
 Magdalena Guerra-Crespo
Magdalena Guerra-Crespo
- Departamento de Neuropatología Molecular, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, México D.F., México
Narcolepsy is a chronic neurodegenerative disease caused by a deficiency of orexin-producing neurons in the lateral hypothalamus. It is clinically characterized by excessive daytime sleepiness and by intrusions into wakefulness of physiological aspects of rapid eye movement sleep such as cataplexy, sleep paralysis, and hypnagogic hallucinations. The major pathophysiology of narcolepsy has been recently described on the bases of the discovery of the neuropeptides named orexins (hypocretins) in 1998; considerable evidence, summarized below, demonstrates that narcolepsy is the result of alterations in the genes involved in the pathology of the orexin ligand or its receptor. Deficient orexin transmission is sufficient to produce narcolepsy, as we describe here, animal models with dysregulated orexin signaling exhibit a narcolepsy-like phenotype. Remarkably, these narcoleptic models have different alterations of the orexinergic circuit, this diversity provide us with the means for making comparison, and have a better understanding of orexin-cell physiology. It is of particular interest that the most remarkable findings regarding this sleep disorder were fortuitous and due to keen observations. Sleep is a highly intricate and regulated state, and narcolepsy is a disorder that still remains as one of the unsolved mysteries in science. Nevertheless, advances and development of technology in neuroscience will provide us with the necessary tools to unravel the narcolepsy puzzle in the near future. Through an evaluation of the scientific literature we traced an updated picture of narcolepsy and orexins in order to provide insight into the means by which neurobiological knowledge is constructed.
Historical Aspects of Narcolepsy
The term narcolepsy derives from the french word “narcolepsie” created by the physician Jean-Baptiste-Édouard Gélineau by combining the Greek νάρκη (narkē, “numbness” or “stupor”), and λάψ⍳ς (lepsis, “attack” or “seizure”). Even though Narcolepsy was first described over 120 years ago (Fisher, 1878; Gélineau, 1880; Westphal, 1887; Löwenfeld, 1902), only in the last decade the underlying neurobiology started to become clear.
Narcolepsy affects approximately 0.2% of the population worldwide and causes disability in 24% of the affected subjects (Longstreth et al., 2007). Narcoleptic patients frequently have severe sleepiness which makes particularly difficult remaining awake during activities that demand an alert state. In addition, these individuals regularly have fragments of rapid eye movement (REM) sleep that intrude into wakefulness, such as dream-like hallucinations (hypnagogic) as they drift off to sleep and cataplexy or have brief episodes of muscle paralysis triggered by strong emotions.
All narcoleptic subjects present chronic sleepiness, but the intensity varies across the day and between individuals. This sleepiness is most troublesome during periods of inactivity, though is often improved temporarily by a brief nap. As a consequence of sleepiness, patients may report inattention, poor memory, blurry vision, diplopia, and automatic behaviors such as driving without awareness (Broughton et al., 1998; Harsh et al., 2000).
Hypnagogic hallucinations are dream-like, often frightening hallucinations that typically occur with drowsiness or the onset of sleep. These hallucinations are usually visual, but also tactile, auditory, or even vestibular with a sense of sudden falling are not uncommon.
Sleep paralysis is profound weakness occuring at the onset of sleep or upon awakening, an intrusion of REM sleep paralysis into wakefulness, and can be associated with a sensation of fear or suffocation. Hypnagogic hallucinations and sleep paralysis are not specific to narcolepsy and can be seen with other conditions of increased sleep pressure such as chronic sleep deprivation or obstructive sleep apnea, and occasionally in normal individuals (Dahlitz and Parkes, 1993; Aldrich, 1996).
Cataplexy is a sudden muscle weakness brought on by strong emotions, particularly joking, laughter, or anger (Guilleminault and Gelb, 1995; Anic-Labat et al., 1999). Most likely, this also is an intrusion of REM sleep atonia into wakefulness, but in contrast with sleep paralysis, cataplexy occurs almost exclusively in narcolepsy. Consciousness is never impaired unless the patient subsequently falls asleep or begins having hypnagogic hallucinations. Episodes of cataplexy may begin with clonic inhibitory movements leading to a fall, followed by a period of atonia and arreflexia usually lasting less than 2 min (Rubboli et al., 2000). Cataplexy may result from excessive activation of descending motor inhibitory pathways that are active during strong emotions or REM sleep (Siegel et al., 1991).
Hypothalamus and Sleep
For years, the notion that narcolepsy was caused by some dysfunction of the hypothalamus or REM sleep-regulating pathways existed, but the fundamental neuropathology has remained a mystery until the last few years.
The first correlation between the hypothalamus and sleep regulation was characterized by the Viennese neurologist Constantin von Economo during the 1915 Spanish influenza epidemic which allowed the pathoanatomical study of brain specimens from patients who had died of encephalitis lethargica (Economo, 1917). The frequent occurrence of pathologically increased sleep in encephalitis lethargica secondary to lesions in the posterior hypothalamus or at the junction of the brainstem and forebrain, prompted him to assume that an ascending arousal system originating in the brainstem that kept the forebrain awake existed.
This belief would be later confirmed by Moruzzi and Magoun (1949), they found that stimulation of the reticular formation of the brain stem evoked changes in the EEG, consisting of abolition of synchronized discharge and introduction of low voltage fast activity in its place, awakening animals from normal sleep; which developed the concept of the “ascending activating reticular system.”
Circling back to Von Economo’s findings, he also established that patients who had experienced sleeplessness before dying had brain lesions in the preoptic area of the anterior hypothalamus, area which came to be known as the “sleep center.”
REM Sleep and Narcolepsy
The dissection of the mechanisms involved in the regulation of sleep-wake cycling provided a better understanding of the nature of narcolepsy.
