 Thomas A. Quinn
Thomas A. Quinn
- F.M. Kirby Center for Molecular Ophthalmology, Department of Ophthalmology, University of Pennsylvania Scheie Eye Institute, Philadelphia, PA, USA
Multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) are neurodegenerative diseases with characteristic inflammatory demyelination in the central nervous system, including the optic nerve. Neuronal and axonal damage is considered to be the main cause of long-term disability in patients with MS. Neuronal loss, including retinal ganglion cell (RGC) apoptosis in eyes with optic neuritis (ON), also occurs in EAE. However, there is significant variability in the clinical course and level of neuronal damage in MS and EAE. The current studies examine the mechanisms and kinetics of RGC loss in C57/BL6 mice immunized with myelin oligodendrocyte glycoprotein to induce a chronic EAE disease. Clinical progression of EAE was scored daily and vision was assessed by optokinetic responses. At various time points, RGCs were counted and optic nerves were examined for inflammatory cell infiltration. Almost all EAE mice develop ON by day 15 post-immunization; however, RGC loss is delayed in these mice. No RGC loss is detected 25 days post-immunization, whereas RGC numbers in EAE mice significantly and progressively decrease compared to controls from 35 to 50 days post-immunization. The delayed time course of RGC loss is in stark contrast to that reported in relapsing EAE, as well as in rats with chronic EAE. Results suggest that different clinical disease courses of optic nerve inflammation may trigger distinct mechanisms of neuronal damage, or RGCs in different rodent strains may have variable resistance to neuronal degeneration.
Introduction
Multiple sclerosis (MS) is an autoimmune-mediated neurodegenerative disease with characteristic inflammatory demyelination in the central nervous system (Constantinescu et al., 1998; Noseworthy et al., 2000). It has become increasingly recognized that MS may not be a single disease entity, but instead involve different immunopathological processes with various clinical phenotypes (Bettelli, 2007). The first clinical presentation in many MS patients is optic neuritis (ON; Arnold, 2005). ON manifests as an acute, self-limited episode of optic nerve inflammation with decreased vision that recovers over several weeks in the majority of patients; however, permanent visual symptoms can persist in 40–60% of patients, and repeat episodes of ON can lead to significant optic nerve atrophy (Beck et al., 2004; Arnold, 2005).
Multiple sclerosis was considered primarily a demyelinating disease in the past, however increasing evidence suggests disease progression and permanent disability in both MS and ON depend upon accumulated axon degeneration (Losseff et al., 1996; Fisher et al., 2006). Axonal degeneration measured by retinal imaging, including thinning of the retinal nerve fiber layer and decreased macular volume, occurs in patients after a single clinical episode of ON and correlates with the level of residual visual dysfunction (Steel and Waldock, 1998; Parisi et al., 1999; Trip et al., 2005; Costello et al., 2006; Fisher et al., 2006). Unfortunately, the precise mechanisms leading to this neurodegeneration remain unknown.
Determining mechanisms underlying neurodegeneration in MS and ON is difficult due to the range of clinical and histological manifestations seen in patients, suggesting that mechanisms may vary between different types of MS. Research on the pathophysiology of MS and associated ON is enhanced by the animal model experimental autoimmune encephalomyelitis (EAE). Different clinical disease courses of EAE that correlate with those seen in MS, including chronic progressive, monophasic, and relapsing/remitting, have been described and depend on the type of immunizing myelin protein and animal strain used (Fritz et al., 1983; Tuohy et al., 1988; Constantinescu et al., 1998; Storch et al., 1998). C57/BL6 mice immunized with myelin oligodendrocyte glycoprotein (MOG) exhibit a chronic EAE course (Mendel et al., 1995). MOG-induced EAE especially reflects some pathophysiological features of MS (Storch et al., 1998), including both an encephalitogenic T-cell response and a demyelinating autoantibody response (Iglesias et al., 2001).
