

- School of Life Sciences, Jawaharlal Nehru University, New Delhi, India
The two major limitations of Golgi–Cox method are that staining takes very long time and it is inconsistent. In this paper we describe a modification of the Golgi–Cox method, in which the tissue blocks were maintained at 37 ± 1°C during chromation for only 24 h and consistent staining of neurons in rat brain sections were observed. The method is simple, reproducible, rapid, inexpensive, and provides uniform staining with very good resolution of neuronal soma, dendrites as well as spines.
Introduction
The classical Golgi method for staining neurons in the brain was first developed by Camillio Golgi (Golgi, 1873 in Mazzarello, 1999). Subsequently, the great neuroanatomist Ramon y Cajal (1909) applied the technique to demonstrate previously unimagined neuronal morphology virtually in all parts of the nervous system. However, the main drawback of the method is inconsistency of impregnation of the stain into the neurons resulting in reduced specificity, reproducibility, and success rate (Globus and Scheibel, 1966; Pasternak and Woolsey, 1975; Zhang et al., 2003). Several modifications of the method have been tried which include variations in pH (Bertram and Ihrig, 1957; Gonzalez-Burgos et al., 1992; Angulo et al., 1994), application of vacuum (Friedland et al., 2006), coating of brain blocks with egg yolk (Zhang et al., 2003), use of microwaves (Armstrong and Parker, 1986; Marani et al., 1987; Zhang et al., 2003) and variations in temperature of the tissue incubating medium (Armstrong and Parker, 1986; Berbel, 1986; Marani et al., 1987; Angulo et al., 1994). These modifications were aimed at faster staining process by decreasing the time for staining, reducing precipitation, promoting uniform crystallization, increasing reliability, and reproducibility of staining neurons by the Golgi method.
The Golgi–Cox method has been frequently used to stain neurons in both less myelinated, younger as well as in more myelinated, older rat brains. As compared to Golgi method the advantage of Golgi–Cox method include increased probability of staining more number of neurons (Scheibel and Tomiyasu, 1978); it is also reported to be better than the rapid Golgi method for studying neuronal dendritic morphology (Buell, 1982). However, the duration of impregnation of stain using Golgi–Cox method has been reported to be between 14 and 80 days (Rutledge et al., 1969; Glaser and Van der Loos, 1981; Zhang et al., 2003), depending on whether whole brain or smaller blocks of brain were used for staining, i.e., primarily depending on the thickness of the tissue block (sample). Thus, apart from inconsistency and lack of uniform, reproducible results, requirement of exceptionally long time to achieve neuronal staining is another major disadvantage of both the Golgi and Golgi–Cox methods, which practically limits the use of these methods.
One of the possibilities to overcome the shortcomings of the Golgi–Cox method mentioned above could be by increasing random motion of the metallic ions in the staining solution facilitating their influx and deposition within the neurons; thus reducing the time required as well as increasing the success rate of more stained neurons and reproducibility of results. Here we describe a simple and inexpensive modification where we raised the temperature of the Golgi–Cox solution in which moderately thick slices of brain were incubated only for 24 h and that gave us significantly more impregnation of stain and consistently excellent results in terms of stained neurons.
Materials and Methods
Experiments were conducted on inbred male wistar rats (250–300 g) maintained in their standard home cages under 12:12 h light/dark cycle with food and water ad libitum. The experiments were approved by the Institutional Animal Ethics Committee (IAEC) and every effort was made to minimize the use of number of animals and their sufferings. Four rats were deeply anesthetized with over dose of ketamine–xylazine (80–32 mg/kg, i.p.) (Chandra Bhagat Pharma Pvt Ltd, India) and then intracardially perfused with 0.1 M phosphate buffered saline (PBS, pH 7.4). For comparison and to differentiate if blood and perfusion play any role in staining, another four rats were sacrificed by cervical dislocation followed by decapitation. All the brains were removed, washed with distilled water followed by with freshly prepared Golgi–Cox solution (Rutledge et al., 1969). The brains were placed in a 1-mm brain slicer (WPI, USA) and three 5 mm thick coronal blocks A, B, and C were prepared (Figure 1). Each of those coronal blocks was further cut into two equal mirror halves by giving a longitudinal cut along the midline. Thus, a total of six blocks labeled as A1, A2, B1, B2, C1, and C2 were obtained (Figure 1).
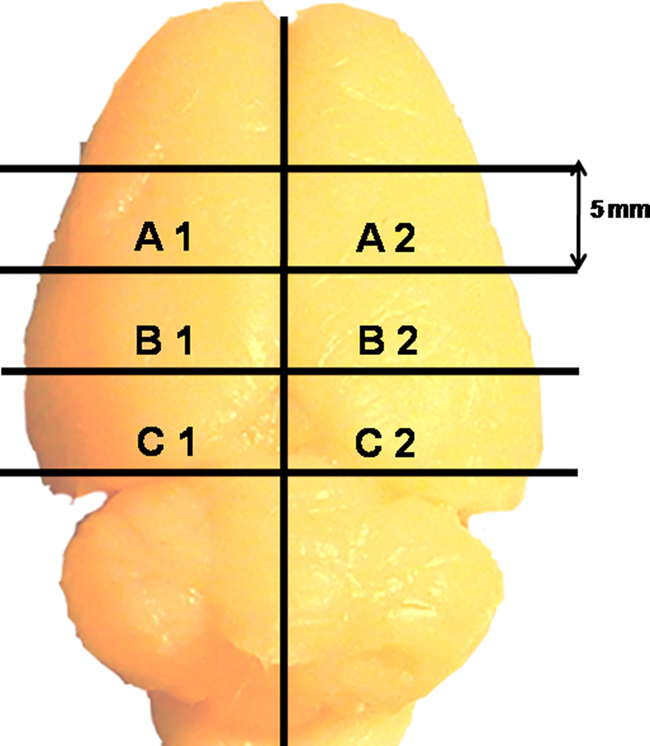
Figure 1. A rat brain showing how four coronal and one sagittal cuts were given to obtain six blocks from each rat brain.
