


- 1 Internal Medicine and Neurology at Culiacan General Hospital, Sinaloa, Mexico
- 2 The University of Texas Health Science Center at Houston, Houston, TX, USA
- 3 The University of Texas Medical Branch at Galveston, Galveston, TX, USA
Therapeutic hypothermia (TH) is nowadays one of the most important methods of neuroprotection. The events that occur after an episode of ischemia are multiple and hypothermia can affect the various steps of this cascade. The mechanisms of action of TH are varied and the possible explanation for the benefits of this therapy is probably the multiple mechanisms of action blocking the cascade of ischemia on many levels. TH can affect many metabolic pathways, reactions of inflammation, apoptosis processes, and promote neuronal integrity. To know the mechanisms of action of TH will allow a better understanding about the indications for this therapy and the possibility of searching for other therapies when used in conjunction with hypothermia will provide a therapeutic synergistic effect.
Introduction
Therapeutic hypothermia (TH) is one of the wonders of medicine. Although TH has gained interest as a therapeutic measure in recent years, it is a very old method. The use of TH in modern clinical medicine has been documented over the past 200 years. Hypothermia was widely used in ancient times; however, due to the ignorance of the adverse effects and the lack of controlled clinical trials, the use of TH began to fall in the late 70s (Varon and Acosta, 2008).
The advent of numerous studies and controlled clinical trials began to emerge later and the interest in the use of hypothermia as a therapeutic measure arouse again (Bernard et al., 2002). TH is nowadays one of the most important methods of neuroprotection. It is well known that the use of TH improves neurological outcome in a variety of clinical scenarios such as post-cardiac arrest (Hypothermia after Cardiac Arrest Study Group, 2002), traumatic brain injury (TBI; The Brain Trauma Foundation, 2007), spinal cord injury (Dietrich, 2009), and it has been used in other diseases where it has proven to be useful as well such as a stroke (Yenari and Hemmen, 2010) and acute liver failure (Stravitz and Larsen, 2009).
Understanding the mechanisms of action through which TH provides neuroprotection will allow a better understanding about the indications for this therapy (Table 1), search for other therapies when used in conjunction with hypothermia will provide a therapeutic synergistic effect and the emergence of more clinical trials demonstrating the utility of this therapy in other diseases as well (Alzaga et al., 2006).

Table 1. The mechanisms of action of therapeutic hypothermia.
The aim of this review is to present the molecular mechanisms of action of TH in the brain, understanding the events that occur after an episode of cerebral ischemia, and how hypothermia can affect the various steps of this cascade.
Cerebral Ischemia, Calcium, and Excitotoxicity
Van Harreveld (1970) and Rothman and Olney (1986) showed a glutamate release during asphyxia and demonstrated that excitatory aminoacids can cause neuronal damage. After asphyxia and release of excitatory neurotransmitters, the reserves of calcium within the endoplasmic reticulum of the cell can be released into the cytoplasm increasing intracellular levels (Taylor, 1987). Calcium imbalance within the cell can lead to cell death (Carafoli, 1987).
In normal situations, endoplasmic reticulum and mitochondria sequesters calcium when intracellular levels increase, but during anoxia or ischemia, calcium levels increase within cells because calcium regulation depends on ATP and oxygen (Siesjo, 1988). Calcium enters the cell through voltage-sensitive calcium channels and agonist-operated calcium channels (Zhang et al., 1993). Agonist-operated calcium channels are activated by glutamate, N-methyl-D-aspartate (NMDA) and kainate, and quisqualate (K/Q). In the K/Q channel, calcium allows the entry of sodium and the output of potassium from the cell. NMDA receptors leads to calcium entry into cells and these are blocked by magnesium and stimulated by glycine (Foster and Fagg, 1987).
When oxygen and blood flow decreases to the brain, an ischemic cascade takes place. ATP decreases, cellular metabolism turns in to anaerobic glycolysis leading to an increase in hydrogen, phosphate, and lactate, culminating in intracellular acidosis. Hydrogen displaces calcium from intracellular proteins and intracellular calcium levels increase. There is a dysfunction of Na/K ATP-dependent pump and ATP-dependent channels, such as calcium, Na, and K channels, stop functioning, this limits the calcium output from the cell and the levels of intracellular calcium further increase. As calcium increases mitochondria sequesters it, leading to mitochondrial dysfunction.
Subsequently calcium activates intracellular proteases and depolarization occurs in the cell membrane, releasing a large number of excitatory neurotransmitters such as glutamate, activating K/Q and NMDA receptors persistently (Leker and Shohami, 2002). The activation of these receptors leads to an influx of sodium and chloride into the postsynaptic cell, causing an intracellular hyperosmolarity, and an influx of water into the cell, resulting in intracellular edema and neuronal death. At the same time, sodium influx inhibits the normal magnesium blockade on NMDA receptors, making them more responsive to glutamate, which stimulates calcium entrance and neuronal death. Furthermore, glutamate has been recognized as a neurotoxin, causes hyperexcitability even after restoring the blood flow after ischemia, this is better known as “Exitotoxic Cascade” (Small et al., 1999).
Mechanisms of Action of Therapeutic Hypothermia
Hypothermia and Neuronal Integrity
There are many mechanisms by which TH maintains the integrity of neurons and promotes the survival of these cells (Figure 1). After cerebral ischemic insult, a significant elevation in extracellular glutamate and other excitatory neurotransmitters such as aspartate occurs, leading to neuronal death (Benveniste et al., 1984). The integrity of neurons depends on the quantities of these neurotransmitters, so that greater amounts, the more neuronal damage (Globus et al., 1988). Hypothermia significantly reduces extracellular levels of excitatory neurotransmitters, including dopamine and glutamate (Okuda et al., 1986). The release of these neurotransmitters is temperature dependent, and it was demonstrated that even mild levels of hypothermia exerts an inhibitory effect (Illievich et al., 1994; Ooboshi et al., 2000).