Significant retinal ganglion cell (RGC) loss by apoptosis has been demonstrated in several experimental models of ON. In a mouse model of relapsing/remitting EAE, RGC loss begins within a few days after onset of optic nerve inflammation (Shindler et al., 2006, 2008), suggesting RGC axonal damage, and cell loss is induced by inflammation. In contrast, RGC loss occurs much earlier in a rat model of chronic EAE (Meyer et al., 2001; Hobom et al., 2004), with an initial wave of RGC loss occurring before optic nerve inflammation is detected, suggesting there may be some direct injury to RGC axons in addition to damage induced secondary to inflammatory cell infiltration.
Although development of ON has been described in C57/BL6 EAE mice immunized with MOG (Shao et al., 2004), RGC loss was not reported. The timing and extent of RGC loss in this chronic mouse EAE model is examined in the current studies, and demonstrates important differences from other experimental ON models.
Materials and Methods
Experimental Animals
Female C57/BL6 mice (6–7 week old) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Housing and treatment of animals conformed to the Institutional Animal Care and Use Committee Guidelines at the University of Pennsylvania.
RGC Labeling
Retrograde labeling of RGCs was performed using the protocol from previous studies (Shindler et al., 2006, 2008). Briefly, mice were anesthetized intraperitoneally with 2 mg ketamine (Sigma, St. Louis, MO, USA) and 1 mg xylazine (Sigma). Heads were shaved, a mediosagittal incision was made and holes were drilled bilaterally through the skull above the superior colliculi. About 2.5 μl of 1.25% hydroxystilbamidine (Fluorogold; Molecular Probes, Eugene, OR, USA) in sterile phosphate buffered saline (PBS) was injected stereotactically into the superior colliculi.
Induction of EAE
Experimental autoimmune encephalomyelitis immunization was performed 1 week after RGC labeling to allow time for retrograde transport of fluorogold into RGC bodies. To induce EAE, mice were anesthetized with ketamine/xylazine solution and injected subcutaneously at two sites along the back with 0.1 ml solution containing 150 μg MOG (Invitrogen, Carlsbad, CA, USA) emulsified with complete Freund’s adjuvant (CFA; Difco, Detroit, MI, USA) containing 2.5 mg/ml mycobacterium tuberculosis (Difco). Control mice were sham immunized with an equal volume of PBS and CFA. MOG and control mice were injected with 200 ng pertussis toxin (List Biological, Campbell, CA, USA) in 0.1 ml PBS on day 0 (day of immunization) and again on day 2. Clinical progression of EAE was scored daily according to the following criteria (Gran et al., 2002): no disease = 0; partial tail paralysis = 0.5; tail paralysis or waddling gait = 1.0; partial tail paralysis and waddling gait = 1.5; tail paralysis and waddling gait = 2.0; partial limb paralysis = 2.5; paralysis of one limb = 3.0; paralysis of one limb and partial paralysis of another = 3.5; paralysis of two limbs = 4.0; moribund state = 4.5; death = 5.0.
Quantification of RGC Numbers
Retinal ganglion cell were counted as in prior studies (Shindler et al., 2006). Mice were sacrificed 15, 25, 35, 40, or 50 days after immunization. Eyes were removed and fixed in 4% paraformaldehyde in PBS. Retinas were dissected, flat mounted on glass slides, and viewed by fluorescent microscopy (Nikon Eclipse E600). Photographs of RGCs were taken in 12 standardized fields (1/6, 3/6, 5/6 of the retinal radius in each quadrant) at 20× magnification. The number of fluorescent-labeled RGCs in each field was counted using Image-Pro Plus 5.0 (Media Cybernetics, Silver Spring, MD, USA) software by an investigator who was blinded to the experimental treatment. Data shown represent the total number of RGCs counted per retina (12 pictures/retina), covering a combined total area of 0.74 mm2.