Each tissue block was placed in separate cotton lined dark colored glass bottle containing 25–30 ml freshly prepared Golgi–Cox solution. Bottles containing blocks A1, B1, and C1 were maintained in an incubator at 37 ± 1°C for 6, 12, and 24 h, respectively, while bottles containing blocks A2, B2, and C2 were incubated at 26 ± 1°C for 6, 12, and 24 h, respectively. Thus, all other conditions, except temperature, were identical in the two groups; hence the groups served as control for each other. At each time point one block (say A1) from 37°C and the corresponding contralateral block (in this case A2) from 26°C were taken and processed simultaneously; blocks B1, C1 and B2, C2 were processed similarly. This strategy ensured comparisons between sections from comparable brain areas from the same animal treated under identical conditions except that of change in the incubation temperature. As it will be seen later, since the results of intracardial perfused brain samples and that of brains taken out without intracardial perfusion were comparable after 24 h incubation, only the latter samples were treated for 6 and 12 h. We treated the three separate blocks of tissues for 6, 12, and 24 h in the Golgi–Cox solution and four such repetitions were conducted.
Further, to confirm if the temperature was affecting primarily the permeability of the stain resulting in increased staining efficiency, we kept separate brain blocks in Golgi–Cox solution containing sodium dodecyl sulfate (SDS) or Triton X-100. In separate sets, brain blocks as mentioned above were incubated at 37 ± 1 and 26 ± 1°C in Golgi–Cox solution containing either 0.1 or 0.2% SDS or 0.5% Triton X-100 (Table 1). Since the results of perfused brain after 24 h incubation in Golgi–Cox solution were similar (Figure 2) to those of non-perfused brain, rest all the studies were conducted on the latter only.
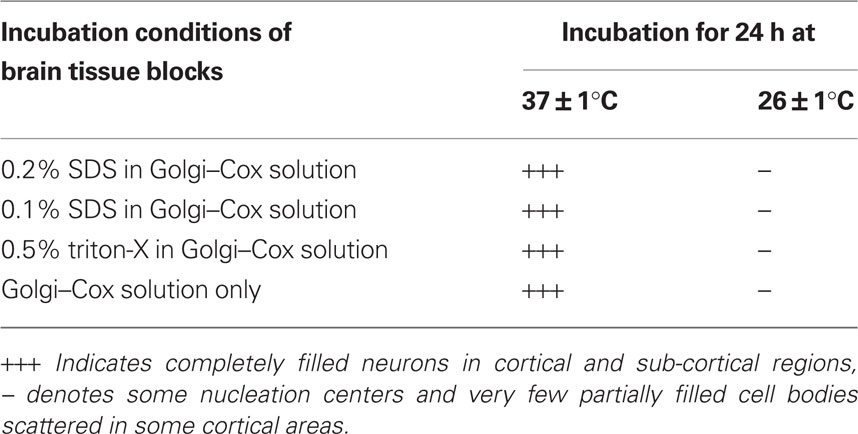
Table 1. Completely filled neurons in 200 μm thick sections prepared from 5 mm brain blocks treated under various conditions.
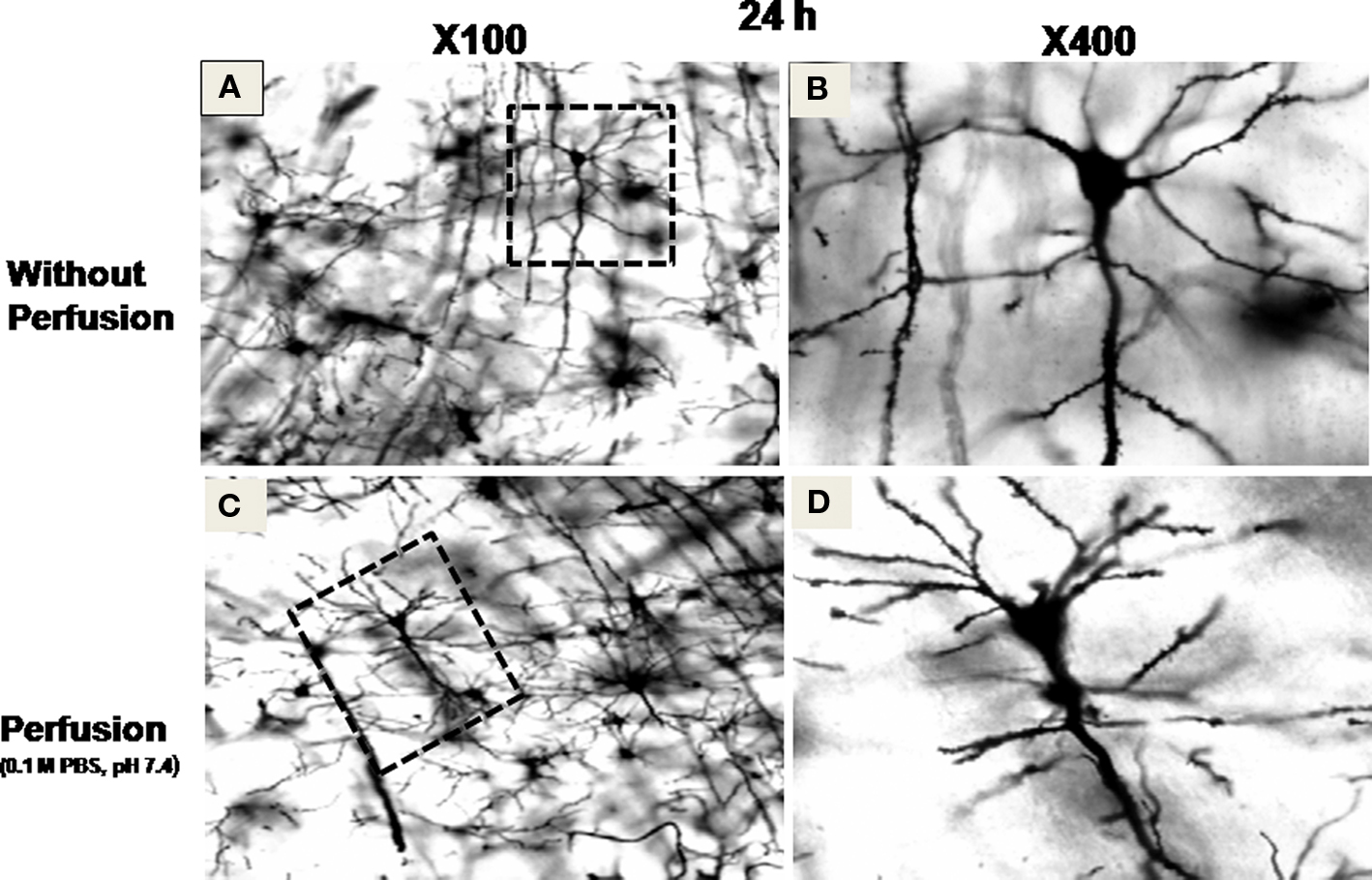
Figure 2. Stained neurons in sections prepared from brain blocks incubated at 37 ± 1°C for 24 h. Sections (A) and (B) were prepared without perfusion of the brain, while sections (C) and (D) were prepared after intracardial perfusion of the brain with 0.1 M phosphate buffered saline (PBS) at pH 7.4. (A) and (C) at ×100 magnification, while (B) and (D) at ×400 magnification of the area marked on (A) and (C), respectively, showing completely filled neurons.
