

- 1 Center for Sleep and Circadian Neurobiology, School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
- 2 Division of Sleep Medicine, School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
Sleep/wake and circadian rest-activity rhythms become irregular with age. Typical outcomes include fragmented sleep during the night, advanced sleep phase syndrome and increased daytime sleepiness. These changes lead to a reduction in the quality of life due to cognitive impairments and emotional stress. More importantly, severely disrupted sleep and circadian rhythms have been associated with an increase in disease susceptibility. Additionally, many of the same brain areas affected by neurodegenerative diseases include the sleep and wake promoting systems. Any advances in our knowledge of these sleep/wake and circadian networks are necessary to target neural areas or connections for therapy. This review will discuss research that uses molecular, behavioral, genetic and anatomical methods to further our understanding of the interaction of these systems.
Studies have shown that sleep and circadian activity rhythms become irregular with age (Morin, 1993; Valentinuzzi et al., 1997; Kendall et al., 2001). Sleep becomes fragmented during the night and daytime sleepiness increases (Carskadon et al., 1982; Huang et al., 2002). The quantity of REM sleep and slow wave sleep (SWS) decreases as well and the normal circadian amplitude of the sleep/wake cycle is dampened and shortened (van Gool et al., 1987; Turek et al., 1995). These changes lead to a reduction in the quality of life due to cognitive impairments and emotional stress. More importantly, severely disrupted sleep and circadian rhythms have been associated with an increase in disease susceptibility (Hastings et al., 2003; Gibson et al., 2009).
The mechanisms responsible for the development of fragmented sleep are not completely understood. However, examining the functional interaction between circadian oscillators and sleep/wake areas has aided in these efforts (Dijk and von Schantz, 2005). With increasing age, circadian and sleep/wake neural areas or connections within the network may compensate for initial dysfunctions (Van Someren et al., 2002). For example, the degeneration of one network component may be deleterious on the systems as a whole, while other components may be more resistant to age-associated degeneration (Naidoo et al., 2011). Discerning how systems change with age is important to understand how they contribute to normal and dysfunctional sleep and wake. Many of the same brain areas in the sleep and wake promoting systems are affected by neurodegenerative diseases. Any advances in our knowledge of these sleep/wake and circadian networks may aid in designing therapies targeting these neural areas or connections. This review will discuss research that uses molecular, behavioral, genetic and anatomical methods to further our understanding of the interaction of these systems.
Aging Circadian Systems
The desynchronization of central and peripheral circadian systems contributes to the decline in optimal functioning of bodily systems. This includes changes in neuroendocrine circadian rhythms, insulin sensitivity, altered thermoregulation and acceleration of tumor growth (Cincotta et al., 1993; Touitou and Haus, 2000; Hastings et al., 2003; Straub and Mocchegiani, 2004; Cretenet et al., 2010; Heller et al., 2011). These are complex interactions and dysfunction can be due to targeted disruption of neurons, neurotransmitter or neuropeptide production, transport or secretion. It is reasonable to expect that the neuronal activity or expression of circadian clock genes be reduced or rhythms phase shifted (Kolker et al., 2003). Additionally, the connections between central and peripheral oscillators may be degraded or less functional (Morales et al., 1987). Any one or a combination of these abnormalities may result in the decoupling of the circadian oscillators and the ensuing pathologies.
The master central pacemaker is the suprachiasmatic nucleus (SCN) which controls circadian rhythmicity. Rhythmicity can dampen and/or elongate/shift with age but the number or cell size of neurons in the SCN does not change. However, the oscillating activity of these neurons does deteriorate (Satinoff et al., 1993; Madeira et al., 1995). Additionally, glucose uptake decreases in the SCN in aged animals (Wise et al., 1988) and the expression of neuropeptides also diminishes. Vasoactive intestinal polypeptide (VIP) expressing neurons of the SCN are retinorecipient and vasopressin (AVP) expressing neurons of the SCN modulate rhythmicity (Wu et al., 2007). In aged male humans, the number of SCN neurons that express VIP decreases and in aged female rats the expression becomes arrhythmic (Zhou et al., 1995; Krajnak et al., 1998). Both genders also express less AVP protein, less mRNA (Roozendaal et al., 1987; Zhou et al., 1995) and the normal daytime peak of AVP is reduced (Hofman and Swaab, 1994; Liu et al., 2000). Decreases in the neuroendocrine output of the SCN may directly or indirectly affect the coupling of central and peripheral oscillators.
In the SCN, clock genes constitute the core clock mechanism of the mammalian timekeeping system. Though the system is fairly complex, for brevity the simplest model is described. BMAL1 and CLOCK proteins dimerize and induce the transcription of Period (Per) and Cryptochrome (Cry) genes. In a negative feedback loop, levels of PER and CRY proteins increase and at a certain threshold form heterodimers which turn off the CLOCK BMAL1 regulated transcription of Per and Cry genes. This process takes roughly 24 h (Ko and Takahashi, 2006).
In aged animals the expression of certain clock genes changes in the SCN. Per1, Per2, and Cry1 expression does not change significantly with age, but the normal photic stimulation of Per1 expression is reduced (Asai et al., 2001; Kolker et al., 2003). Additionally, the free-running period of Per1–luc rhythmicity is shortened in aged animals (Yamazaki et al., 2002) and the amplitude of Clock and Bmal1 expression is decreased (Kolker et al., 2003). Changes in clock gene expression in peripheral tissues do not always reflect what is seen in the SCN (Yamazaki et al., 2000), and may result from a disruption of signals to these tissues or the tissues themselves, which are more susceptible to the aging process.
Furthermore, projections to and from the SCN including peripheral oscillators may change with age. In motoneurons, aging results in a shortening in delay of spike potentials between axon and soma, as well as decreases in axon conduction velocity and increases in input resistance (Morales et al., 1987; Engelhardt et al., 1989). Similar changes may be seen in these circadian projections. Light, food and temperature cues also input to the SCN. The output from circadian and peripheral oscillators do not only influence the sleep/wake cycle, but regulates metabolism and reproduction. Studies indicate that some tissues retain the ability to oscillate, even if connections from the master pacemaker have been degraded. Peripheral tissues in vitro that have become arrhythmic can be chemically induced to oscillate (Yamazaki et al., 2002). However, in aged animals exhibiting a decreased photic response, retinal projections to the SCN are not degraded and must be related to either the retina or SCN clock functions (Zhang et al., 1998). It is increasingly evident that determining the source of age-related sleep/wake or circadian dysfunctions is rather complex.
