

- 1 Department of Neurological Surgery, University of California Davis, Davis, CA, USA
- 2 Department of Neurology, University of California Los Angeles, Los Angeles, CA, USA
Background and Purpose: Moyamoya disease has been classically described by the Asian experience, yet clinical aspects of moyamoya phenomena in the United States remain vastly undefined. The multifocal occlusive arterial disorder may be linked with numerous conditions; however, later stages of this syndrome share common vascular pathophysiology. This study is aimed at characterizing inpatient moyamoya cases in the United States over a broad time span. Methods: A comprehensive analysis of the Nationwide Inpatient Sample of the Healthcare Cost and Utilization Project (Releases 1–13, 1988–2004) based on ICD-9-CM code 437.5 was performed. Annual percentages and trends analyses were conducted for demographic variables, admission characteristics, co-morbidities, and procedures. Result: 2247 admissions of moyamoya cases were analyzed from a wide geographic distribution throughout the United States between 1988 and 2004. Age at admission varied considerably (mean 29.6 ± 18.5 years), affecting women more frequently than men (61.9%). Various racial groups were identified (35.4% White, 19.7% African American, 5.6% Hispanic, 8.3% Asian or Pacific Islander, 1.4% Native American). Admissions were typically emergent (38.8%) or urgent (18.7%), although elective admissions occurred (24.4%). Aside from moyamoya, sickle cell disease was diagnosed in 13.6%, ischemic stroke in 20.7%, intracerebral hemorrhage in 7.4%, transient ischemic attack in 3.4%, and subarachnoid hemorrhage in 3.1%. Cerebral angiography was performed in 24.9% while extracranial–intracranial bypass was done in 8.4% of patients. Conclusion: Moyamoya in the United States is a heterogeneous condition affecting individuals of all ages across a diverse racial spectrum and wide geographic distribution. Further recognition of moyamoya syndrome may facilitate ongoing research and future therapeutic approaches.
Introduction
Moyamoya is a chronic cerebrovascular disease first described in Japan in 1955 (Takeuchi and Shimizu, 1957). It is characterized by the narrowing or occlusion of proximal cerebral arteries, in particularly the distal internal carotid, proximal middle, or anterior cerebral arteries, which often leads to hypertrophy of lenticulostriate arteries and bypassing of the occlusive segment. Moyamoya vasculopathy can be categorized into the syndrome and the disease. Moyamoya syndrome includes the vasculopathic abnormalities in addition to associated conditions, while moyamoya disease has no other associated risk factors. Moyamoya refers specifically to the angiographic findings (Scott and Smith, 2009).
The diagnostic criteria are as follows: (1) stenosis or occlusion of the anterior cerebral, middle cerebral or internal carotid arteries, (2) an abnormality of the vasculature surrounding these arteries, and (3) the presence of bilateral disease seen angiographically. Diagnosis of moyamoya syndrome has been defined based on cases that meet any two of these criteria. Conventional angiography remains the gold-standard imaging technique for evaluation of radiographic moyamoya; however, recent advances in MR angiography are sufficient in specific cases (Burke et al., 2009). While this disease has been reported worldwide, the estimates of incidence in the United States have been limited (Goto and Yonekawa, 1992; Edwards-Brown and Quets, 1997; Graham and Matoba, 1997; Numaguchi et al., 1997; Uchino et al., 2005). Furthermore, there is data that suggests differences in presentation in the North American population compared with the Asian population (Bruno et al., 1988; Numaguchi et al., 1997; Chiu et al., 1998; Cloft et al., 1999; Yilmaz et al., 2001; Hallemeier et al., 2006; Mesiwala et al., 2008).
In this study, we report the inpatient characterization of the moyamoya phenomenon in the United States between 1988 and 2004. Specifically, demographic data, admission diagnoses, and associated procedures are analyzed.
Materials and Methods
The National Inpatient Sample (NIS) database from 1988 to 2004 (Releases 1–13, 1988–2004) was searched for records with a diagnosis of moyamoya using the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) code 437.5. Annual percentages and trends analyses were conducted for demographic variables, admission characteristics, co-morbidities and procedures using descriptive and frequency analysis in SPSS (version 13.0). Admission type and disposition upon discharge was analyzed in addition to the associated diagnoses and procedures during each admission. Using the diagnosis-related group (DRG) codes 001 (craniotomy age >17 except for trauma) or 003 (craniotomy age <17) and the ICD-9-CM procedure code 39.28 (extracranial–intracranial vascular bypass), the incidence of surgical revascularization was calculated (Uchino et al., 2005). Additionally, we reviewed the number of computerized tomography of the head (procedure code 87.03) and magnetic resonance imaging of the brain (procedure code 88.91). Secondary diagnoses were also obtained: sickle cell (diagnosis code 282) and neurofibromatosis (diagnosis code 23771 for specified and 23770 for unspecified). Race and ethnicity were determined based upon the NIS database distinction: White, Hispanic, African American, Asian American (including Pacific Islander), and Native American.