Optic Nerve Histopathology
On the days of sacrifice, optic nerves were removed and fixed in 4% paraformaldehyde in PBS. ONs were embedded in paraffin, cut into 5 μm thick longitudinal sections, and stained with hematoxylin and eosin. Presence of inflammatory cell infiltration in the optic nerves was assessed by a blinded investigator similar to published criteria (Shao et al., 2004): no infiltration = 0; mild cellular infiltration of the optic nerve or optic nerve sheath = 1; moderate infiltration = 2; severe infiltration = 3; massive infiltration = 4.
Optokinetic Responses
Visual function was assessed by the optokinetic tracking response (OKR) using OptoMotry software and apparatus (Cerebral Mechanics), as described previously (Prusky et al., 2004). Briefly, mice are placed on a platform in a closed chamber and a virtual cylinder of a 100% contrast grating is projected at varying spatial frequencies. Mice are observed through a camera, and the investigator assesses whether the mice track the rotating cylinder. Visual function is reflected by the highest spatial frequency at which mice track and is recorded as cycles/degree. Mice only track in one direction with each eye, therefore separate vision measures are recorded for each eye by alternating the direction of rotation of the cylinder.
Statistics
Statistical analyses were performed by ANOVA for comparison of RGC numbers in control eyes versus EAE eyes with and without ON, and ANOVA of repeated measures for OKR responses. To compare RGC numbers in EAE eyes with control eyes, group means were compared by Student’s t-tests. Statistics were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Results
Chronic Progressive Course of EAE Induced by MOG
C57/BL6 mice immunized with MOG developed chronic EAE symptoms (Figure 1), similar to prior studies (Mendel et al., 1995; Gran et al., 2002). Clinical signs of EAE began by day 8 post-immunization, and increased over the next several days before peaking and then reaching a plateau. A similar course of EAE was observed in three additional experiments.
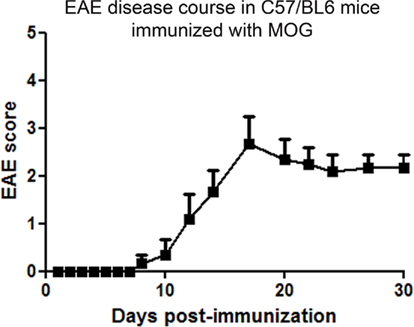
Figure 1. Chronic progressive course of clinical EAE. C57/BL6 mice were immunized with MOG on day 0 and injected with pertussis toxin on day 0 and 2. Mice were observed daily for clinical signs of EAE. Clinical EAE was first detected at day 8 after immunization, and increased through day 17 before reaching a chronic level. One of four experiments is shown. Data represent average (mean ± SEM) EAE score of n = 6 mice.
Optic Neuritis in EAE Mice
Mice were sacrificed 15, 25, 35, 40, or 50 days following immunization with MOG. Optic nerves from control mice demonstrated normal histology without presence of inflammatory cell infiltrates (Figure 2A). Inflammatory cell infiltrates detected in most optic nerves from MOG-immunized EAE mice confirmed the presence of ON (Figure 2B), whereas optic nerves from EAE mice that demonstrated normal histology (Figure 2C) were considered either not to have developed ON, or to have had resolution of inflammation prior to sacrifice. More than 80% of optic nerves developed inflammation by days 15 and 25 post-immunization, with inflammation remaining detectable in more than 60% of optic nerves through day 50 (Figure 2D).
