
- Chief of Neurology, Hines VA Hospital and Neurology Residency Program Director, Loyola University Medical Center, Hines, IL, USA
Muscle diseases can constitute a large variety of both acquired and hereditary disorders. Myopathies in systemic disease results from several different disease processes including endocrine, inflammatory, paraneoplastic, infectious, drug- and toxin-induced, critical illness myopathy, metabolic, and myopathies with other systemic disorders. Patients with systemic myopathies often present acutely or sub acutely. On the other hand, familial myopathies or dystrophies generally present in a chronic fashion with exceptions of metabolic myopathies where symptoms on occasion can be precipitated acutely. Most of the inflammatory myopathies can have a chance association with malignant lesions; the incidence appears to be specifically increased only in patients with dermatomyositis. In dealing with myopathies associated with systemic illnesses, the focus will be on the acquired causes. Management is beyond the scope of this chapter. Prognosis is based upon the underlying cause and, most of the time, carries a good prognosis. In order to approach a patient with suspected myopathy from systemic disease, a stepwise approach is utilized.
Step 1: Overview and Understanding of Myopathies in Systemic Illnesses
Muscle diseases represent a wide variety of both acquired and hereditary disorder. The underlying disease can involve muscles at both structural and functional levels including metabolism as well as muscle ion channel. The primary care physician is most likely to be involved in the initial diagnosis of inflammatory myopathy and of myopathy that results from systemic disease or toxic exposure (Srinivasan and Amato, 2003; Jackson, 2008). Please see Table 1 for classification of myopathies with systemic disorders.

Table 1. Classification of myopathies with systemic disorders.
Endocrine Myopathies
Overall, muscle fatigue is more common than true weakness. The cause of weakness in these disorders is not well-defined. It also remains unclear that weakness results from disease of muscle as opposed to another part of the motor unit. The serum CK level is often normal (except in hypothyroidism). Nearly all endocrine myopathies respond to treatment (Horak and Pourmand, 2000).
Thyroid disorders
Abnormalities of thyroid function can cause a wide array of muscle disorders. This is very obvious from the important role of thyroid hormones in regulating the metabolism of carbohydrates, proteins, and lipids. In addition, thyroid hormones also stimulate calorigenesis in muscle, increase muscle demand for vitamins, and enhance muscle sensitivity to circulating catecholamines.
Hypothyroidism
Patients with hypothyroidism have frequent muscle complaints, and proximal muscle weakness occurs in about one-third of them. Muscle cramps, pain, and stiffness are common. Features of slow muscle contraction and relaxation occur in 25% of patients; the relaxation phase of muscle stretch reflexes is characteristically prolonged and best observed at the ankle or biceps brachii reflexes. The serum CK level is often elevated (up to 10 times normal), even when there is minimal clinical evidence of muscle disease. Hoffman’s syndrome results in prominent muscle enlargement and weakness with muscle stiffness (Horak and Pourmand, 2000; Turker et al., 2008).
Hyperthyroidism
Patients who are thyrotoxic commonly have proximal muscle weakness and atrophy on examination, but they rarely complain of the deficit. Muscle stretch reflexes are usually preserved and often brisk. Bulbar, respiratory, and even esophageal muscles may occasionally be affected, causing dysphagia, dysphonia, and aspiration. Other neuromuscular disorders occur in association with hyperthyroidism, including hypokalemic periodic paralysis, myasthenia gravis, and a progressive ocular myopathy. Serum CK levels are not elevated in thyrotoxic myopathy. The muscle biopsy reveals atrophy of muscle fibers (Kung, 2007; Tsuda et al., 2008; Mansourian, 2010).
Hypoparathyroidism
An overt myopathy due to hypocalcemia rarely occurs. Neuromuscular symptoms are usually related to localized or generalized tetany. Serum CK levels may be increased secondary to muscle damage from sustained tetany. Hyporeflexia or areflexia is usually present and contrasts with the hyperreflexia in hyperparathyroidism (Syriou et al., 2005).
Hyperparathyroidism
Proximal muscle weakness, muscle wasting, and brisk muscle stretch reflexes. Serum CK levels are usually normal or slightly elevated. Serum calcium and phosphorus levels show no correlation with the clinical neuromuscular manifestations. Muscle biopsy reveals only varying degrees of atrophy without muscle fiber degeneration (Horak and Pourmand, 2000; Habib and Camacho, 2010).
Adrenal disorders
Can lead to variety of problems from either excess or deficiency. Glucocorticoid excess, either endogenous or exogenous, produces various degrees of proximal limb weakness in the form of myopathy. Steroid myopathy is the most commonly diagnosed endocrine muscle disease. Muscle wasting may be striking. A cushingoid appearance usually accompanies clinical signs of myopathy. Muscle biopsy demonstrates muscle type II fiber atrophy (Kanda et al., 2001).
Adrenal insufficiency commonly causes muscle fatigue. The degree of weakness may be difficult to assess but is typically mild. In primary hyperaldosteronism (Conn’s syndrome), neuromuscular complications with proximal muscle weakness are due to potassium depletion. Serum CK levels may be elevated, and a muscle biopsy may demonstrate degenerating fibers, some with vacuoles. These changes relate to hypokalemia and are not a direct effect of aldosterone on skeletal muscle (Horak and Pourmand, 2000; Cicala and Mantero, 2010).
Pituitary disorders
Patients with acromegaly usually have mild proximal weakness without muscle atrophy. Muscles often appear enlarged but exhibit decreased force generation. The duration of acromegaly, rather than the serum growth hormone levels, correlates with the degree of myopathy. A multitude of treatment options exist for patients with acromegaly including surgery, radiotherapy, somatostatin analogs, dopamine agonists, and growth hormone receptor antagonists (Horak and Pourmand, 2000; Del Porto et al., 2011).
Diabetes mellitus
Neuromuscular complications of diabetes mellitus are most often related to neuropathy – cranial, peripheral nerve, or lumbosacral plexus. The only notable myopathy of diabetes mellitus is ischemic infarction of thigh muscles. This condition occurs in patients with poorly controlled diabetes and presents with abrupt onset of pain, tenderness, and edema of one thigh. The area of muscle infarction is hard and indurated. The muscles most often affected include the vastus lateralis, thigh adductors, and biceps femoris. CT or MRI can demonstrate focal abnormalities in the affected muscle. Diagnosis by imaging is preferable to muscle biopsy, if possible (Yildirim Donmez and Feldman, 2008).
Inflammatory Myopathies: Association with Malignancies
Most of the inflammatory myopathies can have a chance association with malignant lesions. The incidence of malignant conditions appears to be specifically increased only in patients with Dermatomyositis, and not in Polymyositis or Inclusion body myositis. The more common tumors associated with dermatomyositis are ovarian cancer, breast cancer, melanoma, colon cancer, and non-Hodgkin’s lymphoma. The extent of the search that should be conducted for an occult neoplasm in adults with dermatomyositis depends on the clinical circumstances. Tumors in these patients are usually uncovered by abnormal findings in the medical history and physical examination. The weight of evidence argues against performing expensive, invasive, and non-directed tumor searches. Nishigori et al. (2009) have emphasized higher malignancy in patients with vesiculobullous dermatomyositis. Vigilance is required with a complete annual examination with pelvic, breast (mammogram, if indicated), and rectal examinations (with colonoscopy according to age and family history); urine analysis; complete blood count; complete metabolic profile; chest x-ray film should be adequate in most cases. In Asians, nasopharyngeal cancer is common, and a careful examination of ears, nose, and throat is indicated (Kawachi et al., 2008; Nishigori et al., 2009).
