

- 1 Department of Physiology, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
- 2 Department of Physical Performance, Norwegian School of Sport Sciences, Oslo, Norway
Sympathetic hyperactivity and parasympathetic insufficiency characterize blood pressure (BP) control in genetic hypertension. This shift is difficult to investigate in anesthetized rats. Here we present a pharmacological approach to simultaneously provoke sympathetic and parasympathetic transmitter release, and identify their respective roles in the concomitant cardiovascular response. To stimulate transmitter release in anesthetized normotensive (WKY) and spontaneously hypertensive rats (SHR), we injected intravenously 4-aminopyridine (4-AP), a voltage-sensitive K+ channel (KV) inhibitor. A femoral artery catheter monitored BP, an ascending aorta flow-probe recorded cardiac output and heart rate (HR). Total peripheral vascular resistance (TPVR) was calculated. 4-AP-induced an immediate, atropine (muscarinic antagonist)- and hexamethonium (ganglion blocker)-sensitive bradycardia in WKY, and in both strains, a subsequent, sustained tachycardia, and norepinephrine but not epinephrine release. Reserpine (sympatholytic), nadolol (β-adrenoceptor antagonist) or right vagal nerve stimulation eliminated the late tachycardia, adrenalectomy, scopolamine (central muscarinic antagonist) or hexamethonium did not. 4-AP increased TPVR, transiently in WKY but sustained in SHR. Yohimbine (α2-adrenoceptor antagonist) prevented the TPVR down-regulation in WKY. Reserpine and prazosin (α1-adrenoceptor antagonist) eliminated the late vasoconstriction in SHR. Plasma epinephrine overflow increased in nadolol-treated SHR. Through inhibition of KV, 4-AP activated parasympathetic ganglion transmission and peripheral, neuronal norepinephrine release. The sympathetic component dominated the 4-AP–HR-response in SHR. α2-adrenoceptor-dependent vasodilatation opposed norepinephrine-induced α1-adrenergic vasoconstriction in WKY, but not SHR. A βAR-activated, probably vagal afferent mechanism, hampered epinephrine secretion in SHR. Thus, 4-AP activated the autonomic system and exposed mechanisms relevant to hypertensive disease.
Introduction
The autonomic nervous system controls blood pressure (BP) through its influence on the heart and vascular smooth muscle cells (VSMC). Sympathetic nerves, through β-adrenoceptors (AR), and vagal, parasympathetic nerves, through muscarinic acetylcholine (ACh) receptors (mAChR), are the main, acute, peripheral regulators of heart rate (HR), with a positive and negative chronotropic effect, respectively. VSMC tension is maintained by sympathetic nerve activity, and opposed by endothelial relaxing factors such as nitric oxide (NO). Although ACh is a potent stimulator of NO synthesis in vitro, parasympathetic nerves do not innervate the vascular endothelium. However, vagal nerve ACh release may influence endothelial function through cholinergic inhibition of inflammatory cell cytokine production (Borovikova et al., 2000). In addition, ACh may influence norepinephrine release through presynaptic receptors (Boehm and Kubista, 2002). Sympathetic and parasympathetic control of BP exists in a homeostatic balance, integrated in the central nervous system (CNS) after afferent signals from peripheral receptors, among them the baroreceptors. The baroreflex has long been known to be impaired in hypertension (Thrasher, 2005). The disorder is regarded as an important factor in the pathogenesis of the hypertensive disease, precipitating an elevated resting BP, HR, and total peripheral vascular resistance (TPVR; Esler et al., 2001; Palatini and Julius, 2009). A high resting HR has been shown to be the most reliable predictor of cardiovascular morbidity (Palatini and Julius, 1997; Tjugen et al., 2010).
The sympathetic system mostly elicits a positive response, such as an increase in BP, HR, or vascular tension. These are easily measured. The function of the parasympathetic system is generally to hamper the adrenergic response. Parasympathetic influence for instance on HR can therefore be recorded only through its inhibitory effect on an adrenergically stimulated tachycardia. Thus, identification of pathogenic factors that cause a shift in the autonomic control of cardiac function or vascular tension, requires an experimental condition where both the parasympathetic and sympathetic components are active.