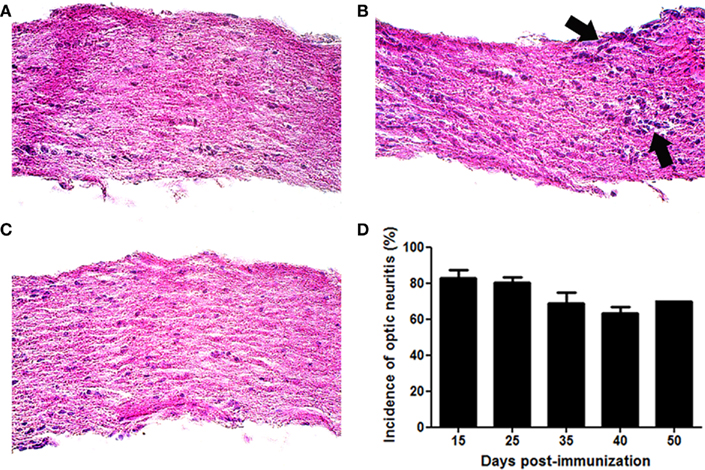
Figure 2. Experimental autoimmune encephalomyelitis eyes develop optic neuritis. C57/BL6 mice were immunized with MOG and sacrificed 15, 25, 35, 40, or 50 days later. Longitudinal sections of isolated optic nerves were stained with H&E and examined for the presence of inflammatory cell infiltrates. (A) Control optic nerve showing normal histology. (B) Foci of increased cellularity (arrows) show mild inflammation observed in an EAE eye with ON. (C) EAE optic nerve without ON. (D) Most optic nerves develop inflammation by day 15 post-immunization. The percentage of eyes with optic nerve inflammation is shown. Data represent the average (mean ± SEM) incidence of ON at each time point (n = 3–4 separate experiments/time point, except day 50 which represents a single experiment). Photographs (A–C) shown at original magnification ×40.
EAE Induces RGC Loss
Significant loss of RGCs was observed in eyes from EAE mice sacrificed 50 days post-immunization (Figure 3), the latest time point examined. Retinas from control mice had an average of 960.1 ± 91.5 RGCs/eye (n = 8). Significantly fewer RGCs were present in the retinas of EAE mice, both from eyes with histological evidence of ON (592.6 ± 128.4 RGCs, n = 7; p = 0.00003), and from eyes without ON (720.3 ± 77.4 RGCs, n = 3; p = 0.006). Although a trend toward fewer RGCs was observed in the retinas of mice of EAE with ON compared to EAE eyes without ON, the difference was not significant.
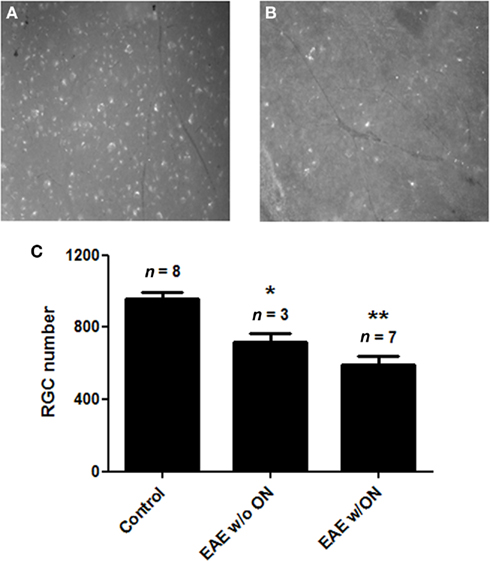
Figure 3. Retinal ganglion cell numbers decreased in EAE eyes 50 days after immunization. RGCs of C57/BL6 mice were retrogradely labeled with fluorogold by stereotactic injection into the superior colliculi. Mice were immunized with MOG or sham injected with PBS, and were sacrificed 50 days post-immunization. Dissected retina from each eye was viewed by fluorescent microscopy. Optic nerve from each eye was examined histologically for presence of inflammatory cell infiltrates. (A) Representative photograph shown at original magnification ×20 demonstrates numerous fluorescent-labeled RGCs observed in retina from a control (non-EAE) mouse eye. (B) Fewer RGCs found in the retina from an EAE mouse eye. (C) RGC numbers were counted in 12 standardized fields from each retina. The number of RGCs present in control eyes was significantly higher than the number of RGCs in eyes from EAE mice that did not contain histologic evidence of ON (*p = 0.006) and EAE eyes with ON (**p = 0.00003).