Inflammatory myopathies: Overlap syndromes
The association of inflammatory myopathies with connective tissues disease defines the overlap syndromes. Inflammatory myopathies do not commonly occur as a part of well-defined connective tissue disorders like SLE or RA but can be a part of overlap syndromes such as mixed connective tissue disease. A well-characterized overlap syndrome occurs in patients with dermatomyositis who also have manifestations of systemic sclerosis or mixed connective tissue disease including: sclerotic thickening of the dermis, contractures, esophageal hypomotility, microangiopathy, and calcium deposits. Blood vessels in muscles can be involved as part of a systemic vasculitis. Patients with the overlap syndrome of dermatomyositis and systemic sclerosis may have a specific antinuclear antibody, the anti-PM/Scl, directed against a nucleolar-protein complex. The treatment for inflammatory myopathies is similar to that for polymyositis and dermatomyositis (Kawachi et al., 2008; Nishigori et al., 2009).
Paraneoplastic Myopathy
Patients with paraneoplastic syndrome develop myalgias and rapid progression of weakness involving the extremities, pharyngeal and respiratory muscles, often resulting in death. Serum muscle enzymes are elevated, and muscle biopsy shows extensive necrosis with minimal or absent inflammation and sometimes deposits of complement. The disorder occurs as a paraneoplastic manifestation of a variety of cancers including small cell lung cancer and cancer of the gastrointestinal tract, breast, kidney, and prostate, among others. Glucocorticoids or treatment of the underlying tumor rarely control the disorder (Ramchandren and Dalmau, 2005; Antoine and Camdessanché, 2007; Kawachi et al., 2008). Apostolidis et al. (2009) have reported a rare case with dermatomyositis and non-hepatitis-associated advanced hepatocellular carcinoma with metastases. The dermatomyositis responded well to treatment with prednisolone and azathioprine (Apostolidis et al., 2009).
Myopathy from Infectious Disease
When acute weakness is associated with painful muscle cramps, rhabdomyolysis, and myoglobinuria, it may be due to metabolic disorders or infectious illness. Among the infectious etiologies – viral infections are more common. Influenza virus myositis is characterized by severe pain, tenderness and swelling, predominantly affecting the calves and to some extent may involve the thighs also. Myalgia is the most common symptoms and usually begins a week after the onset of influenza infections and can last for another 2–3 weeks. The disorder is usually self limited but severe cases are at risk for myoglobinuria and renal failure. Epidemics of Coxsackie virus group B can lead to self-limited acute inflammatory myopathy (Kumar et al., 2011).
The clinical and histopathologic features of HIV-associated myopathy are indistinguishable from those of idiopathic polymyositis, and the condition is often referred to as HIV polymyositis. It may occur at any stage of HIV infection, but is rarely the first clinical presentation of HIV. HIV polymyositis may range in severity from an asymptomatic elevation in creatine kinase levels to a subacute syndrome characterized by proximal weakness. HIV-associated inclusion body myositis as well as nemaline rod myopathy has been described as well (Dalakas et al., 2007; de Sanctis et al., 2008). Characteristics of polymyositis in HIV versus non-HIV infected patients have been compared. It has been observed that HIV polymyositis is treatable but may present with less than twofold CK elevation above normal (Heckmann et al., 2010). The treatment of HIV polymyositis is similar to that of idiopathic polymyositis and includes glucocorticoids, azathioprine, cyclophosphamide, and intravenous immunoglobulin (IVIgG). Immunosuppressants must be used cautiously in HIV patients as prolonged administration of oral glucocorticoids has been associated with an increased risk of opportunistic infections, and pulse intravenous glucocorticoids may be a more attractive alternative (Tien-Auh Chan et al., 2007; Kung et al., 2011).
Several animal parasites, such as protozoa (toxoplasma, trypanosoma), cestodes (cysticerci), and nematodes (trichinae), may produce a focal or diffuse inflammatory myopathy known as parasitic polymyositis. Kung et al. (2011) have recently reported a case of AIDS and toxoplasmosis presenting initially with myelitis in the absence of encephalitis and subsequently progressing to myositis despite antiparasitic treatment. American trypanosomiasis (Chagas disease) is caused by Trypanosoma cruzi and can cause inflammatory myopathy as well superimposed neuropathy. African trypanosomiasis on the other hand can manifest with myocarditis, polymyositis, and encephalopathy. Cysticercosis results from infection by Cysticercus cellulosae, the larval form of the pork tapeworm Taenia solium. The encysted parasite may be found in the skeletal and heart muscle, as well as the eye and brain. The clinical features may vary based upon the number and location of the cysts. Trichinosis is caused by larva of Trichinella spiralis, and is associated with fever, myalgia, proximal muscle weakness, periorbital, and facial edema. Trichinosis on occasion may mimic mild dermatomyositis.
Staphylococcus aureus, Yersinia, Streptococcus, or other anaerobic bacteria like clostridia may produce a suppurative myositis, known as topical polymyositis, or pyomyositis. Pyomyositis, previously rare in the west, is now occasionally seen in AIDS patients. Other bacteria, such as Borrelia burgdorferi (Lyme disease) and Legionella pneumophila (Legionnaires’ disease) may infrequently cause myositis (Crum-Cianflone, 2008; Al-Najar et al., 2010; Kung et al., 2011).
Fungal myositis is uncommon and may be seen in immunocompromised patients (e.g., AIDS or with malignancies). Sporotrichosis, histoplasmosis, mucormycosis, cryptococcosis, and candidiasis are all associated with myositis. Mucormycosis can spread to the orbit producing ophthalmoplegia, proptosis, and eyelid edema. Sporotrichosis and histoplasmosis can involve a single muscle or a group of muscles with abscess formation. Patients with disseminated candidiasis can develop papular rash, myalgia, and diffuse muscle weakness (Al-Najar et al., 2010).
Drugs- and Toxin-Induced Myopathies
Drug-induced myopathies are relatively uncommon in the clinical practice with the exception of those caused by the cholesterol-lowering agents and glucocorticoids. The knowledge of these myopathies remains critical since timely diagnosis allows for recovery (Kuncl, 2009; Abd and Jacobson, 2011).
Myopathy from lipid-lowering agents
All classes of lipid-lowering agents have been implicated in muscle toxicity including fibrates (clofibrate, gemfibrozil), HMG-CoA reductase inhibitors (statins), and niacin (nicotinic acid). Myalgia, malaise, and muscle tenderness are the most common manifestations. Muscle pain may be related to exercise. Patients may exhibit proximal weakness. Varying degrees of muscle necrosis are seen, and in severe reactions there are rhabdomyolysis and myoglobinuria. Toxicity is dose and time related, secondary to initiation of HMG-CoA reductase inhibitor (Kuncl, 2009). Concomitant use of statins with fibrates and cyclosporine is more likely to cause adverse reactions than use of one agent alone. Elevated serum CK is an important indication of toxicity. Muscle weakness is accompanied by a myopathic EMG, and muscle necrosis is observed by muscle biopsy (Findling et al., 2008; Radcliffe and Campbell, 2008). Myopathic reactions are indications for stopping the drug and patients improve with drug cessation. Most recently, switching to lower dose or even non-daily doses of long acting stating has been recommended as an alternate to discontinuing statins (Abd and Jacobson, 2011).