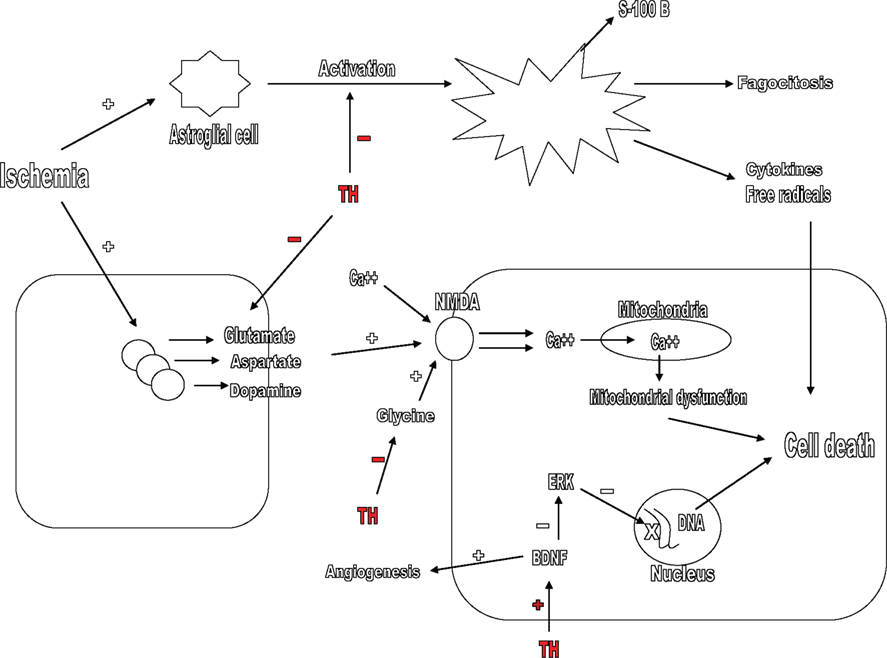
Figure 1. Mechanisms by which therapeutic hypothermia (TH) maintains neuronal integrity after cerebral ischemia. TH blocks the release of excitatory neurotransmitters such as glutamate, aspartate, and dopamine, inhibits the action of glycine in the NMDA receptor, increases angiogenesis, and inhibits ERK pathway by stimulating the formation of BDNF. TH also avoids the transformation and activation of astroglial cells after ischemia. TH, therapeutic hypothermia; NMDA, N-methyl-D-aspartate; BDNF, brain-derived neurotrophic factor.
Another mechanism by which hypothermia promotes survival of neurons is through an interaction on glycine. Excitatory aminoacids act on the NMDA receptor, increasing calcium influx into the cell. The NMDA receptor requires the presence of glycine to be activated, the presence of glycine accelerates the function of these receptors (Johnson and Ascher, 1987). TH significantly decreases brain glycine levels after ischemia, thus decreasing hyperexcitability by glutamate (Baker et al., 1991; Kvrivishvili, 2002).
Glycine and glycine receptor are fields of therapeutic action that have been studied previously. Glycine receptor antagonist drugs have been developed with poor results (Russell and Laverty, 2001). Nanoparticle formulations to antagonize the glycine site on the NMDA receptor have been created and may be a promising therapeutic measure and the role they can play as a combined therapy with TH is a very interesting field to explore (Sopala et al., 2002).
The interaction TH plays with nerve growth factors is another mechanism by which TH promotes neuronal survival. Neurotrophins are a group of neuronal growth and survival factors and brain-derived neurotrophic factor (BDNF) is the major neurotrophin studied. BDNF affects a known mitogen-activated enzymatic pathway protein kinase, the ERK, and through this enzymatic pathway, neurotrophins preserve neuronal integrity after the ischemic insult (Kokaia et al., 1993). Hypothermia reduces neuronal damage because significantly increases levels of BDNF and other neurotrophins after ischemic injury (D’Cruz et al., 2002; Vosler et al., 2005; Huang et al., 2008). Hypothermia may also stimulate angiogenesis in ischemic areas, a beneficial effect due to the activation of BDNF (Xie et al., 2007).
The field of action of nerve growth factors has been previously measured in experimental studies as a therapeutic target. BDNF has been used intravenously and intraventricular in focal and global ischemia models (Schabitz et al., 1997, 2000). The results have been favorable, BDNF decreases the degree of neuronal damage and the infarction size. Finally, experimental reports have tested the synergy of hypothermia and BDNF in ischemia models. The results have been impressing, leading to a reduction of the infarct size up to 40% (Berger et al., 2004).
Finally, it is important to mention that TH also promotes neuronal integrity through a mechanism of action on astroglial cells. After an ischemic insult, while neurons perform cell necrosis, glial cells survive to ischemia, experience hypertrophy and an activation process making them more responsive to immunologic stimuli. Astroglial cells proliferate, transform in to phagocytes, migrate, and release pro-inflammatory cytokines and free radicals. These cells can be cytotoxic (Gehrmann et al., 1992). Protein S-100B, which is found in astroglial cells, is a serum marker of poor neurologic outcome in patients undergoing hypothermia after cardiac arrest (Rundgren et al., 2009). Hypothermia avoids the proliferation, migration, transformation, and activation of astroglial cells after the ischemic insult (Hachimi-Idrissi et al., 2004).
Hypothermia and Apoptosis
Two types of cell death exist: necrotic cell death and programmed cell death, also known as apoptosis. Necrosis is a form of cell death where edema and cellular inflammatory response occur, leading to sudden death. Apoptosis is caused by a connection between intracellular enzyme pathways; it is the culmination of an interaction between pro-apoptotic and antiapoptotic proteins (Mattson et al., 2000). It is a programmed intracellular process which leads to cell death. TH inhibits apoptotic neuronal death because decreases p53 protein levels in the brain, a cellular transcription factor which activates genes of apoptosis and pro-apoptotic proteins, including Bak, Bax, and PUMA (Bargonetti and Manfredi, 2002; Ji et al., 2007; Figure 2).