Results
We analyzed 2247 inpatient admissions with moyamoya between 1988 and 2004. In this cohort, the mean age of the patients was 29.6 ± 18.5, ranging from 0 to 101 years in age. Women were admitted for moyamoya more often than men (61.9%, Figure 1). Based upon the categories determined by the NIS database, the distribution of moyamoya disease among ethnicities during this time period is as follows: Whites 35.4%, African American 19.7%, Asian or Pacific Islanders 8.3%, Hispanics 5.6%, and Native Americans 1.4%. When comparing gender and ethnicity, all ethnicities except African Americans displayed a female predominance (Figure 2).
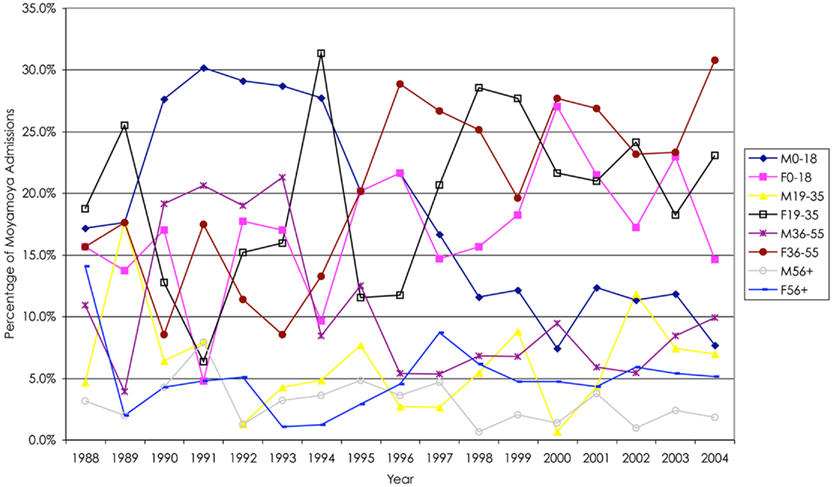
Figure 1. Overall, there was a higher percentage of female relative to male hospital admissions for moyamoya; however, in general, there were fewer males and females over 55 years of age who presented as hospital admissions. Of note after 1994, there was a decline in male hospital admissions in patients less than 19 years of age.
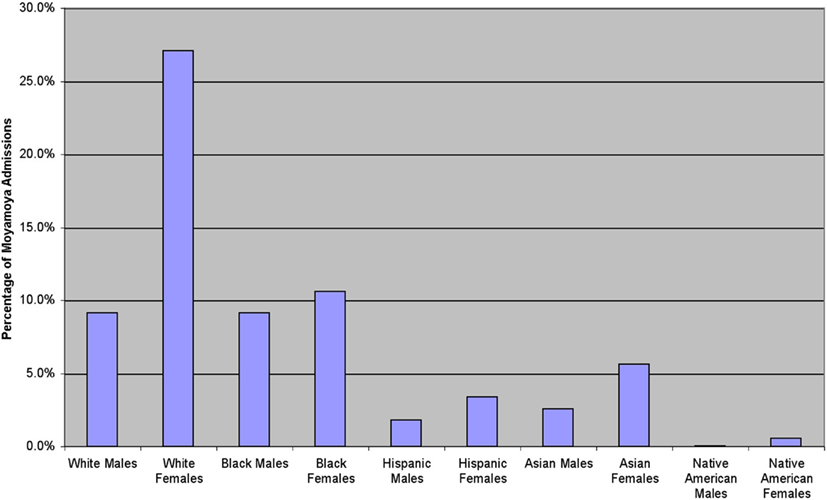
Figure 2. In the United States, there is a predominance of female-to-male admissions for moyamoya. Over this time period, there is a similar number of White and African American males, while more White than African American females were admitted to the hospital with moyamoya during that time.