Discovery of REM sleep by Aserinsky and Kleitman (1953) was one of the most valuable breakthroughs regarding sleep research. They described that this novel sleep stage had an EEG pattern that resembled the vigilance state, occurred in a recurrent interval and presented characteristic REM with muscle atonia, suggesting that these physiological phenomena, and probably dreaming, were very likely all manifestations of a particular level of cortical activity which is encountered normally during sleep (Aserinsky and Kleitman, 1953). With this finding, they placed the last piece of the puzzle of sleep architecture.
The early descriptions of REM sleep and its associated atonia led multiple investigators to study sleep onset in narcolepsy. Vogel was the first to report REM sleep at sleep onset in a narcoleptic patient, establishing the close relationship between REM sleep and narcolepsy (Vogel, 1976). However, regardless of having mapped the structural design of the sleep-wake cycle (SWC), the factual nature of narcolepsy was not established hitherto.
The Midbrain Reticular Formation and its Chemical Nature in Sleep
The consensus notion that sleep resulted from active inhibition of the vigilance midbrain reticular neurons located in the rostral part of the brainstem gave Hernández–Peón’s group the notion that presynaptic hypnogenic inhibitory terminals were located in the same anatomical region.
By administering a perfused substance from a sleep-deprived donor cat obtained from the midbrain reticular formation (MRF) into the MRF of an awaked recipient cat, the latter began to show the typical behavioral preparatory attitude of sleep (Drucker-Colín et al., 1970; Drucker-Colín, 1973). The results showed that the MRF was a specific area of the brain involved in the regulation of the SWC and gave the stepping stone to the discovery of the neurotransmitters involved in the maintenance of sleep and vigilance. Nowadays, we know that an alert, active, and responsive awake state depends upon influences ascending from the brainstem to the cerebral cortex to stimulate cortical activation and descending from the brainstem to the spinal cord to stimulate behavioral arousal with muscle tone (Olszewski and Baxter, 1954; Butler and Hodos, 1996; Jones, 2008).
Amongst the neurotransmitters that regulate the SWC, acetylcholine (ACh) has been known to play a critical role in stimulating cortical activation since very early pharmacological studies (Holmes and Jones, 1994), during waking and also in REM sleep. Cholinergic neurons can therefore stimulate cortical activation irrespective of behavioral arousal (Gnadt and Pegram, 1986; Hallanger et al., 1987).
Another major group is the locus coeruleus (LC) noradrenergic neurons, which differ from neurons of the MRF and from the cholinergic neurons of the brainstem and basal forebrain in that they have either excitatory or inhibitory actions upon postsynaptic neurons, and have the capacity to directly excite the thalamo-cortical relay neurons and the cholinergic basal–cortical neurons, as well as cortical pyramidal neurons through α1-adrenergic receptors (AR), while inhibiting sleep-promoting neurons through α2-ARs (McCormick, 1992). Hence, when LC neurons are active, they can simultaneously stimulate cortical activation and behavioral arousal. In contrast to cholinergic neurons, LC noradrenergic neurons discharge selectively during waking and cease firing during sleep to be completely off during REM sleep (Hobson et al., 1975; Aston-Jones and Bloom, 1981).
Albeit having a general knowledge of the principal chemical substrates that regulate the SWC, it appeared that arousal systems, which also include histaminergic and serotonergic neurons are highly redundant and it appears that none of them is necessary for the maintenance of a waking state (Jones, 2003).
The Discovery of Orexins
One of the most important breakthroughs came with the coincidently discovery by two different groups of two neuropeptides residing in the lateral hypothalamus (LH); one group named this peptide orexin for its presumed role in appetite whereas another group named it hypocretin for its possible resemblance to secretin, found by orphan receptor technologies and subtractive cDNA cloning, respectively (de Lecea et al., 1998; Sakurai et al., 1998).
Orexins are exclusively synthesized in the LH, derived from a single protein precursor named prepro-orexin. Prepro-orexin is enzymatically matured into two peptides, orexin-A and orexin-B which are 33- and 28-amino-acid peptides, respectively. Two cloned orexin receptors OX1R and OX2R are serpentine G-protein-coupled receptors, both of which bind orexins and are coupled to Ca2+ mobilization (Sakurai et al., 1998). Orexins neurons project within the LH throughout the central nervous system to nuclei involved in the control of feeding, sleep-wakefulness, neuroendocrine homeostasis, and autonomic regulation (Peyron et al., 1998; Marcus et al., 2001).
Orexin neurons are active during wakefulness as indicated by the expression of Fos (Estabrooke et al., 2001), and extracellular concentrations of orexin are higher during periods of wakefulness (Yoshida et al., 2001). Electrophysiological recordings from the orexin neuron region identified many wake-active neurons, with particularly high firing rates when an animal is physically active (Steininger et al., 1999; Alam et al., 2002).
In vitro, orexin excites the LC and other aminergic neurons (Hagan et al., 1999; Bayer et al., 2001; Brown et al., 2001). In vivo, injections of orexin into the lateral ventricles or near specific arousal regions such as the LC increase wakefulness and markedly suppress REM sleep (Hagan et al., 1999; Bourgin et al., 2000).
Interestingly, the orexin neuron population was previously described by Jouvet’s group in 1989 as prolactin-secreting neurons (Paut-Pagano et al., 1989), later on it was described that the prolactin antiserum was recognizing an epitope carried by a particular fragment of the prepro-orexin gene (Risold et al., 1999).