Glucocorticoid-related myopathies
Glucocorticoid myopathy is the most common type of drug-induced myopathy. It occurs with chronic treatment with proximal weakness accompanied by cushingoid manifestations. The chronic use of prednisone at a daily dose of ≥ 30 mg/day is most often associated with toxicity. Patients taking fluorinated glucocorticoids (triamcinolone, betamethasone, dexamethasone) appear to be especially high risk for myopathy. Patients receiving high-dose, intravenous glucocorticoids for status asthmaticus, chronic obstructive pulmonary disease, or other indications may develop severe generalized weakness. In chronic steroid myopathy the serum CK is usually normal but serum potassium may be low. In acute cases with quadriplegic myopathy, the muscle biopsy is abnormal and shows a distinctive loss of thick filaments (Myosin). The muscle biopsy in chronic cases shows preferential type 2 muscle fiber atrophy. EMG is usually normal as it measures type I fiber function only. Given the treatment is primarily reduction or discontinuation of steroids, it remains very essential to distinguish myopathy from glucocorticoid versus muscle inflammatory disease. In acute quadriplegic myopathy, recovery is slow and patients require supportive care and rehabilitation (Horak and Pourmand, 2000; Pereira and Freire de Carvalho, 2011).
Alcohol and drugs of abuse related myopathies
Myotoxicity is a potential consequence of addiction to alcohol and illicit drugs. Ethanol is one of the most commonly abused substances with potential to damage muscle. Direct muscle damage is less certain, since toxicity usually occurs in the setting of poor nutrition and possible contributing factors such as hypokalemia and hypophosphatemia. Acute muscle weakness with myoglobinuria may occur with prolonged severe hypokalemia, hypophosphatemia, or hypomagnesemia that is seen in chronic alcoholics and patients on nasogastric suction receiving parenteral hyperalimentation. Low vitamin D levels are related to muscle atrophy while changes in muscle antioxidant enzymes may play a role in alcoholic myopathy. Chronic alcoholic may develop painful myopathy with myoglobinuria after a bout of heavy drinking or present with a painless, acute hypokalemic myopathy that is completely reversible with replacement therapy (Noordzij et al., 2007; González-Reimers et al., 2010).
Drugs of abuse and focal myopathies
Other potential toxins include cocaine, heroin, and amphetamines. Coma and seizures, causing rhabdomyolysis, myoglobinuria, and renal failure are some complications (Gutiérrez-Gutiérrez, 2010). Focal myopathies from self-administration of meperidine, heroin, and pentazocine can cause pain, swelling, muscle necrosis, and hemorrhage. The cause is multifactorial including needle trauma, direct toxicity of the drug or vehicle, infection, non-granulomatous or granulomatous disorders such as sarcoidosis. When severe, there is skin induration and contractures with replacement of muscle by connective tissue can occur. Elevated serum CK is characteristic and peak value for blood myoglobin might be a good indicator of rhabdomyolysis induced acute renal failure. EMG is significant for myopathic findings and muscle biopsy reveals focal areas of necrosis. In focal myositis, ESR and CK are always within normal range. Histologically, hypertrophy of muscle fibers and internalization of nuclei are pronounced in focal myositis, but not in polymyositis. Focal myositis is self limited and usually regresses spontaneously (Chawla et al., 2005). Treatment is focused on rhabdomyolysis which require adequate hydration to reduce serum myoglobin and protection of renal function. In all of these conditions, counseling is essential to limit drug abuse (Streichenberger et al., 2004; Chawla et al., 2005; O’Connor and McMahon, 2008; Brancaccio et al., 2010; Kasaoka et al., 2010).
Other drug-induced myopathies
D-Penicillamine and procainamide may produce a true myositis resembling polymyositis. In 1989 an epidemic of eosinophilia–myalgia syndrome (dermatomyositis like illness) in the United States was caused by L-tryptophan, a contaminant in the product from one manufacturer. The incidence of this inflammatory muscle disease is about 1% (Silver et al., 1992; Klopstock, 2008; Rothschild, 2010). Myasthenia gravis is also induced by D-penicillamine, with a higher incidence estimated at 7%. These disorders resolve with drug withdrawal, although immunosuppressive therapy may be required in severe cases. Reports of other drugs causing an inflammatory myopathy include cimetidine, phenytoin, procainamide, and propylthiouracil. In most cases, a cause-and-effect relationship is uncertain.
Certain drugs produce painless, largely proximal, muscle weakness (please see Table 2). These drugs include the amphophilic cationic drugs (amiodarone, chloroquine, hydroxychloroquine) and antimicrotubular drugs (colchicine). Muscle biopsy can be useful in the identification of toxicity since autophagic vacuoles are prominent pathologic features of these toxins (Klopstock, 2008; Cantarini et al., 2010; Rothschild, 2010).
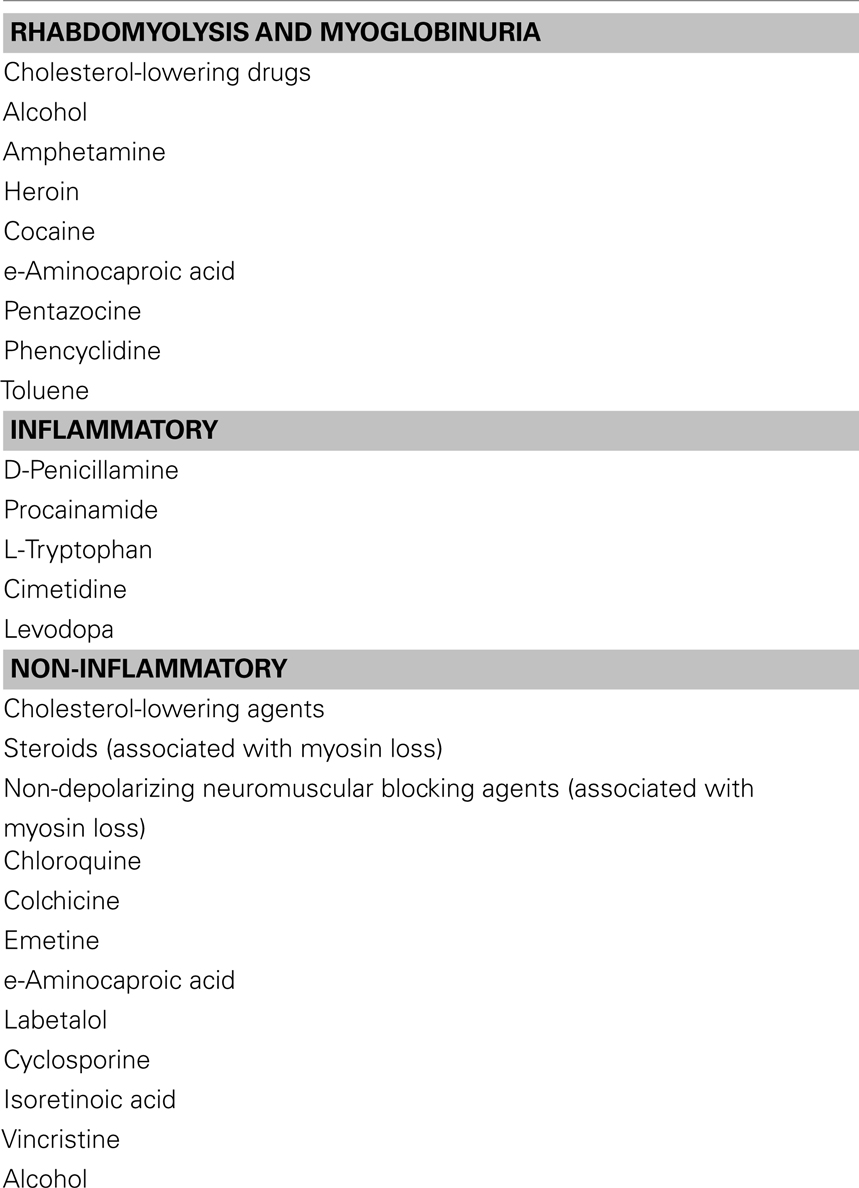
Table 2. Drugs induced toxic myopathies.