The onset of RGC loss in chronic EAE mice was evaluated by quantifying RGC numbers at earlier time points; 15, 25, 35, and 40 days post-immunization. No difference in RGC numbers was found between control mouse eyes and EAE mouse eyes with ON at either 15 (Figure 4A) or 25 (Figure 4B) days post-immunization. By 35 days post-immunization, significant RGC loss was detected in EAE eyes with ON as compared to controls (Figure 4C), with similar RGC loss present at day 40 (Figure 4D).
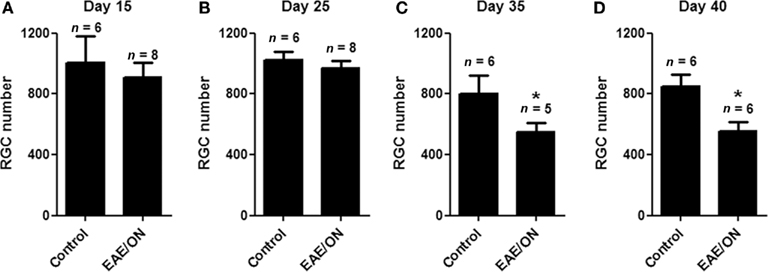
Figure 4. Retinal ganglion cell loss occurs more than 25 days after immunization to induce EAE. RGCs were labeled with fluorogold, and mice were immunized with MOG or sham injected with PBS. Mice were euthanized 15, 25, 35, or 40 days post-immunization and isolated retina from each eye was viewed by fluorescent microscopy. Optic nerves were examined histologically for presence of inflammatory cell infiltrates. (A) No difference in RGC numbers was found between control eyes and EAE eyes with ON 15 days post-immunization. (B) No difference in RGC numbers was observed at day 25. (C) At day 35, EAE ON eyes had fewer remaining RGCs than control eyes (*p = 0.048). (D) At day 40, similar RGC loss was found in EAE ON eyes (*p = 0.026).
Vision loss in EAE mice
To examine whether experimental ON leads to significant vision loss, and determine where in the disease course vision loss occurs, visual function was measured by OKR in a group of control and EAE mice. Baseline OKR responses were recorded prior to immunization, then repeated at several time points through day 35 post-immunization (Figure 5). OKR responses were equivalent between control and EAE mouse eyes prior to day 30 post-immunization, but then showed a significant drop in vision in EAE mouse eyes compared to control mouse eyes at both day 30 (p = 0.05) and day 35 (p = 0.019).
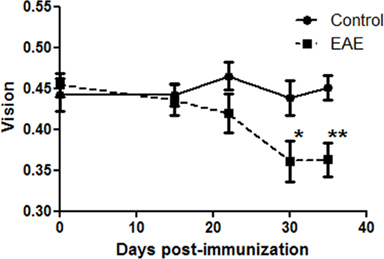
Figure 5. Vision decreased in EAE mice by 30 days post-immunization. OKR responses were recorded in C57/BL6 mice prior to immunization (time 0). Vision is reported as the threshold cycles/degree tracked on a rotating cylinder. EAE mice were immunized with MOG, and control mice were sham injected with PBS, on day 0, and OKR responses were recorded 15, 22, 30, and 35 days later. OKR responses were significantly reduced in EAE eyes as compared to control eyes at day 30 (*p = 0.05) and day 35 (**p = 0.019) post-immunization.
Discussion
Results of the current studies show that immunization of C57/BL6 mice with MOG induces a chronic EAE disease course with a high incidence of ON, consistent with prior studies (Mendel et al., 1995; Shao et al., 2004). While Shao et al. (2004) previously described the development of ON in these mice, RGC degeneration was not investigated. Our results demonstrate that there is also a significant component of neuronal degeneration in this experimental ON model. Interestingly, RGC loss was observed in EAE eyes with or without histological evidence of ON. This reflects the likelihood that almost all eyes did indeed develop ON, but active inflammation can resolve prior to sacrifice in some animals and therefore is missed on histological analysis.
