


- 1 Department of Neurology, Memory and Aging Center, University of California at San Francisco, San Francisco, CA, USA
- 2 Brazilian Aging Brain Study Group-LIM 22, Department of Pathology, University of São Paulo Medical School, São Paulo, Brazil
- 3 Dr. Senckenberg Chronomedical Institute, Goethe-University, Frankfurt/Main, Germany
- 4 Laboratory of Morphological Brain Research, Psychiatrics Clinics, University of Wuerzburg, Wuerzburg, Germany
The most frequent neurodegenerative diseases (NDs) are Alzheimer’s disease (AD), Parkinson’s disease (PD), and frontotemporal lobar degeneration associated with protein TDP-43 (FTLD–TDP). Neuropathologically, NDs are characterized by abnormal intracellular and extra-cellular protein deposits and by disease-specific neuronal death. Practically all terminal stages of NDs are clinically associated with dementia. Therefore, major attention was directed to protein deposits and neuron loss in supratentorial (telencephalic) brain regions in the course of NDs. This was also true for PD, although the pathological hallmark of PD is degeneration of pigmented neurons of the brainstem’s substantia nigra (SN). However, PD pathophysiology was explained by dopamine depletion in the telencephalic basal ganglia due to insufficiency and degeneration of the projection neurons located in SN. In a similar line of argumentation AD- and FTLD-related clinical deficits were exclusively explained by supratentorial allo- and neo-cortical laminar neuronal necrosis. Recent comprehensive studies in AD and PD early stages found considerable and unexpected involvement of brainstem nuclei, which could have the potential to profoundly change our present concepts on origin, spread, and early clinical diagnosis of these diseases. In contrast with PD and AD, few studies addressed brainstem involvement in the course of the different types of FTLD–TDP. Some of the results, including ours, disclosed a higher and more widespread pathology than anticipated. The present review will focus mainly on the impact of brainstem changes during the course of the most frequent NDs including PD, AD, and FTLD–TDP, with special emphasis on the need for more comprehensive research on FTLDs.
Introduction
Increased longevity in industrial and post-industrial countries is inevitably associated with an increased prevalence of neurodegenerative diseases (NDs). Clinically, NDs implicate devastating conditions causing enormous suffering of the affected individuals, and they impose a heavy burden on the families and society. Therefore, considerable efforts are underway to improve diagnosis and to gain insight into the etiology and pathogenesis of these diseases.
Neurodegenerative diseases share common characteristics: (1) non-random anatomical progression associated with neuron loss, (2) disease-specific intra- and extra-cellular protein deposits. The most frequent NDs are: Alzheimer’s disease (AD), Parkinson’s disease (PD), and frontotemporal lobar degeneration (FTLD). AD is characterized by intracellular tau and extra-cellular amyloid-β proteins, PD by intracellular positive α-synuclein deposits, and FTLD by deposits of different proteins. Over 50% of the FTLD cases are characterized by TAR DNA-binding protein-43 (TDP-43) positive inclusion.
The relationship between these deposits and neuronal loss is a matter of dispute. However, most neuropathologists agree that deposits are, at least, surrogate markers for specific NDs, since mild to moderate neuronal loss is difficult to assess in routine paraffin sections.
Progress in immunohistochemical methods, standardization of the protocols, as well as using thick histological sections are mandatory for establishing staging systems that describe the progression of the disease and correlate staging with clinical manifestations. The final goal behind these efforts is to uncover the initial stages in individual NDs in order to develop therapies in the earlier stages of the diseases and to validate therapeutic interventions during disease progression.
Several staging systems have already been developed and are in use for AD and PD (Braak and Braak, 1991; Mirra et al., 1991; National Institute on Aging–Reagan Institute Working Group, 1997; Braak et al., 2003). There is no universally accepted staging system for FTLDs yet, since this group of diseases has been described only recently. Recent results indicate that the currently used staging systems do not cover all aspects of central nervous system (CNS) pathology. This is especially true for the involvement of the brainstem in the course of AD and FTLD–TDP.
This review will focus on the characteristic, non-random anatomical sequence of lesions in the early stages of AD, PD, and FTLD with special emphasis on the brainstem. In addition, we will point out gaps in our knowledge on brainstem involvement in these conditions.
The Human Brainstem
The brainstem is the central axis of the brain, and is considered an intermediate regulatory system that links the spinal cord with the superordinate prosencephalon. It represents an integration center of responses directly aimed at the survival of the individual by means of its somato-, branchio-, and visceromotor nuclei. The brainstem is divided rostrocaudally into midbrain, metencephalon (pons and cerebellum), and medulla oblongata (Figure 1). The brainstem is furthermore subdivided from dorsal to ventral into a roof, a tegmentum, and a neencephalic basal part. The roof consists of the midbrain quadrigeminal lamina and the metencephalic cerebellum. The tegmentum represents the most conservative part of the brainstem, comprising the sensory and motor nuclei of the cranial nerves III through XII and the reticular formation. The pyramidal tract, pontine nuclei, and the inferior olives compose the neencephalic parts of the brainstem, which have considerably enlarged relatively and absolutely in primates. Neencephalic components have attained their greatest expansion in humans. The reticular formation of the brainstem is an extended ill-defined network of interconnected neurons comprising the complete spectrum of neurons, from small- to giant-sized. In terms of comparative anatomy, it is an old center of coordination that integrates motor and sensory nuclei with cortical and spinal cord afferents. Some of the reticular formation nuclei are believed to be involved in primary homeostatic mechanisms, including gastrointestinal regulation, pain perception, mood control, and sleep–wake cycles. Consequently, the reticular formation is included into the greater limbic system (Nieuwenhuys et al., 2008). In addition, specific reticular formation nuclei produce aminergic transmitters such as serotonin in the raphe nuclei, norepinephrine in the locus ceruleus (LC), and dopamine in the substantia nigra (SN). The aminergic nuclei project diffusely into the brain. Of particular interest, aminergic nuclei are believed to transmit information via “volume transmission.” Volume transmission suggests that the terminals of the respective neuronal axons do not have a classical synaptic conformation. Therefore, the neurotransmitter is released into the intercellular space and can affect a higher number of receptors (Mountcastle, 1998). This kind of modulation is likely to impact several different areas of the brain at once. No less important, cholinergic cell groups are also found in the brainstem (Mesulam et al., 1983). Thus, the brainstem represents an intersection where mind and body could interact, making it paramount to place it within an etiopathogenic context.