Sleep/Wake Systems Age
Another source of sleep/wake changes can likely be attributed to age-related neuronal dysfunction in the arousal and sleep promoting areas of the brain. The SCN directly or indirectly communicates with the sleep and wake promoting systems (Abrahamson et al., 2001; Aston-Jones et al., 2001; Chou et al., 2002). Orexinergic (or hypocretinergic) neurons are known to stabilize or maintain wake (Saper et al., 2001, 2005). These cells receive input from the SCN via the dorsomedial hypothalamus (DMH) and are localized in the perifornical and lateral hypothalamus (LH). There are two forms of the neuropeptide orexin/hypocretin (A and B) and two receptors. Though the neurons are limited to a discrete area, both orexinergic fibers and receptors are widely distributed throughout the brain. In diurnal and nocturnal rodents, orexinergic neurons are most active during the active phase (Martinez et al., 2002). Hypothalamic microdialysate analysis shows orexin-1 levels increase during wake and REM in adult animals (Kiyashchenko et al., 2002).
Mammals with no orexin or dysfunctional orexin/hypocretin receptors have disrupted sleep/wake cycles and narcoleptic symptoms. When orexin is decreased, the circadian rhythm of the sleep/wake cycle is disrupted. The flip–flop model of sleep/wake control suggests that there is a mutual inhibition between the areas that control sleep and the areas that control the wake state (Saper et al., 2001). In short, the ventrolateral preoptic area (VLPO) controls sleep and the brainstem cholinergic and monoaminergic systems control waking. Flipping weight between these areas controls the wake and sleep states. One input that stabilizes this switch is from the orexin neurons. Blocking or destroying these neurons or the orexin receptor 2 may flip the animal’s state quickly from waking to sleep and vice versa, such as what occurs in narcoleptic individuals, orexin knockout mice, and canine narcolepsy (Chemelli et al., 1999; Lin et al., 1999; Peyron et al., 2000; Thannickal et al., 2000).
A disruption in orexin function or a reduction in orexin levels leads to less stable sleep/wake cycles such as that seen in many elderly patients with sleep disorders (Porkka-Heiskanen, 2003). Additionally, decreases in excitatory orexin innervation to the noradrenergic locus coeruleus (LC) is thought to be a contributing factor of poor sleep/wake quality in aged cats (Zhang et al., 2002). Orexin B immunoreactive (-ir) axon density was determined to be significantly lower in the LC of aged macaques than that observed in the young or adult animals (43 and 35% decrease respectively; Downs et al., 2007). Real time PCR studies showed that preprohypocretin mRNA does not change in the aged hypothalamus (Terao et al., 2002) but in situ hybridization studies show that at the single cell level, preproorexin gene expression does decrease in cell count and optical density (Porkka-Heiskanen et al., 2004). Furthermore, orexin A and B protein expression as measured by radioimmunoassay was decreased in the LH (Porkka-Heiskanen et al., 2004). The number of orexinergic-ir neurons as well as the optical density of respective fibers in the LH is reduced in aged animals (Brownell and Conti, 2010; Sawai et al., 2010). It is interesting to note that orexinergic innervation of the cholinergic basal forebrain, which modulates wake and REM sleep, is reduced in aged guinea pigs (Zhang et al., 2005). Orexin/hypocretin receptor mRNA expression is also decreased in aged animals. Hypocretin receptor 1 mRNA is reduced in the hippocampus and hypocretin receptor 2 mRNA is significantly reduced in thalamic areas, hippocampus, and the brainstem (Terao et al., 2002). Neural activity measured by c-fos immunoreactivity is reduced in orexinergic neurons of mice at 24 months (Naidoo et al., 2011).
Changes are also seen in the cholinergic and monoaminergic wake active areas of aged animals. Nicotinic and muscarinic receptors of the acetylcholinergic system decrease in the SCN with age (van der Zee et al., 1991). In young animals, the noradrenergic neurons of the LC are important in wake promotion, receive direct input from the SCN and follow a circadian pattern of activation (Aston-Jones et al., 2001). In aged rats, LC projections to the frontal cortex and dentate gyrus decrease but axonal branching increases depending on the target and age. This is suggested to be a compensatory mechanism (Shirokawa et al., 2000). In the ventral periaqueductal gray (vPAG) the wake active dopaminergic neurons have recently been identified (Lu et al., 2006). We have recently reported a reduction in the neural activity of these dopaminergic neurons of the vPAG and the noradrenergic neurons of the LC in aged mice (Naidoo et al., 2009, 2011).
The wake active histaminergic system originates in the tuberomammillary nucleus (TMN) and sends widespread projections to areas that include the cortex, thalamus and brainstem. Histamine levels were found to be increased in middle aged rats when compared to young, and the level of histamine methyl transferase was decreased (Mazurkiewicz-Kwilecki and Prell, 1984). The histamine receptor mRNA levels also change with age. There are four types of histaminergic receptors located throughout the brain and body. Histamine H1, H2 and H3 receptor mRNA is decreased in the aging brain (Terao et al., 2004). Given these changes in the aging wake promoting neurotransmitter systems as well as wake maintaining systems, it is clear that therapies to alleviate or attenuate these changes need to be developed.
Sleep promoting areas may show a reduction in function with age. The VLPO neurons are active during SWS (Sherin et al., 1996; Szymusiak et al., 1998) and when lesioned results in insomnia (Lu et al., 2000). GABAergic and galaninergic inhibitory neurons from this area project to wake active histaminergic neurons (Sherin et al., 1996). Interestingly, the number of activated VLPO neurons during sleep does not change in old rats (Shiromani et al., 2000) although connections between these areas may become dysfunctional or degraded with age. The SCN has a minor input into the VLPO, but substantial direct and indirect inputs to the DMH (Novak and Nunez, 2000; Chou et al., 2002). The DMH heavily inputs the VLPO and it would be beneficial if these pathways were examined during aging.
Age-associated changes in the serotonergic system affect the function of respiratory motor output during sleep. Serotonergic input to the hypoglossal nucleus decreases, which is thought to lead to a decline in upper airway muscle performance (Behan and Brownfield, 1999). In aged rhesus monkeys, serotonin receptor 2 density reduces in the occipital and parietal cortex including the deep layers of the motor cortex (Wenk et al., 1989; Bigham and Lidow, 1995). Serotonin levels also decrease in the occipital areas but do not change in the cingulate cortex in aged monkeys (Beal et al., 1991). It is likely with normal aging that changes in any neurotransmitter system affecting sleep vary across the brain. This presents a difficult task to fully determine the interaction of aging and sleep.
