto, F. J., Szklo, M., Chambless, L. E., and Bond, G. (1993). Carotid artery intimal-medial wall thickening and plasma homocysteine in asymptomatic adults. The Atherosclerosis risk in communities study. Circulation 87, 1107–1113.
McNamara, R. L., Lima, J. A. C., Whelton, P. K., and Powe, N. R. (1997). Echocardiographic identification of cardiovascular sources of emboli to guide clinical management of stroke: a cost-effectiveness analysis. Ann. Intern. Med. 127, 775–787.
Montgomery. D. H., Ververis, J. J., McGorisk, G., Frohwein, S., and Martin, R. P. (1996). Natural history of severe atheromatous disease of the thoracic aorta: a transesophageal echocardiographic study. J. Am. Coll. Cardiol. 27, 95–101.
Olin, J. W., Fonseca, C., Childs, M. B., Piedmonte, M. R., Hertzer, N. R., and Young J. R. (1998). The natural history of asymptomatic moderate internal carotid artery stenosis by duplex ultrasound. Vasc. Med. 3, 101–108.
Pistavos, C. E., Aggeli, K. I., Barbetseas, J. D., Skoumas, I. N., Lambrou, S. G., Frogoudaki, A. A., Stefanadis, C. I., and Toutouzas, P. K. (1998). Effect of pravastatin on thoracic aortic atherosclerosis in patients with heterozygous familial hypercholesterolemia. Am. J. Cardiol. 82, 1484–1488.
Rumberger, J. A. (2001). Tomographic (plaque) imaging: state of the art. Am. J. Cardiol. 88, 66E–69E.
Schminke, U., Motsch, L., Griewing, B., Gaull, M., and Kessler, C. (2000). Three-dimensional power-mode ultrasound for quantification of progression of carotid artery atherosclerosis. J. Neurol. 247, 106–111.
Sen, S., Hinderliter, A., Sen, P. K., Simmons, J. W., Beck, J., Offenbacher, S., Ohman, M. E., and Oppenheimer, S. M. (2007). Aortic arch atheroma progression and recurrent vascular events in stroke/TIA patients. Circulation 116, 928–935.
Sen, S., Oppenheimer, S. M., Lima, J. A. C., and Cohen, B. (2002). Risk factors for progression of aortic atheroma in stroke/TIA patient. Stroke 33, 930–935.
Sen, S., Wu, K., McNamara, R., Lima, J., Piantadosi, S., and Oppenheimer, S. M. (2000). Distribution, severity and risk factors for aortic atherosclerosis in cerebral ischemia. Cerebrovasc. Dis. 10, 102–109.
Shimojo, M., Tsuda, N., Iwasaka, T., and Inada, M. (1991). Age-related changes in aortic elasticity determined by gated radionucleotide angiography in patients with systemic hypertension or healed myocardial infarcts and in normal subjects. Am. J. Cardiol. 68, 950–953.
Spence, J. D., Malinow, M. R., Barnett, P. A., Marian, A. J., Freeman, D., and Hegele R. A. (1999). Plasma homocyst(e)ine concentration, but not MTHFR genotype, is associated with variation in carotid plaque area. Stroke 30, 969–973.
Stampfer, M. J., Malinow, M. R., Willett, W. C., Newcomer, L. M., Upson, B., Ullman, D., Tishler, P. V., and Hennekens, C. H. (1992). A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA 268, 877–881.
Subar, A. F., Thompson, F. E., Kipnis, V., Midthune, D., Hurwitz, P., McNutt, S., McIntosh, A., and Rosenfeld, S. (2001). Comparative validation of the Block, Willett, and National Cancer Institute food frequency questionnaires: the Eating at America’s Table Study. Am. J. Epidemiol. 154, 1089–1099.
Takahashi, K., and Stanford, W. (2005). Multidetector CT of the thoracic aorta. Int. J. Cardiovasc. Imaging 21, 141–153.
The French Study of Aortic Plaques in Stroke Group. (1996). Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N. Engl. J. Med. 334, 1121–1221.
Ti, L. K., Mackensen, G. B., Grocott, H. P., Laskowitz, D. T., Phillips-Bute, B. G., Milano, C. A., Hilton, A. K., Newman, M. F., Mathew, J. P., and Neurologic Outcome Research Group. (2003). Neurologic outcome research group. Apolipoprotein E4 increases aortic atheroma burden in cardiac surgical patients. J. Thorac. Cardiovasc. Surg. 125, 211–213.
Toole, J. F., Malinow, M. R., Chambless, L. E., Spence, J. D., Pettigrew, L. C., Howard, V. J., Sides, E. G., Wang, C. H., and Stampfer, M. (2004). Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the vitamin intervention for stroke prevention (VISP) randomized controlled trial. JAMA 291, 565–575.
Tribouilloy, C. M., Peltier, M., Iannetta Peltier, M. C., Trojette, F., Andrejak, M., and Lesbre, J. M. (2000). Plasma homocysteine and severity of thoracic aorta atherosclerosis. Chest 118, 1685–1689.
Tunick, P. A., and Kronzon, I. (2000). Atheromas of the thoracic aorta: clinical and therapeutic update. J. Am. Coll. Cardiol. 35, 545–554.
U.S. Department of Agriculture, Agricultural Research Service. (2001). USDA Nutrient Database for Standard Reference, Release 14. Nutrient Data Laboratory Home Page. Available at: http://www.nal.usda.gov/fnic/foodcomp
Yonemura, A., Momiyama, Y., Fayad, Z. A., Ayaori, M., Ohmori, R., Higashi, K., Kihara, T., Sawada, S., Iwamoto, N., Ogura, M., Taniguchi, H., Kusuhara, M., Nagata, M., Nakamura, H., Tamai, S., and Ohsuzu F. (2005). Effect of lipid-lowering therapy with atorvastatin on atherosclerotic aortic plaques detected by noninvasive magnetic resonance imaging. J. Am. Coll. Cardiol. 45, 733–742.
Keywords: cerebrovascular disease, stroke, cardiac embolism, aorta, homocysteine
Citation: Sen S, Reddy PL, Grewal RP, Busby M, Chang P and Hinderliter A (2010) Hyperhomocysteinemia is associated with aortic atheroma progression in stroke/TIA Patients. Front. Neur. 1:131. doi: 10.3389/fneur.2010.00131
Received: 18 April 2010;
Paper pending published: 17 August 2010;
Accepted: 13 September 2010;
Published online: 26 November 2010.
Edited by:
Scott Silliman, University of Florida, USACopyright: © 2010 Sen, Reddy, Grewal, Busby, Chang and Hinderliter. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Souvik Sen, Department of Neurology, University of South Carolina School of Medicine, 8 Medical Park Drive, Suite 420, Columbia, SC 29203, USA. e-mail:c291dmlrLnNlbkB1c2NtZWQuc2MuZWR1