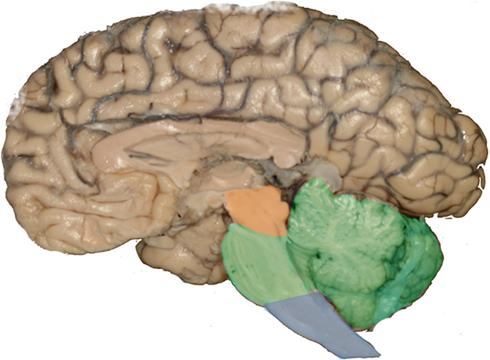
Figure 1. Human brainstem. Sagittal view. The brainstem is divided rostrocaudally into midbrain (orange), metencephalon (green), and medulla oblongata (blue).
Most of the brainstem nuclei are long and narrow. Therefore, the use of conventional horizontally cut histological slides may cause the false impression that these nuclei are inconspicuous. However, a closer look at 3D reconstructions invariably eliminates this biased impression (Grinberg et al., 2010; Figure 2).
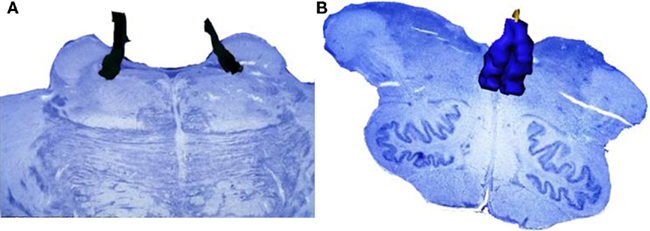
Figure 2. Most of the brainstem nuclei are narrow in a dorsoventral aspect, however, they extend for a considerable distance in a rostrocaudal direction. Routinely used histological sections are cut in a horizontal plan and just display the dorsoventral profile of these nuclei causing the impression that the these nuclei are inconspicuous. However, 3D reconstructions of the nuclei demonstrate their real significant volume. (A) Horizontally cut section at the level of the rostral pons. The black structure is a reconstruction of the locus ceruleus. Note the small profile at the intersection between the reconstruction and the horizontally cut histological slide. (B) Horizontally cut section at the level of the medulla oblongata. The blue structure is a reconstruction of the raphe magnus nucleus. As in the previous example, this structure looks inconspicuous in a horizontally cut section.
Brainstem in Early Stages of Parkinson’s Disease
Parkinson’s disease is the second most common neurodegenerative disorder after AD, with an estimated prevalence of 1% in people over 60 years of age. PD has been considered one of the best examples of pure movement-disorder for almost 150 years. Its hallmark is the degeneration of the pigmented neurons of the midbrain part of the brainstem. The pigmented neurons of the SN are supplying the basal ganglia with dopamine. From 30 to 70% of neuronal loss is considered to cause clinical relevant dopamine depletion in basal ganglia circuits (Cheng et al., 2010). Therefore, although the SN belongs to the brainstem, the disease manifestation was mainly associated to supratentorial disturbances.
Many non-motor symptoms were described in PD patients, but they were mostly regarded as unassociated to PD lesions in the brain. This scenario changed dramatically from 2003, when Braak et al. (2003) put findings in context by conflating and extending various previous studies in PD. They provided a chronological scheme for the spread of Lewy bodies during the disease course, and demonstrated that several brainstem nuclei are involved by PD-type changes even before the SN (Braak et al., 2003). The dorsal motor nucleus of the vagus (DNX), located in the caudal medulla oblongata, and the olfactory bulb, are the two areas thought to be first affected in the brain (Braak et al., 2003). In addition, all serotoninergic nuclei, select cholinergic, noradrenergic, some precerebellar, and cranial nerve nuclei, and the reticular formation, are vulnerable to PD. It is now accepted that the SN is affected only in stage III, and the first motor symptoms are usually detected in stage IV, by which time most of the SN has already degenerated. In addition, they showed that a number of non-vulnerable neurons keep their integrity throughout the disease. It suggests that neuronal damage is not arbitrary in PD (Braak et al., 2003), although the reason for this selective vulnerability is still unclear.
After these findings, it was possible to correlate many of the non-motor, non-specific symptoms – such as autonomic dysfunction, depression, and sleep disorders – to the biological changes occurring in the brainstem (Grinberg et al., 2010). For instance, the general visceroefferent DNX controls the postganglionic parasympathetic nerve cells of the enteric and cardiac system. Symptoms such as constipation, dysphagia, and drooling, which may precede the motor PD features for years (Pfeiffer, 2003; Kaufmann et al., 2004), are believed to be partly caused by DNX degeneration.
Another example was the conclusion that a specific and devastating sleep disorder, the REM-sleep behavior disorder (RBD), may arise as a direct manifestation of PD damage in the chief brainstem nuclei (Ondo et al., 2001). Actually, RBD is likely to be a better marker of a synucleinopathy than parkinsonism is. RBD is characterized by the loss of normal skeletal muscle atonia during REM-sleep, with prominent motor activity and dreaming enactment (Boeve et al., 2007). The cholinergic neurons of the pedunculopontine nucleus, degenerated in PD stage III, are directly involved in promoting REM-sleep. In addition, the LC and the dorsal raphe (DR) nucleus, affected respectively in stages II and III, also participate in the regulation of sleep (Saper et al., 2001), whereas the caudal raphe participates in suppressing the locomotor activity during sleep (Lai and Siegel, 1999). For additional information about the brainstem nuclei involved in early PD stages and their clinical counterpart (see Grinberg et al., 2010).
These findings have dramatically changed our view, and PD was redefined in the last few years as a multisystem disorder involving general visceromotor system, reticular formation, and isodendritic brainstem core and thus, several neurotransmitter systems, and it is characterized by motor and non-motor symptoms. Patients now benefit from improved management of formerly neglected symptoms (Obeso et al., 2010).
However, the precise pathways linking the chronologically and topographically early affected centers with later affected cortical and subcortical structures need to be unraveled. What is the relationship between peripheral ganglia α-synucleinopathy and the involvement of the DNX and intermediate reticular formation? Is there a direct pathway involved in propagating α-synuclein deposits between the olfactory bulb and the DNX, or do they suffer independent involvement? In case there is a direct pathway, is the spread antero- or retro-grade? Which pathways are related to involvement of the isodendritic core (IC), including the SN, LC, and DR nucleus?