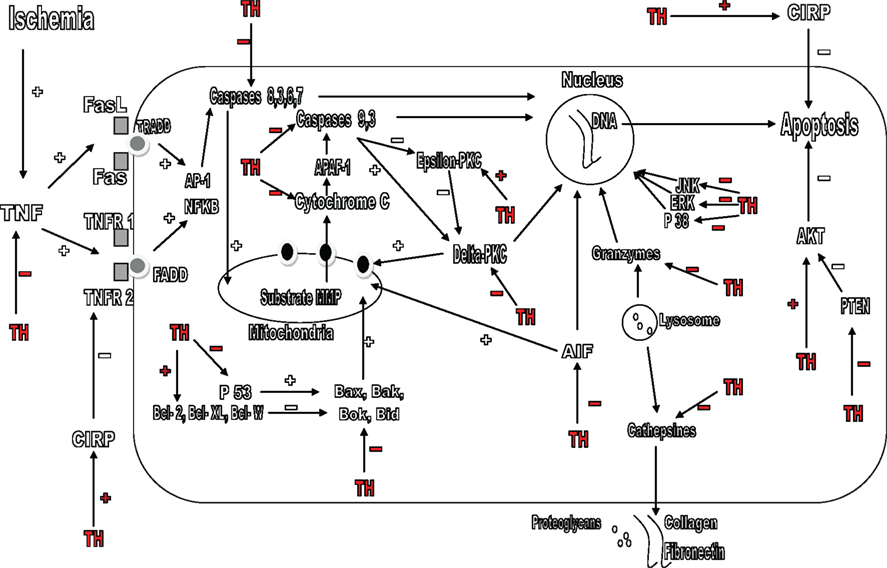
Figure 2. Mechanisms by which therapeutic hypothermia (TH) avoids apoptosis. TH blocks the TNF pathway and, increases the levels of CIRP; it inhibits the P38, JNK, and ERK pathways. TH avoids apoptosis directly by inhibition of the caspases pathway, blocking caspases 8, 3, 6, 7, 9, and cytochrome C. TH also blocks the P53 protein and affects the Bcl-2 family, blocking pro-apoptotic proteins such as Bax, Bak, Bok, Bid, Delta-PKC, PTEN, AIF, P38, JNK, ERK, granzymes, and reinforcing Epsilon-PKC, Bcl-2, Bcl-XL and Bcl-W, and AKT pathway. TH also avoids degradation of collagen, fibronectin, and proteoglycans by blocking cathepsines. TH, therapeutic hypothermia; TNF, tumor necrosis factor; CIRP, cold-inducible RNA-binding protein; TRADD, TNF receptor-associated death domain; FADD, Fas-associated protein with death domain; ERK, extracellular signal-regulated kinase; JNK, C-Jun-N-terminal protein kinase; AIF: apoptosis inducing factor.
The Bcl-2 family is a group of apoptotic proteins. Some of them such as Bcl-XL, Bcl-W, and Bcl-2 have antiapoptotic properties, whereas others, such as Bak, Bax, Bok, and Bid have pro-apoptotic properties (Reed, 1998). If pro-apoptotic proteins are activated, they lead to the formation of pores in the mitochondria’s membrane, allowing the release of cytochrome C to the cytosol, activating caspases, culminating in neuronal death. Hypothermia beneficially affects the levels of proteins Bcl-2 and cytochrome C after the ischemic insult, notwithstanding there is few other reports that suggest otherwise (Bossenmeyer-Pourie et al., 2000; Khar et al., 2003; Zhao et al., 2004).
Another pathway that also triggers the process of apoptosis is the TNF pathway. TNF through its receptors can initiate several routes (such as caspases) and culminate in apoptosis. These receptors are the Fas ligand (FasL), Fas (CD95), the complex of TNFR1 and 2, and many others (Brunner et al., 1995). TNF receptor-associated death domain (TRADD) and Fas-associated protein with death domain (FADD) are adapter proteins which can interact with TNF receptors allowing the apoptotic signal to continue (Zhang and Winoto, 1996). The induction of TNF apoptotic pathway has been described in TBI and is one of the most important pathway mediating neuronal apoptosis. TNF receptors can trigger apoptosis because of an activation of pro-apoptotic transcription factors such as NFkB and AP-1 (Morganti-Kossmann et al., 1997). There are experimental studies in where TH has demonstrated to block the TNF pathway of apoptosis, its receptors such as FasL, Fas, TNFR1, and the following events that culminate in cell death (Liu et al., 2008). The TNF pathway of apoptosis is a promising area of study.
Another important pathway in neuronal apoptosis is the stress-activated signaling pathways in which hypothermia exerts also a mechanism of action. Many regulatory pathways within the cell exist, working as intermediaries to control processes of proliferation, differentiation, and apoptosis. Protein kinases are the most recognized group of these proteins. They can travel to the nucleus modifying the expression of genes in DNA. Three groups of protein kinases are the most important: mitogen- activated protein kinase (MAPK), mitogen-activated protein kinase kinase (MAPKK), and mitogen-activated protein kinase kinase kinase (MAPKKK; Pawton, 1995). Each of these proteins are constituted by sub-families; for example in the MAPK, there are the extracellular signal-regulated kinase (ERK), c-Jun-N-terminal protein kinase (JNK), and p38. These proteins constitutes the major stress-activated signaling pathways and the role of these intracellular regulators is very important, trying to maintain cellular homeostasis (Schaeffer and Weber, 1999). There are experimental studies in where hypothermia affects these routes, avoiding consequently cell apoptosis. The stress-activated signaling pathways are an important target of study (Schmitt et al., 2007; Yang et al., 2009).