Sample Preparation
At the end of incubation of the brain tissue in the staining solution for varying period, 200 μm thick sections were prepared from each of the treated and control blocks using a vibratome (3000 series, Evergreen Blvd-St. Louis, MO, USA; Gibb and Kolb, 1998). It may also be noted that in pilot studies uneven hand cut (90–350 μm) sections also gave us comparable staining. The, sections were processed as follows:
1. Rinsed twice (5 min each) in distilled water to remove traces of impregnating solution.
2. Dehydrated in 50% alcohol for 5 min.
3. Kept in ammonia solution (3:1, ammonia:distilled water) for 5–10 min.
4. Rinsed twice (5 min each) in distilled water.
5. Kept in 5% sodium thiosulfate for 10 min in dark.
6. Rinsed twice for 2 min each in distilled water.
7. Dehydrated twice (5–10 min each) in 70, 80, 95% ethanol and 99% 1-butanol, cleared in toluene and mounted in DPX on gelatinized slides.
The slides were allowed to dry at room temperature and were observed under a microscope (Nikon Eclipse E400, Japan and Olympus BX51, Japan) at low and high magnifications. Images were captured using a charged coupled device (CCD) digital camera (JVC, Tokyo, Japan and MBF CX9000) attached to the microscope using dedicated software Image-Pro Plus 5.1.1 (Media Cybernetics, Silver Spring, USA) and/or Neurolucida 9 (MBF Biosciences, USA).
The intensity of black staining of neurons in the sections was visually compared under microscope. In our pilot studies no nucleation of black stain was observed until 4 h incubation; however, significant number of completely filled neurons was seen after 24 h of incubation at 37°C. Hence, in this study we chose to incubate the brain slices at 37°C and 26°C for 6, 12 and 24 h in the staining solution. The following criteria were adopted for identification of staining for evaluation.
i) Staining was considered to have been initiated if visible black spot (nucleation) could be distinctly identified inside the neurons without spilling over to the surrounding; the latter (not many though) was considered due to neuronal damage or non-specific artifact.
ii) Neurons were considered completely stained if the soma, axons, and dendrites could be seen well demarcated by the impregnated black stain without spilling outside the neurons.
For quantitative estimation, five to six sections from each category (6, 12, 24 h in 26 and 37°C) were observed under microscope (×100 magnification). Every section was divided into two equal zones from the center; outer one was considered cortical while adjacent inner part was considered as sub-cortical (Figure 3). In each section four to six frames (Figure 3) were taken at random from cortical and sub-cortical regions (640 × 480 pixels or 220 × 170 μm) using CCD camera (JVC, Japan) attached to the microscope (Nikon Eclipse E400). On an average about 26 ± 2 images were taken from sections of each category. We counted the stained neurons under various conditions as defined above using dedicated software Image-Pro Plus 5.1.1 (Media Cybernetics, Silver Spring, USA). Stereoscopic view of sample neurons was also observed by tracing soma, dendrites, and axons in 3-Dimension (3-D) using Neurolucida software (MBF Biosciences, USA). The neurons were viewed in different planes and were captured using a digital camera (MBF CX9000) attached to the microscope (Olympus BX51, Japan). Dedicated software Neurolucida 9 and Neurolucida Explorer (MBF Biosciences, USA) were used to reconstruct 3-D view (X1000) of the neurons from the captured pictures, which may be conveniently used for qualitative and quantitative analysis.
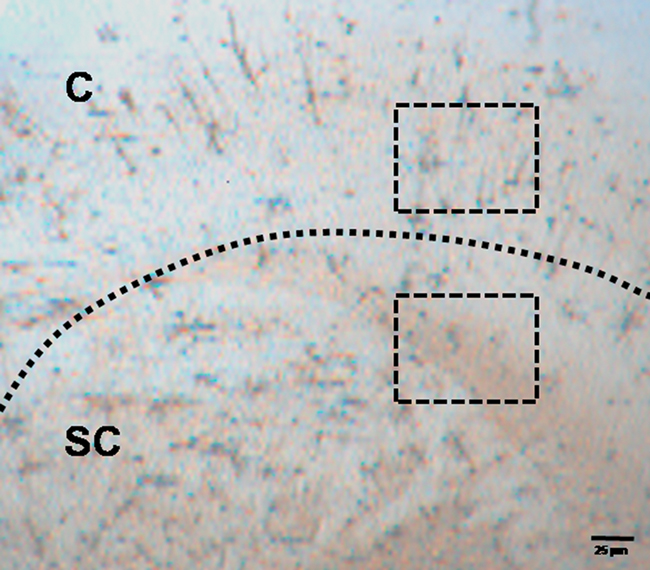
Figure 3. Photomicrograph (×20) of Golgi–Cox stained section prepared from a block incubated for 24 h at 37 ± 1°C. Dashed line defines the boundary between Cortical (C) and sub-cortical (SC) zones. Stained neurons were counted within randomly selected rectangular areas as shown in the figure and explained in the text.