Brainstem in Early Stages of Alzheimer’s Disease
Like PD, AD is a progressive and, so far, incurable dementia. It accounts for 50 to 70% of all dementia cases. In the same fashion as all NDs, AD is characterized neuropathologically by accumulative lesions (protein deposits) and degenerative lesions (neuronal loss). AD accumulative lesions constitute its two neuropathological hallmarks: (1) accumulation of extra-cellular β-amyloid in the form of neuritic plaques, and (2) intracellular accumulation of phosphorylated-tau in the form of neurofibrillary tangles (NFT), neuropil threads, and dystrophic neurites. Cognitive decline in AD correlates best with neuronal loss, followed by NFT spread, whereas the neuritic plaque burden is only slightly associated with cognitive decline (Berg et al., 1998; Giannakopoulos et al., 2003).
As with Lewy bodies in PD, NFT-pathology in AD follows a characteristic, non-random anatomical progression. This stereotypical progression, demonstrated by detailed neuropathological studies, is the base for the currently used Braak and Braak staging system (Braak and Braak, 1991). According to this system, NFTs progression can be staged in six grades. NFTs are confined to the transentorhinal region in stage I, and they will reach the primary neocortical areas in stage VI. However, the Braak and Braak staging is confined to allo- and neo-cortical fields, and thus does not address brain changes in the brainstem. Probably, this may partly explain why AD has been featured as a basically supratentorial disease. Numerous studies confirmed regular brainstem involvement in AD, but this involvement was mostly regarded as secondary to supratentorial changes (Hardy et al., 1986).
Finally in 2009, using a postmortem brain collection focused on normal aging, it was demonstrated that the midbrain DR nucleus shows NFTs before the Braak and Braak I’s transentorhinal region. In this study of 118 cases, 100% of the 80 subjects in which transentorhinal region was affected by NFTs also showed NFTs in the DR. In addition, 22% of the 38 subjects who had no cortical NFTs (Braak 0) showed NFTs in the DR (Grinberg et al., 2009). Interestingly, the NFT burden in the DR is proportional to the Braak and Braak stage (Rub et al., 2000; Grinberg et al., 2009). This finding suggests that brainstem areas are affected by AD before the supratentorial regions. The demonstration that the DR is likely to be affected by AD in its earliest stages drew new attention to the brainstem, and motivated new studies and revived old hypotheses (Simic et al., 2009). In addition, because the DR produces a great part of the brain serotonin, its degeneration may explain the depressive symptoms experienced by many AD patients, years before the cognitive changes’ onset (Panza et al., 2010).
However, the DN is not the sole brainstem nucleus involved in AD early stages. The DN, together with the LC, the SN, and the telencephalic cholinergic nucleus basalis of Meynert (NbM) form the IC (Figure 3), an interconnected network, whose neurons share morphological features, such as big somata, overlapping dendritic fields, predominantly poorly myelinated axons that extend to distant projection sites, and aminergic/cholinergic volume transmission. The IC is involved in modulation of many basic physiologic processes (Ramon-Moliner and Nauta, 1966) and it is intimately connected with those areas of the cerebral cortex that undergo early neurofibrillary changes in AD (Insausti et al., 1987). Several studies reported the early existence of NFTs and/or neuronal loss in the LC, the SN, and the cholinergic NbM (Ohm et al., 1989; Sassin et al., 2000; Lyness et al., 2003; Geula et al., 2008). However, very few previous studies addressed these four nuclei as a network, which is necessary for effectively studying possible interactions of all major pathogenic factors of this process in the same brain (Simic et al., 2009). The great majority of the studies focused either on neuronal loss or on protein inclusion. Although heavily explored, the relationship between neuronal loss and NFT- and NP-burden in AD is yet to be settled. Recent evidence suggests that this relationship is not as simple as it was originally considered (Arendt et al., 1985; Cras et al., 1995; Kril et al., 2002). For instance, out of the IC nuclei, the NbM and LC show the greatest neuronal loss, but not the greatest inclusion burden (Lyness et al., 2003). Furthermore, there are many disparities among studies on individual nuclei of the IC, which may be attributed to the use of biased morphological methods and the use of outdated pathological criteria (Khachaturian, 1985). This implies that many of erstwhile control cases would be considered early state AD by current standards (see Introduction).
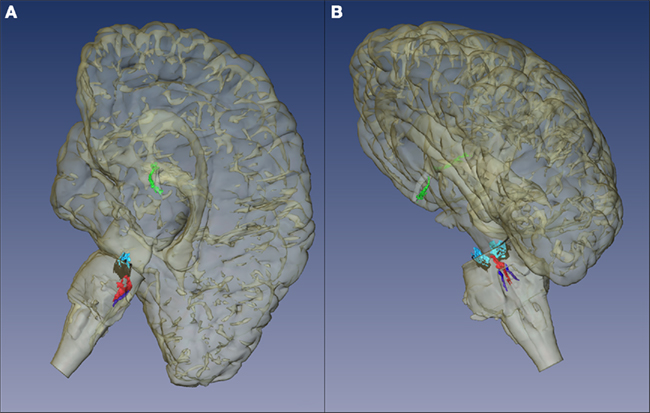
Figure 3. Isodendritic core. The components of this network share morphological features, such as neurons with big somata, overlapping dendritic fields, predominantly poorly myelinated axons that extend to distant projection sites, and aminergic/cholinergic volume transmission. The isodendritic core is involved in modulation of many basic physiologic processes (Ramon-Moliner and Nauta, 1966). This figure shows the representation of the Isodendritic core components: dorsal nucleus of the raphe (red), locus ceruleus (dark blue), substantia nigra (pars compacta in black and pars diffusa in light blue), and the telencephalic cholinergic nucleus basalis of Meynert (green). (A) Medial view. (B) Dorsomedial view.
Other limitations were the lack of age matched control groups, and the use of cases with long postmortem interval, which causes autolysis of the neuronal cells (Lyness et al., 2003). Regardless of the difficulties, due to the evidence of early involvement of the IC in AD, it is worthwhile to make an effort to clarify its role in this disease. Further studies are sought to determine at which AD stage the other components of the IC become affected, and how the involvement of each of these nuclei reflects into the other. If it can be proven that the IC is affected early in AD, strategies to protect these neurons may delay the progression of the disease to supratentorial regions. In addition, this knowledge may help to improve clinicopathological correlations, especially concerning non-specific symptoms seen in AD prodromal stages. Over 90% of AD patients experience behavioral and psychological symptoms before onset of cognitive decline (Devanand et al., 1997), and some of these symptoms may be related to IC degeneration. Even today, the most used drugs for symptomatic treatment of AD, such as acetylcholinesterase inhibitors, aim to compensate for symptoms related to IC abnormalities.