Narcolepsy and Orexins: The Missing Element
The interest of investigators in orexins has focused on narcolepsy, this initiated with a bright but incidental observation. The general rationale was that genes expressed in the hypothalamus might have medically useful effects due to the role of the hypothalamus in control of appetite, thirst, pituitary hormones and many other autonomic and arousal functions. In order to determine orexin peptides role in food consumption Yanagisawa’s group constructed a mice with a deletion of the gene coding for prepro-orexin, as they were originally believed to be primarily important in the regulation of appetite. Although they did not observe significant changes in food consumption between wildtype and orexin knockout mice, they did scrutinize with video recording analysis that orexin knockout mice displayed severe sleepiness and cataplexy like events (Chemelli et al., 1999). This association was coincidently confirmed in the same year when Mignot’s group sought to identify the gene that caused canine narcolepsy by a systematic chromosomal analysis. They identified that dogs with inherited narcolepsy had an exon-skipping mutation in the type OX2R gene (Lin et al., 1999).
In follow up work, Yanagisawa’s team created mice lacking either of the two identified orexin receptors. They found that knockout mice of the OX2R experienced cessations of activity which they concluded were sleep attacks. They also found that mice deficient of the OX1R had disrupted sleep but showed fewer of the signs of narcolepsy than the OX2R knockout mice (Chemelli et al., 2000; Willie et al., 2003).
In order to confirm if specific orexin-cell loss derives in narcolepsy, Shiromani’s team suggested that since orexin neurons are located only in the LH but neither electrolytic nor pharmacological lesions of this region have produced narcoleptic-like sleep, specific neurons need to be destroyed. They specifically damaged orexin neurons in the LH by means of the endogenous ligand orexin-B conjugated to the ribosome-inactivating protein saporin. The lesioned rats presented an increase in slow-wave sleep, REM sleep, and REM sleep intrusions during wakefulness, characteristic of a narcoleptic phenotype (Gerashchenko et al., 2001). However, in the vast majority of the cases, human narcolepsy is not secondary to lesions as in saporin-lesioned rats.
Meanwhile, Yanagisawa’s group where trying to deal with this problem, to come up with an animal model that strictly resembled human narcolepsy. They generated transgenic mice in which orexin-containing neurons are ablated by orexinergic-specific expression of a truncated Machado–Joseph disease gene product (ataxin-3) with an expanded polyglutamine stretch. These mice presented a gradual orexin-cell neurodegeneration and showed a phenotype strikingly similar to human narcolepsy, including behavioral arrests, premature entry into REM sleep and poorly consolidated sleep patterns (Hara et al., 2001).
Orexins and Human Narcolepsy
Even though murine and canine narcolepsy were demonstrated to be caused by alterations of the orexinergic circuit, most human narcolepsy is not familial, is discordant in identical twins, and has not been linked to mutations of the orexin system, Mignot’s group sought to make this association, they measured immunoreactive orexin in cerebrospinal fluid (CSF) and reported major deficiencies in orexin CSF levels among narcoleptic patients (110 pg/ml, one-third of mean control value; Nishino et al., 2000) implying either a large orexin-cell loss or a complete cessation of orexin production in these cells.
In a parallel work they also described the virtual absence of orexin in narcoleptic brains by means of in situ hybridization and peptide radioimmunoassay techniques (Peyron et al., 2000). In the same year, while Mignot’s team reported a total loss of orexinergic neurons, Siegel reported an 85–95% decrease in orexin-containing neurons in brains of patients with narcolepsy (Thannickal et al., 2000). This finding was quite selective, as the MCH neurons, which are intermingled with the orexin cells, were completely spared, and it probably represented cell loss rather than downregulation of orexin expression as there was concomitant loss of other markers (dynorphin and neuronal activity-related pentraxin) of the orexin-cell population.
Regardless of having elucidated the association of human narcolepsy and a deficit in orexin peptide production, only in a single narcoleptic patient has been reported a mutation of the gene responsible for orexin production (Peyron et al., 2000). Hence, the most widely accepted view regarding the mechanism of the orexin deficiency is that a degenerative process, probably immune in origin, selectively destroys orexin-releasing neurons, thereby producing the clinical syndrome of narcolepsy.
Loss of Orexin Neurons: The Unanswered Mystery
Facts have now established that narcolepsy is caused by a selective loss of orexin signaling in the brain; however, loss of orexin signaling might not be due to a simple genetic abnormality, as narcolepsy is in most of the cases acquired during young adulthood, and the vast majority of people with narcolepsy do not have mutations of the genes encoding the orexin peptides or their OX1 or OX2 receptors (Guilleminault and Anognos, 2000). Loss of orexin signal is most likely not a simple failure in producing orexin peptides alone.
The question that arose from this particular riddle was: what other genes might be altered and cause orexin neuron deficiency in narcolepsy? Approximately 90% of patients with narcolepsy have human leukocyte antigen DQB1 * 0602 and DQA1 * 0102 (Mignot et al., 2001; Scammell, 2003), leading many researchers to hypothesize that the loss of orexin neurons may be immune-mediated.
In order to analyze this, Mignot’s team performed gene expression profiling in the hypothalamus to identify novel genes dysregulated in a rodent model of narcolepsy (orexin–ataxin-3 mice), as these could be the target of autoimmune attack or modulate orexin gene expression. They found a novel gene which colocalized in orexin cells named insulin-like growth factor binding protein 3 (IGFBP3) that was dysregulated in the orexin–ataxin-3 mice and colocalized in the remaining orexin neurons. They postulated that excessive IGFBP3 expression might initiate orexin-cell death and cause narcolepsy (Honda et al., 2009).