Hypothermia exerts also has a mechanism of action on one of the most important pathways in the process of apoptosis, the caspases pathway. This pathway is the point in where many of the other vias meet, caspases are the final pathway which directly “executes” the cell. Caspases are among the largest and most important pathways responsible for apoptosis. There are two principal apoptotic pathways mediated by caspases: Intrinsic and Extrinsic pathway. The intrinsic pathway is known to be activated by cellular stress such as radiation, hypoxia, and substrate deprivation. When the cell is subjected to one of these stressors, the substrate MMP is formed, this substrate acts at the mitochondria allowing the release of Cytochrome C, which interacts with another substrate, the Apaf-1, eventually leading to the activation of caspases 9 and 3, culminating in cell death (Kuida, 2000). The extrinsic pathway is activated by Fas-FasL, TNF-TNFR1 and leads to the activation of caspase-8, 3, 6, and 7. They stimulate release of cytochrome C from mitochondria leading to apoptosis (Muzio et al., 1998). Experimental models have shown that TH strongly inhibits the caspases pathway preventing apoptosis. The effect of hypothermia in theses pathways is such that makes it probably the therapeutic measure with the most benefit preventing neuronal apoptosis (Zhao et al., 2004; López-Hernández et al., 2006; Schmitt et al., 2007).
Other pathways and proteins cause apoptosis without directly involving the caspases, for example, a protein called apoptosis inducing factor (AIF) located in the mitochondrial membrane. When this protein is released by the mitochondria, travels to the nucleus and performs a DNA fragmentation culminating in cell death (Susin et al., 1999). There are other families of proteins called calpaines, cathepsines, and granzymes. Located inside the lysosomes, cathepsin degrades collagen, fibronectin, and proteoglycans. Granzymes can lead to DNA fragmentation (Shi et al., 1992). TH blocks all these proteins responsible for mediating the caspase-independent apoptosis and this is also an interesting field of study (Zhao et al., 2007).
After cerebral ischemia there is an important increase in some pro-apoptotic proteins such as the delta-PKC. This is a protein that belongs to the protein kinase C (PKC) family and plays an important role in neuronal death (Bright et al., 2004). Delta-PKC is activated by caspase-3 after which induces mitochondrial dysfunction, or travels to the nucleus to induce apoptosis (Raval et al., 2005). Epsilon-PKC unlike delta-PKC is another protein which shows antiapoptotic properties, but is degraded by caspases after the ischemic insult. Hypothermia acts in both proteins, blocking the translocation to the mitochondria and the nucleus of delta-PKC and stimulating the preservation of the action of Epsilon-PKC after ischemia (Shimohata et al., 2007a,b).
Another important antiapoptotic molecule is the Akt. In order to exerts an antiapoptotic function, this molecule needs to be phosphorylated, but PTEN which is a pro-apoptotic molecule, exerts its action by preventing the phosphorylation of Akt. Interestingly, these reactions can be regulated by free radicals (Zhao et al., 2006). Experimental models of ischemia have shown that hypothermia reduces the infarct size in experimental models of ischemia after reinforcement of Akt pathway, and can also carry out structural alterations in PTEN, decreasing apoptosis, thus protecting the cell (Zhao et al., 2005; Lee et al., 2009).
Finally some proteins, including cold-inducible RNA-binding protein (CIRP), are induced in the cells by a cold stimulus and have the capacity to stop apoptosis at many levels, providing cellular protection (Jones and Inouye, 1994). Mild hypothermia induces their formation and deep hypothermia avoids it. They act primarily in the processes of RNA transcription, but can also act in DNA and prevent free radical’s damage and can block apoptosis pathways such as the TNF-alpha pathway (Sakurai et al., 2006).
Hypothermia and Metabolism
After the cerebral blood flow decreases, metabolism turns in to anaerobic metabolism and intracellular levels of hydrogen, phosphate, and lactate increase creating intra and extracellular acidosis. In the absence of ATP, there is mitochondrial dysfunction and cell death. Hypothermia is likely to lower lactate levels and other wastes from anaerobic metabolism decreasing cellular acidosis (Jiang et al., 2004; Figure 3).
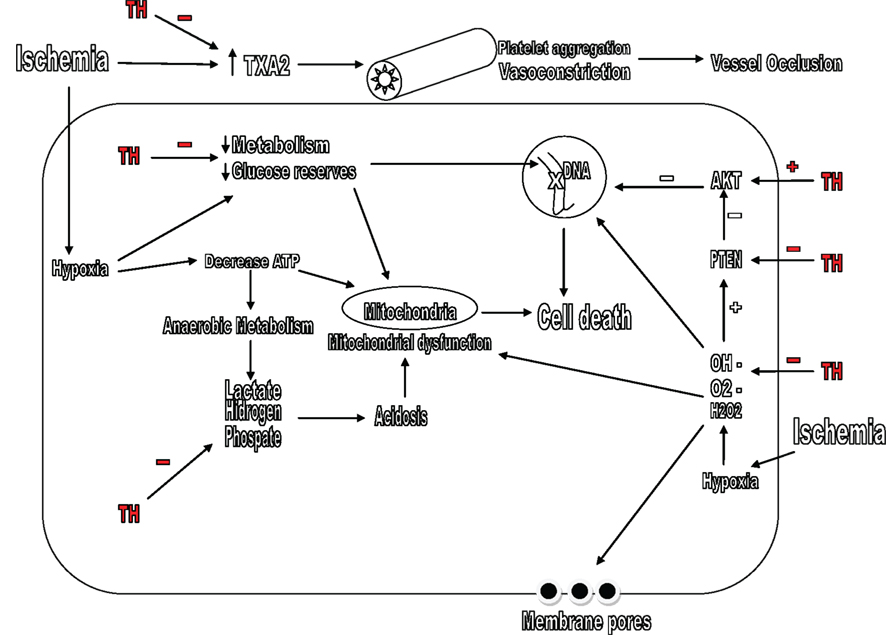
Figure 3. Mechanisms by which therapeutic hypothermia (TH) affects the metabolism and oxidative stress. TH inhibits the formation of lactate, hydrogen, and phosphate, increases glucose reserves and metabolism of the neurons. TH also decreases the levels of TXA2 and prevents vessel occlusion. TH, therapeutic hypothermia; TXA2, thromboxane A2; OH–, hydroxyl radicals; O2–, superoxide; H2O2: hydrogen peroxide.