Besides the IC, other brainstem structures were reported as early involved in AD, such as the tegmentopontine reticular nucleus (nucleus of Bechterew), which is part of the premotor oculomotor circuit (Rub et al., 2001). However, further studies are needed to determine how early and consistently these nuclei are affected.
The widely used Braak and Braak staging system gives an impressive example of how pathological intraneuronal tau fibrils will accumulate first in mesocortical, afterward in allocortical, and finally in isocortical neurons in a unvaried and layer-specific manner. Presently we do not know the mechanism that lies behind this systemic spread of pathologic neuronal changes. However, it is likely to use major pathways including the trisynaptic neuronal circuit and cortical primary, unimodal, and polymodal associational layer 3 and layer 5 pyramidal cell interconnections. The emergence of pathological tau fibrils within neurons of these interconnected circuits is reversed compared to the flow of information processing via primary sensory, unimodal, and polymodal association cortices to limbic regions (Mesulam, 1998) and thus, considered to progresses in a feedback manner. At first sight, early Alzheimer’s-related tau pathology in brainstem nuclei does not fit into this concept of telencephalic feedback propagating pathology. However, both the DR nucleus and the LC are connected to the prefrontal cortex, and thus are supplied with highly processed polymodal information. By virtue of these afferents, the brainstem nuclei could represent final targets of information processing. By means of their widespread afferent projections, they are in a position to modulate disparate brain regions including the telencephalon, diencephalon, and the brainstem in a way to generate integrated somatomotor, visceromotor, and endocrine responses. Consequently, the IC represents a major component of limbic brainstem core and paracore zones, as proposed by Nieuwenhuys (1996).
Frontotemporal Dementias
Frontotemporal dementias (FTDs) comprise a major group of incurable and incapacitating NDs. In the last few years, they have been transformed from a secondary entity into a major focus of dementia neuroscience research. They are estimated to affect 400,000 Americans younger than 65 years of age (Maslow, 2006), making it as prevalent as AD in this age group. Furthermore, recent evidence suggests that FTDs are frequent also in older populations (Ratnavalli et al., 2002; Rosso et al., 2003).
The entities belonging to the FTD group have overlapping clinical and pathologic features. They usually present with marked declines in socioemotional functioning, and behavioral or language disturbances (Kramer et al., 2003). The current FTD research criteria define three clinical syndromes: (1) frontotemporal dementia or behavioral variant FTD, (2) semantic dementia or temporal variant FTD, and (3) progressive non-fluent aphasia (Neary et al., 1998). Recently, the definition and diagnostic criteria for the language variants (semantic dementia and progressive non-fluent aphasia) have been revised. The new criteria recognized three subtypes of primary progressive aphasia (PPA): non-fluent or agrammatic variant, semantic variant, and logopenic variant (Gorno-Tempini et al., 2011). In addition, some cases may present as progressive supranuclear palsy (PSP), or corticobasal syndrome, and even concomitant motor neuron disease (Kertesz and Munoz, 2004; Liscic et al., 2008). Traditionally, damage to frontal and temporal lobe cortical regions are accounted as the cause of the majority of the clinical abnormalities characteristic of FTDs. Thus, FTDs would predominantly represent telencephalic NDs.
The FTDs may be underlaid by different neuropathological entities. The overarching name for these neuropathological entities is FTLD. Therefore, FTLD will be used here as a neuropathological term, while FTD is used as a clinical term. All these entities share some neuropathological features, such as vacuolation, gliosis, and neuronal loss of the superficial cortical layers, especially in frontal and temporal areas. However, as in the other NDs, classification of the heterogeneous group of FTLD disorders is based on the protein composition of intracellular abnormal deposits (Cairns et al., 2007). The most frequent form of FTLD is associated with deposits composed of the TDP-43, and it is called FTLD–TDP. Recent evidence suggests that FTLD–TDP can be further subdivided into at least three histologic variants, based on the patterns of TDP-43 immunoreactivity (Mackenzie et al., 2006; Sampathu et al., 2006).
The second most common FTLD form is characterized by phospho-tau deposits, both in neurons and glia. They are called tauopathies, or more recently FTLD-tau (Mackenzie et al., 2010).
The FTD clinical picture is indicative of the affected brain region, either orbitofrontal, dorsolateral prefrontal, temporal, and so on. However, a brain region can be hit by different FTLD entities, and therefore it is not possible to determine the biochemical signature of a FTD case based exclusively on the clinical and imaging features. This review will focus only in FTLD–TDP, the most prevalent form of FTLD.
Brainstem in Early Stages of FTLD–TDP
Macroscopically, FTLD–TDP is characterized by peculiar and asymmetric widened sulci and flattened gyri in the frontal and temporal region. Therefore, FTLD–TDPs were initially considered to be confined to the telencephalic cortex. However, more comprehensive studies have also demonstrated some significant brainstem changes in the FTLD–TDP. These studies were either performed prior to the modern biochemical characterization of FTLDs or using a limited number of cases (Lüers and Spatz, 1957; Jakob, 1960, 1979; Yang and Schmitt, 2001). Degeneration of brainstem nuclei was considered as a consequence of anterograde degeneration due to death of subcortically projecting isocortical pyramidal cells. In contrast to this prevailing conjecture, Poppe and Tennstedt (1964) suggested early brainstem involvement in describing a patient presenting what would be classified as a behavioral variant FTD. Their hypothesis challenged contemporary beliefs. However, for historical and technical reasons it is not possible to reclassify the Poppe and Tennstedt case into the presently used diagnostic system.
So far, only a few recent studies addressed the neuropathological alterations in the brainstem in context with TDP-43. These studies focused mainly on motor-related nuclei. Not surprisingly, most of the changes were found in cases with coincident motor neuron disease (FTLD–MND; Dickson et al., 2007; Pikkarainen et al., 2008; Davidson et al., 2009). Unexpectedly, Davidson et al. (2009) explicitly stated that deposits were absent in the pontine nuclei in all FTLD–TDP subtypes. Other non-motor nuclei were practically neglected in the recent studies.