A myopathy similar to HIV polymyositis can be seen in patients following prolonged treatment with the Zidovudine (AZT). The toxic side effect of the drug is dose-dependent and is related to its ability to interfere with the function of mitochondrial polymerases. The complication occurs in about one fifth of patients treated with doses of 1200 mg/day for 6 months. It is usually reversible following discontinuation of the drug. Patients present with progressive proximal weakness and profound muscle wasting, often with muscle pain. Serum levels of CK are usually elevated, and EMG shows non-specific myopathic changes. Muscle biopsy reveals the histological hallmark of red ragged fibers. The introduction of protease inhibitors for treatment of HIV infection has led to lower doses of zidovudine therapy and a decreased incidence of myopathy (Scruggs and Dirks Naylor, 2008).
Critical Illness Myopathy
An acute myopathy affecting critically ill patients has been identified by different terms; including acute quadriplegic myopathy or critical illness myopathy (CIM). Unlike inflammatory myopathies, myalgias are less commonly seen in CIM. Patients may receive non-depolarizing neuromuscular blocking agents such as pancuronium, with or without glucocorticoids, may cause the acute CIM. The clinical features are identical to acute quadriplegic myopathy secondary to glucocorticoids. Major criteria include a critical illness (sepsis, multiple organ failure), difficulties with ventilator weaning, and possible limb weakness (Chawla, 2007; Chawla and Gruener, 2010; Fernández-Lorente et al., 2010). CIM may appear in various forms including thick filament myopathy, acute necrotizing myopathym, or cachectic myopathy.
Thick filament myopathy is mainly observed in patients with severe asthma often requiring ventilator support, high-dose corticosteroids, or neuromuscular blocking agents. Serum CK levels are mildly elevated. Muscle biopsy shows destruction of thick myosin filaments. Acute Necrotizing Myopathy is often precipitated by multiple etiologies causing myoglobinuria. Serum CK levels are markedly elevated. There is myoglobulinuria. Electrophysiological studies demonstrate a severe myopathy. Muscle biopsies show widespread necrosis of muscle fibers. Cachectic Myopathy is also known as disuse atrophy, it is responsible for marked wasting and weakness of muscles. Laboratory studies are generally non-specific. Muscle biopsy may show type 2 fiber atrophy. Cachectic myopathy remains a diagnosis of exclusion.
Muscle biopsy is occasionally indicated for exact treatment as well as prognosis in order to differentiate CIM from other myopathies or from critical illness polyneuropathy. Management of CIM requires avoiding corticosteroids and neuromuscular blocking agents or use the lowest possible dosages, and only for a short period of time. Prognosis is good with full recovery in mild to moderate cases. In severe cases, recovery may be longer or incomplete. Severe cases especially with necrotizing myopathy are associated with a high mortality rate (Chawla and Gruener, 2010).
Metabolic Myopathies
Evaluation of patients with metabolic causes of muscle weakness remains a challenge due to frequent lack of clinical symptoms. Typical manifestations include exercise induced myalgias, exercise intolerance, and cramps. Myopathies are caused by either excess or deficiencies of some of these minerals including potassium, calcium, phosphorous, magnesium, and vitamin D (Volpi et al., 2011).
Metabolic myopathies: Vitamin D deficiency
Vitamin D deficiency can occur from decreased intake, decreased absorption, or impaired vitamin D metabolism (as occurs in renal disease). Clinical manifestations include ataxic neuropathy due to loss of proprioception and myopathy with proximal weakness. Pain reflects the underlying bone disease (osteomalacia). Progressive external ophthalmoplegia is a distinctive finding. It has not been established that deficiency of other vitamins causes a myopathy. Screening for vitamin D deficiency is essential as one of the treatable causes of acquired myopathies (Prabhala et al., 2000; Noordzij et al., 2007; Al-Said et al., 2009).
Metabolic myopathies: Potassium and sodium related
Hypokalemia often causes proximal or generalized weakness that is usually painless. With acute hypokalemic paralysis, serum potassium is well below 3.0 mEq/L; weakness improves within 1–2 h and resolves over several hours or days with potassium replacement. If hypokalemia has been severe or associated with alcoholism, an acute necrotizing myopathy with rhabdomyolysis and renal failure may result, which improves partially or completely over days to weeks (Kotsaftis et al., 2009).
Hyperkalemia has severe and acute effects on both skeletal and cardiac muscle, causing rapidly ascending quadriparesis, frequent respiratory failure, and cardiac arrest. Serum potassium is usually > 7.5 mEq/L. CK and muscle biopsy are usually normal. Skeletal muscle weakness usually resolves within hours, even if serum potassium has not changed. The common presence of paresthesias and Chvostek’s signs suggests that hyperkalemia causes weakness more through its effects on nerve than muscle (Adeniyi et al., 2004).
Hypernatremia is rare but known to cause proximal muscle weakness. Serum CK and sodium level are elevated with myogenic changes on the EMG study. Muscle weakness improves while the hypernatremia is getting corrected. The pathophysiology is likely due to depletion of the intramuscular energy stores from an overworking Na–K pump to correct the intracellular electrolyte imbalance (Hiromatsu et al., 1994).
Metabolic myopathies: Calcium and phosphorous related
Abnormalities of calcium and phosphorus homeostasis and bone metabolism in chronic renal failure result from a reduction in 1,25-dihydroxyvitamin D, leading to decreased intestinal absorption of calcium.
Hypocalcemia causes paresthesias and motor dysfunction from tetany more than weakness with presence of Chvostek’s and Trousseau’s signs; thus its symptoms and signs are better explained by spontaneous peripheral nerve activity than by myopathy.
Hypercalcemia causes weakness with hyperreflexia, so this may relate more to central nervous system effects than those on muscle.
Severe hypophosphatemia (serum phosphate less than 0.4 mM/L) causes acute and severe areflexic weakness with rhabdomyolysis. Because mild hypophosphatemia can be caused by hyperventilation, only serum phosphorus levels less than 1.0 mM/L should be considered as severe enough to cause weakness or rhabdomyolysis (Amanzadeh and Reilly, 2006; Assadi, 2010).
The clinical picture of the myopathy of chronic renal failure is identical to that of primary hyperparathyroidism and osteomalacia. There is proximal limb weakness with bone pain. Gangrenous calcification, extensive skin necrosis, painful myopathy, and even myoglobinuria may occur (Srinivasan and Amato, 2003; Adeniyi et al., 2004; Ning et al., 2009).
Metabolic myopathies: Magnesium related
Hypomagnesemia is often associated with hypocalcemia and may contribute to signs of tetany with weakness.
Hypermagnesemia is rarely encountered and usually is due to exogenous administration of magnesium; if serum magnesium levels exceed 9.0 mEq/L, severe generalized weakness with respiratory failure usually develops (Adeniyi et al., 2004).