Results
Early sign of nucleation (filling of neurons with black products) started appearing after 6 h of incubation of the blocks at 37 ± 1°C, while no such sign was visible in the sections prepared from blocks incubated at 26 ± 1°C (Figure 4).
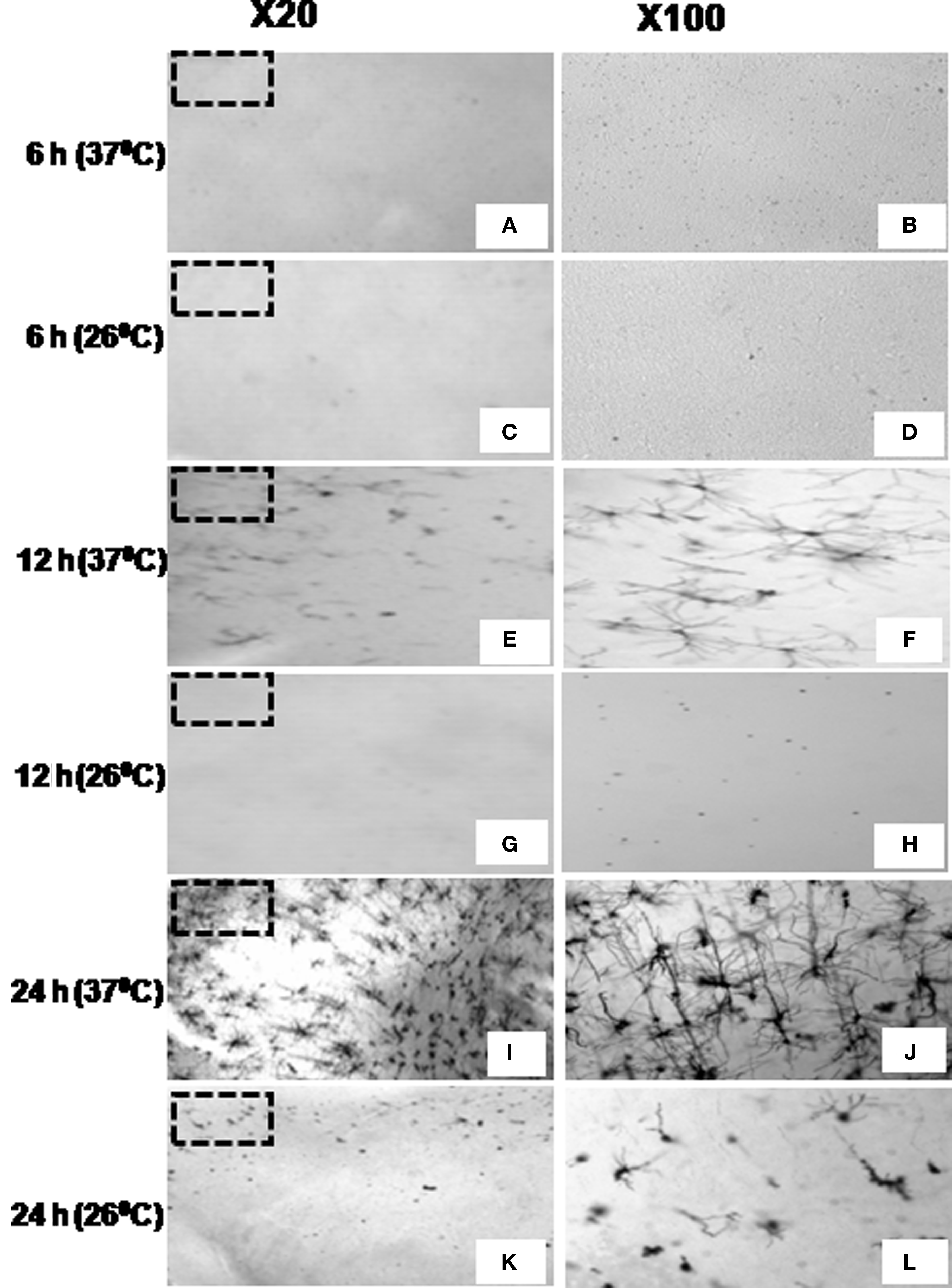
Figure 4. Stained neurons in cortical sections at ×20 (left panel) and ×100 (right panel, magnified view of the marked area on ×20) prepared from brain blocks incubated in Golgi–Cox solution for 6 h (A–D), 12 h (E–H), or 24 h (I–L) at 37 ±?1°C (A,B,E,F,I,J) or at 26 ± 1°C (C,D,G,H,K,L) showing temperature and duration of incubation are critical for staining.
By 12 h of incubation at 37 ± 1°C although many significantly filled (black) neurons could be seen in the cortical region of the sections; in very few sections partially filled neurons could be occasionally seen in the sub-cortical regions only. After 24 h of incubation almost all the stained neurons were completely impregnated with black stain in both the cortical and the sub-cortical regions of the sections prepared from tissue blocks incubated at 37 ± 1°C. However, the number of completely filled neurons in the sub-cortical region was significantly lesser than those in the cortical region (Figures 4–6). Under higher magnification (×1000), nice and clear dendritic spines could be seen against a clear background in sections obtained from the blocks incubated at 37 ± 1°C for 24 h (Figure 6). Such results from blocks incubated at 37 ± 1°C were uniformly distributed throughout the sections and were consistent in all the sets of experiments irrespective of intracardial perfusion or not. Detail morphology of stained neurons in 3-D is shown in Figure 7.