Regarding neuronal loss in brainstem of FTLD–TDP cases, Yang and Schmitt (2001) compared the amount of neuronal loss and ubiquitin deposits in selected brainstem nuclei of 12 FTLD without tau-positive deposits, 30 AD and 35 controls. They found significant neuronal loss in the DR nucleus and nucleus centralis superior of the pons, but none in the LC in FTLD cases. Deposits were not found in any of the aminergic nuclei examined. Similarly to PD and AD, the serotonergic system is apparently susceptible to FTLD–TDP. At variance with PD and AD, the LC seems to be spared in FTLD–TDP.
In order to fill existing gaps concerning brainstem pathology during the course of all FTLD–TDP subtypes, we conducted a screening study using 14 cases belonging to the UCSF Neurodegenerative Disease Brain Bank. The cases comprised all clinical and pathological spectra of FTLD–TDP (Table 1). Five TDP-43 immunostained horizontal sections at the level of the midbrain, rostral pons, caudal pons, and medulla oblongata were screened in each case. The microscopic assessment was done using a high magnification objective, and covered the whole slide. This strategy aimed to prevent topographical mapping during the screening. The exact location of each inclusion found was recorded with the aid of an optical marker attached to the microscope. All deposits were photographed and categorized. At the end of each screening, a map was generated and used to locate the deposits in each section. Later on, clinical and pathological information were added.
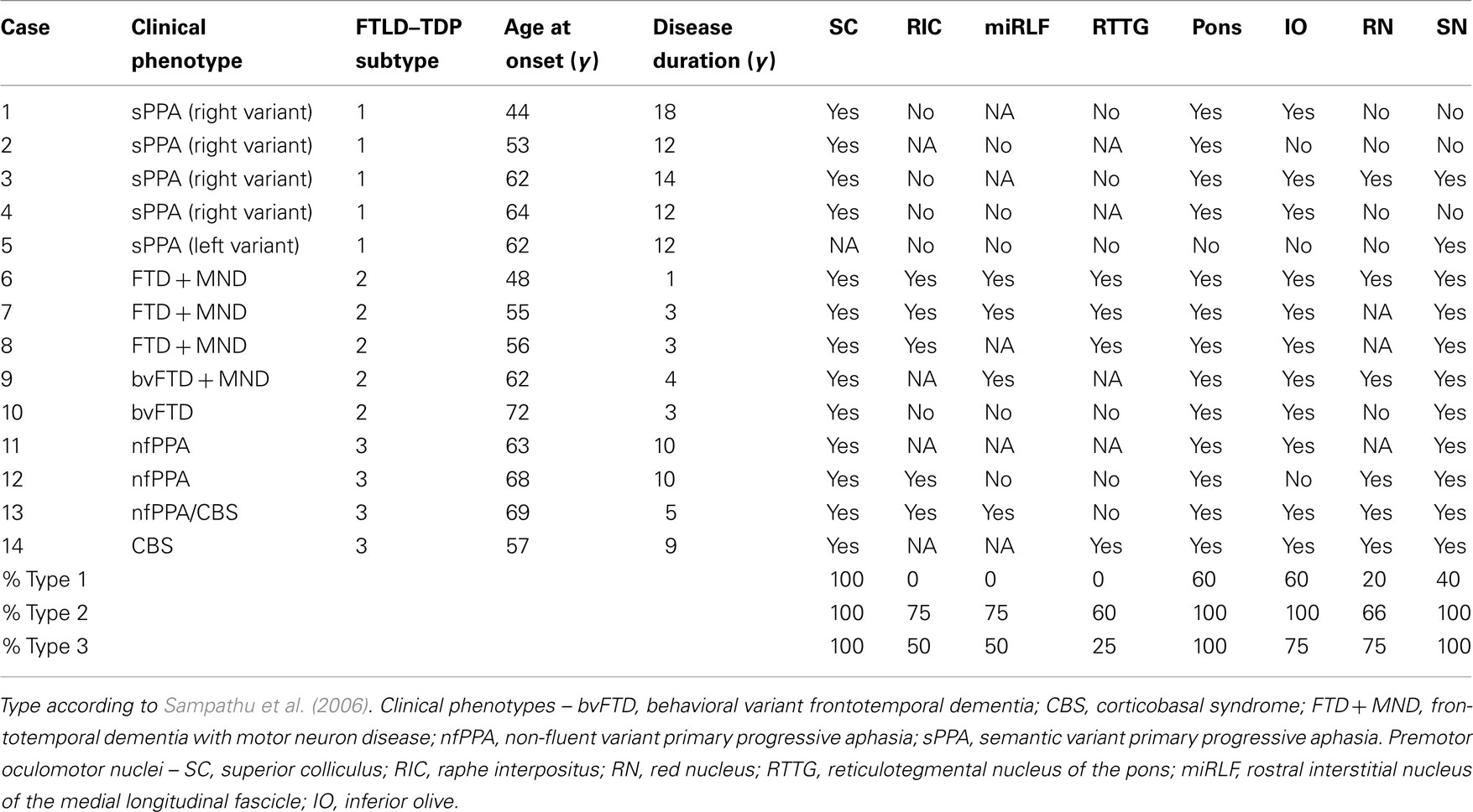
Table 1. Summary of cases screened for FTLD–TDP.
TAR DNA-binding protein-43 deposits were present in the brainstems of all cases (Table 1). Size, shape, and intracellular location of the deposits predicted the individual cortical subtype, suggesting that every FTLD–TDP subtype has a fingerprint also in the brainstem. Strikingly, the superior colliculus (SC) showed deposits in all the cases analyzed. The SC is a layered structure located at the midbrain tectum and it is interconnected with several cortical and subcortical structures. The SC plays several roles in modulating eye movements (e.g., generating the orienting response to visual or auditory stimuli) and is involved in the initiation of saccades. It is also important for the suppression of unwanted saccades when steady fixation is necessary (Nieuwenhuys et al., 2008). In fact, an imaging study by Cardenas et al. (2007) demonstrated atrophy of the midbrain tectum, in which the SC is located, in FTLD cases.