Myopathies in other systemic illness
Systemic illnesses such as chronic respiratory, cardiac, or hepatic failure are frequently associated with severe muscle wasting and complaints of weakness. Fatigue is usually a more significant problem than weakness, which is typically mild (Srinivasan and Amato, 2003; Adeniyi et al., 2004; Ning et al., 2009).
Step 2: Clinical Approach to a Patient with Muscle Weakness
A successful clinical approach to a patient with a suspected myopathy is based on a thorough medical history and neurological examination (Srinivasan and Amato, 2003; Jackson, 2008; Weis and Nolte, 2009; Selcen, 2011). The following systemic symptoms and signs are extremely helpful in narrowing the differential diagnosis.
• History and physical examination are critical components to appropriate diagnosis in any patient. It is a true saying – eyes cannot see those things if brain doesn’t know what to look for. Therefore, it remains essential to start knowing the pattern of the disease while establishing your differential diagnosis. Please see Table 3 for symptoms and signs associated with systemic myopathies.
• Neurological History should include at the least the following aspects:
◦ Fatigue is a very non-specific complaint. Many patients who complain of diffuse global weakness or fatigue do not have a disorder of muscle, particularly if the neurological examination is normal. On the other hand, abnormal fatigability after exercise can result from certain metabolic and mitochondrial myopathies in addition to neuromuscular junction disorders. In addition, duration and intensity of exercise which precipitates the fatigue is important (DiMauro, 2010; Schwager et al., 2010).
◦ Myalgias are episodic in metabolic myopathies or persistent in various inflammatory muscle disorders (Table 4 lists muscle disorders with associated myalgias). Cramps are usually benign and can occur with multiple conditions like dehydration, hyponatremia, azotemia, myxedema, etc. Contractures differ from cramps in that they usually last longer and are electrically silent with needle electromyography examination. Myoglobinuria is caused by the excessive release of myoglobin from muscle during periods of muscle destruction (rhabdomyolysis). Severe myoglobinuria (Table 5 lists acquired causes of myoglobinuria) can result in renal failure due to acute tubular necrosis. Patients generally complain of coke-colored or red urine during or after the exercise. Recurrent myoglobinuria is usually due to an underlying metabolic myopathy, whereas isolated episodes can occur in several conditions like rigorous exercise, infections, drugs and toxins, inflammatory myopathies, and malignant hyperthermia (Cervellin et al., 2010).
◦ Temporal evolutions of symptoms help you to narrow down your differential diagnosis. Know if the symptom presentation is acute, sub acute, or chronic, whether the symptoms are constant or episodic, and if they progressive or non-progressive. Identifying the age that symptoms began can provide crucial information leading to the correct diagnosis (please see Table 6 for age of onset and the diagnosis of myopathy). Myopathies related to systemic diseases generally present with either constant weakness unlike episodic periods of weakness with normal strength interictally (periodic paralysis, metabolic myopathies due to certain glycolytic pathway disorders). Ask for common precipitating factors including any relationship with exercise, any association with high fever or exposure to cold, any Critical Illness, and ICU stay, exposure to drugs or toxin or any correlation with carbohydrate meal (Srinivasan and Amato, 2003; Jackson, 2008).
• Neuromuscular Examination should look into and focus on the following aspects:
◦ Start with identification of the pattern of weakness. Look for if symmetrical or asymmetrical, whether the involvement of muscles is proximal > distal, distal > proximal or equal in both proximal and distal muscles. Don’t forget the involvement of cranial nerves with ocular, pharyngeal, and facial. Most common pattern of weakness in myopathies is symmetric, proximal > distal weakness in the upper and lower extremities. Neck muscles are frequently involved. Differential diagnosis of proximal acquired weakness consists of inflammatory myopathy, including polymyositis and dermatomyositis, endocrine myopathy, toxic- or drug-induced as well as CIM (Jackson, 2008; Distad et al., 2011).
◦ Proximal muscle weakness is associated with difficulty climbing stairs, arising from a chair, combing hair or lifting objects overhead. On the other hand, distal muscle weakness can lead to difficulty opening or closing jars, foot drop. Always make an attempt to look for cranial muscle weakness with ptosis, facial weakness, dysphagia, or any dysarthria.
◦ Look for other associated signs including myotonia, paramyotonia, and muscle hypertrophy to exclude other non-systemic causes of muscle weakness. Myotonia can be elicited by percussion, occurs by impaired relaxation of muscle due to repetitive depolarization of the muscle. Both myotonia and paramyotonia are worsened by exposure to cold. Myotonia improves with repeated exercise and paramyotonia is worsened by exercise. Also look for muscle hypertrophy which is seen with hypothyroid myopathy, amyloidosis, sarcoidosis, neoplastic, and inflammatory process.
◦ Please keep in consideration other system involvement: cardiac disease remains as the most important cause of mortality. Therefore identifying conditions with associated cardiac disease is mandatory in patient care. The clinical presentation of cardiac disease in these patients is either congestive heart failure or cardiac arrhythmias. Most are seen with genetic rather than acquired cause’s, e.g., polymyositis. Presence of hepatomegaly, optic atrophy, and/or pigmentary retinopathy should lead the examiner to explore causes of myopathies other than systemic disorders. The presence of a rash is extremely helpful in confirming the diagnosis of dermatomyositis and vasculitis. Family history is important to know and is actually important for genetic causes and less likely for myopathies with systemic illnesses (Jackson, 2008).
• Establishment of Differential Diagnosis based upon the pattern of disease process:
◦ Differential diagnosis remains a very crucial step in coming to the diagnosis and ultimate decisive process in the management of your patient. Understanding of the prior steps is essential as how to approach your next patient with muscle weakness and helps you identify a pattern of the disease (Srinivasan and Amato, 2003; Weis and Nolte, 2009; Marie, 2011; Selcen, 2011). The differential diagnosis of any systemic myopathy (please also see Table 1) should include the following:
• Endocrine myopathies
• Inflammatory myopathies
• Paraneoplastic myopathy
• Infectious myopathies
• Drug- and toxin-induced myopathies
• Critical illness myopathy
• Metabolic and myopathies with other systemic disorders

Table 3. Symptoms and signs associated with systemic myopathies.

Table 4. Muscle disorders associated with myalgias.

Table 5. Acquired causes of myoglobinuria.

Table 6. Differential diagnosis of systemic myopathy based on age of onset.
Step 3: Utility of Laboratory Testing Including Muscle Biopsy
Once you have established an appropriate differential diagnosis, the next step is to order the necessary testing’s. This is similar to establishing a hypothesis and ruling in and ruling out your diagnosis. Cost effective tests can be ordered by the priority of differential diagnosis. Commonly used tests in a patient with suspected myopathy include Blood: CK, Aldolase, SGOT, SGPT, and LDH, Neurophysiology: EMG/NCS, Muscle Biopsy, and Genetic testing. Genetic testing is not relevant for acquired systemic causes of muscle disorders.
The blood test of higher yield is CK although SGOT, SGPT, and LDH should also be tested. CK elevation can occur with or without muscle weakness, In addition, CK elevation is seen with certain medications and occasion without any known cause. Measurement of aldolase may be helpful in patient with mild weakness and normal CK’s (Nozaki and Pestronk, 2009).