Retinal ganglion cell loss in this chronic EAE model is delayed by 2 weeks beyond the onset of optic nerve inflammation, making it distinct from other reported experimental ON models. In relapsing/remitting mouse EAE, significant RGC loss occurs within 4–5 days of the onset of optic nerve inflammation (Shindler et al., 2006, 2008). Differences in the timing of RGC loss may represent differences in the magnitude or phenotype of the inflammatory response induced by distinct antigens. Alternatively, different mouse strains may have varying levels of resistance to neurodegeneration based on differential genetic backgrounds or specific genetic mutations. Mice with the Wallerian degeneration slow mutation, for example, demonstrate delayed axonal degeneration in injured peripheral nerves (Lunn et al., 1989) and in RGCs in a model of glaucoma (Beirowski et al., 2008). Following optic nerve crush injury in 15 different mouse strains, strain significantly affected the time necessary to observe RGC loss (Li et al., 2007). The current results are consistent with the hypothesis that genetic background controls inherent mechanisms of resistance to axonal degeneration, and suggest that this relative resistance delays the inflammation-induced degeneration of RGCs in C57/BL6 mice.
Differences in the timing of RGC loss exist in other experimental ON models as well. In a chronic rat EAE model, significant RGC loss was measured prior to the onset of optic nerve inflammation (Hobom et al., 2004). Similarly, in another mouse EAE model, an increase in reactive oxygen species was detected in optic nerve as early as 3–6 days post-immunization, well before inflammation is detectable by histology (Qi et al., 2007). These studies suggest that in some cases ON can induce oxidative stress that may lead to direct axonal injury, but our results suggest that such direct axonal injury is likely limited or absent in C57/BL6 MOG EAE mice.
Since the mechanisms that trigger MS and ON in patients is not known, the variety of mechanisms occurring in the different available experimental ON models is important to consider when evaluating potential new therapies. If a treatment is designed to prevent injury by blocking inflammation, for example, then it may be less likely to work in a model where injury occurs prior to onset of inflammation. We and others have often used the relapsing/remitting EAE model to study ON because this represents the most common clinical disease course of MS (Noseworthy et al., 2000). However, the delayed RGC loss reported here in chronic murine EAE may be more similar to what occurs in ON patients, as significant RGC axonal loss marked by retinal nerve fiber layer thinning does not occur until about 3 months after onset of ON (Costello et al., 2006). Our results suggest that there may be an extended window of opportunity to initiate therapy with a neuroprotective agent in this model, and therefore this is an important ON model to consider for such studies. Interestingly, in addition to delayed RGC loss, OKR responses demonstrated that vision loss is delayed as well. This was not necessarily expected, since optic nerve inflammation alone could have interfered with visual function. Results suggest that the level of inflammation present was insufficient to reduce vision, and instead visual loss correlates better with the timing of RGC loss in this experimental ON model.
While the exact timing of RGC loss in MOG-induced EAE in C57/BL6 mice has not been reported previously, at least one study has used this model to evaluate a potential neuroprotective therapy to prevent RGC loss (Guo et al., 2011). They demonstrate that spermidine prevents loss of cells in the RGC layer 25 days after onset of EAE, but did not examine whether RGCs were lost at earlier time points. Their results are consistent with ours, with cell loss occurring at a similarly late time, as EAE begins 8–10 days after immunization, suggesting they examined RGC loss between 30 and 40 days post-immunization. Importantly, while they showed protective effects of spermidine, mice were treated beginning at the time of immunization, prior to onset of optic nerve inflammation. Because our results show that RGC loss is delayed, it will be important to go back and examine whether spermidine can be administered after onset of optic nerve inflammation and exert similar neuroprotective effects. Potential future therapies will need to be examined in this paradigm, as patients with ON will not present before developing symptoms associated with optic nerve inflammation.