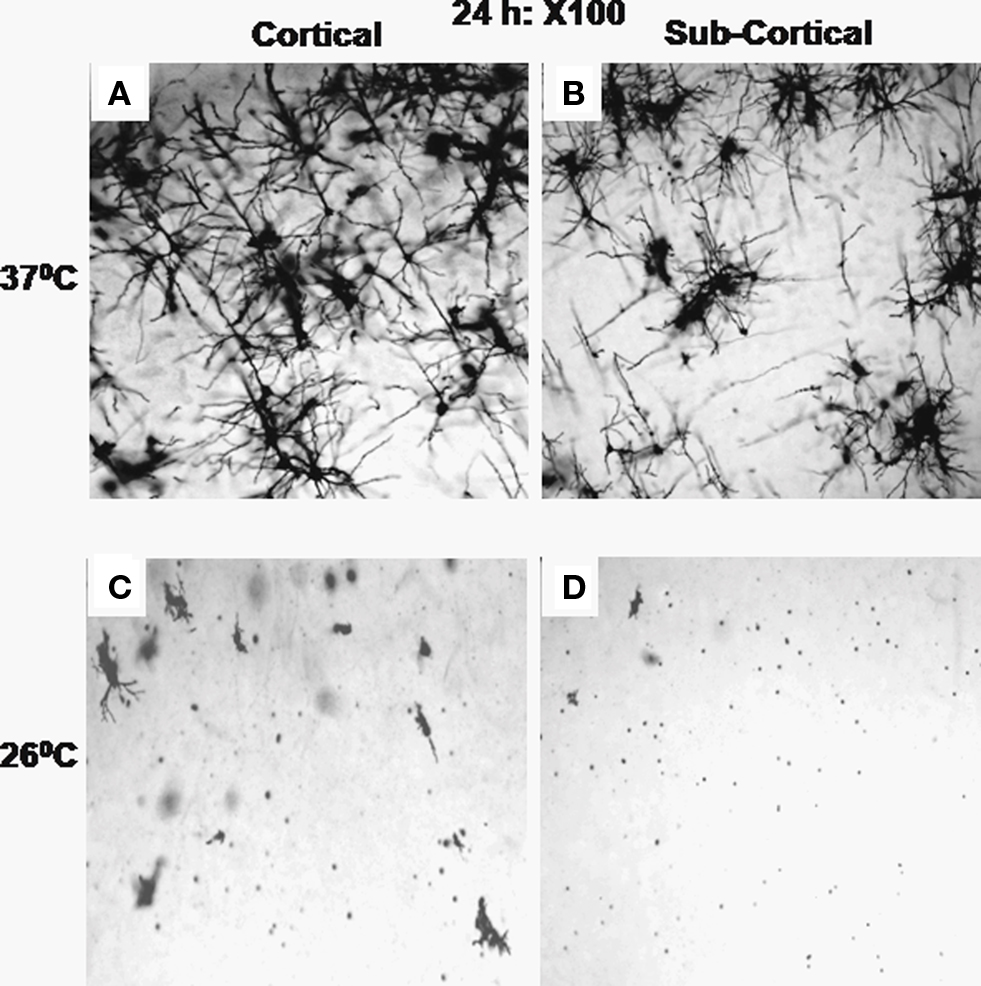
Figure 5. Stained neurons (×100) in sections through cortical (A and C) and sub-cortical (B and D) areas after incubation of the brain blocks for 24 h at 37 ± 1°C (A,B) and at 26 ± 1°C (C,D) are shown. All the sections showed completely filled neurons at the end of 24 h incubation at 37 ± 1°C; however, very few neurons can be seen after incubation at 26 ± 1°C.

Figure 6. Stained neurons or parts in sections from cortical areas of brain blocks incubated at 37 ± 1°C for 24 h are shown under various magnification in (A–C); while after incubation for 24 h at 26 ± 1°C are shown in (D–F). (A) and (D) are at ×100; the areas marked on (A) and (D) are shown at ×400 in (B) and (E), respectively. The areas marked on (B) and (E) have been shown at ×1000 in (C) and (F), respectively; spines can be clearly seen in (C) but not in (F).
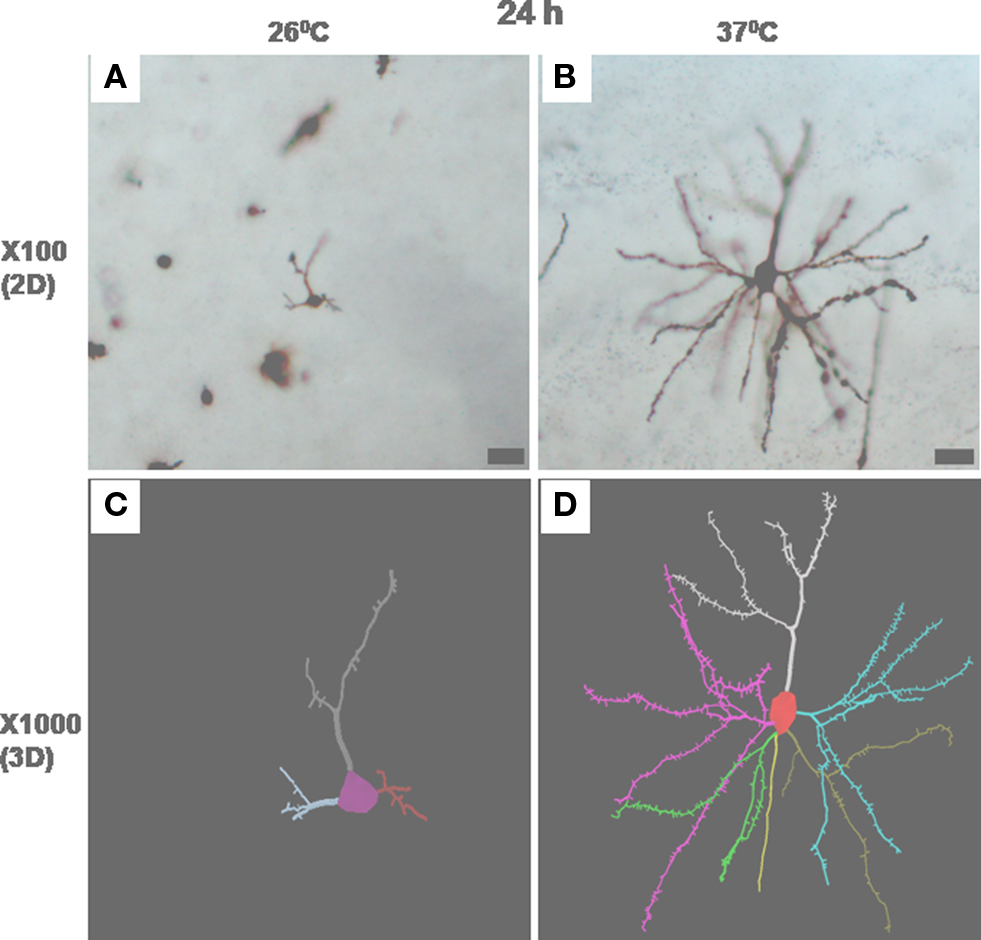
Figure 7. Photomicrographs in one focal plane at ×100 of stained neurons incubated at 26 ± 1°C (A) and 37 ± 1°C (B) for 24 h are shown. Several such traces taken in different planes have been reconstructed automatically by Neurolucida 9 and Neurolucida Explorer 9 and are shown in (C) and (D) as 3-D solid images. Magnified view of neuron in (A) and (B) are shown in (C) and (D), respectively. Scale bar = 25 μm.