Rosomoff and Holaday (1954) first described that hypothermia decreased cerebral blood flow and oxygen consumption, he also showed this effect was proportional to the depth of hypothermia. It is now established that hypothermia decreases cerebral blood flow along with cerebral oxygen consumption, glucose utilization, and brain metabolism, so this decrease is not deleterious. Brain metabolism decreases in range of 5–6% or even up to 10% for every degree decrease in temperature (Hagerdal et al., 1975). Hypothermia can also improve brain glucose metabolism and preserves glucose reserves in the brain, so they can be used even days after brain damage (Kimura et al., 2002).
Finally, after ischemia or TBI, there is an increased production of thromboxane A2 (TXA2) which is a potent vasoconstrictor, stimulates platelet aggregation and could lead to vessel occlusion (Dogné et al., 2004). Hypothermia decreases the concentrations of thromboxane A2 in experimental models of head trauma and ischemia. This may be a potential experimental target specially in patients under TH after TBI (Aibiki et al., 2000).
Hypothermia, Oxidative Stress, and Inflammation
Free radicals are oxygen derived compounds. They are extremely reactive and can produce cellular damage when acts in DNA, lipid membranes, proteins and enzymes. These compounds play an important role in brain damage after ischemia and TBI (Peters et al., 1998). Free radicals can come from arachidonic acid, nitric oxide, catecholamines, glutamate, and activation of NMDA receptors. Examples of these radicals are hydroxyl radicals (OH–), superoxide (O2–), and hydrogen peroxide (H2O2; Beckman et al., 1990; Globus et al., 1995a). Globus was the first to demonstrate the formation of free radicals after ischemic damage and that this production was temperature dependent. Hypothermia significantly reduces free radical levels after neuronal damage (Figure 3). The combination of specific free radicals antagonists and hypothermia may be a potential synergistic combination and thus a field of study (Globus et al., 1995b; Horiguchi et al., 2003).
Along with free radicals production, after an ischemic insult, inflammatory mechanisms emerge and may last hours to several days. This is a normal physiological response; there are cytokines that have been shown to be protective and other cytokines that are deleterious such as TNF-alpha and IL-1Beta (Merrill and Benveniste, 1996). Hypothermia decreases the production of inflammatory cytokines, leukotrienes, and inflammatory cells function such as macrophages (Kimura et al., 2002; Xiong et al., 2009).
Hypothermia, Intracranial Hypertension, and Brain Electrical Activity
After ischemia or TBI there is a disruption in the integrity of cell membranes and an increased permeability of endothelial cells, so the blood–brain barrier is lost in some level. This allows the development of cerebral edema leading to intracranial hypertension (Stocchetti et al., 2007). When intracranial hypertension develops it is important to treat the cause behind this rather than the result. Hypothermia decreases the cytotoxic edema formed after TBI, reduces disruption of the blood–brain barrier, and decreases the damage of the endothelial vasculature. Some authors claim that hypothermia is probably nowadays the best measure for intracranial hypertension after TBI. The combination of other available measures for intracranial hypertension with TH as a synergistic therapy is another potential field of action and research (Chi et al., 2001; McIntyre et al., 2003; González et al., 2009).
Another complication after stroke or TBI is non-convulsive status epilepticus (NCSE). NCSE is an electroencephalographic evidence of continuous seizure activity in a patient without motor convulsions. The presence of NCSE is associated with high morbidity and mortality, independently of diagnosis (Vespa et al., 1999). Hypothermia can suppress epileptogenic electrical activity and is effective in both the NCSE and in cases of generalized tonic–clonic seizures. The combination of other available measures for NCSE and generalized tonic–clonic seizures with TH as a synergistic therapy after stroke or TBI is another interesting field research (Corry et al., 2008).
Recommendations for Future Studies
Knowing the multiple mechanisms of action of TH at the molecular level has a very ample clinical relevance because we strongly believe it would allow in the future, firstly the emergence of more controlled clinical trials that combine other existing therapies for neuroprotection with hypothermia searching for synergies and favorable neurological outcomes. Secondly the development of new drugs for some specific pathways of the cascade of ischemia that may in the same way exert a synergistic effect with hypothermia and provide neuroprotection.
Some therapies have been tested previously in experimental models. For example the combination of TH with neuronal growth factors, especially BDNF. This combination therapy has shown impressive results in animal models (Berger et al., 2004).
Among other possible combination therapies to find a synergistic therapeutic effect we can mention the use of the nanoparticle formulations to antagonize the glycine site on the NMDA receptor or glycine receptor antagonist along with TH.
Other possible synergistic combinations are the association of TH with TNF antagonists, free radical and thromboxane A2 antagonists, drugs developed specifically to block several pathways of apoptosis such as caspases pathway, among others. Similarly, the combination of TH with other therapeutic measures commonly used to reduce intracranial pressure after TBI and antiepileptic drugs used in seizures or NCSE after TBI or stroke.
Conclusion
Therapeutic hypothermia is one of the most important therapies for providing neuroprotection. Its use in different clinical scenarios such as post-cardiac arrest, TBI, stroke, among others is increasingly being more used. The mechanisms of action of TH are multiple and varied. TH can affect many metabolic pathways, reactions of inflammation, apoptosis processes, among many other pathways. The possible explanation for the benefits and the success of this therapy is probably the multiple mechanisms of action blocking the cascade of ischemia on many levels.
We strongly believe that the use of TH will continue to increase in various clinical settings. Although most of the studies supporting the benefits of TH are experimental studies in animal models, the emergence of more controlled clinical trials evaluating synergistic effects of TH in association with other therapies will allow a widespread use of this therapy. The field of research of TH is very ample.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aibiki, M., Maekawa, S., and Yokono, S. (2000). Moderate hypothermia improves imbalances of thromboxane A2 and prostaglandin I2 production after traumatic brain injury in humans. Crit. Care Med. 28, 3902–3906.
Alzaga, A. G., Cerdan, M., and Varon, J. (2006). Therapeutic hypothermia. Resuscitation 70, 369–380.
Baker, A. J., Zornow, M. H., Grafe, M. R., Scheller, M. S., Skilling, S. R., Smullin, D. H., and Larson, A. A. (1991). Hypothermia prevents ischemia-induced increases in hippocampal glycine concentrations in rabbits. Stroke 22, 666–673.