Almost simultaneously, our group was working with a mouse carrying a deletion of the gene coding for the transcription factor O/E3 (also called ebf2), which is normally highly expressed in the central nervous system and regulates neuronal differentiation in the hypothalamus. While determining the role of O/E3 in olfactory circuit architecture; we observed that these O/E3-null mice presented what appeared to be a sudden and transient episode of loss of muscle tone during an active period of vigilance, which indicated it could be a direct transition from vigilance to REM sleep and that O/E3-null mice might have a narcoleptic phenotype. By means of electrophysiological analysis of the SWC and immunostaining against orexin, we found that O/E3 knockout mice presented direct transitions from vigilance to REM sleep characteristic of a narcoleptic phenotype (Figures 1D,G) and a fragmented SWC (Figure 1F), whereas their wildtype littermates presented a normal sleep pattern (Figure 1E) and normal sleep transitions (from nREM to REM, Figure 1C). Additionally, OE/3-null mice presented a loss of approximately 80% of orexin neurons (Figure 1B) compared to their wildtype littermates (Figure 1A; De la Herrán-Arita et al., 2011).
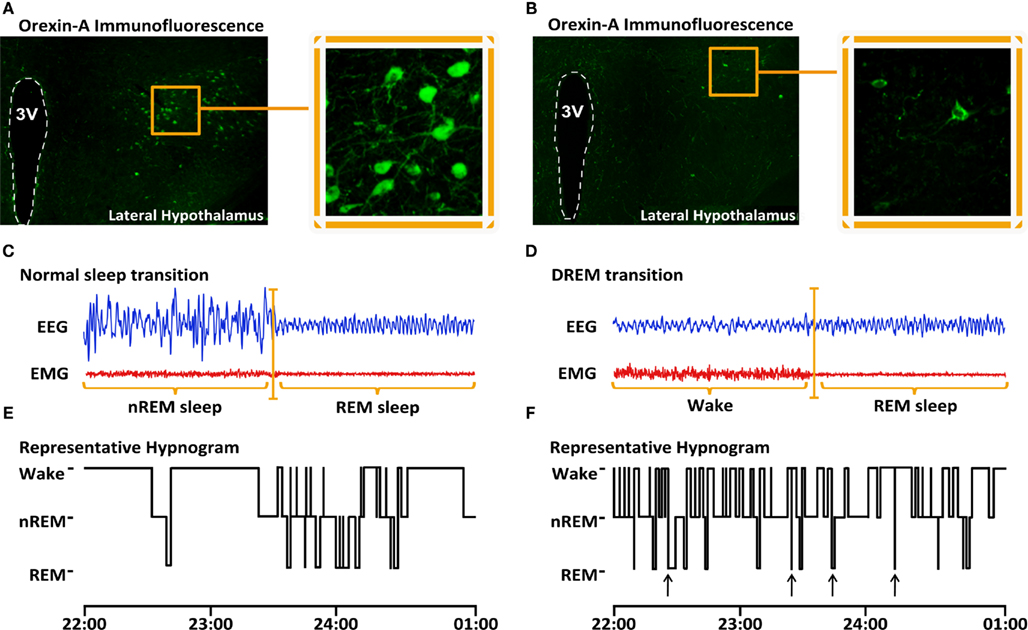
Figure 1. Decreased number of orexin-producing cells in the LH and narcoleptic phenotype of O/E3-null mice. Orexin-A immunostaining of brain coronal sections show orexin-producing cell bodies in the lateral hypothalamic region of Wt animals [(A), bregma −1.70 mm, 3rdV denotes the lumen of the 3rd ventricle). In O/E3-null narcoleptic mice, orexin-A-positive cell bodies are drastically reduced in number and are restricted to a smaller region of the LH (B). Representative EEG/EMG recordings of transitions to REM sleep obtained during the dark period from Wt mice reveal that REM sleep is preceded by nREM sleep (C). Representative recordings from O/E3-null animals show direct transitions to REM sleep (DREM), where REM sleep is immediately preceded by an epoch of wake (D). Representative hypnograms derived from the EEG/EMG recordings show multiple DREM episodes occurring in O/E3-null mice (G), while none can be detected in Wt animals (F).
Worthy of notice, O/E3 was one of the downregulated genes in the narcoleptic orexin–ataxin-3 mice in the work published by Mignot’s group (Honda et al., 2009), confirming that O/E3 is expressed in the LH, where it might regulate prepro-orexin gene expression. By using a sequence alignment approach we were able to identify two conserved sequences within the promoter regions of the prepro-orexin genes of four different species, which appear to correspond to putative olf-1-responsive sequences (De la Herrán-Arita et al., 2011). Olf-1 binding sites have been demonstrated to serve as targets for members of the O/E transcription factor family (Wang et al., 1993). One of the olf-1 sites in the prepro-orexin gene is superposed to the OE1 element, which suggests that O/E3 could directly regulate prepro-orexin gene expression (Figure 2).

Figure 2. Presence of olf-1 (O/E3 binding site) like elements in the promoter region of prepro-orexin genes. Alignment of the upstream (283–300 bp) sequences of the prepro-orexin (prepro-hypocretin) locus (Hcrt) from four separate mammals shows two highly conserved olf-1-like (olf-1/ebf, bold face) elements. The consensus olf-1 sequence, depicted under the alignments, was identified as an OE1 binding site in genes expressed in the olfactory epithelium. One of these olf-1-like elements overlaps with one of the regions (OE1, underlined) that appear to restrict prepro-orexin expression in the lateral hypothalamus. The second olf-1-like element lies at position-7 from the putative transcription start site (arrow indicating direction of transcription). A conserved TATA box can also be identified at position-24 in all these genes. Translation start site (ATG) is indicated in italics.