Age-Associated Neurodegenerative Diseases and Sleep
In many patients afflicted with neurodegenerative diseases the physical and mental consequences lead to sleep disorders (Table 1). For example, sleep fragmentation can occur if the patient cannot move well or insomnia may develop due to depression or feelings of helplessness. Medications used to alleviate some of the motor or cognitive symptoms such as levodopa in Parkinson’s disease (PD) can also contribute to disruptions in normal sleep/wake behaviors. Sleep disorders may occur secondarily or due to concurrent or related neurodegenerative pathologies. However, some research indicates that sleep disturbances may predict manifestation of neurodegenerative diseases (Postuma and Montplaisir, 2009). Sleep disturbance or loss also affects metabolic and immune function (Krueger et al., 1998; Knutson et al., 2007). Chronic sleep loss could lead to neuronal damage resulting in altered hypothalamic pituitary adrenal axis function, cognitive deficits and memory loss. Increases in the number of patients with neurodegenerative diseases may be related to or the result of a society that does not sleep.
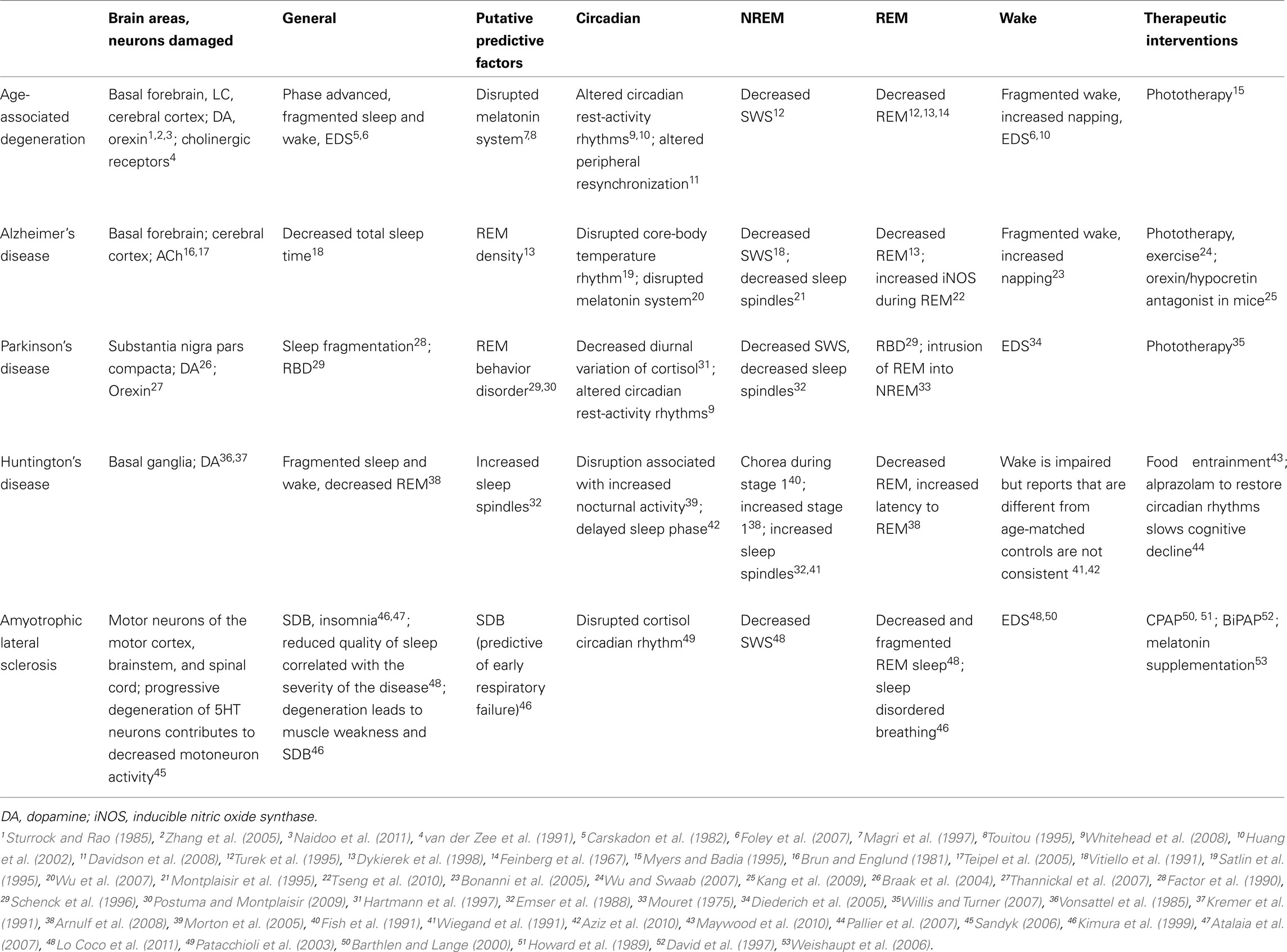
Table 1. Common sleep and wake characteristics of neurodegenerative diseases.
In Alzheimer’s disease (AD), sleep disturbances increase with the severity of the disease. Initially there is an increase in nighttime arousals and a decrease in SWS (Vitiello et al., 1991). In the later stages, circadian disruption, severe daytime wakefulness and a reduction in REM sleep occurs, likely due to a reduction in acetylcholine (Dykierek et al., 1998). Circadian rhythm dysfunction has been proposed to be due to changes in SCN and pineal functions (Wu et al., 2007). Degeneration of cholinergic input from the nucleus basalis of Meynert to the cortex may be responsible for some of the sleep/wake changes (Montplaisir et al., 1995). Neurofibrillary tangles found in the histaminergic TMN of AD patients and amyloid-β peptide (Aβ) aggregation also contributes to the AD pathology. Normally in the interstitial fluid Aβ has a diurnal fluctuation with low levels during sleep and peak levels during wake. Recently one study showed that prolonged wake and/or orexin administration increased levels of the Aβ in the interstitial fluid of the brain in mice (Kang et al., 2009). Administration of an orexin antagonist reduced amyloid deposits in several brain areas suggesting that manipulating sleep or the orexin system in AD patients could improve symptoms (Kang et al., 2009). Although the research is sparse, melatonin, phototherapy and exercise have all had positive effects in the treatment of circadian and sleep/wake disorders of AD patients (Wu and Swaab, 2007). As one in three Americans develops AD, there is a crucial need for more research in these therapies.
REM sleep behavior disorder (RBD) has been associated with PD and thought to be an early manifestation (Schenck et al., 1996; Boeve et al., 2003; Postuma and Montplaisir, 2009). Sleep attacks and excessive daytime sleepiness (EDS) are also commonly seen in patients with PD (Factor et al., 1990; Diederich et al., 2005). The degeneration begins at the brainstem and progresses rostrally, although degeneration of the dopaminergic neurons of the substantia nigra pars compacta is the main contributor to PD characteristics (Braak et al., 2004). RBD results from pedunculopontine dysfunction and likely explains RBD manifesting previous to PD (Rye, 1997; Boeve et al., 2007). Some studies have successfully seen bright light therapy or sleep modifications reduce the symptoms of PD (Hogl et al., 1998; Willis and Turner, 2007).