The SC is part of the premotor oculomotor network (Figure 4). This network is mostly located in the brainstem tegmentum, generates and modulates several eye movements, and coordinates the activity of the three oculomotor brainstem nuclei (Leigh and Zee, 2006; Rub et al., 2009). In our screening study, the components of the premotor oculomotor network were variably involved. Neuropathologically, premotor ocular network abnormalities were found in all FTLD–TDP subtypes, but clinical studies showed absence of oculomotor abnormalities in semantic dementia patients (Boxer et al., 2006; Garbutt et al., 2008; Sturm et al., 2011). Therefore, it was surprising to find neuropathological changes in the preocular motor nuclei of the semantic dementia cases we analyzed. The meaning of these deposits should be further elucidated.
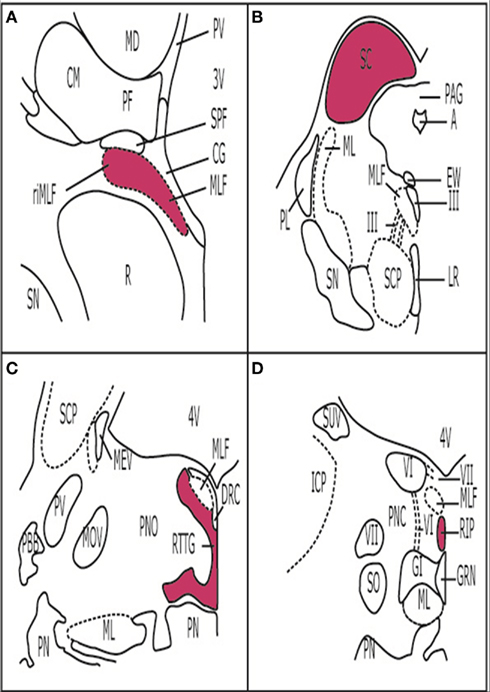
Figure 4. Premotor oculomotor nuclei. (A) Brain stem scheme of a frontal section cut perpendicular to the intercommissural axis of Forel showing the rostral midbrain. The Rostral interstitial nucleus of the medial longitudinal fascicle is part of the premotor oculomotor system. These cells, which are embedded in MLF showTDP-43 inclusions in FTLD-TDP cases. (B) Brain stem scheme of a horizontal section through the caudal midbrain with the superior colliculus (SC). TDP-43 inclusions were found in the SC in all the cases examined. (C) Brain stem scheme of a horizontal section through the mid-level of the pons with the reticulotegmental nucleus of the pons (RTTG; nucleus Bechterew) and the medial longitudinal fascicle (MLF). (D) Brain stem scheme of a horizontal section through the caudal pons with the raphe interpositus nucleus (RIP), and the MLF. Abbreviations: A, aqueduct; CG, central gray; CM, centromedian nucleus of the thalamic central complex; DRC, dorsal raphe nucleus, caudal part; EW, Edinger-Westphal nucleus; GI, gigantocellular reticular nucleus; GRN, great raphe nucleus; ICP, inferior cerebellar peduncle; LR, linear raphe nucleus; MD, mediodorsal nucleus of the thalamus; MEV, mesencephalic trigeminal tract; ML, medial lemniscus; MLF, medial longitudinal fascicle; MOV, motor trigeminal nucleus; PAG, periaqueductal gray; PBB, pontobulbar body; PF, parafascicular nucleus of the thalamic central complex; PL, paralemniscal nucleus; PN, pontine nuclei; PNC, pontine reticular formation, caudal nucleus; PNO, pontine reticular formation, oral nucleus; PV, paraventricular nuclei of the thalamus; R, red nucleus; RIP, raphe interpositus nucleus; ri MLF, rostral interstitial nucleus of the medial longitudinal fascicle; RTTG, reticulotegmental nucleus of the pons (nucleus Bechterew); SC, superior colliculus; SCP, superior cerebellar peduncle; SN, substantia nigra; SO, superior olive; SCP, superior cerebellar peduncle; SPF, subparafascicular nucleus of the thalamic central complex; SUV, superior vestibular nucleus; III, oculomotor nucleus; III, oculomotor nerve; VI, abducens nucleus; VI, abducens nerve; VII, facial nucleus; VII, facial nerve 3V, third ventricle; 4V, fourth ventricle.
Furthermore, the pontine nuclei, inferior olive, and the SN were affected in all subtypes. Changes to these nuclei are associated with motor abnormalities.
In summary, the research on brainstem changes in FTLD–TDP by means of state-of-the-art methods is still incipient and partly controversial. Most investigators obviously consider the involvement of brainstem nuclei in context with an anterograde degeneration and, therefore, as a logical consequence of primary cortical neuron loss. Different subtypes of FTLD–TDP are associated with a highly characteristic pattern of brainstem neuropathology that leaves a kind of fingerprint which could help in understanding the etiopathogenesis of these diseases. Furthermore, the SC is invariably affected in all FTLD–TDP subtypes, indicating a special vulnerability of the oculomotor system in the course of this disease. Given the unexpected previous and recent findings in the brainstems of AD and PD patients, it is plausible and it seems rewarding to subject the brainstem of FTLD–TDP patients to a more comprehensive investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Lea Tenenholz Grinberg is supported by the grant P50 AG23501, John Douglas French Alzheimer Foundation, and Alzheimer Association.
References
Arendt, T., Bigl, V., Tennstedt, A., and Arendt, A. (1985). Neuronal loss in different parts of the nucleus basalis is related to neuritic plaque formation in cortical target areas in Alzheimer’s disease. Neuroscience 14, 1–14.
Berg, L., Mckeel, D. W. Jr., Miller, J. P., Storandt, M., Rubin, E. H., Morris, J. C., Baty, J., Coats, M., Norton, J., Goate, A. M., Price, J. L., Gearing, M., Mirra, S. S., and Saunders, A. M. (1998). Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch. Neurol. 55, 326–335.
Boeve, B. F., Dickson, D. W., Olson, E. J., Shepard, J. W., Silber, M. H., Ferman, T. J., Ahlskog, J. E., and Benarroch, E. E. (2007). Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Med. 8, 60–64.
Boxer, A. L., Garbutt, S., Rankin, K. P., Hellmuth, J., Neuhaus, J., Miller, B. L., and Lisberger, S. G. (2006). Medial versus lateral frontal lobe contributions to voluntary saccade control as revealed by the study of patients with frontal lobe degeneration. J. Neurosci. 26, 6354–6363.