Taken together, the data demonstrated that O/E3 is the newest discovered member in the regulation of orexin-cell physiology. Interestingly, the narcoleptic phenotype of O/E3-null mice differs from orexin-null (Chemelli et al., 1999) and orexin–ataxin-3 transgenic mice (Hara et al., 2001), since O/E3-null mice have less wakefulness, more nREM sleep and an increase in REM sleep during the dark period (Table 1). These differences could be partially due to the genetic background in which the distinct mutations are maintained: while the orexin-modified mouse lines are C57BL/6 hybrids, crossed to either DBA/1 (orexin–ataxin) or 129/SvEv (orexin knockout) strains, O/E3-null mice are kept within a 129S2/Sv background. It has been demonstrated that even a single chromosome substitution within a given background can alter the SWC parameters of mouse strains (Yang et al., 2009).
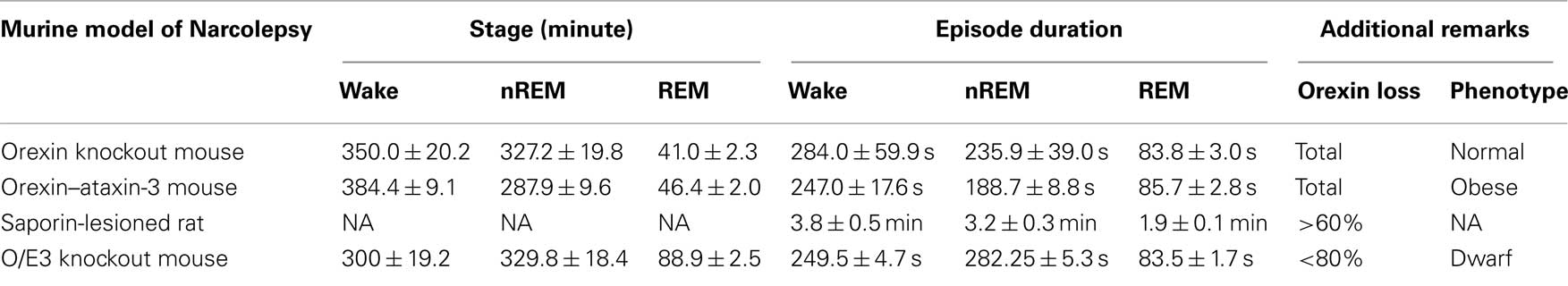
Table 1. Time during the dark phase.
This finding brings us closer to determine the factors involved in the regulation of the SWC and the basis of the narcolepsy–cataplexy syndrome, which in the near future might help the development of a proper therapeutic solution for narcolepsy.
The Struggle Against Orexin Deficit
Narcolepsy is now identified to be a neurodegenerative disease characterized by a massive specific loss of orexin neurons in the LH. Even though the cause of orexin neuronal loss is still unknown, different approaches to revert the absence of these neuropeptides have been attempted.
Gene transfer has proven to be an effective neurobiological tool in a number of neurodegenerative diseases; since narcolepsy is a disease where the orexin precursor prepro-orexin is missing, a viral vector was constructed to transfer the gene to the LH of orexin knockout mice. During the 4-day life-span of the vector, the incidence of cataplexy declined by 60%, and the levels of REM sleep were similar to wildtype mice, indicating that narcoleptic sleep–wake behavior in orexin knockout mice can be improved by targeted gene transfer (Liu et al., 2008).
In another effort to reverse the narcoleptic phenotype in a transgenic animal, Mieda et al. (2004, 2005) produced a transgenic mouse that overexpress a prepro-orexin transgene under the control of a β-actin/cytomegalovirus hybrid promoter (CAG-orexin transgenic mice), and crossed them to orexin–ataxin-3 mice to produce offspring that carry both transgenes (double hemizygous mice). Their results showed that orexin–ataxin-3/CAG–orexin double transgenic littermates exhibited longer, more consolidated bouts of wakefulness and normalized amounts of REM sleep during the dark phase than their orexin–ataxin-3 littermates. Most outstandingly, no direct transitions from vigilance to REM sleep were ever detected in any double transgenic mice. However, chronic overproduction of orexins in CAG–orexin mice caused destabilization of sleep.
In a different experimental approach, they examined whether acute intra-cerebroventricular administrations of orexin peptide might also rescue the narcoleptic phenotype. When orexin–ataxin-3 mice were administered orexin-A peptide, the frequency of arrests and overall time spent in cataplexy were significantly reduced in a 3-h period (Mieda et al., 2004). The result was later confirmed by our research in a similar experiment where O/E3-null mice were given the same dose of orexin-A peptide (De la Herrán-Arita et al., 2011), however, O/E3-null mice presented a total REM sleep abolition, probably due to the different genetic background in which the distinct mutations were maintained. Thus, administration of orexin-A is sufficient to acutely suppress behavioral arrests in narcoleptic mice, but a simpler method for administration should be developed. With these results, it is demonstrated that high level of orexin peptides, as well as a lack of orexins, results in an inadequate regulation of the SWC.
Given that narcolepsy is secondary to a loss of orexin-producing neurons, a logical inference would be to replace the missing neurons in order to reverse the symptoms. Following that thought, our group decided to evaluate the survival of orexin neuron grafts, based on the premise that studies made by others and us have demonstrated that grafted tissue survives, integrates within the host brain, and provides functional recovery following brain injury in various animal models (Perlow et al., 1979; Drucker-Colín et al., 1984; Björklund and Lindvall, 2000). By analyzing the survival of orexin-containing neurons grafted into the pontine reticular formation, a region traditionally regarded to be essential for REM sleep generation; our group found that orexin-immunoreactive somata with processes and varicosities were present in the graft zone 36 days after the implant (Arias-Carrión et al., 2004). However, efforts should be made to increase survival of these neurons and to evaluate the effect of these grafts in narcoleptic animals.