Differential Diagnosis of CK Elevation with Weakness
• CK elevation can occur with weakness in myopathies of Inflammatory, metabolic, endocrine, drug- or toxin-induced, and viral illnesses. In addition, CK elevation can occur with weakness in other disorders including channelopathies, neuropathies (like GBS, CIDP), and motor neuron disease. Metabolic myopathy should be suspected in patients with sole exercise-related muscle pain and CK seven times higher than the normal (Filosto et al., 2007). Please see Table 7 for differential diagnosis of CK elevation.
Differential Diagnosis of CK Elevation without Weakness
• CK elevation is occasionally encountered in patients without any clinical weakness or any other expression of a neuromuscular disorder. It is still unresolved whether extensive investigations and specifically a muscle biopsy should be performed in clinically normal individuals with elevated CK levels. CK elevation without weakness can occur in strenuous exercise, after EMG studies, trauma including surgeries, intramuscular injections, carrier state (dystrophinopathy), “Idiopathic HyperCKemia,” metabolic and congenital myopathy, medications as well as African American’s. Please see Table 8 for effect of race and gender on CK measurement. Abnormal muscle biopsies were found in over half the patients with asymptomatic or minimally symptomatic hyperCKemia (Johnson et al., 1971; Joy and Oh, 1989; Dabby et al., 2006).
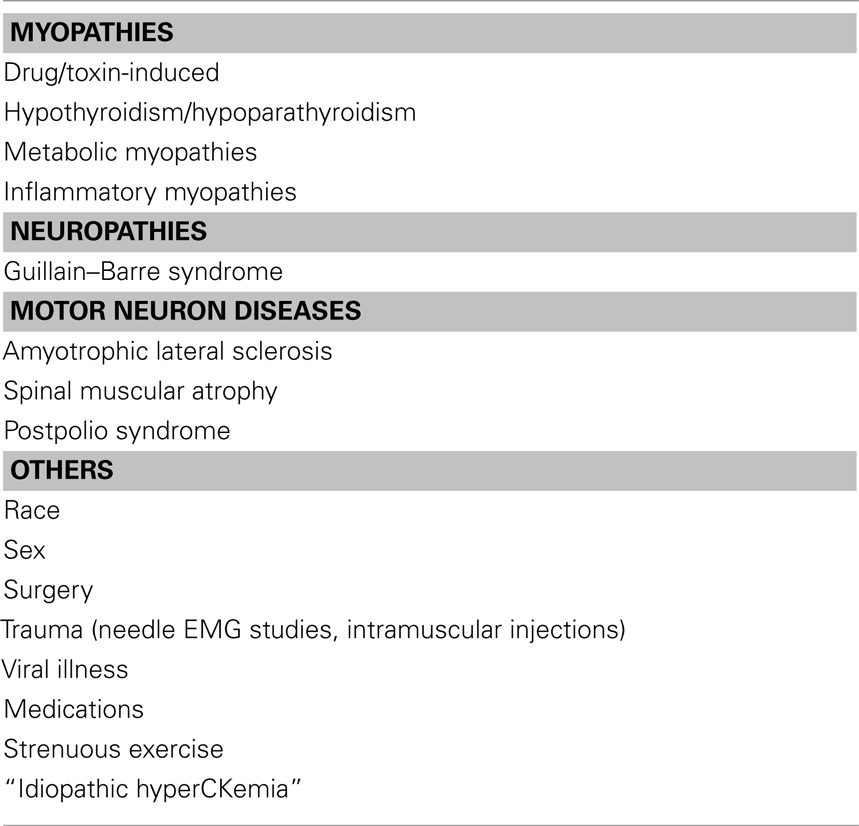
Table 7. Differential diagnosis of acquired causes of creatine kinase elevation.

Table 8. Effect of race and gender on creatine kinase measurements.
Medications and CK Elevations
• In addition, note should be made of certain medications elevating CK’s. These medications are Lipid-lowering drugs like HMG-CoA reductase inhibitors (Statins), Fibric acid derivatives (gemfibrozil), and Niacin. In addition, AZT, Cyclosporine, Chloroquine, and Colchicine are also found to increase the CK’s (Abdel-Hamid et al., 2008; Baker et al., 2008).
Role of EMG and Muscle Biopsy in Systemic Illnesses
• Nerve conduction studies and EMG examination are an extension of the neurological examination. The study is individualized based upon the neurological examination and differential diagnosis, and modified in real time as the study progresses to obtain further information. Localization of the disorder is the remains the major aim. In patients with pure weakness, electrodiagnostic studies can be useful to localize the disorder to the motor neuron/axon, neuromuscular junction or the muscle. The pattern of nerve conduction and more importantly EMG abnormalities usually can differentiate among these possibilities and help guide subsequent laboratory testing’s. Electrodiagnostic studies are also able to help determine the problem with sensory, motor or mixed fibers. It is also able to distinguish axonal versus demyelinating nerve pathology as well as temporal course of the disease including hyperacute, acute, subacute as well as chronic.
• Generally muscle biopsy is not required in myopathy associated with systemic illnesses. Most of the times a diagnosis can be made based upon – history, examination, blood tests as well as EMG’s. Genetic testing is mainly reserved for hereditary muscle disorders. Similar to the use of electrophysiological studies, biopsy results must be interpreted in the light of the clinical history and other laboratory studies.
• Muscle biopsy should least likely be considered in patients with normal strength and normal EMG. In one of the studies of patients with only muscle pain and a normal neurologic examination, the probability of having definite muscle pathology was found to be 2%. It is not recommended to biopsy muscles that are MRC grade ≤ 3 since results likely to show “end-stage” muscle. Last but not the least; avoid muscles that have been studied by EMG due to risk of false positive inflammatory results (Engel, 1967; Dabby et al., 2006; Filosto et al., 2007).
• Muscle biopsy remains essential to distinguish polymyositis from dermatomyositis. Given, dermatomyositis is primarily a B-cell disorder, there is 30% incidence of associated malignancies and therefore it remains practical to do muscle biopsy for appropriate, diagnosis, management, and counseling. Muscle biopsy is occasionally required to differentiate CIM from other myopathies or from critical illness polyneuropathy. It is also helpful for prognosis as severe cases especially with necrotizing myopathy are associated with a high mortality rate (Chawla and Gruener, 2010).
• Viral infections lead the volumes among infectious disorders and fortunately, the disorders are usually self limited without the need for a muscle biopsy (Kumar et al., 2011). On the other hand, the clinical and histopathologic features of HIV-associated myopathy are indistinguishable from those of idiopathic polymyositis, and the condition is often referred to as HIV polymyositis. HIV-associated inclusion body myositis as well as nemaline rod myopathy has been described as well Given the treatment of HIV polymyositis is similar to that of idiopathic polymyositis, in view of the increased risk of opportunistic infections with immunosuppressant’s, muscle biopsy can sometimes be helpful (Dalakas et al., 2007; de Sanctis et al., 2008).
• Several animal parasites including toxoplasma, trypanosome, cysticerci, and trichinae, may produce a focal or diffuse inflammatory myopathy known as parasitic polymyositis. Trichinosis on occasion may mimic mild dermatomyositis and may require muscle biopsy. Generally bacterial and fungal infections have other associated local and systemic symptoms that patients rarely need a muscle biopsy for the diagnosis (Al-Najar et al., 2010; Kung et al., 2011).
• Therefore, it remains very prudent for a meticulous selection of patients before performing a muscle biopsy. Although muscle biopsy remains a useful tool, molecular genetic studies are now available for non-invasive diagnosis of muscle diseases with a suspicion of non-systemic or hereditary disorders (Dabby et al., 2006; Filosto et al., 2007; Jackson, 2008; Nadaj-Pakleza et al., 2010; Selcen, 2011).