Huntington’s disease (HD) is a genetic disorder characterized by a polyglutamine (CAG) repeat (Scherzinger et al., 1999). Neurodegeneration is extensive throughout the brain, affecting cortical and subcortical areas but primarily affects the basal ganglia (Vonsattel et al., 1985). Sleep and wake regions of the brain including the brainstem, thalamus, hypothalamus and cortex are also affected in HD (Kremer et al., 1991). The SCN pacemaker is functional in mouse models of HD, so a dysfunction of the circadian circuitry is proposed to contribute to circadian abnormalities (Pallier et al., 2007). Central and peripheral clock gene expression is altered as well (Morton et al., 2005; Maywood et al., 2010). The sleep/wake cycle is disrupted in HD patients characterized by self-reported EDS, sleep fragmentation at night, and delayed sleep phase (Arnulf et al., 2008; Videnovic et al., 2009; Aziz et al., 2010). Sleep is lighter with an increase in Stage 1 and a decrease in REM sleep (Arnulf et al., 2008). Disruptions in the circadian and sleep/wake cycles of these patients exacerbate symptoms, increasing depression, cognitive deficits and metabolic dysfunctions (Aziz et al., 2010). It is important to note that pharmacological and behavioral manipulation of sleep and wake reduces disease progression and improves cognitive function and circadian gene expression in a mouse model of HD (Hockly et al., 2002; Pallier et al., 2007; Pallier and Morton, 2009; Maywood et al., 2010).
Amyotrophic lateral sclerosis (ALS) is considered an age-associated neurodegenerative disease with the age of onset ranging from 40 to 70. Although onset can occur in children this is rare. Most ALS cases are sporadic and about 10% are familial. Also called Lou Gehrig’s or motor neuron disease (Boillee et al., 2006), both upper and lower motor neurons are affected. Motor neurons of the motor cortex, brainstem and spinal cord gradually degenerate leading to muscle weakness, sleep disordered breathing (SDB) and paralysis (Kimura et al., 1999). Additionally, sleep is reduced in both REM and SWS stages with resulting EDS (Barthlen and Lange, 2000; Lo Coco et al., 2011). Some patients find relief using assisted breathing such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BiPAP; Howard et al., 1989; David et al., 1997; Barthlen and Lange, 2000).
Amyotrophic lateral sclerosis is a complex disease of many subtypes with various genetic and environmental contributing factors. One such factor is glutamate toxicity which is decreased using the drug Riluzole (Shaw and Ince, 1997). Alternative therapies are also being considered including the regulation of the serotonin system. Levels of serotonin are decreased in ALS patients and compensatory increases in glutamate lead to excitotoxicity. It has been suggested that motor neurons with a high density of serotonergic innervation are more susceptible to degeneration (Sandyk, 2006). Serotonin is the precursor to melatonin, which is also likely to be decreased. As melatonin has antioxidant properties and inhibits glutamate release, this reduction would further exacerbate degeneration. Indeed, melatonin supplements slowed disease progression when given to a mouse model of familial ALS (Weishaupt et al., 2006).
Plasticity and Compensation
Normally the SCN is coupled to peripheral oscillators, although studies have shown that SCN control is not necessary for sustaining oscillatory activity. If signals from central oscillators reduce in strength due to age or neurodegeneration, other cues may entrain the peripheral oscillators (Weinert, 2005). Unmasking mechanisms within sleep/wake systems is difficult due to the many checks and balances that ensure homeostasis. Although compensation and plasticity occurs to a lesser extent in older animals, a relatively high degree is preserved (Van Someren et al., 2002). This may not always be advantageous as epigenetic methylation of circadian genes has been associated with dementia (Liu et al., 2008). Understanding how the aging brain can compensate and remain plastic will be beneficial to focus on more effective treatments for sleep/wake and neurodegenerative disorders.
Discussion
Many neurodegenerative diseases result from targeted destruction of neurotransmitter systems. The co-morbidity of sleep disorders with neurodegenerative diseases suggests that changes in many of these neural areas manifests in sleep/wake and circadian dysfunction. Effects on the sleep/wake and circadian systems may result from, or contribute to, the increasing pathology. Some research has shown the benefit of pharmacologically or behaviorally restoring rhythms and sleep/wake for delaying pathologies (Table 1). This is important to understand in a society where sleep is not considered a priority. A few points worth considering are as follows: Sleep is a basic need that is made secondary to work schedules and some leisure activities for many adults. In developing children and adolescents, early school start times and late night extracurricular meetings contribute to a culture of sleep deprived, cognitively unhealthy Americans. If restoring our circadian and sleep/wake cycles can ward off the deterioration of the brain, it is imperative to educate the public about the very real damage of abnormal sleep/wake cycles not only in aging individuals but at every age.
The fragmented sleep/wake pattern seen in aging individuals can be due to the degeneration or dysfunction of the circadian and sleep/wake networks. Uncoupling of the central and peripheral oscillators may exacerbate dysfunction via altered feedback signals or signaling pathways. It is likely that several brain regions are affected and that there are individual differences in how the sleep/wake and circadian networks degrade. Additionally there may be differential plasticity and compensation in the integration of these neural systems, making the identification of applicable therapies very difficult. However, if mechanisms contributing to the normal aging process of these networks are identified, this may elucidate a general therapy for restoring sleep/wake and circadian homeostasis. Ultimately this could also reduce the onset or improve the symptoms of neurodegenerative diseases. It is crucial that we immediately invest our energies and resources in understanding these mechanisms as well as in the dissemination and implementation of current knowledge and therapies to the public.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by AG23500.
References
Abrahamson, E. E., Leak, R. K., and Moore, R. Y. (2001). The suprachiasmatic nucleus projects to posterior hypothalamic arousal systems. Neuroreport 12, 435–440.
Arnulf, I., Nielsen, J., Lohmann, E., Schiefer, J., Wild, E., Jennum, P., Konofal, E., Walker, M., Oudiette, D., Tabrizi, S., and Durr, A. (2008). Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 65, 482–488.
Asai, M., Yoshinobu, Y., Kaneko, S., Mori, A., Nikaido, T., Moriya, T., Akiyama, M., and Shibata, S. (2001). Circadian profile of Per gene mRNA expression in the suprachiasmatic nucleus, paraventricular nucleus, and pineal body of aged rats. J. Neurosci. Res. 66, 1133–1139.
Aston-Jones, G., Chen, S., Zhu, Y., and Oshinsky, M. L. (2001). A neural circuit for circadian regulation of arousal. Nat. Neurosci. 4, 732–738.