Discussion
Key elements of narcolepsy have been revealed in the last decade. Sleep research is an intricate area; unraveling and identifying the mechanisms involved in the regulation of the SWC has been a difficult attempt, due its nature as an intricate system.
Identifying the cause of a sleep disorder is a complex task. Narcolepsy may be one of the few neurological disorders in which the pathological process has been identified.
A giant leap in the way to unravel the mystery of this disease was taken with the finding of orexin peptides and its association with SWC regulation. Novel evidence demonstrates that impaired orexin signaling is the major, if not the sole cause of the pathogenesis of Narcolepsy, thus contributing to the idea that it is actually a neurodegenerative disease.
Clearly, alterations in the orexin system are fundamental for this disorder. Nonetheless, clarification of the mechanisms responsible for the loss of orexinergic neurons still needs to be determined; this may shed light on new tools for diagnosing and treating this disease and will have important implications for the comprehension of the orexin system and analysis of human narcolepsy. Narcoleptic animal models will aid in solving this dilemma, the variations between the murine narcolepsy models offer us a broad perspective of the Narcolepsy syndrome. The difference in the time spent in each stage of the SWC in these narcoleptic animals is caused by exclusive alterations of the orexinergic circuit. This divergence opens new ways to fully understand how the orexinergic circuit is formed and provides the means for a deeper analysis of the genes that are altered in narcolepsy or implicated in orexin regulation.
Genetic profile in narcolepsy may have implications for future therapies, which might involve orexin gene therapy, transplantation of orexin neurons, stem cell precursors, or engineered cells to produce orexin peptides. Moreover, narcolepsy can be added to the list of the few neurologic disorders which arise from the selective loss of a single population of neurons.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by UNAM-DGAPA-PAPIIT grants IN225209 to R. Drucker-Colín. We would like to thank Diana Millán-Aldaco, Marcela Palomero-Rivero, Teresa Torres-Peralta, and Francisco Pérez-Eugenio for their technical assistance and support in the reviewing of this manuscript. We would also like to thank the Doctorado en Ciencias Biomédicas of the Universidad Nacional Autónoma de México and the DGIP of the Universidad Autónoma de Sinaloa for their support.
References
Alam, M. N., Gong, H., Alam, T., Jaganath, R., McGinty, D., and Szymusiak, R. (2002). Sleep-waking discharge patterns of neurons recorded in the rat perifornical lateral hypothalamic area. J. Physiol. 538, 619–631.
Aldrich, M. S. (1996). The clinical spectrum of narcolepsy and idiopathic hypersomnia. Neurology 46, 393–401.
Anic-Labat, S., Guilleminault, C., Kraemer, H. C., Meehan, J., Arrigoni, J., and Mignot, E. (1999). Validation of a cataplexy questionnaire in 983 sleep-disorders patients. Sleep 22, 77–87.
Arias-Carrión, O., Murillo-Rodriguez, E., Xu, M., Blanco-Centurion, C., Drucker-Colín, R., and Shiromani, P. J. (2004). Transplantation of hypocretin neurons into the pontine reticular formation: preliminary results. Sleep 27, 1465–1470.
Aserinsky, E., and Kleitman, N. (1953). Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science 118, 273–274.
Aston-Jones, G., and Bloom, F. E. (1981). Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1, 876–886.
Bayer, L., Eggermann, E., Serafin, M., Saint-Mleux, B., Machard, D., Jones, B., and Mühlethaler, M. (2001). Orexins (hypocretins) directly excite tuberomammillary neurons. Eur. J. Neurosci. 14, 1571–1575.
Björklund, A., and Lindvall, O. (2000). Cell replacement therapies for central nervous system disorders. Nat. Neurosci. 3, 537–544.
Bourgin, P., Huitrón-Résendiz, S., Spier, A. D., Fabre, V., Morte, B., Criado, J. R., Sutcliffe, J. G., Henriksen, S. J., and de Lecea, L. (2000). Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J. Neurosci. 20, 7760–7765.
Broughton, R., Dunham, W., Newman, J., Lutley, K., Duschesne, P., and Rivers, M. (1998). Ambulatory 24 hour sleep-wake monitoring in narcolepsy-cataplexy compared to matched controls. Electroencephalogr. Clin. Neurophysiol. 70, 473–481.
Brown, R. E., Sergeeva, O., Eriksson, K. S., and Haas, H. L. (2001). Orexin A excites serotonergic neurons in the dorsal raphe nucleus of the rat. Neuropharmacology 40, 457–459.
Butler, A. B., and Hodos, W. (1996). The Reticular Formation. Comparative Vertebrate Neuroanatomy. Evolution and Adaptation. New York: Wiley-Liss, 165
Chemelli, R. M., Sinton, C. M., and Yanagisawa, M. (2000). Polysomnographic characterization of orexin-2 receptor knockout mice. Sleep 23, 296–297.
Chemelli, R. M., Willie, J. T., Sinton, C. M., Elmquist, J. K., Scammell, T., Lee, C., Richardson, J. A., Williams, S. C., Xiong, Y., Kisanuki, Y., Fitch, T. E., Nakazato, M., Hammer, R. E., Saper, C. B., and Yanagisawa, M. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451.
De la Herrán-Arita, A. K., Zomosa-Signoret, V. C., Millán-Aldaco, D. A., Palomero-Rivero, M., Guerra-Crespo, M., Drucker-Colín, R., and Vidaltamayo, R. (2011). Aspects of the narcolepsy–cataplexy syndrome in O/E3 (Ebf2)-null mutant mice. Neuroscience. doi: 10.1016/j.neuroscience.2011.03.029. [Epub ahead of print]
de Lecea, L., Kilduff, T. S., Peyron, C., Gao, X., Foye, P. E., Danielson, P. E., Fukuhara, C., Battenberg, E. L., Gautvik, V. T., Bartlett, F. S. II, Frankel, W. N., van den Pol, A. N., Bloom, F. E., Gautvik, K. M., and Sutcliffe, J. G. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 322–327.