Braak, H., and Braak, E. (1991). Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Cairns, N. J., Bigio, E. H., Mackenzie, I. R., Neumann, M., Lee, V. M., Hatanpaa, K. J., White, C. L. III, Schneider, J. A., Grinberg, L. T., Halliday, G., Duyckaerts, C., Lowe, J. S., Holm, I. E., Tolnay, M., Okamoto, K., Yokoo, H., Murayama, S., Woulfe, J., Munoz, D. G., Dickson, D. W., Ince, P. G., Trojanowski, J. Q., and Mann, D. M. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 114, 5–22.
Cardenas, V. A., Boxer, A. L., Chao, L. L., Gorno-Tempini, M. L., Miller, B. L., Weiner, M. W., and Studholme, C. (2007). Deformation-based morphometry reveals brain atrophy in frontotemporal dementia. Arch. Neurol. 64, 873–877.
Cheng, H. C., Ulane, C. M., and Burke, R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725.
Cras, P., Smith, M. A., Richey, P. L., Siedlak, S. L., Mulvihill, P., and Perry, G. (1995). Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol. 89, 291–295.
Davidson, Y., Amin, H., Kelley, T., Shi, J., Tian, J., Kumaran, R., Lashley, T., Lees, A. J., Duplessis, D., Neary, D., Snowden, J., Akiyama, H., Arai, T., Hasegawa, M., Bandopadhyay, R., Sikkink, S., Pickering-Brown, S., and Mann, D. M. (2009). TDP-43 in ubiquitinated inclusions in the inferior olives in frontotemporal lobar degeneration and in other neurodegenerative diseases: a degenerative process distinct from normal ageing. Acta Neuropathol. 118, 359–369.
Devanand, D. P., Folz, M., Gorlyn, M., Moeller, J. R., and Stern, Y. (1997). Questionable dementia: clinical course and predictors of outcome. J. Am. Geriatr. Soc. 45, 321–328.
Dickson, D. W., Josephs, K. A., and Amador-Ortiz, C. (2007). TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 114, 71–79.
Garbutt, S., Matlin, A., Hellmuth, J., Schenk, A. K., Johnson, J. K., Rosen, H., Dean, D., Kramer, J., Neuhaus, J., Miller, B. L., Lisberger, S. G., and Boxer, A. L. (2008). Oculomotor function in frontotemporal lobar degeneration, related disorders and Alzheimer’s disease. Brain 131, 1268–1281.
Geula, C., Nagykery, N., Nicholas, A., and Wu, C. K. (2008). Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67, 309–318.
Giannakopoulos, P., Herrmann, F. R., Bussiere, T., Bouras, C., Kovari, E., Perl, D. P., Morrison, J. H., Gold, G., and Hof, P. R. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500.
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., Ogar, J. M., Rohrer, J. D., Black, S., Boeve, B. F., Manes, F., Dronkers, N. F., Vandenberghe, R., Rascovsky, K., Patterson, K., Miller, B. L., Knopman, D. S., Hodges, J. R., Mesulam, M. M., and Grossman, M. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014.
Grinberg, L. T., Rub, U., Ferretti, R. E., Nitrini, R., Farfel, J. M., Polichiso, L., Gierga, K., Jacob-Filho, W., and Heinsen, H. (2009). The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol. Appl. Neurobiol. 35, 406–416.
Grinberg, L. T., Rueb, U., Alho, A. T., and Heinsen, H. (2010). Brainstem pathology and non-motor symptoms in PD. J. Neurol. Sci. 289, 81–88.
Hardy, J. A., Mann, D. M., Wester, P., and Winblad, B. (1986). An integrative hypothesis concerning the pathogenesis and progression of Alzheimer’s disease. Neurobiol. Aging 7, 489–502.
Insausti, R., Amaral, D. G., and Cowan, W. M. (1987). The entorhinal cortex of the monkey: III. Subcortical afferents. J. Comp. Neurol. 264, 396–408.
Jakob, H. (1960). On the pathological anatomy of Pick’s disease. I. Comparative research on the extent and critical areas of atrophy. Arch. Psychiatr. Nervenkr. Z. Gesamte Neurol. Psychiatr. 201, 269–297.
Jakob, H. (1979). “Die Picksche Krankheit. Eine neuropathologisch-anatomisch-klinische Studie,” in Monographien Aus Dem Gesamtgebiete Der Psychiatrie, eds H. Hippius, W. Janzarik, and C. Müller (Berlin: Springer-Verlag), 1–110.
Kaufmann, H., Nahm, K., Purohit, D., and Wolfe, D. (2004). Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology 63, 1093–1095.
Kertesz, A., and Munoz, D. (2004). Relationship between frontotemporal dementia and corticobasal degeneration/progressive supranuclear palsy. Dement. Geriatr. Cogn. Disord. 17, 282–286.
Kramer, J. H., Jurik, J., Sha, S. J., Rankin, K. P., Rosen, H. J., Johnson, J. K., and Miller, B. L. (2003). Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn. Behav. Neurol. 16, 211–218.
Kril, J. J., Patel, S., Harding, A. J., and Halliday, G. M. (2002). Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 103, 370–376.
Lai, Y., and Siegel, J. (1999). ”Muscle atonia and REM sleep,” in Rapid Eye Movement Sleep, eds B. Mallick and S. Inoue (New York: Marcel Dekker), 69–90.
Leigh, R. J., and Zee, D. S. (2006). The Neurology of Eye Movements. Oxford: Oxford University Press.
Liscic, R. M., Grinberg, L. T., Zidar, J., Gitcho, M. A., and Cairns, N. J. (2008). ALS and FTLD: two faces of TDP-43 proteinopathy. Eur. J. Neurol. 15, 772–780.
Lüers, T., and Spatz, H. (1957). “Picksche Krankheit. (Progressive umschriebene Groß hirnatrophie),” in Hanbuch der speziellen pathologischen Anatomie und Histologie, eds O. Lubarsch, F. Henke, and R. Hoeslle (Berlin: Springer), 614–715.
Lyness, S. A., Zarow, C., and Chui, H. C. (2003). Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol. Aging 24, 1–23.
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., Kovacs, G. G., Ghetti, B., Halliday, G., Holm, I. E., Ince, P. G., Kamphorst, W., Revesz, T., Rozemuller, A. J., Kumar-Singh, S., Akiyama, H., Baborie, A., Spina, S., Dickson, D. W., Trojanowski, J. Q., and Mann, D. M. (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4.