Atalaia, A., De Carvalho, M., Evangelista, T., and Pinto, A. (2007). Sleep characteristics of amyotrophic lateral sclerosis in patients with preserved diaphragmatic function. Amyotroph. Lateral Scler. 8, 101–105.
Aziz, N. A., Anguelova, G. V., Marinus, J., Lammers, G. J., and Roos, R. A. (2010). Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat. Disord. 16, 345–350.
Barthlen, G. M., and Lange, D. J. (2000). Unexpectedly severe sleep and respiratory pathology in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 7, 299–302.
Beal, M. F., Walker, L. C., Storey, E., Segar, L., Price, D. L., and Cork, L. C. (1991). Neurotransmitters in neocortex of aged rhesus monkeys. Neurobiol. Aging 12, 407–412.
Behan, M., and Brownfield, M. S. (1999). Age-related changes in serotonin in the hypoglossal nucleus of rat: implications for sleep-disordered breathing. Neurosci. Lett. 267, 133–136.
Bigham, M. H., and Lidow, M. S. (1995). Adrenergic and serotonergic receptors in aged monkey neocortex. Neurobiol. Aging 16, 91–104.
Boeve, B. F., Silber, M. H., Parisi, J. E., Dickson, D. W., Ferman, T. J., Benarroch, E. E., Schmeichel, A. M., Smith, G. E., Petersen, R. C., Ahlskog, J. E., Matsumoto, J. Y., Knopman, D. S., Schenck, C. H., and Mahowald, M. W. (2003). Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology 61, 40–45.
Boeve, B. F., Silber, M. H., Saper, C. B., Ferman, T. J., Dickson, D. W., Parisi, J. E., Benarroch, E. E., Ahlskog, J. E., Smith, G. E., Caselli, R. C., Tippman-Peikert, M., Olson, E. J., Lin, S. C., Young, T., Wszolek, Z., Schenck, C. H., Mahowald, M. W., Castillo, P. R., Del Tredici, K., and Braak, H. (2007). Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130(Pt 11), 2770–2788.
Boillee, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59.
Bonanni, E., Maestri, M., Tognoni, G., Fabbrini, M., Nucciarone, B., Manca, M. L., Gori, S., Iudice, A., and Murri, L. (2005). Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J. Sleep Res. 14, 311–317.
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H., and Del Tredici, K. (2004). Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134.
Brownell, S. E., and Conti, B. (2010). Age- and gender-specific changes of hypocretin immunopositive neurons in C57Bl/6 mice. Neurosci. Lett. 472, 29–32.
Brun, A., and Englund, E. (1981). Regional pattern of degeneration in Alzheimer’s disease: neuronal loss and histopathological grading. Histopathology 5, 549–564.
Carskadon, M. A., Brown, E. D., and Dement, W. C. (1982). Sleep fragmentation in the elderly: relationship to daytime sleep tendency. Neurobiol. Aging 3, 321–327.
Chemelli, R. M., Willie, J. T., Sinton, C. M., Elmquist, J. K., Scammell, T., Lee, C., Richardson, J. A., Williams, S. C., Xiong, Y., Kisanuki, Y., Fitch, T. E., Nakazato, M., Hammer, R. E., Saper, C. B., and Yanagisawa, M. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451.
Chou, T. C., Bjorkum, A. A., Gaus, S. E., Lu, J., Scammell, T. E., and Saper, C. B. (2002). Afferents to the ventrolateral preoptic nucleus. J. Neurosci. 22, 977–990.
Cincotta, A. H., Schiller, B. C., Landry, R. J., Herbert, S. J., Miers, W. R., and Meier, A. H. (1993). Circadian neuroendocrine role in age-related changes in body fat stores and insulin sensitivity of the male Sprague-Dawley rat. Chronobiol. Int. 10, 244–258.
Cretenet, G., Le Clech, M., and Gachon, F. (2010). Circadian clock-coordinated 12 Hr period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 11, 47–57.
David, W. S., Bundlie, S. R., and Mahdavi, Z. (1997). Polysomnographic studies in amyotrophic lateral sclerosis. J. Neurol. Sci. 152(Suppl. 1), S29–S35.
Davidson, A. J., Yamazaki, S., Arble, D. M., Menaker, M., and Block, G. D. (2008). Resetting of central and peripheral circadian oscillators in aged rats. Neurobiol. Aging 29, 471–477.
Diederich, N. J., Vaillant, M., Mancuso, G., Lyen, P., and Tiete, J. (2005). Progressive sleep “destructuring” in Parkinson’s disease. A polysomnographic study in 46 patients. Sleep Med. 6, 313–318.
Dijk, D. J., and von Schantz, M. (2005). Timing and consolidation of human sleep, wakefulness, and performance by a symphony of oscillators. J. Biol. Rhythms 20, 279–290.
Downs, J. L., Dunn, M. R., Borok, E., Shanabrough, M., Horvath, T. L., Kohama, S. G., and Urbanski, H. F. (2007). Orexin neuronal changes in the locus coeruleus of the aging rhesus macaque. Neurobiol. Aging 28, 1286–1295.
Dykierek, P., Stadtmüller, G., Schramm, P., Bahro, M., van Calker, D., Braus, D. F., Steigleider, P., Löw, H., Hohagen, F., Gattaz, W. F., Berger, M., and Riemann, D. (1998). The value of REM sleep parameters in differentiating Alzheimer’s disease from old-age depression and normal aging. J. Psychiatr. Res. 32, 1–9.
Emser, W., Brenner, M., Stober, T., and Schimrigk, K. (1988). Changes in nocturnal sleep in Huntington’s and Parkinson’s disease. J. Neurol. 235, 177–179.
Engelhardt, J. K., Morales, F. R., Yamuy, J., and Chase, M. H. (1989). Cable properties of spinal cord motoneurons in adult and aged cats. J. Neurophysiol. 61, 194–201.
Factor, S. A., McAlarney, T., Sanchez-Ramos, J. R., and Weiner, W. J. (1990). Sleep disorders and sleep effect in Parkinson’s disease. Mov. Disord. 5, 280–285.
Feinberg, I., Koresko, R. L., and Heller, N. (1967). EEG sleep patterns as a function of normal and pathological aging in man. J. Psychiatr. Res. 5, 107–144.
Fish, D. R., Sawyers, D., Allen, P. J., Blackie, J. D., Lees, A. J., and Marsden, C. D. (1991). The effect of sleep on the dyskinetic movements of Parkinson’s disease, Gilles de la Tourette syndrome, Huntington’s disease, and torsion dystonia. Arch. Neurol. 48, 210–214.
Foley, D. J., Vitiello, M. V., Bliwise, D. L., Ancoli-Israel, S., Monjan, A. A., and Walsh, J. K. (2007). Frequent napping is associated with excessive daytime sleepiness, depression, pain, and nocturia in older adults: findings from the National Sleep Foundation “2003 Sleep in America” Poll. Am. J. Geriatr. Psychiatry 15, 344–350.