In contrast, the brain tissue blocks kept at 26 ± 1°C showed none or negligible staining after 12 h of incubation. However, after 24 h incubation at 26 ± 1°C, inconsistent between experiments and incomplete staining of a few neurons could be seen occasionally, discretely spread mostly in the cortical regions of the sections; insignificant number of partially stained neurons could be seen in the sub-cortical region (Figures 4–6). Qualitative comparison between control and experimental sections are shown in Tables 1 and 2. Quantitative analysis of number of neurons stained in different brain regions under various conditions has been shown in Figure 8. Stereoscopic 3-D view of a neuron with its morphology and projections are shown in Figure 7, which may be conveniently used for further analysis.

Table 2. Effects of varying duration and temperature of incubation of the brain tissues on impregnation of Golgi–Cox stain into the neurons.
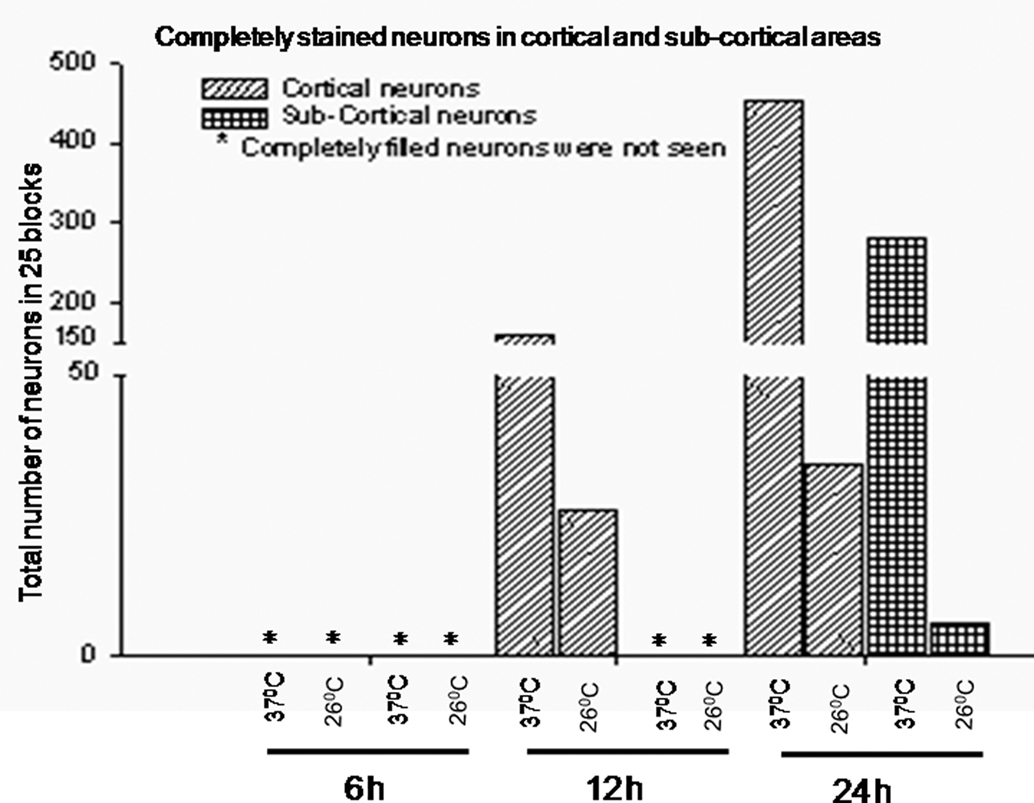
Figure 8. Total number of completely filled neurons in cortical and sub-cortical brain areas in 26 ± 2 frames (as described in text) after incubation of the brain blocks at 37 ± 1°C and 26 ± 1°C for 6, 12, and 24 h are shown.
Neither SDS nor Triton X-100 in the incubating medium improved the staining quality. The staining quality was comparable when the tissues were incubated at higher (37 ± 1°C) or lower (26 ± 1°C) temperature as described above, irrespective of whether they were incubated in medium containing SDS or Triton X-100 (Table 1). Similarly, perfusion of the brain with PBS was also of little consequences and gave comparable results when incubated at the higher (37 ± 1°C) or lower (26 ± 1°C) temperature. These observations suggested that the improvement of staining was primarily due to the increased temperature of the tissue incubation medium.
Discussion
Golgi–Cox stain is one of the powerful techniques to study the neuronal morphology in brain sections (Ramon y Cajal, 1909). However, its major drawbacks are it is much time consuming to get the results, taking several weeks to more than a month; the results are not uniform, inconsistent and prone to high failure rates (Globus and Scheibel, 1966; Zhang et al., 2003; Friedland et al., 2006). In this study we incubated the brain tissues in classical Golgi–Cox solution at different temperatures and observed that incubation of the tissues at normal body temperature 37 ± 1°C overcame those drawbacks. The effects due to the presence of blood and its constituents or due to perfusion with PBS were ruled out by using the brain taken out with or without perfusion with PBS. Similarly, to rule out the effect of membrane damage the brain tissues were incubated at various temperatures in medium with or without SDS and Triton-X 100 and the results were comparable. Thus, the findings suggest that the only factor to reduce the time for the stain to impregnate into the neurons was the incubation temperature at 37 ± 1°C.