Conclusion
The step wise approach to a patient with muscle weakness can be extremely helpful in narrowing the differential diagnosis with a myopathy of systemic diseases. Without compromising the patient care, the bottom line remains in practicing cost effective medicine to confirm the diagnosis. Recognition is extremely important as most of these are reversible when diagnosed and managed in a timely fashion and thus carry a good prognosis.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abd, T. T., and Jacobson, T. A. (2011). Statin-induced myopathy: a review and update. Expert Opin. Drug Saf. 10, 373–387.
Abdel-Hamid, H., Oddis, C. V., and Lacomis, D. (2008). Severe hydroxychloroquine myopathy. Muscle Nerve 38, 1206–1210.
Adeniyi, O., Agaba, E. I., King, M., Servilla, K. S., Massie, L., and Tzamaloukas, A. H. (2004). Severe proximal myopathy in advanced renal failure. Diagnosis and management. Afr. J. Med. Med. Sci. 3, 385–388.
Al-Najar, M., Obeidat, F., Ajlouni, J., Mithqal, A., and Hadidy, A. (2010). Primary extensive pyomyositis in an immunocompetent patient: case report and literature review. Clin. Rheumatol. 29, 1469–1472.
Al-Said, Y. A., Al-Rached, H. S., Al-Qahtani, H. A., and Jan, M. M. (2009). Severe proximal myopathy with remarkable recovery after vitamin D treatment. Can. J. Neurol. Sci. 36, 336–339.
Amanzadeh, J., and Reilly, R. F. Jr. (2006). Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol. 2, 136–148.
Antoine, J. C., and Camdessanché, J. P. (2007). Peripheral nervous system involvement in patients with cancer. Lancet Neurol. 6, 75–86.
Apostolidis, L., Kahlert, C., Siegmund, A., Thom, R., Horstmann, S., Jäger, D., and Lordick, F. (2009). Remission of paraneoplastic dermatomyositis associated with hepatocellular carcinoma under prednisolone and azathiopin, and concommittant sorafenib. Onkologie 32, 50–53.
Assadi, F. (2010). Hypophosphatemia: an evidence-based problem-solving approach to clinical cases. Iran. J. Kidney Dis. 4, 195–201.
Baker, S. K., Vladutiu, G. D., Peltier, W. L., Isackson, P. J., and Tarnopolsky, M. A. (2008). Metabolic myopathies discovered during investigations of statin myopathy. Can. J. Neurol. Sci. 35, 94–97.
Brancaccio, P., Lippi, G., and Maffulli, N. (2010). Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 48, 757–767.
Cantarini, L., Volpi, N., Galeazzi, M., Giani, T., Fanti, F., Lucherini, O. M., Aglianò, M., Alessandrini, C., Giannini, F., Magi, S., Greco, G., Baldari, C. T., and Cimaz, R. (2010). Colchicine myopathy and neuromyopathy: two cases with different characteristics. J. Clin. Rheumatol. 16, 229–232.
Cervellin, G., Comelli, I., and Lippi, G. (2010). Rhabdomyolysis: historical background, clinical, diagnostic and therapeutic features. Clin. Chem. Lab. Med. 48, 749–56.
Chawla, J. (2007). “Critical illness polyneuropathy and myopathy,” in The Interface of Neurology and Internal Medicine, ed. J. Biller (Philadelphia, PA: Lippincott Williams & Wilkins), 809–812.
Chawla, J., and Gruener, G. (2010). Management of critical illness polyneuropathy and myopathy. Neurol. Clin. 28, 961–977.
Chawla, J., Thomas, C., and Fisher, M. (2005). Focal nodular sarcoid myositis: case report and literature review. Case Rep. Clin. Pract. Rev. 6, 131–135.
Cicala, M. V., and Mantero, F. (2010). Primary aldosteronism: what consensus for the diagnosis. Best Pract. Res. Clin. Endocrinol. Metab. 24, 915–921.
Crum-Cianflone, N. F. (2008). Bacterial, fungal, parasitic, and viral myositis. Clin. Microbiol. Rev. 21, 473–494.
Dabby, R., Sadeh, M., Herman, O., Berger, E., Watemberg, N., Hayek, S., Jossiphov, J., and Nevo, Y. (2006). Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr. Med. Assoc. J. 8, 110–113.
Dalakas, M. C., Rakocevic, G., Shatunov, A., Goldfarb, L., Raju, R., and Salajegheh, M. (2007). Inclusion body myositis with human immunodeficiency virus infection: four cases with clonal expansion of viral-specific T cells. Ann. Neurol. 61, 466–475.
de Sanctis, J. T., Cumbo-Nacheli, G., Dobbie, D., and Baumgartner, D. (2008). HIV-associated nemaline rod myopathy: role of intravenous immunoglobulin therapy in two persons with HIV/AIDS. AIDS Read. 18, 90–94.
Del Porto, L. A., Liubinas, S. V., and Kaye, A. H. (2011). Treatment of persistent and recurrent acromegaly. J. Clin. Neurosci. 18, 181–190.
DiMauro, S. (2010). Pathogenesis and treatment of mitochondrial myopathies: recent advances. Acta Myol. 29, 333–338.
Distad, B. J., Amato, A. A., and Weiss, M. D. (2011). Inflammatory myopathies. Curr. Treat. Options Neurol. 13, 119–130.
Engel, W. K. (1967). Focal myopathic changes produced by electromyographic and hypodermic needles. “Needle myopathy.” Arch. Neurol. 16, 509–511.
Fernández-Lorente, J., Esteban, A., Salinero, E., Traba, A., Prieto, J., and Palencia, E. (2010). Critical illness myopathy. Neurophysiological and muscular biopsy assessment in 33 patients. Rev. Neurol. 50, 718–726.
Filosto, M., Tonin, P., Vattemi, G., Bertolasi, L., Simonati, A., Rizzuto, N., and Tomelleri, G. (2007). The role of muscle biopsy in investigating isolated muscle pain. Neurology 68, 181–186.
Findling, O., Meier, N., Sellner, J., Nedeltchev, K., and Arnold, M. (2008). Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 71, e34–e37.
González-Reimers, E., Durán-Castellón, M. C., López-Lirola, A., Santolaria-Fernández, F., Abreu-González, P., Alvisa-Negrín, J., and Sánchez-Pérez, M. J. (2010). Alcoholic myopathy: vitamin D deficiency is related to muscle fibre atrophy in a murine model. Alcohol Alcohol. 45, 223–230.
Gutiérrez-Gutiérrez, G. (2010). Toxic myopathies caused by industrial, animal and vegetal toxins. Rev. Neurol. 51, 360–366.
Habib, Z., and Camacho, P. (2010). Primary hyperparathyroidism: an update. Curr. Opin. Endocrinol. Diabetes Obes. 17, 554–560.
Heckmann, J. M., Pillay, K., Hearn, A. P., and Kenyon, C. (2010). Polymyositis in African HIV-infected subjects. Neuromuscul. Disord. 20, 735–739.
Hiromatsu, K., Kobayashi, T., Fujii, N., Itoyama, Y., Goto, I., and Murakami, J. (1994). Hypernatremic myopathy. J. Neurol. Sci. 122, 144–147.
Johnson, E. W., Braddom, R., and Watson, R. (1971). Electromyographic abnormalities after intramuscular injections. Arch. Phys. Med. Rehabil. 52, 250–252.