Gibson, E. M., Williams, W. P. III, and Kriegsfeld, L. J. (2009). Aging in the circadian system: considerations for health, disease prevention and longevity. Exp. Gerontol. 44, 51–56.
Hartmann, A., Veldhuis, J. D., Deuschle, M., Standhardt, H., and Heuser, I. (1997). Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol. Aging 18, 285–289.
Hastings, M. H., Reddy, A. B., and Maywood, E. S. (2003). A clockwork web: circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 4, 649–661.
Heller, H. C., Edgar, D. M., Grahn, D. A., and Glotzbach, S. F. (2011). Sleep, thermoregulation and circadian rhythms. Compr. Physiol. 1361–1374.
Hockly, E., Cordery, P. M., Woodman, B., Mahal, A., van Dellen, A., Blakemore, C., Lewis, C. M., Hannan, A. J., and Bates, G. P. (2002). Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. Ann. Neurol. 51, 235–242.
Hofman, M. A., and Swaab, D. F. (1994). Alterations in circadian rhythmicity of the vasopressin-producing neurons of the human suprachiasmatic nucleus (SCN) with aging. Brain Res. 651, 134–142.
Hogl, B. E., Gómez-Arévalo, G., García, S., Scipioni, O., Rubio, M., Blanco, M., and Gershanik, O. S. (1998). A clinical, pharmacologic, and polysomnographic study of sleep benefit in Parkinson’s disease. Neurology 50, 1332–1339.
Howard, R. S., Wiles, C. M., and Loh, L. (1989). Respiratory complications and their management in motor neuron disease. Brain 112(Pt 5), 1155–1170.
Huang, Y. L., Liu, R. Y., Wang, Q. S., Van Someren, E. J., Xu, H., and Zhou, J. N. (2002). Age-associated difference in circadian sleep-wake and rest-activity rhythms. Physiol. Behav. 76, 597–603.
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., Fujiki, N., Nishino, S., and Holtzman, D. M. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007.
Kendall, A. R., Lewy, A. J., and Sack, R. L. (2001). Effects of aging on the intrinsic circadian period of totally blind humans. J. Biol. Rhythms 16, 87–95.
Kimura, K., Tachibana, N., Kimura, J., and Shibasaki, H. (1999). Sleep-disordered breathing at an early stage of amyotrophic lateral sclerosis. J. Neurol. Sci. 164, 37–43.
Kiyashchenko, L. I., Mileykovskiy, B. Y., Maidment, N., Lam, H. A., Wu, M. F., John, J., Peever, J., and Siegel, J. M. (2002). Release of hypocretin (orexin) during waking and sleep states. J. Neurosci. 22, 5282–5286.
Knutson, K. L., Spiegel, K., Penev, P., and Van Cauter, E. (2007). The metabolic consequences of sleep deprivation. Sleep Med. Rev. 11, 163–178.
Ko, C. H., and Takahashi, J. S. (2006). Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 15, R271–R277.
Kolker, D. E., Fukuyama, H., Huang, D. S., Takahashi, J. S., Horton, T. H., and Turek, F. W. (2003). Aging alters circadian and light-induced expression of clock genes in golden hamsters. J. Biol. Rhythms 18, 159–169.
Krajnak, K., Kashon, M. L., Rosewell, K. L., and Wise, P. M. (1998). Aging alters the rhythmic expression of vasoactive intestinal polypeptide mRNA but not arginine vasopressin mRNA in the suprachiasmatic nuclei of female rats. J. Neurosci. 18, 4767–4774.
Kremer, H. P., Roos, R. A., Dingjan, G. M., Bots, G. T., Bruyn, G. W., and Hofman, M. A. (1991). The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington’s disease. Neurosci. Lett. 132, 101–104.
Krueger, J. M., Fang, J., Taishi, P., Chen, Z., Kushikata, T., and Gardi, J. (1998). Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann. N. Y. Acad. Sci. 856, 148–159.
Lin, L., Faraco, J., Li, R., Kadotani, H., Rogers, W., Lin, X., Qiu, X., de Jong, P. J., Nishino, S., and Mignot, E. (1999). The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376.
Liu, H. C., Hu, C. J., Tang, Y. C., and Chang, J. G. (2008). A pilot study for circadian gene disturbance in dementia patients. Neurosci. Lett. 435, 229–233.
Liu, R. Y., Zhou, J. N., Hoogendijk, W. J., van Heerikhuize, J., Kamphorst, W., Unmehopa, U. A., Hofman, M. A., and Swaab, D. F. (2000). Decreased vasopressin gene expression in the biological clock of Alzheimer disease patients with and without depression. J. Neuropathol. Exp. Neurol. 59, 314–322.
Lo Coco, D., Mattaliano, P., Spataro, R., Mattaliano, A., and La Bella, V. (2011). Sleep-wake disturbances in patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatr. 82, 839–842.
Lu, J., Greco, M. A., Shiromani, P., and Saper, C. B. (2000). Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J. Neurosci. 20, 3830–3842.
Lu, J., Jhou, T. C., and Saper, C. B. (2006). Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J. Neurosci. 26, 193–202.
Madeira, M. D., Sousa, N., Santer, R. M., Paula-Barbosa, M. M., and Gundersen, H. J. (1995). Age and sex do not affect the volume, cell numbers, or cell size of the suprachiasmatic nucleus of the rat: an unbiased stereological study. J. Comp. Neurol. 361, 585–601.
Magri, F., Locatelli, M., Balza, G., Molla, G., Cuzzoni, G., Fioravanti, M., Solerte, S. B., and Ferrari, E. (1997). Changes in endocrine circadian rhythms as markers of physiological and pathological brain aging. Chronobiol. Int. 14, 385–396.
Martinez, G. S., Smale, L., and Nunez, A. A. (2002). Diurnal and nocturnal rodents show rhythms in orexinergic neurons. Brain Res. 955, 1–7.
Maywood, E. S., Fraenkel, E., McAllister, C. J., Wood, N., Reddy, A. B., Hastings, M. H., and Morton, A. J. (2010). Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J. Neurosci. 30, 10199–10204.
Mazurkiewicz-Kwilecki, I. M., and Prell, G. D. (1984). Age-related changes in brain histamine. Agents Actions 14, 554–557.
Montplaisir, J., Petit, D., Lorrain, D., Gauthier, S., and Nielsen, T. (1995). Sleep in Alzheimer’s disease: further considerations on the role of brainstem and forebrain cholinergic populations in sleep-wake mechanisms. Sleep 18, 145–148.