The majority of admissions were for emergent care (38.8%), followed by the elective admission type (24.4%) and urgent admissions (18.7%). Inpatient stays were on average 7.75 days. In addition to moyamoya, the patients also presented with other diagnoses upon admission. All of the patients were coded as having some sort of cerebrovascular disease, while 20.7% of patients were admitted with the diagnosis of an ischemic stroke (Goldstein, 1998). Intracerebral hemorrhage occurred in 7.4% of cases, subarachnoid hemorrhage in 3.1% or another unspecific intracranial hemorrhage in 0.6% of admissions. The diagnosis of a transient ischemic attack (TIA) was present in 3.4% of admissions, while anemia was coded in 13.5% of this cohort. Of note, sickle cell disease or trait was noted in 13.6% of all moyamoya cases, while only 51 cases were concomitantly diagnosed with neurofibromatosis. The principal diagnosis in patients admitted for moyamoya follows a similar order and included ischemic stroke (9.2%), anemia (7.6%), intracerebral hemorrhage (5.5%), subarachnoid hemorrhage (2.4%), and TIA (1.8%).
Cerebral angiography was the most common procedure performed in these patients (24.9%). Surgical revascularization was performed in 23.7% of patients. Specifically, craniotomies were done on patients greater than 17 years of age in 10.1% of patients, while patients between the ages of 0 and 17 received craniotomies in 5.2% of the cases. Cerebral artery bypasses were also performed in these patients (9.7%) with the extracranial–intracranial vascular bypass being the most common (8.4%). Packed red cell transfusions were indicated in 11.0% of this population. The principal procedures followed a similar distribution: cerebral angiography (13.1%), cerebrovascular bypass (7.9%), and packed red cell transfusion (4.3%, Figure 3). Over the 17-year period, computerized tomography of the head was only reported in 183 cases, while MRI was reported in 178 cases.
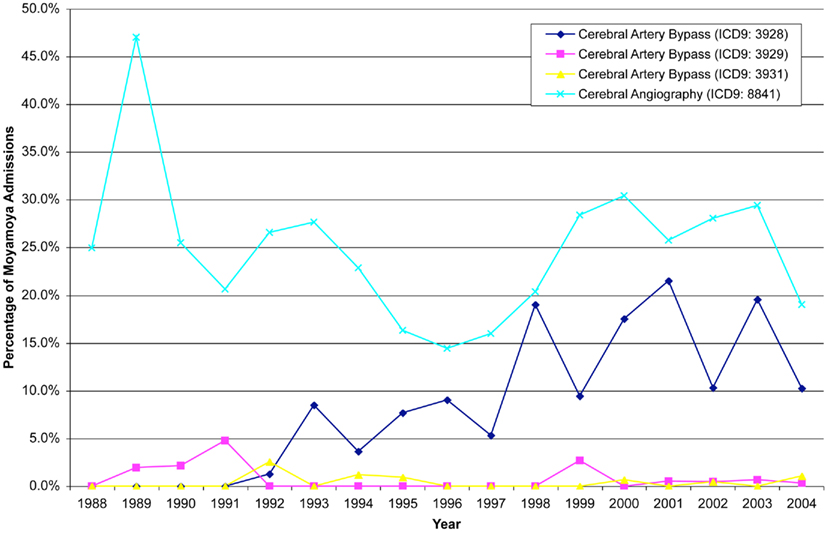
Figure 3. Angiography remains the most common procedure associated with patients admitted for moyamoya, while there has been progressively more cerebral artery bypasses from 1992–2004.
To determine the outcomes of patients after admission, we analyzed the inpatient deaths and discharge dispositions. There was a 3.0% mortality rate during hospitalization, while the majority of patients were routinely discharged (76.8%). Patients were also discharged to short-term hospital facilities (4.6%), home health care (4.3%), or other type of facility (9.4%).
Discussion
Moyamoya in the US affects a broad spectrum of individuals from diverse backgrounds and age groups. According to the NIS database, the number of inpatient moyamoya cases has been increasing; however, this number is deceptive because the number of hospitals sampled has also increased. Moyamoya is a lifelong process and has been seen in patients ranging from newborn individuals to those in their ninth decade. Since these patients are relatively stable, this chronic disorder is often seen with other co-morbidities, and patients are often subject to developing other pathologies.
In general, there were more moyamoya admissions in the female cohort (3:2 female-to-male), consistent with previous data suggesting a 2:1 ratio (Scott et al., 2004; Baba et al., 2008). While females generally had more admissions, there was a greater difference between female and male admissions between the ages of 19 and 55 years of age. Over the 17-year period based upon age quartiles (0–18, 19–35, 36–55, greater than 55 years of age), female admissions for moyamoya were greater in the 19–35 age group (3:1 female-to-male), and 36–55 age group (2:1) compared with the more extreme age groups (1:1). There was variability among the years. For instance, males less than 18 years of age were admitted for moyamoya more often than females less than 18 years of age prior to 1998 at which point females in this age group were more prevalent. Furthermore, there was a significant increase in females between the ages of 36 and 55 years of age after 1994.