Mackenzie, I. R. A., Baborie, A., Pickering-Brown, S., Du Plessis, D., Jaros, E., Perry, R. H., Neary, D., Snowden, J. S., and Mann, D. M. A. (2006). Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 112, 539–549.
Maslow, K. (2006). Early Onset Dementia. A National Challenge, A Future Crisis. Washington, DC: Alzheimer’s Association.
Mesulam, M. M., Mufson, E. J., Wainer, B. H., and Levey, A. I. (1983). Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience 10, 1185–1201.
Mirra, S. S., Heyman, A., Mckeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., Vogel, F. S., Hughes, J. P., Van Belle, G., and Berg, L. (1991). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486.
Mountcastle, V. B. (1998). Perceptual Neuroscience: The Cerebral Cortex. Cambridge, MA: Harvard University Press.
National Institute on Aging–Reagan Institute Working Group. (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol. Aging 18, S1–S2.
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., Freedman, M., Kertesz, A., Robert, P. H., Albert, M., Boone, K., Miller, B. L., Cummings, J., and Benson, D. F. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554.
Nieuwenhuys, R. (1996). The greater limbic system, the emotional motor system and the brain. Prog. Brain Res. 107, 551–580.
Nieuwenhuys, R., Voogd, J., and Van Huijzen, C. (2008). “Topography of spinal cord, brain stem and cerebellum,” in The Human Central Nervous System, 4th Edn, eds R. Nieuwenhuys, J. Voogd, and C. Van Huijzen (Berlin: Spinger), 177–245.
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., Hirsch, E. C., Farrer, M., Schapira, A. H., and Halliday, G. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661.
Ohm, T. G., Heilmann, R., and Braak, H. (1989). The human oral raphe system. Architectonics and neuronal types in pigment-Nissl preparations. Anat. Embryol. 180, 37–43.
Ondo, W. G., Dat Vuong, K., Khan, H., Atassi, F., Kwak, C., and Jankovic, J. (2001). Daytime sleepiness and other sleep disorders in Parkinson’s disease. Neurology 57, 1392–1396.
Panza, F., Frisardi, V., Capurso, C., D’Introno, A., Colacicco, A. M., Imbimbo, B. P., Santamato, A., Vendemiale, G., Seripa, D., Pilotto, A., Capurso, A., and Solfrizzi, V. (2010). Late-life depression, mild cognitive impairment, and dementia: possible continuum? Am. J. Geriatr. Psychiatry 18, 98–116.
Pfeiffer, R. F. (2003). Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2, 107–116.
Pikkarainen, M., Hartikainen, P., and Alafuzoff, I. (2008). Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J. Neuropathol. Exp. Neurol. 67, 280–298.
Poppe, W., and Tennstedt, A. (1964). Die Verbindung der Pickschen Atrophie mit Hirnnervenkernveranderungen und Pyramidenbahnsymptomatik. Acta Neuropathol. 4, 169–174.
Ramon-Moliner, E., and Nauta, W. J. (1966). The isodendritic core of the brain stem. J. Comp. Neurol. 126, 311–335.
Ratnavalli, E., Brayne, C., Dawson, K., and Hodges, J. R. (2002). The prevalence of frontotemporal dementia. Neurology 58, 1615–1621.
Rosso, S. M., Landweer, E. J., Houterman, M., Donker Kaat, L., Van Duijn, C. M., and Van Swieten, J. C. (2003). Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J. Neurol. Neurosurg. Psychiatr. 74, 1574–1576.
Rub, U., Del Tredici, K., Schultz, C., Thal, D. R., Braak, E., and Braak, H. (2000). The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol. Appl. Neurobiol. 26, 553–567.
Rub, U., Heinsen, H., Brunt, E. R., Landwehrmeyer, B., Den Dunnen, W. F., Gierga, K., and Deller, T. (2009). The human premotor oculomotor brainstem system – can it help to understand oculomotor symptoms in Huntington’s disease? Neuropathol. Appl. Neurobiol. 35, 4–15.
Rub, U., Schultz, C., Del Tredici, K., and Braak, H. (2001). Early involvement of the tegmentopontine reticular nucleus during the evolution of Alzheimer’s disease-related cytoskeletal pathology. Brain Res. 908, 107–112.
Sampathu, D. M., Neumann, M., Kwong, L. K., Chou, T. T., Micsenyi, M., Truax, A., Bruce, J., Grossman, M., Trojanowski, J. Q., and Lee, V. M. (2006). Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 169, 1343–1352.
Saper, C. B., Chou, T. C., and Scammell, T. E. (2001). The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 24, 726–731.
Sassin, I., Schultz, C., Thal, D. R., Rüb, U., Arai, K., Braak, E., and Braak, H. (2000). Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 100, 259–269.
Simic, G., Stanic, G., Mladinov, M., Jovanov-Milosevic, N., Kostovic, I., and Hof, P. R. (2009). Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 35, 532–554.
Sturm, V. E., Mccarthy, M. E., Yun, I., Madan, A., Yuan, J. W., Holley, S. R., Ascher, E. A., Boxer, A. L., Miller, B. L., and Levenson, R. W. (2011). Mutual gaze in Alzheimer’s disease, frontotemporal and semantic dementia couples. Soc. Cogn. Affect Neurosci. 6, 359–367.
Keywords: brainstem, human, pathology, neurodegenerative diseases, dementia, Alzheimer’s disease, frontotemporal lobar degeneration, Parkinson’s disease
Citation: Grinberg LT, Rueb U and Heinsen H (2011) Brainstem: neglected locus in neurodegenerative diseases. Front. Neur. 2:42. doi: 10.3389/fneur.2011.00042
Received: 04 March 2011;
Paper pending published: 13 April 2011;
Accepted: 13 June 2011;
Published online: 11 July 2011.
Edited by:
Martin Rhys Farlow, Indiana University School of Medicine, USAReviewed by:
Stefano F. Cappa, Vita-Salute San Raffaele University, ItalyRichard Mohs, Eli Lilly and Company, USA
Copyright: © 2011 Grinberg, Rueb and Heinsen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Helmut Heinsen, Laboratory of Morphological Brain Research, University of Wuerzburg, Oberdürrbacher Strasse 6, 97080 Wuerzburg, Germany. e-mail:aGVpbnNlbkB1bmktd3VlcnpidXJnLmRl