Morales, F. R., Boxer, P. A., Fung, S. J., and Chase, M. H. (1987). Basic electrophysiological properties of spinal cord motoneurons during old age in the cat. J. Neurophysiol. 58, 180–194.
Morin, L. P. (1993). Age, but not pineal status, modulates circadian periodicity of golden hamsters. J. Biol. Rhythms 8, 189–197.
Morton, A. J., Wood, N. I., Hastings, M. H., Hurelbrink, C., Barker, R. A., and Maywood, E. S. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163.
Mouret, J. (1975). Differences in sleep in patients with Parkinson’s disease. Electroencephalogr. Clin. Neurophysiol. 38, 653–657.
Myers, B. L., and Badia, P. (1995). Changes in circadian rhythms and sleep quality with aging: mechanisms and interventions. Neurosci. Biobehav. Rev. 19, 553–571.
Naidoo, N., Galante, R., Lian, J., Zhu, Y., and Veasey, S. (2009). “ER dyshomeostasis occurs in key sleep-wake control regions of aging,” in 2009 Neuroscience Meeting Planner, 376.24, Chicago, IL: Society for Neuroscience.
Naidoo, N., Zhu, J., Zhu, Y., Fenik, P., Lian, J., Galante, R., and Veasey, S. (2011). Endoplasmic reticulum stress in wake active neurons progresses with aging. Aging Cell 10, 640–649.
Novak, C. M., and Nunez, A. A. (2000). A sparse projection from the suprachiasmatic nucleus to the sleep active ventrolateral preoptic area in the rat. Neuroreport 11, 93–96.
Pallier, P. N., Maywood, E. S., Zheng, Z., Chesham, J. E., Inyushkin, A. N., Dyball, R., Hastings, M. H., and Morton, A. J. (2007). Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J. Neurosci. 27, 7869–7878.
Pallier, P. N., and Morton, A. J. (2009). Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res. 1279, 90–98.
Patacchioli, F. R., Monnazzi, P., Scontrini, A., Tremante, E., Caridi, I., Brunetti, E., Buttarelli, F. R., and Pontieri, F. E. (2003). Adrenal dysregulation in amyotrophic lateral sclerosis. J. Endocrinol. Invest. 26, RC23–R25.
Peyron, C., Faraco, J., Rogers, W., Ripley, B., Overeem, S., Charnay, Y., Nevsimalova, S., Aldrich, M., Reynolds, D., Albin, R., Li, R., Hungs, M., Pedrazzoli, M., Padigaru, M., Kucherlapati, M., Fan, J., Maki, R., Lammers, G. J., Bouras, C., Kucherlapati, R., Nishino, S., and Mignot, E. (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997.
Porkka-Heiskanen, T. (2003). Gene expression during sleep, wakefulness and sleep deprivation. Front. Biosci. 8, s421–s437.
Porkka-Heiskanen, T., Alanko, L., Kalinchuk, A., Heiskanen, S., and Stenberg, D. (2004). The effect of age on prepro-orexin gene expression and contents of orexin A and B in the rat brain. Neurobiol. Aging 25, 231–238.
Postuma, R. B., and Montplaisir, J. (2009). Predicting Parkinson’s disease – why, when, and how? Parkinsonism Relat. Disord. 15(Suppl. 3), S105–S109.
Roozendaal, B., van Gool, W. A., Swaab, D. F., Hoogendijk, J. E., and Mirmiran, M. (1987). Changes in vasopressin cells of the rat suprachiasmatic nucleus with aging. Brain Res. 409, 259–264.
Rye, D. B. (1997). Contributions of the pedunculopontine region to normal and altered REM sleep. Sleep 20, 757–788.
Sandyk, R. (2006). Serotonergic mechanisms in amyotrophic lateral sclerosis. Int. J. Neurosci. 116, 775–826.
Saper, C. B., Chou, T. C., and Scammell, T. E. (2001). The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 24, 726–731.
Saper, C. B., Scammell, T. E., and Lu, J. (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263.
Satinoff, E., Li, H., Tcheng, T. K., Liu, C., McArthur, A. J., Medanic, M., and Gillette, M. U. (1993). Do the suprachiasmatic nuclei oscillate in old rats as they do in young ones? Am. J. Physiol. 265(Pt 2), R1216–R1222.
Satlin, A., Volicer, L., Stopa, E. G., and Harper, D. (1995). Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol. Aging 16, 765–771.
Sawai, N., Ueta, Y., Nakazato, M., and Ozawa, H. (2010). Developmental and aging change of orexin-A and -B immunoreactive neurons in the male rat hypothalamus. Neurosci. Lett. 468, 51–55.
Schenck, C. H., Bundlie, S. R., and Mahowald, M. W. (1996). Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 46, 388–393.
Scherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R., Hasenbank, R., Bates, G. P., Lehrach, H., and Wanker, E. E. (1999). Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609.
Shaw, P. J., and Ince, P. G. (1997). Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J. Neurol. 244(Suppl. 2), S3–S14.
Sherin, J. E., Shiromani, P. J., McCarley, W., and Saper, C. B. (1996). Activation of ventrolateral preoptic neurons during sleep. Science 271, 216–219.
Shirokawa, T., Ishida, Y., and Isobe, K. I. (2000). Age-dependent changes in axonal branching of single locus coeruleus neurons projecting to two different terminal fields. J. Neurophysiol. 84, 1120–1122.
Shiromani, P. J., Lu, J., Wagner, D., Thakkar, J., Greco, M. A., Basheer, R., and Thakkar, M. (2000). Compensatory sleep response to 12 h wakefulness in young and old rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R125–R133.
Straub, R. H., and Mocchegiani, E. (2004). The Neuroendocrine Immune Network in Ageing. Amsterdam: Elsevier.
Sturrock, R. R., and Rao, K. A. (1985). A quantitative histological study of neuronal loss from the locus coeruleus of ageing mice. Neuropathol. Appl. Neurobiol. 11, 55–60.
Szymusiak, R., Alam, N., Steininger, T. L., and McGinty, D. (1998). Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 803, 178–188.
Teipel, S. J., Flatz, W. H., Heinsen, H., Bokde, A. L., Schoenberg, S. O., Stöckel, S., Dietrich, O., Reiser, M. F., Möller, H. J., and Hampel, H. (2005). Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain 128(Pt 11), 2626–2644.
Terao, A., Apte-Deshpande, A., Morairty, S., Freund, Y. R., and Kilduff, T. S. (2002). Age-related decline in hypocretin (orexin) receptor 2 messenger RNA levels in the mouse brain. Neurosci. Lett. 332, 190–194.
Terao, A., Steininger, T. L., Morairty, S. R., and Kilduff, T. S. (2004). Age-related changes in histamine receptor mRNA levels in the mouse brain. Neurosci. Lett. 355, 81–84.