Based upon the NIS classification, the White ethnicity consisted of the majority of moyamoya admissions. However, for patients under 19 years of age, the African American ethnicity was the majority (Whites 9.9%, African Americans 11.7%, Asian or Pacific Islanders 2.4%, Hispanics 2.4%, and Native Americans 0.3% of the total admission population). This finding may be due to an association between moyamoya and sickle cell disease (Pegelow, 2001; Riebel et al., 2003; Gebreyohanns and Adams, 2004), as sickle cell disease is more prevalent in the African American population. Not all of the African American population has been concomitantly diagnosed with sickle cell disease, as only 1.3% of the entire population was diagnosed with a red cell blood disorder between the ages of 0 and 17 years.
While this study does not have the ability to comment on medical treatment, the percentage of bypasses coded increased over time and peaking in the year 2001. In 1988, of the cases coded none of the patients underwent a bypass, while in 2001, 21.5% of patients with moyamoya underwent bypass. After 2001, there was a decline in the percentage of patients who underwent bypass as evidenced by only 10.3% of patients undergoing bypass in 2004. The incidence of cerebral angiography varied over time, ranging from 13.9 to 47.1%; however, there was no particular trend in the incidence of this procedure.
Moyamoya in the US may be distinct from Asian descriptions. It is important to note that defining moyamoya is important when analyzing the NIS database. The NIS does not distinguish between moyamoya disease and moyamoya syndrome. It is likely that the majority of the cases are likely moyamoya syndrome due to the presence of various pathologies; however, this information is unable to be ascertained based upon this data alone. Although moyamoya may be rare, it is important to study these cases not only for the care of these patients, but also because moyamoya syndrome may provide insight on the chronic development and role of collaterals over time. Although surgical and nascent endovascular techniques, such as stenting, may be utilized to revascularize these individuals, the role and impact of these interventions needs to be compared with natural history in the US. While it has not been shown that indiscriminate moyamoya screening is beneficial, there has been a correlation with early screening for unilateral moyamoya onset and decreased stroke burden and improved clinical outcome (Scott and Smith, 2009).
The moyamoya phenomenon is a varied disease, and this coding system does not differentiate between moyamoya disease and moyamoya syndrome; however, it is more likely to actually be a variant of the moyamoya phenomenon because it is such a rare and specific condition. It is difficult to be certain that the same patients are not being counted more than once due to a second need for admission. As noted by Uchino et al. (2005) there may be a selection and ascertainment bias as patients of Asian origin may be given the diagnosis of moyamoya more commonly.
Due to the fact that this is a national database, this study is restricted to the information inputted by the various hospitals throughout the nation. It is only a study of inpatient hospitalizations and does not include outpatient data. Unfortunately, it does not elucidate how these patients were diagnosed. Typically, this disease process is diagnosed through angiography, and only 24.9% of the patients reported here had this procedure. Thus, patient likely were admitted with a prior diagnosis of moyamoya. An additional possibility is that a particular patient may have been admitted multiple times, suggesting a selection bias. Still, the numbers presented here likely are underestimating the total number of patients across the United States. The data gathered is approximates 20% of the stratified sample of community hospitals across the United States. During each year period, there are a different number of states participating in the database, ranging from 8 states in 1988 to 37 states in 2004. Thus, even in the latest version not all states are represented. Furthermore, the sensitivity and specificity of the coding system is unknown. However, this is the most comprehensive database in the United States and is specific for moyamoya, providing an objective measure. Moyamoya is a relatively rare phenomenon; thus, it is unlikely that one would positively code moyamoya, leading to relatively high specificity. This does suggest that the number of cases reported in this study is an underestimate of the cases in the United States.
Conclusion
Moyamoya in the United States remains a heterogeneous disease process that affects a diverse population of individuals over a wide geographic distribution. Continued cataloging of specific aspects of moyamoya disease and syndrome may improve the characterization of this disease process and foster improved research and therapeutic approaches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Baba, T., Houkin, K., and Kuroda, S. (2008). Novel epidemiological features of moyamoya disease. J. Neurol. Neurosurg. Psychiatr. 79, 900–904.