Experimental models of ON represent important tools in advancing our understanding and developing new therapies for ON and MS. Various models demonstrate distinct disease courses with differences in the timing of RGC loss, but share the common feature of recapitulating the neurodegenerative aspect of this disorder. The current results demonstrate the relatively slow degeneration of RGCs in MOG-induced chronic EAE. This model will likely prove important for evaluation of potential neuroprotective agents, as treatment can be initiated well after optic nerve inflammation begins, but still before RGCs have been lost.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by NIH Grants EY015098 and EY019014; National Multiple Sclerosis Society Grant RG 4214-A-1; Research to Prevent Blindness; and the F. M. Kirby Foundation.
References
Arnold, A. C. (2005). Evolving management of optic neuritis and multiple sclerosis. Am. J. Ophthalmol. 139, 1101–1108.
Beck, R. W., Gal, R. L., Bhatti, M. T., Brodsky, M. C., Buckley, E. G., Chrousos, G. A., Corbett, J., Eggenberger, E., Goodwin, J. A., Katz, B., Kaufman, D. I., Keltner, J. L., Kupersmith, M. J., Miller, N. R., Moke, P. S., Nazarian, S., Orengo-Nania, S., Savino, P. J., Shults, W. T., Smith, C. H., Trobe, J. D., Wall, M., Xing, D., and the Optic Neuritis Study Group. (2004). Visual function more than 10 years after optic neuritis: experience of the optic neuritis treatment trial. Am. J. Ophthalmol. 137, 77–83.
Beirowski, B., Babetto, E., Coleman, M. P., and Martin, K. R. (2008). The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 28, 1166–1179.
Bettelli, E. (2007). Building different mouse models for human MS. Ann. N. Y. Acad. Sci. 1103, 11–18.
Constantinescu, C. S., Hilliard, B., Fujioka, T., Bhopale, M. K., Calida, D., and Rostami, A. M. (1998). Pathogenesis of neuroimmunologic diseases. Immunol. Res. 17, 217–227.
Costello, F., Coupland, S., Hodge, W., Lorello, G. R., Koroluk, J., Pan, Y. I., Freedman, M. S., Zackon, D. H., and Kardon, R. H. (2006). Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann. Neurol. 59, 963–969.
Fisher, J. B., Jacobs, D. A., Markowitz, C. E., Galetta, S. L., Volpe, N. J., Nano-Schiavi, M. L., Baier, M. L., Frohman, E. M., Winslow, H., Frohman, T. C., Calabresi, P. A., Maguire, M. G., Cutter, G. R., and Balcer, L. J. (2006). Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology 113, 324–332.
Fritz, R. B., Chou, J., and McFarlin, D. E. (1983). Relapsing murine experimental allergic encephalomyelitis induced by myelin basic protein. J. Immunol. 130, 1024–1026.
Gran, B., Zhang, G.-X., Yu, S., Li, J., Chen, X. H., Ventura, E. S., Kamoun, M., and Rostami, A. (2002). IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J. Immunol. 169, 7104–7110.
Guo, X., Harada, C., Namekata, K., Kimura, A., Mitamura, Y., Yoshida, H., Matsumoto, Y., and Harada, T. (2011). Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Invest. Ophthalmol. Vis. Sci. [Epub ahead of print].
Hobom, M., Storch, M., Weissert, R., Maier, K., Radhakrishnan, A., Kramer, B., Bahr, M., and Diem, R. (2004). Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol. 14, 148–157.
Iglesias, A., Bauer, J., Litzenburger, T., Schubart, A., and Linington, C. (2001). T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia 36, 220–234.
Li, Y., Semaan, S. J., Schlamp, C. L., and Nickells, R. W. (2007). Dominant inheritance of retinal ganglion cell resistance to optic nerve crush in mice. BMC Neurosci. 8, 19. doi: 10.1186/1471-2202-8-19
Losseff, N. A., Webb, S. L., O’Riordan, J. I., Page, R., Wang, L., Barker, G. J., Tofts, P. S., McDonald, W. I., Miller, D. H., and Thompson, A. J. (1996). Spinal cord atrophy and disability in multiple sclerosis: a new reproducible and sensitive MRI method with potential to monitor disease progression. Brain 119, 701–708.