Incubating brain blocks in the Golgi–Cox solution at 37°C dramatically reduced the time required to achieve complete and uniform staining of neurons. Sections obtained from contralateral blocks of the same brain kept in Golgi–Cox solution at 26°C were used as control. While excellent staining was achieved within 24 h in blocks incubated at 37°C, very few completely stained neurons could be seen in homotopic areas in blocks kept at room temperature (26 ± 1°C). Since perfusion of the brain with PBS, and incubation of the tissues in medium containing SDS or Triton-X100 produced comparable results when incubated at the same temperature, neuronal membrane damage causing influx of the stain may be ruled out. A possible rationale for such efficient staining is likely to be due to faster movement of the metallic molecules present in the Golgi–Cox stain solution at the higher incubation temperature. At higher temperature the rate of movement and kinetic energy of the metallic ions in the Golgi–Cox stain solution would facilitate to increase staining in reduced time. Variations in incubating temperature have been tried earlier but those did not yield better results. Earlier studies used various temperatures in the form of heat (Berbel, 1986; Angulo et al., 1994), microwave (Armstrong and Parker, 1986; Marani et al., 1987; Zhang et al., 2003), etc; however, temperature around 37°C for shorter time as that of in this study was not tested. Although a critical temperature (37°C) improved staining, we cannot comment from this study what specific role the temperature might be playing in this process. It may be noted that the body fluids are composed of a large number of metallic and non-metallic ions, which exchange through the cellular membranes. Also, the body temperature in the homeotherms is set at a unique (magic) number close to 37°C. Whether optimum ion transport through biological membrane has any bearing on evolutionary and physiological significance with the set temperature for thermoregulation in warm blooded animals is an open question.
Several methodological modifications of Golgi–Cox staining process have been attempted in the past, e.g., varying duration of incubation of the tissues in the Golgi solution and multiple impregnations (Millhouse, 1969), changes in pH of chromatin solutions (Bertram and Ihrig, 1957; Gonzalez-Burgos et al., 1992; Angulo et al., 1994), vacuum immersion of the tissues (Friedland et al., 2006), use of single sections for staining (Landas and Phillips, 1982; Gabbott and Somogyi, 1984), use of microwave energy (Armstrong and Parker, 1986; Marani et al., 1987; Zhang et al., 2003), and changes in temperature (Berbel, 1986; Angulo et al., 1994). Instead of blocks, use of individual sections (Landas and Phillips, 1982; Gabbott and Somogyi, 1984) are tedious and suffer from additional drawback that neurons stained in one section can not be followed in the next section as they may remain unimpregnated. In other methods (Table 3), the earliest complete staining of blocks could be obtained after at least 5 days unlike within 24 h in this study. Although in one study using solution (not Golgi–Cox) at 60°C, staining within 24 h has been reported (Marani et al., 1987) with some other shortcomings though, since, it was not physiological temperature, it has more chance to alter architecture of neurons after staining. In all those methods up to 3 mm thick brain blocks were incubated in the staining solution, where as in our study we have used 5 mm thick blocks. Further, in most of those modified methods 80–150 μm sections were studied under microscope. However, in our study we could use 200 μm or thicker sections, which allow successfully following the extent of projections of stained neurons to increased depth. Thus, our method is significantly more advantageous for tracing neuronal projections and to study their synaptic connections for longer depth in 3-D. These properties and advantages could be used to study the connections between neurons located in areas which have been physiologically identified, e.g., in sleep, wake, and REM sleep modulation.
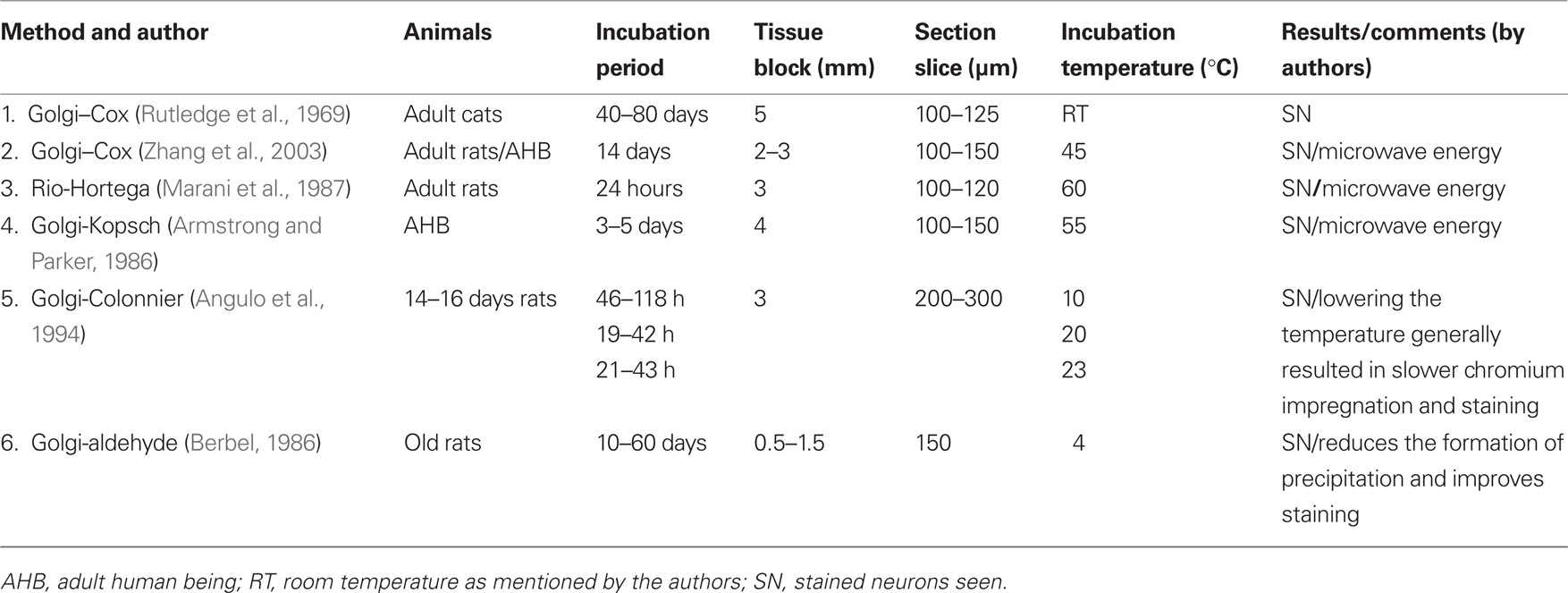
Table 3. Comparison of impregnation of Golgi stain into the neurons under various conditions used by different authors.