Bruno, A., Adams, H. P. Jr., Biller, J., Rezai, K., Cornell, S., and Aschenbrener, C. A. (1988). Cerebral infarction due to moyamoya disease in young adults. Stroke 19, 826–833.
Burke, G. M., Burke, A. M., Sherma, A. K., Hurley, M. C., Batjer, H. H., and Bendok, B. R. (2009). Moyamoya disease: a summary. Neurosurg. Focus 26, E11.
Chiu, D., Shedden, P., Bratina, P., and Grotta, J. C. (1998). Clinical features of moyamoya disease in the United States. Stroke 29, 1347–1351.
Cloft, H. J., Kallmes, D. F., Snider, R., and Jensen, M. E. (1999). Idiopathic supraclinoid and internal carotid bifurcation steno-occlusive disease in young American adults. Neuroradiology 41, 772–776.
Edwards-Brown, M. K., and Quets, J. P. (1997). Midwest experience with moyamoya disease. Clin. Neurol. Neurosurg. 99(Suppl. 2), S36–S38.
Gebreyohanns, M., and Adams, R. J. (2004). Sickle cell disease: primary stroke prevention. CNS Spectr. 9, 445–449.
Goldstein, L. B. (1998). Accuracy of ICD-9-CM coding for the identification of patients with acute ischemic stroke: effect of modifier codes. Stroke 29, 1602–1604.
Goto, Y., and Yonekawa, Y. (1992). Worldwide distribution of moyamoya disease. Neurol. Med. Chir. (Tokyo) 32, 883–886.
Graham, J. F., and Matoba, A. (1997). A survey of moyamoya disease in Hawaii. Clin. Neurol. Neurosurg. 99(Suppl. 2), S31–S35.
Hallemeier, C. L., Rich, K. M., Grubb, R. L. Jr., Chicoine, M. R., Moran, C. J., Cross, D. T. III, Zipfel, G. J., Dacey, R. G. Jr., and Derdeyn, C. P. (2006). Clinical features and outcome in North American adults with moyamoya phenomenon. Stroke 37, 1490–1496.
Mesiwala, A. H., Sviri, G., Fatemi, N., Britz, G. W., and Newell, D. W. (2008). Long-term outcome of superficial temporal artery-middle cerebral artery bypass for patients with moyamoya disease in the US. Neurosurg. Focus 24, E15.
Numaguchi, Y., Gonzalez, C. F., Davis, P. C., Monajati, A., Afshani, E., Chang, J., Sutton, C. L., Lee, R. R., and Shibata, D. K. (1997). Moyamoya disease in the United States. Clin. Neurol. Neurosurg. 99(Suppl. 2), S26–S30.
Pegelow, C. H. (2001). Stroke in children with sickle cell anaemia: aetiology and treatment. Paediatr. Drugs 3, 421–432.
Riebel, T., Kebelmann-Betzing, C., Gotze, R., and Overberg, U. S. (2003). Transcranial Doppler ultrasonography in neurologically asymptomatic children and young adults with sickle cell disease. Eur. Radiol. 13, 563–570.
Scott, R. M., and Smith, E. R. (2009). Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 360, 1226–1237.
Scott, R. M., Smith, J. L., Robertson, R. L., Madsen, J. R., Soriano, S. G., and Rockoff, M. A. (2004). Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J. Neurosurg. 100, 142–149.
Takeuchi, K., and Shimizu, K. (1957). Hypoplasia of the bilateral internal carotid arteries. Brain Nerve 9, 37–43.
Uchino, K., Johnston, S. C., Becker, K. J., and Tirschwell, D. L. (2005). Moyamoya disease in Washington State and California. Neurology 65, 956–958.
Keywords: moyamoya, collateral, stroke, cerebral ischemia
Citation: Lee DJ and Liebeskind DS (2011) Characterization of Inpatient Moyamoya in the United States: 1988–2004. Front. Neur. 2:43. doi: 10.3389/fneur.2011.00043
Received: 26 February 2011;
Accepted: 21 June 2011;
Published online: 04 July 2011.
Edited by:
Dileep Yavagal, University of Miami, USAReviewed by:
Angelos A. Konstas, Massachusetts General Hospital, USAElias A. Giraldo, Drexel University College of Medicine, USA
Copyright: © 2011 Lee and Liebeskind. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: David S. Liebeskind, Department of Neurology, University of California Los Angeles, 300 UCLA Medical Plaza Suite B200, Los Angeles, CA 90095, USA. e-mail:ZGF2aWRsaWViZXNraW5kQHlhaG9vLmNvbQ==