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33.
Mendel, I., Kerlero de Rosbo, N., and Ben-Nun, A. (1995). A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V g expression of encephalitogenic T cells. Eur. J. Immunol. 25, 1951–1959.
Meyer, R., Weissert, R., Diem, R., Storch, M. K., de Graaf, K. L., Kramer, B., and Bahr, M. (2001). Acute neuronal apoptosis in a rat model of multiple sclerosis. J. Neurosci. 21, 6214–6220.
Noseworthy, J. H., Lucchinetti, C., Rodriguez, M., and Weinshenker, B. G. (2000). Multiple sclerosis. N. Engl. J. Med. 343, 938–952.
Parisi, V., Manni, G., Spadaro, M., Colacino, G., Restuccia, R., Marchi, S., Bucci, M. G., and Pierelli, F. (1999). Correlation between morphological and functional retinal impairment in multiple sclerosis patients. Invest. Ophthalmol. Vis. Sci. 40, 2520–2527.
Prusky, G. T., Alam, N. M., Beekman, S., and Douglas, R. M. (2004). Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest. Ophthalmol. Vis. Sci. 45, 4611–4616.
Qi, X., Lewin, A. S., Sun, L., Hauswirth, W. H., and Guy, J. (2007). Suppression of mitochondrial oxidative stress provides long-term neuroprotection in experimental optic neuritis. Invest. Ophthalmol. Vis. Sci. 48, 681–691.
Shao, H., Huang, Z., Sun, S. L., Kaplan, H. J., and Sun, D. (2004). Myelin/oligodendrocyte glycoprotein-specific T-cells induce severe optic neuritis in the C57BL/6 mouse. Invest. Ophthalmol. Vis. Sci. 45, 4060–4065.
Shindler, K. S., Guan, Y., Ventura, E., Bennett, J., and Rostami, A. (2006). Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Mult. Scler. 12, 526–532.
Shindler, K. S., Ventura, E., Dutt, M., and Rostami, A. (2008). Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp. Eye Res. 87, 208–213.
Steel, D. H. W., and Waldock, A. (1998). Measurement of the retinal nerve fiber layer with scanning laser polarimetry in patients with previous demyelinating optic neuritis. J. Neurol. Neurosurg. Psychiatr. 64, 505–509.
Storch, M. K., Stefferl, A., Brehm, U., Weissert, R., Wallstrom, E., Kerchensteiner, M., Olsson, T., Linington, C., and Lassmann, H. (1998). Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 8, 681–694.
Trip, S. A., Schlottmann, P. G., Jones, S. J., Altmann, D. R., Garway-Heath, D. F., Thompson, A. J., Plant, G. T., and Miller, D. H. (2005). Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann. Neurol. 58, 383–391.
Keywords: optic neuritis, multiple sclerosis, EAE, RGC, neurodegeneration
Citation: Quinn TA, Dutt M and Shindler KS (2011) Optic neuritis and retinal ganglion cell loss in a chronic murine model of multiple sclerosis. Front. Neur. 2:50. doi: 10.3389/fneur.2011.00050
Received: 03 June 2011;
Accepted: 22 July 2011;
Published online: 02 August 2011.
Edited by:
Eric Eggenberger, Michigan State University, USAReviewed by:
Ashref J. AlMoosa, Michigan State University, USARatna Karuna Bitra, Michigan State University, USA
Copyright: © 2011 Quinn, Dutt and Shindler. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Kenneth S. Shindler, F.M. Kirby Center for Molecular Ophthalmology, Department of Ophthalmology, University of Pennsylvania Scheie Eye Institute, Stellar-Chance Laboratories, 3rd Floor, 422 Curie Boulevard, Philadelphia, PA 19104, USA. e-mail:a2VubmV0aC5zaGluZGxlckB1cGhzLnVwZW5uLmVkdQ==