Bargonetti, J., and Manfredi, J. J. (2002). Multiple roles of the tumor suppressor p53. Curr. Opin. Oncol. 14, 86–91.
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624.
Benveniste, H., Drejer, J., Schousboe, A., and Diemer, N. H. (1984). Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 43, 1369–1374.
Berger, C., Schabitz, W. R., Wolf, M., Mueller, H., Sommer, C., and Schwab, S. (2004). Hypothermia and brain-derived neurotrophic factor reduce glutamate synergistically in acute stroke. Exp. Neurol. 185, 305–312.
Bernard, S. A., Gray, T. W., Buist, M. D., Jones, B. M., Silvester, W., Gutteridge, G., and Smith, K. (2002). Treatment of comatose survivors of out-of-the hospital cardiac arrest with induced hypothermia. N. Engl. J. Med. 346, 557–563.
Bossenmeyer-Pourie, C., Koziel, V., and Daval, J. L. (2000). Effects of hypothermia on hypoxia-induced apoptosis in cultured neurons from developing rat forebrain: comparison with preconditioning. Pediatr. Res. 47, 385–391.
Bright, R., Raval, A. P., Dembner, J. M., Pérez-Pinzón M. A., Steinberg G. K., Yenari M. A., and Mochly-Rosen, D. (2004). Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. 24, 6880–6888.
Brunner, T., Mogil, R. J., LaFace, D., Yoo, N. J., Mahboubi, M., Echeverri, F., Martin, S. J., Force, W. R., Lynch, D. H., Ware, C. F., and Green, D. R. (1995). Cell-autonomous Fas (CD95)/Fas ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373, 441–444.
Chi, O. Z., Liu, X., and Weiss, H. R. (2001). Effects of mild hypothermia on blood – brain barrier disruption during isoflurane or pentobarbital anesthesia. Anesthesiology 95, 933–938.
Corry, J. J., Dhar, R., Murphy, T., and Diringer, M. N. (2008). Hypothermia for refractory status epilepticus. Neurocrit. Care 9, 189–197.
D’Cruz, B. J., Fertig, K. C., Filiano, A. J., Hicks, S. D., DeFranco, D. B., and Callaway, C. W. (2002). Hypothermic reperfusion after cardiac arrest augments brain-derived neurotrophic factor activation. J. Cereb. Blood Flow Metab. 22, 843–851.
Dietrich, W. D. III. (2009). Therapeutic hypothermia for spinal cord injury. Crit. Care Med. 37(Suppl. 7), S238–S242.
Dogné, J. M., Hanson, J., de Leval, X., Masereel, B., Kolh, P., and Pirotte, B. (2004). New developments on thromboxane modulators. Mini Rev. Med. Chem. 4, 649–657.
Gehrmann, J., Bonnekoh, P., Miyazawa, T., Hossmann, K. A., and Kreutzberg, G. W. (1992). Immunocytochemical study of an early microglial activation in ischemia. J. Cereb. Blood Flow Metab. 12, 257–269.
Globus, M. Y., Alonso, O., Dietrich, W. D., Busto, R., and Ginsberg, M. D. (1995a). Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J. Neurochem. 65, 1704–1711.
Globus, M. Y., Busto, R., Lin, B., Schnippering, H., and Ginsberg, M. D. (1995b). Detection of free radical activity during transient global ischemia and recirculation: effects of intra-ischemic brain temperature modulation. J. Neurochem. 65, 1250–1256.
Globus, M. Y-T., Busto, R., Dietrich, W. D., Martinez, E., Valdes, I., and Ginsberg, M. D. (1988). Effect on ischemia on the in vivo release of striatal dopamine, glutamate, and gamma-aminobutyric acid studied by intracerebral microdialysis. J. Neurochem. 51, 1455–1464.
González, I. F. P., Martínez, F. J. I., and García, T. E. (2009). Therapeutic hypothermia in traumatic brain injury. A. S. Sin. 3, 21–26.
Hachimi-Idrissi, S., Van Hemelrijck, A., Michotte, A., Smolders, I., Sarre, S., Ebinger, G., Huyghens, L., and Michotte, Y. (2004). Postischemic mild hypothermia reduces neurotransmitter release and astroglial cell proliferation during reperfusion after asphyxial cardiac arrest in rats. Brain Res. 1019, 217–225.
Hagerdal, M., Harp, J., Nilsson, L., and Siesjo, B. K. (1975). The effect of induced hypothermia upon oxygen consumption in the rat brain. J. Neurochem. 24, 311–316.
Horiguchi, T., Shimizu, K., Ogino, M., Suga, S., Inamasu, J., and Kawase, T. (2003). Postischemic hypothermia inhibits the generation of hydroxyl radical following transient forebrain ischemia in rats. J. Neurotrauma 20, 511–520.
Huang, X. C., Xu, W., and Jiang, J. Y. (2008). Effect of resuscitation after selective cerebral ultraprofound hypothermia on expressions of nerve growth factor and glial cell line-derived neurotrophic factor in the brain of monkey. Neurosci. Bull. 24, 150–154.
Hypothermia after Cardiac Arrest Study Group. (2002). Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 346, 549–556.
Illievich, U. M., Zornow, M. H., Choi, K. T., Scheller, M. S., and Strnat, M. A. (1994). Effects of hypothermic metabolic suppression on hippocampal glutamate concentrations after transient global cerebral ischemia. Anesth. Analg. 78, 905–911.
Ji, X., Luo, Y., Ling, F., Stetler, R. A., Lan, J., Cao, G., and Chen, J. (2007). Mild hypothermia diminishes oxidative DNA damage and pro-death signaling events after cerebral ischemia: a mechanism for neuroprotection. Front. Biosci. 12, 1737–1747.
Jiang, J. Y., Liang, Y. M., Luo, Q. Z., and Zhu, C. (2004). Effect of mild hypothermia on brain dialysate lactate after fluid percussion brain injury in rodents. Neurosurgery 54, 713–717.