Drucker-Colín, R. (1973). Crossed perfusion of a sleep inducing brain tissue substance in conscious cats. Brain Res. 56, 123–134.
Drucker-Colín, R., Aguilar-Roblero, R., García-Hernández, F., Fernández-Cancino, F., and Bermúdez-Rattoni, F. (1984). Fetal suprachiasmatic nucleus transplants: diurnal rhythm recovery of lesioned rats. Brain Res. 311, 353–357.
Drucker-Colín, R., Rojas-Ramírez, J. A., Vera-Trueba, J., Monroy-Ayala, G., and Hernandez-Peón, R. (1970). Effect of crossed-perfusion of the midbrain reticular formation upon sleep. Brain Res. 23, 269–273.
Estabrooke, I. V., McCarthy, M. T., Ko, E., Chou, T. C., Chemelli, R. M., Yanagisawa, M., Saper, C. B., and Scammell, T. E. (2001). Fos expression in orexin neurons varies with behavioral state. J. Neurosci. 21, 1656–1662.
Gerashchenko, D., Kohls, M. D., Greco, M., Waleh, N. S., Salin-Pascual, R., Kilduff, T. S., Lappi, D. A., and Shiromani, P. J. (2001). Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J. Neurosci. 21, 7273–7283.
Gnadt, J. W., and Pegram, G. V. (1986). Cholinergic brainstem mechanisms of REM sleep in the rat. Brain Res. 384, 29–41.
Guilleminault, C., and Anognos, A. (2000). “Narcolepsy,” in Principles and Practice of Sleep Medicine, eds M. H. Kryger, T. Roth, and W. C. Dement (Philadelphia: W.B. Saunders), 676–686.
Guilleminault, C., and Gelb, M. (1995). Clinical aspects and features of cataplexy. Adv. Neurol. 67, 65–77.
Hagan, J. J., Leslie, R. A., Patel, S., Evans, M. L., Wattam, T. A., Holmes, S., Benham, C. D., Taylor, S. G., Routledge, C., Hemmati, P., Munton, R. P., Ashmeade, T. E., Shah, A. S., Hatcher, J. P., Hatcher, P. D., Jones, D. N., Smith, M. I., Piper, D. C., Hunter, A. J., Porter, R. A., and Upton, N. (1999). Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. U.S.A. 96, 10911–10916.
Hallanger, A. E., Levey, A. I., Lee, H. J., Rye, D. B., and Wainer, B. H. (1987). The origins of cholinergic and other subcortical afferents to the thalamus in the rat. J. Comp. Neurol. 262, 105–124.
Hara, J., Beuckmann, C. T., Nambu, T., Willie, J. T., Chemelli, R. M., Sinton, C. M., Sugiyama, F., Yagami, K., Goto, K., Yanagisawa, M., and Sakurai, T. (2001). Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354.
Harsh, J., Peszka, J., Hartwig, G., and Mitler, M. (2000). Night-time sleep and daytime sleepiness in narcolepsy. J. Sleep Res. 9, 309–316.
Hobson, J. A., McCarley, R. W., and Wyzinski, P. W. (1975). Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science 189, 55–58.
Holmes, C. J., and Jones, B. E. (1994). Importance of cholinergic, GABAergic, serotonergic and other neurons in the medullary reticular formation for sleep-wake states studied by cytotoxic lesions in the cat. Neuroscience 62, 1179–1200.
Honda, M., Eriksson, K. S., Zhang, S., Tanaka, S., Lin, L., Salehi, A., Hesla, P. E., Maehlen, J., Gaus, S. E., Yanagisawa, M., Sakurai, T., Taheri, S., Tsuchiya, K., Honda, Y., and Mignot, E. (2009). IGFBP3 colocalizes with and regulates hypocretin (orexin). PLoS ONE 4, e4254. doi: 10.1371/journal.pone.0004254
Jones, B. E. (2008). Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann. N. Y. Acad. Sci. 1129, 26–34.
Lin, L., Faraco, J., Kadotani, H., Rogers, W., Lin, X., Qui, X., de Jong, P., Nishino, S., and Mignot, E. (1999). The REM sleep disorder canine narcolepsy is caused by a mutation in the hypo-cretin (orexin) receptor gene. Cell 98, 365–376.
Liu, M., Thankachan, S., Kaur, S., Begum, S., Blanco-Centurion, C., Sakurai, T., Yanagisawa, M., Neve, R., and Shiromani, P. J. (2008). Orexin (hypocretin) gene transfer diminishes narcoleptic sleep behavior in mice. Eur. J. Neurosci. 7, 1382–1393.
Longstreth, W. T. Jr., Koepsell, T. D., Ton, T. G., Hendrickson, A. F., and van Belle, G. (2007). The epidemiology of narcolepsy. Sleep 30, 13–26.
Marcus, J. N., Aschkenasi, C. J., Lee, C. E., Chemelli, R. M., Saper, C. B., Yanagisawa, M., and Elmquist, J. K. (2001). Differential expression of orexin receptors 1 and 2 in the rat brain. J. Comp. Neurol. 435, 6–25.
McCormick, D. A. (1992). Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog. Neurobiol. 39, 337–388.
Mieda, M., Willie, J. T., Hara, J., Sinton, C. M., Sakurai, T., and Yanagisawa, M. (2004). Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc. Natl. Acad. Sci. U.S.A. 13, 4649–4654.
Mieda, M., Willie, J. T., and Sakurai, T. (2005). Rescue of narcoleptic orexin neuron-ablated mice by ectopic overexpression of orexin peptides. Contemporary Clin. Neurosci. V 359–366.