The results obtained in this study were reproducible and were not seen in the controls. Further, the findings were independent of tissue, animal, and area of the brain tissue being stained; hence, they cannot be due to non-specific artifacts. Another possibility of faster staining of neurons could be due to longitudinal sectioning of the brain tissue. However, such argument cannot be supported by the observation that the cortical neurons were stained first than the sub-cortical neurons; this study does not allow us to comment on this aspect.
Conclusion
Incubating the brain tissue around body temperature at 37 ± 1°C improved Golgi–Cox staining efficiency and reduced the time of staining, which were the primary drawbacks of using Golgi–Cox method. This modified protocol should allow the researchers to significantly increase their turnover in studying the anatomical connections between neurons located in different anatomical regions and whose functions might have been known. It will facilitate studying arborizations as well as spine counting on neurons, including whose anatomical connections and physiological identifications, e.g., wake, sleep, REM sleep, memory, etc has been established. Additional significant advantage of this modified method lies in the fact that it does not require any specialized equipment or chemicals.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding from Indian agencies viz. Council of Scientific and Industrial Research, Department of Science and Technology and UGC-Capacity Build Up are acknowledged.
References
Angulo, A., Merchan, J. A., and Molina, M. (1994). Golgi-Colonnier method: correlation of the degree of chromium reduction and pH change with quality of staining. J. Histochem. Cytochem. 42, 393–403.
Armstrong, E., and Parker, B. (1986). A new Golgi method for adult human brains. J. Neurosci. Methods 17, 247–254.
Berbel, P. J. (1986). Chromation at low temperatures improves impregnation of neurons in Golgi-aldehyde methods. J. Neurosci. Methods 17, 255–259.
Bertram, E. G., and Ihrig, H. K. (1957). Improvement of the Golgi method by pH control. Stain Technol. 32, 87–94.
Buell, S. J. (1982). Golgi-Cox and rapid golgi methods as applied to autopsied human brain tissue: widely disparate results. J. Neuropathol. Exp. Neurol. 41, 500–507.
Friedland, D. R., Los, J. G., and Ryugo, D. K. (2006). A modified Golgi staining protocol for use in the human brain stem and cerebellum. J. Neurosci. Methods 150, 90–95.
Gabbott, P. L., and Somogyi, J. (1984). The “single” section Golgi-impregnation procedure: methodological description. J. Neurosci. Methods 11, 221–230.
Gibb, R., and Kolb, B. (1998). A method for vibratome sectioning of Golgi-Cox stained whole rat brain. J. Neurosci. Methods 79, 1–4.
Glaser, E. M., and Van der Loos, H. (1981). Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J. Neurosci. Methods 4, 117–125.
Globus, A., and Scheibel, A. B. (1966). Loss of dendrite spines as an index of pre-synaptic terminal patterns. Nature 212, 463–465.
Gonzalez-Burgos, I., Tapia-Arizmendi, G., and Feria-Velasco, A. (1992). Golgi method without osmium tetroxide for the study of the central nervous system. Biotech. Histochem. 67, 288–296.
Landas, S., and Phillips, M. I. (1982). Staining of human and rat brain Vibratome sections by a new Golgi method. J. Neurosci. Methods 5, 147–151.
Marani, E., Guldemond, J. M., Adriolo, P. J., Boon, M. E., and Kok, L. P. (1987). The microwave Rio-Hortega technique: a 24 hour method. Histochem. J. 19, 658–664.
Millhouse, O. E. (1969). A Golgi study of the desending medial forebrain bundle. Brain Res. 15, 341–363.
Pasternak, J. F., and Woolsey, T. A. (1975). On the “selectivity” of the Golgi-Cox method. J. Comp. Neurol. 160, 307–312.
Ramon y Cajal, R. (1909). Histologie du System Nerveux de I’Homme et des Veertebres. Madrid: Instituto Ramon y Cajal, 774–838.
Rutledge, L. T., Duncan, J., and Beatty, N. (1969). A study of pyramidal cell axon collaterals in intact and partially isolated adult cerebral cortex. Brain Res. 16, 15–22.
Scheibel, A. B., and Tomiyasu, U. (1978). Dendritic sprouting in Alzheimer’s presenile dementia. Exp. Neurol. 60, 1–8.
Keywords: Golgi–Cox stain, incubation temperature, normal body temperature, reduced incubation period, thick section, thick tissue block
Citation: Ranjan A and Mallick BN (2010) A modified method for consistent and reliable Golgi–Cox staining in significantly reduced time. Front. Neur. 1:157. doi: 10.3389/fneur.2010.00157
Received: 27 July 2010;
Accepted: 14 December 2010;
Published online: 28 December 2010.
Edited by:
René Drucker-Colin, Universidad Nacional Autónoma de México, MexicoReviewed by:
Raul Aguilar-Roblero, Universidad Nacional Autónoma de México, MexicoJoaquin Reyes Tellez Giron, Universidad Nacional Autonoma de Mexico, Mexico
Copyright: © 2010 Ranjan and Mallick. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Birendra Nath Mallick, School of Life Sciences, Jawaharlal Nehru University, New Delhi, India. e-mail:cmVtc2JubUB5YWhvby5jb20=