Johnson, J. W., and Ascher, P. (1987). Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531.
Jones, P. G., and Inouye, M. (1994). The cold-shock response: a hot topic. Mol. Microbiol. 11, 811–818.
Khar, A., Pardhasaradhi, B. V., Ali, A. M., and Kumari, A. L. (2003). Protection conferred by Bcl-2 expression involves reduced oxidative stress and increased glutathione production during hypothermia-induced apoptosis in AK-5 tumor cells. Free Radic. Biol. Med. 35, 949–957.
Kimura, A., Sakurada, S., Ohkuni, H., Todome, Y., and Kurata, K. (2002). Moderate hypothermia delays proinflammatory cytokine production of human peripheral blood mononuclear cells. Crit. Care Med. 30, 1499–1502.
Kimura, T., Sako, K., and Tanaka, K. (2002). Effect of mild hypothermia on energy state recovery following transient forebrain ischemia in the gerbil. Exp. Brain Res. 145, 83–90.
Kokaia, Z., Bengzon, J., Metsis, M., Kokaia, M., Persson, H., and Lindvall, O. (1993). Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 90, 6711–6715.
Kvrivishvili, G. (2002). Glycine and neuroprotective effect of hypothermia in hypoxic-ischemic brain damage. Neuroreport 13, 1995–2000.
Lee, S. M., Zhao, H., Maier, C. M., and Steinberg, G. K. (2009). The protective effect of early hypothermia on PTEN phosphorylation correlates with free radical inhibition in rat stroke. J. Cereb. Blood Flow Metab. 29, 1589–1600.
Leker, R. R., and Shohami, E. (2002). Cerebral ischemia and trauma – different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res. Rev. 39, 55–73.
Liu, L., Kim, J. Y., Koike, M. A., Yoon, Y. J., Tang, X. N., Ma, H., Lee, H., Steinberg, G. K., Lee, J. E., and Yenari, M. A. (2008). FasL shedding is reduced by hypothermia in experimental stroke. J. Neurochem. 106, 541–550.
López-Hernández, F. J., Ortiz, M. A., and Piedrafita, F. J. (2006). The extrinsic and intrinsic apoptotic pathways are differentially affected by temperature upstream of mitochondrial damage. Apoptosis 11, 1339–1347.
Mattson, M. P., Culmsee, C., and Yu, Z. F. (2000). Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 301, 173–187.
McIntyre, L. A., Fergusson, D. A., and Hebert, P. C. (2003). Prolonged therapeutic hypothermia after traumatic brain injury in adults. JAMA 289, 2992–2999.
Merrill, J. E., and Benveniste, E. N. (1996). Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 19, 331–338.
Morganti-Kossmann, M. C., Lenzlinger, P. M., Hans, V., Stahel, P., Csuka, E., Ammann, E., Stocker, R., Trentz, O., and Kossmann, T. (1997). Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry 2, 133–136.
Muzio, M., Stockwell, B. R., Stennicke, H. R., Salvesen, G. S., and Dixit, V. M. (1998). An induced proximity model for caspase-8 activation. J. Biol. Chem. 273, 2926–2930.
Okuda, C., Saito, A., Miyazaki, M., and Kuriyama, K. (1986). Alterations of the turnover of dopamine and 5-hydroxytryptamine in rat brain associated with hypothermia. Pharmacol. Biochem. Behav. 25, 79–83.
Ooboshi, H., Ibayashi, S., Takano, K., Sadoshima, S., Kondo, A., Uchimura, H., and Fujishima, M. (2000). Hypothermia inhibits ischemia-induced efflux of amino acids and neuronal damage in the hippocampus of aged rats. Brain Res. 884, 23–30.
Peters, O., Back, T., Lindauer, U., Busch, C., Megow, D., Dreier, J., and Dirnagl, U. (1998). Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J. Cereb. Blood Flow Metab. 18, 196–205.
Raval, A. P., Dave, K. R., Prado, R., Katz, L. M., Busto, R., Sick, T. J., Ginsberg, M. D., Mochly-Rosen, D., and Pérez-Pinzón, M. A. (2005). Protein kinase C delta cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation. J. Cereb Blood Flow Metab. 25, 730–741.
Rosomoff, H. L., and Holaday, D. A. (1954). Cerebral blood flow and cerebral oxygen consumption during hypothermia. Am. J. Physiol. 179, 85–88.
Rothman, S. M., and Olney, J. W. (1986). Glutamate and the pathophysiology of hypoxicic-ischemic brain damage. Ann. Neurol. 19, 105–111.
Rundgren, M., Karlsson, T., Nielsen, N., Cronberg, T., Johnsson, P., and Friberg, H. (2009). Neuron specific enolase and S-100B as predictors of outcome after cardiac arrest and induced hypothermia. Resuscitation 80, 784–789.
Russell, B. R., and Laverty, R. (2001). The effect of (R)-HA966 or ACEA 1021 on dexfenfluramine or (S)-MDMA-induced changes in temperature, activity, and neurotoxicity. Pharmacol. Biochem. Behav. 68, 565–574.
Sakurai, T., Itoh, K., Higashitsuji, H., Nonoguchi, K., Liu, Y., Watanabe, H., Nakano, T., Fukumoto, M., Chiba, T., and Fujita, J. (2006). CIRP protects against tumor necrosis factor-alpha-induced apoptosis via activation of extracellular signal-regulated kinase. Biochim. Biophys. Acta 1763, 290–295.
Schabitz, W. R., Schwab, S., Spranger, M., and Hacke, W. (1997). Intraventricular brain-derived neurotrophic factor reduces infarct size after focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 17, 500–506.
Schabitz, W. R., Sommer, C., Zoder, W., Kiessling, M., Schwaninger, M., and Schwab, S. (2000). Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke 9, 2212–2217.
Schaeffer, H. J., and Weber, M. J. (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell Biol. 19, 2435–2444.
Schmitt, K. R., Diestel, A., Lehnardt, S., Schwartlander, R., Lange, P. E., Berger, F., Ullrich, O., and Abdul-Khaliq, H. (2007). Hypothermia suppresses inflammation via ERK signaling pathway in stimulated microglial cells. J. Neuroimmunol. 189, 7–16.