Thannickal, T. C., Lai, Y. Y., and Siegel, J. M. (2007). Hypocretin (orexin) cell loss in Parkinson’s disease. Brain 130(Pt 6), 1586–1595.
Thannickal, T. C., Moore, R. Y., Nienhuis, R., Ramanathan, L., Gulyani, S., Aldrich, M., Cornford, M., and Siegel, J. M. (2000). Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474.
Touitou, Y. (1995). Effects of ageing on endocrine and neuroendocrine rhythms in humans. Horm. Res. 43, 12–19.
Touitou, Y., and Haus, E. (2000). Alterations with aging of the endocrine and neuroendocrine circadian system in humans. Chronobiol. Int. 17, 369–390.
Tseng, I. J., Liu, H. C., Yuan, R. Y., Sheu, J. J., Yu, J. M., and Hu, C. J. (2010). Expression of inducible nitric oxide synthase (iNOS) and period 1 (PER1) clock gene products in different sleep stages of patients with cognitive impairment. J. Clin. Neurosci. 17, 1140–1143.
Turek, F. W., Penev, P., Zhang, Y., van Reeth, O., and Zee, P. (1995). Effects of age on the circadian system. Neurosci. Biobehav. Rev. 19, 53–58.
Valentinuzzi, V. S., Scarbrough, K., Takahashi, J. S., and Turek, F. W. (1997). Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. Am. J. Physiol. 273(Pt 2), R1957–R1964.
van der Zee, E. A., Streefland, C., Strosberg, A. D., Schröder, H., and Luiten, P. G. (1991). Colocalization of muscarinic and nicotinic receptors in cholinoceptive neurons of the suprachiasmatic region in young and aged rats. Brain Res. 542, 348–352.
van Gool, W. A., Witting, W., and Mirmiran, M. (1987). Age-related changes in circadian sleep-wakefulness rhythms in male rats isolated from time cues. Brain Res. 413, 384–387.
Van Someren, E. J., Riemersma, R. F., and Swaab, D. F. (2002). Functional plasticity of the circadian timing system in old age: light exposure. Prog. Brain Res. 138, 205–231.
Videnovic, A., Leurgans, S., Fan, W., Jaglin, J., and Shannon, K. M. (2009). Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat. Disord. 15, 471–474.
Vitiello, M. V., Poceta, J. S., and Prinz, P. N. (1991). Sleep in Alzheimer’s disease and other dementing disorders. Can. J. Psychol. 45, 221–239.
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Weinert, D. (2005). The temporal order of mammals. Evidence for multiple central and peripheral control mechanisms and for endogenous and exogenous components: some implications for research on aging. Biol. Rhythm Res. 36, 293–308.
Weishaupt, J. H., Bartels, C., Pölking, E., Dietrich, J., Rohde, G., Poeggeler, B., Mertens, N., Sperling, S., Bohn, M., Hüther, G., Schneider, A., Bach, A., Sirén, A. L., Hardeland, R., Bähr, M., Nave, K. A., and Ehrenreich, H. (2006). Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 41, 313–323.
Wenk, G. L., Pierce, D. J., Struble, R. G., Price, D. L., and Cork, L. C. (1989). Age-related changes in multiple neurotransmitter systems in the monkey brain. Neurobiol. Aging 10, 11–19.
Whitehead, D. L., Davies, A. D., Playfer, J. R., and Turnbull, C. J. (2008). Circadian rest-activity rhythm is altered in Parkinson’s disease patients with hallucinations. Mov. Disord. 23, 1137–1145.
Wiegand, M., Möller, A. A., Lauer, C. J., Stolz, S., Schreiber, W., Dose, M., and Krieg, J. C. (1991). Nocturnal sleep in Huntington’s disease. J. Neurol. 238, 203–208.
Willis, G. L., and Turner, E. J. (2007). Primary and secondary features of Parkinson’s disease improve with strategic exposure to bright light: a case series study. Chronobiol. Int. 24, 521–537.
Wise, P. M., Cohen, I. R., Weiland, N. G., and London, E. D. (1988). Aging alters the circadian rhythm of glucose utilization in the suprachiasmatic nucleus. Proc. Natl. Acad. Sci. U.S.A. 85, 5305–5309.
Wu, Y. H., and Swaab, D. F. (2007). Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer’s disease. Sleep Med. 8, 623–636.
Wu, Y. H., Zhou, J. N., Van Heerikhuize, J., Jockers, R., and Swaab, D. F. (2007). Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol. Aging 28, 1239–1247.
Yamazaki, S., Numano, R., Abe, M., Hida, A., Takahashi, R., Ueda, M., Block, G. D., Sakaki, Y., Menaker, M., and Tei, H. (2000). Resetting central and peripheral circadian oscillators in transgenic rats. Science 288, 682–685.
Yamazaki, S., Straume, M., Tei, H., Sakaki, Y., Menaker, M., and Block, G. D. (2002). Effects of aging on central and peripheral mammalian clocks. Proc. Natl. Acad. Sci. U.S.A. 99, 10801–10806.
Zhang, J. H., Sampogna, S., Morales, F. R., and Chase, M. H. (2002). Age-related changes in hypocretin (orexin) immunoreactivity in the cat brainstem. Brain Res. 930, 206–211.
Zhang, J. H., Sampogna, S., Morales, F. R., and Chase, M. H. (2005). Age-related changes of hypocretin in basal forebrain of guinea pig. Peptides 26, 2590–2596.
Zhang, Y., Brainard, G. C., Zee, P. C., Pinto, L. H., Takahashi, J. S., and Turek, F. W. (1998). Effects of aging on lens transmittance and retinal input to the suprachiasmatic nucleus in golden hamsters. Neurosci. Lett. 258, 167–170.
Keywords: aging, neurodegenerative, sleep, wake, circadian, disease
Citation: Singletary KG and Naidoo N (2011) Disease and degeneration of aging neural systems that integrate sleep drive and circadian oscillations. Front. Neur. 2:66. doi: 10.3389/fneur.2011.00066
Received: 24 May 2011;
Accepted: 28 September 2011;
Published online: 24 October 2011.
Edited by:
Birendra N. Mallick, Jawaharlal Nehru University, IndiaReviewed by:
Luigi Ferini-Strambi, San Raffaele Scientific Institute, ItalyEdgar Garcia-Rill, University of Arkansas for Medical Sciences, USA
Claude Gottesmann, University of Nice Sophia Antipolis, France
Copyright: © 2011 Singletary and Naidoo. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Kristan G. Singletary, Center for Sleep and Circadian Neurobiology, University of Pennsylvania, TRL Suite 2100, 125 South 31st Street, Philadelphia, PA 19104, USA. e-mail:a3Jpc3RhbnNAbWFpbC5tZWQudXBlbm4uZWR1