Joy, J. L., and Oh, S. J. (1989). Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 12, 206–209.
Kanda, F., Okuda, S., Matsushita, T., Takatani, K., Kimura, K. I., and Chihara, K. (2001). Steroid myopathy: pathogenesis and effects of growth hormone and insulin-like growth factor-I administration. Horm. Res. 56(Suppl. 1), 24–28.
Kasaoka, S., Todani, M., Kaneko, T., Kawamura, Y., Oda, Y., Tsuruta, R., and Maekawa, T. (2010). Peak value of blood myoglobin predicts acute renal failure induced by rhabdomyolysis. J. Crit. Care. 25, 601–604.
Kawachi, Y., Koike, Y., Kano, T., Furuta, J., Fujisawa, Y., Nakamura, Y., Ishii, Y., Takahashi, T., and Otsuka, F. (2008). Paraneoplastic dermatomyositis triggered and exacerbated by oral 5-fluorouracil administration. Eur. J. Dermatol. 18, 195–196.
Kotsaftis, P., Savopoulos, C., Agapakis, D., Ntaios, G., Tzioufa, V., Papadopoulos, V., Fahantidis, E., and Hatzitolios, A. (2009). Hypokalemia induced myopathy as first manifestation of primary hyperaldosteronism – an elderly patient with unilateral adrenal hyperplasia: a case report. Cases J. 2, 6813.
Kumar, K., Guirgis, M., Zieroth, S., Lo, E., Menkis, A. H., Arora, R. C., and Freed, D. H. (2011). Influenza myocarditis and myositis: case presentation and review of the literature. Can. J. Cardiol. 27, 514–522.
Kung, A. W. (2007). Neuromuscular complications of thyrotoxicosis. Clin. Endocrinol. (Oxf.) 67, 645–650.
Kung, D. H., Hubenthal, E. A., Kwan, J. Y., Shelburne, S. A., Goodman, J. C., and Kass, J. S. (2011). Toxoplasmosis myelopathy and myopathy in an AIDS patient: a case of immune reconstitution inflammatory syndrome? Neurologist 17, 49–51.
Mansourian, A. R. (2010). A review on hyperthyroidism: thyrotoxicosis under surveillance. Pak. J. Biol. Sci. 13, 1066–1076.
Nadaj-Pakleza, A., Kierdaszuk, B., and Kaminska, A. (2010). The role of skeletal muscle biopsy in the diagnosis of neuromuscular disorders. Neurol. Neurochir. Pol. 44, 481–491.
Ning, L., Sippel, R., Schaefer, S., and Chen, H. (2009). What is the clinical significance of an elevated parathyroid hormone level after curative surgery for primary hyperparathyroidism? Ann. Surg. 249, 469–472.
Nishigori, K., Yamamoto, T., and Yokozeki, H. (2009). Vesiculo-bullous dermatomyositis: report of three cases. Dermatol. Online J. 15, 6.
Noordzij, M., Boeschoten, E. W., Bos, W. J., Dekker, F. W., Bossuyt, P. M., Krediet, R. T., Krediet, R. T., Korevaar, J. C., and for the NECOSAD Study Group. (2007). Disturbed mineral metabolism is associated with muscle and skin complaints in a prospective cohort of dialysis patients. Nephrol. Dial. Transplant. 22, 2944–2949.
Nozaki, K., and Pestronk, A. (2009). High aldolase with normal creatine kinase in serum predicts a myopathy with perimysial pathology. J. Neurol. Neurosurg. Psychiatr. 80, 904–908.
O’Connor, G., and McMahon, G. (2008). Complications of heroin abuse. Eur. J. Emerg. Med. 15, 104–106.
Pereira, R. M., and Freire de Carvalho, J. (2011). Glucocorticoid-induced myopathy. Joint Bone Spine 78, 41–44.
Prabhala, A., Garg, R., and Dandona, P. (2000). Severe myopathy associated with vitamin D deficiency in western New York. Arch. Intern. Med. 160, 1199–1203.
Radcliffe, K. A., and Campbell, W. W. (2008). Statin myopathy. Curr. Neurol. Neurosci. Rep. 8, 66–72.
Ramchandren, S., and Dalmau, J. (2005). Metastases to the peripheral nervous system. J. Neurooncol. 75, 101–110.
Schwager, S., Hochuli, M., and Jung, H. H. (2010). Muscle weakness: differential diagnosis and assessment. Praxis (Bern 1994). 99, 1121–1129; quiz 1129–1130.
Scruggs, E. R., and Dirks Naylor, A. J. (2008). Mechanisms of zidovudine-induced mitochondrial toxicity and myopathy. Pharmacology 82, 83–88.
Silver, R. M., Sutherland, S. E., Carreira, P., and Heyes, M. P. (1992). Alterations in tryptophan metabolism in the toxic oil syndrome and in the eosinophilia-myalgia syndrome. J. Rheumatol. 19, 69–73.
Streichenberger, N., Meyronet, D., Fiere, V., Pellissier, J. F., and Petiot, P. (2004). Focal myositis associated with S-1 radiculopathy: report of two cases. Muscle Nerve 29, 443–446.
Syriou, V., Kolitsa, A., Pantazi, L., and Pikazis, D. (2005). Hypoparathyroidism in a patient presenting with severe myopathy and skin rash. Case report and review of the literature. Hormones (Athens) 4, 161–164.
Tien-Auh Chan, A., Kirton, C., Estanislao, L., and Simpson, D. M. (2007). Myopathy in HIV infection. Handb. Clin. Neurol. 85, 139–145.
Tsuda, E., Imai, T., Matsumura, A., Hisahara, S., Nonaka, M., Shiraishi, H., Motomura, M., and Shimohama, S. (2008). Thyrotoxic myopathy mimicking myasthenic syndrome associated with thymic hyperplasia. Intern. Med. 47, 445–457.
Turker, H., Bayrak, O., Gungor, L., Yilmaz, A., Terzi, M., Turker, C. M., Onar, M. K., and Kahraman, H. (2008). Hypothyroid myopathy with manifestations of Hoffman’s syndrome and myasthenia gravis. Thyroid 18, 259–262.
Volpi, L., Ricci, G., Orsucci, D., Alessi, R., Bertolucci, F., Piazza, S., Simoncini, C., Mancuso, M., and Siciliano, G. (2011). Metabolic myopathies: functional evaluation by different exercise testing approaches. Musculoskelet. Surg. 95, 59–67.
Weis, J., and Nolte, K. (2009). Inflammatory and other myopathies and skeletal muscle vasculitis: the role of muscle and nerve biopsy. Z. Rheumatol. 68, 459–464.
Keywords: CK, EMG, HyperCKemia, IVIgG, CIM, HMG-CoA, HIV
Citation: Chawla J (2011) Stepwise approach to myopathy in systemic disease. Front. Neur. 2:49. doi: 10.3389/fneur.2011.00049
Received: 13 March 2011;
Accepted: 14 July 2011;
Published online: 05 August 2011.
Edited by:
Guy Rouleau, University of Montreal, CanadaReviewed by:
Nicolas Dupre, Laval University, CanadaWalter George Bradley, University of Miami Miller School of Medicine, USA
Copyright: © 2011 Chawla. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jasvinder Chawla, Chief of Neurology, Hines VA Hospital and Neurology Residency Program Director, Loyola University Medical Center, Hines, IL, USA. e-mail:amFzdmluZGVyLmNoYXdsYTJAdmEuZ292