Shi, L., Kraut, R. P., Aebersold, R., and Greenberg, A. H. (1992). A natural killer cell granule protein that induces DNA fragmentation and apoptosis. J. Exp. Med. 175, 553–566.
Shimohata, T., Zhao, H., and Steinberg, G. K. (2007a). Epsilon PKC may contribute to the protective effect of hypothermia in a rat focal cerebral ischemia model. Stroke 38, 375–380.
Shimohata, T., Zhao, H., Sung, J. H., Sun, G., Mochly-Rosen, D., and Steinberg, G. K. (2007b). Suppression of delta-PKC activation after focal cerebral ischemia contributes to the protective effect of hypothermia. J. Cereb. Blood Flow Metab. 27, 1463–1475.
Siesjo, B. K. (1988). Historical overview. Calcium, ischemia, and death of brain cells. Ann. N.Y. Acad. Sci. 522, 638–661.
Small, D. L., Morley, P., and Buchan, A. M. (1999). Biology of ischemic cerebral cell death. Prog. Cardiovasc. Dis. 42, 185–207.
Sopala, M., Schweizer, S., Schäfer, N., Nürnberg, E., Kreuter, J., Seiller, E., and Danysz, W. (2002). Neuroprotective activity of a nanoparticulate formulation of the glycineB site antagonist MRZ 2/576 in transient focal ischaemia in rats. Arzneimittelforschung 52, 168–174.
Stocchetti. N., Colombo, A., Ortolano, F., Videtta, W., Marchesi, R., Longhi, L., and Zanier, E. R. (2007). Time course of intracranial hypertension after traumatic brain injury. J. Neurotrauma 24, 1339–1346.
Stravitz, R. T., and Larsen, F. S. (2009). Therapeutic hypothermia for acute liver failure. Crit. Care Med. 37(Suppl. 7), S258–S264.
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M., and Kroemer, G. (1999). Molecular characterization of mitochondrial apoptosis- inducing factor. Nature 397, 441–446.
The Brain Trauma Foundation. (2007). The guidelines for the management of severe traumatic brain injury. J. Neurotrauma 24, S21–S25.
Van Harreveld, A. (1970). A mechanism for fluid shifts specific for the central nervous system. Bibl. Psychiatr. 143, 62–70.
Varon, J., and Acosta, P. (2008). Therapeutic hypothermia. Past, present, and future. Chest 133, 1267–1274.
Vespa, P. M., Nuwer, M. R., Nenov, V., Ronne-Engstrom, E., Hovda, D. A., Bergsneider, M., Kelly, D. F., Martin, N. A., and Becker, D. P. (1999). Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic head injury as detected by continuous EEG in the intensive care unit. J. Neurosurg. 91, 750–760.
Vosler, P. S., Logue, E. S., Repine, M. J., and Callaway, C. W. (2005). Delayed hypothermia preferentially increases expression of brain-derived neurotrophic factor exon III in rat hippocampus after asphyxial cardiac arrest. Brain Res. Mol. Brain Res. 135, 21–29.
Xie, Y. C., Li, C. Y., Li, T., Nie, D. Y., and Ye, F. (2007). Effect of mild hypothermia on angiogenesis in rats with focal cerebral ischemia. Neurosci. Lett. 422, 87–90.
Xiong, M., Yang, Y., Chen, G. Q., and Zhou, W. H. (2009). Post-ischemic hypothermia for 24h in P7 rats rescues hippocampal neuron: association with decreased astrocyte activation and inflammatory cytokine expression. Brain Res. Bull. 79, 351–357.
Yang, D., Guo, S., Zhang, T., and Li, H. (2009). Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett. 583, 2500–2506.
Yenari, M. A., and Hemmen, T. M. (2010). Therapeutic hypothermia for brain ischemia: where have we come and where do we go? Stroke 41(Suppl. 10), S72–S74.
Zhang, J., and Winoto, A. (1996). A mouse Fas-associated protein with homology to the human MORT1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell Biol. 16, 2756–2763.
Zhang, J. F., Randall, A. D., Ellinor, P. T., Horne, W. A., Sather, W.A., Tanabe, T., Schwarz, T. L., and Tsien, R. W. (1993). Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology 32, 1075–1088.
Zhao, H., Sapolsky, R. M., and Steinberg, G. K. (2006). Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 34, 249–270.
Zhao, H., Shimohata, T., Wang, J. Q., Sun, G., Schaal, D. W., Sapolsky, R. M., and Steinberg, G. K. (2005). Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J. Neurosci. 25, 9794–9806.
Zhao, H., Wang, J. Q., Shimohata, T., Sun, G., Yenari, M. A., Sapolsky, R. M., and Steinberg, G. K. (2007). Conditions of protection by hypothermia and effects on apoptotic pathways in a rat model of permanent middle cerebral artery occlusion. J. Neurosurg. 107, 636–641.
Keywords: therapeutic hypothermia, neuronal death, cerebral ischemia, mechanism of action, excitatory neurotransmitters, neuronal acidosis
Citation: González-Ibarra FP, Varon J and López-Meza EG (2011) Therapeutic hypothermia: critical review of the molecular mechanisms of action. Front. Neur. 2:4. doi: 10.3389/fneur.2011.00004
Received: 09 June 2010;
Paper pending published: 15 June 2010;
Accepted: 14 January 2011;
Published online: 03 February 2011.
Edited by:
Oscar Prospéro-García, Universidad Nacional Autónoma de México, MexicoReviewed by:
H. Aslami, Academic Medical Center, NetherlandsSamuel A. Tisherman, University of Pittsburgh, USA
Copyright: © 2011 González-Ibarra, Varon and López-Meza. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Fernando Pavel Gonzalez-Ibarra, Internal Medicine and Neurology, Culiacan General Hospital, Sinaloa, Mexico. e-mail:ZmVybmFuY2hpdG9AaG90bWFpbC5jb20=