Mignot, E., Lin, L., Rogers, W., Honda, Y., Qiu, X., Lin, X., Okun, M., Hohjoh, H., Miki, T., Hsu, S., Leffell, M., Grumet, F., Fernandez-Vina, M., Honda, M., and Risch, N. (2001). Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am. J. Hum. Genet. 68, 686–699.
Moruzzi, G., and Magoun, H. W. (1949). Brain stem reticular formation and activation of the EEG. Electroencephalogr. Clin. Neurophysiol. 1, 455–473.
Nishino, S., Ripley, B., Overeem, S., Lammers, G. J., and Mignot, E. (2000). Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355, 39–40.
Olszewski, J., and Baxter, D. (1954). Cytoarchitecture of the Human Brain Stem. Philadelphia: Lippincot
Paut-Pagano, L., Valatx, J. L., Kitahama, K., and Jouvet, M. (1989). Prolactin-secreting neurons in the dorsolateral hypothalamus in Sprague-Dawley rats. C. R. Acad. Sci. III, Sci. Vie 309, 369–376.
Perlow, M., Freed, W. J., Hoffer, B., Seiger, A., Olson, L., and Wyatt, R. J. (1979). Brain grafts reduce motor abnormalities produced by destruction of nigrostriatal dopamine system. Science 204, 643–647.
Peyron, C., Faraco, J., Rogers, W., Ripley, B., Overeem, S., Charnay, Y., Nevsimalova, S., Aldrich, M., Reynolds, D., Albin, R., Li, R., Hungs, M., Pedrazzoli, M., Padigaru, M., Kucherlapati, M., Fan, J., Maki, R., Lammers, G. J., Bouras, C., Kucherlapati, R., Nishino, S., and Mignot, E. (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997.
Peyron, C., Tighe, D. K., van den Pol, A. N., de Lecea, L., Heller, H. C., Sutcliffe, J. G., and Kilduff, T. S. (1998). Neurons containing hypocretin (orexin) project to multiple neuronal systems. J. Neurosci. 18, 9996–10015.
Risold, P. Y., Griffond, B., Kilduff, T. S., Sutcliffe, J. G., and Fellmann, D. (1999). Preprohypocretin (orexin) and prolactin-like immunoreactivity are coexpressed by neurons of the rat lateral hypothalamic area. Neurosci. Lett. 259, 153–156.
Rubboli, G., d’Orsi, G., Zaniboni, A., Gardella, E., Zamagni, M., Rizzi, R., Meletti, S., Valzania, F., Tropeani, A., and Tassinari, C. A. (2000). A video-polygraphic analysis of the cataplectic attack. Clin. Neurophysiol. 111(Suppl. 2), S120–S128.
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., Williams, S. C., Richarson, J. A., Kozlowski, G. P., Wilson, S., Arch, J. R., Buckingham, R. E., Haynes, A. C., Carr, S. A., Annan, R. S., McNulty, D. E., Liu, W. S., Terrett, J. A., Elshourbagy, N. A., Bergsma, D. J., and Yanagisawa, M. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585.
Scammell, T. E. (2003). The neurobiology, diagnosis, and treatment of narcolepsy. Ann. Neurol. 53, 154–166.
Siegel, J. M., Nienhuis, R., Fahringer, H. M., Paul, R., Shiromani, P., Dement, W. C., Mignot, E., and Chiu, C. (1991). Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science 252, 1315–1318.
Steininger, T. L., Alam, M. N., Gong, H., Szymusiak, R., and McGinty, D. (1999). Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 840, 138–147.
Thannickal, T. C., Moore, R. Y., Nienhuis, R., Ramanathan, L., Gulyani, S., Aldrich, M., Cornford, M., and Siegel, J. M. (2000). Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474.
Wang, M. M., Tsai, R. Y., Schrader, K. A., and Reed, R. R. (1993). Genes encoding components of the olfactory signal transduction cascade contain a DNA binding site that may direct neuronal expression. Mol. Cell. Biol. 13, 5805–5813.
Willie, J. T., Chemelli, R. M., Sinton, C. M., Tokita, S., Williams, S. C., Kisanuki, Y. Y., Marcus, J. N., Lee, C., Elmquist, J. K., Kohlmeier, K. A., Leonard, C. S., Richardson, J. A., Hammer, R. E., and Yanagisawa, M. (2003). Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of Non-REM and REM sleep regulatory processes. Neuron 38, 715–730.
Yang, H. S., Vitaterna, M. H., Laposky, A. D., Shimomura, K., and Turek, F. W. (2009). Genetic analysis of daily physical activity using a mouse chromosome substitution strain. Physiol. Genomics 39, 47–55.
Keywords: narcolepsy, orexins, hypothalamus, sleep
Citation: De la Herrán-Arita AK, Guerra-Crespo M and Drucker-Colín R (2011) Narcolepsy and orexins: an example of progress in sleep research. Front. Neur. 2:26. doi: 10.3389/fneur.2011.00026
Received: 24 February 2011; Paper pending published: 08 March 2011;
Accepted: 04 April 2011; Published online: 18 April 2011.
Edited by:
Michael J. Thorpy, Albert Einstein College of Medicine of Yeshiva University, USAReviewed by:
Michael J. Thorpy, Albert Einstein College of Medicine of Yeshiva University, USAThomas C. Thannickal, University of California Los Angeles, USA
Copyright: © 2011 De la Herrán-Arita, Guerra-Crespo and Drucker-Colín. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: René Drucker-Colín, Departamento de Neuropatología Molecular, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Apdo. Postal 70-600, 04510 México D.F., México. email:ZHJ1Y2tlckBzZXJ2aWRvci51bmFtLm14