Active dual autonomic control is present in conscious individuals, and the parasympathetic/sympathetic balance can be evaluated by studying autonomic reflexes, such as in baroreflex sensitivity studies or respiratory mediated HR variability studies (Freeman, 2006). Such and similar studies make use of the baroreceptor reflex, where an increase in BP will stimulate the baroreceptors and increase parasympathetic inhibition of sympathetic BP or HR control. Vice versa, a fall in BP reduces baroreceptor signaling, and sympathetic activity is increased due to reduced parasympathetic inhibition. Such tests may not function in anesthetized animals, since anesthetics often disturb reflex activation. In Nembutal-anesthetized rats, large changes in BP elicited hardly any change in HR (Bjørnstad-Østensen and Berg, 1994; Berg et al., 2009). Still, anesthesia must be used in studies where acute surgery is implemented in the experimental design. However, the absence of reflexes may in fact be advantageous since baroreflex activation will not interfere with the cardiovascular response to a pharmacologically stimulated transmitter release. We know of no method, which will simultaneously activate both the parasympathetic and sympathetic nervous system in the anesthetized rat.
The purpose of the present investigation was therefore to establish a method to achieve dual activation of both branches of the autonomic nervous system in the anesthetized rat, so that factors, which influence their balance, can be studied. We have previously used 4-aminopyridine (4-AP) to study the role of voltage-sensitive K+ channels (KV) in vascular tension control (Berg, 2002). By inhibiting these channels, 4-AP induces depolarization and opening of voltage-sensitive Ca2+ channels. In VSMC, the entry of Ca2+ elicits vasoconstriction, while it in neurons activates vesicular release of transmitters such as norepinephrine and ACh (Glover, 1982). Thus, apart from inducing VSMC constriction in isolated aortic rings as well as in vivo, 4-AP also induced a reserpine-sensitive tachycardia in both normotensive (WKY) and spontaneously hypertensive rats (SHR; Berg, 2002, 2003). Furthermore, 4-AP-induced an atropine-sensitive salivation (Berg, 2002). It therefore seemed that 4-AP might activate transmitter release from both branches of the autonomic nervous system in the anesthetized rat. However, before 4-AP could be used as a tool to analyze the altered autonomic control in hypertension, the involvement of autonomic nerve transmitter release in the cardiovascular response to 4-AP needed verification. In the present study, we tested if 4-AP indeed induced dual autonomic nerve transmitter release, and if the impact of both the parasympathetic and the sympathetic branch on the cardiovascular system could be separated and identified. We also tested if 4-AP revealed changes in autonomic control of HR and TPVR in SHR similar to that known to be present in hypertensive disease.
Materials and Methods
Experimental Procedure
All experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and were approved by the institutional ethics committee. The study included 130 male WKY (Wistar Kyoto, 284 ± 3 g) and 131 male SHR (NHsd strain, 286 ± 2 g; 12–14 weeks) on conventional rat chow diet (0.7% NaCl) and water ad lib. The rats were anesthetized with Nembutal (70–75 mg/kg, i.p.), and connected to a positive-pressure respirator, ventilated with air. Blood gas parameters were maintained in both strains (Berg, 2002, 2003). Systolic (SBP) and diastolic BP (DBP) were monitored through a catheter in the femoral artery, and cardiac output (CO) and HR by a flow-probe on the ascending aorta (Berg et al., 2010). Mean arterial BP [MBP = (SBP−DBP/3) + DBP] and TPVR (MBP/CO) were calculated. Body temperature was monitored by a thermo sensor inserted ingenually into the abdominal cavity, and kept at 37–38°C by external heating. Drugs were dissolved in phosphate buffered saline (PBS: 0.01 M Na-phosphate, pH 7.4, 0.14 M NaCl) and administered through the femoral vein (0.6–1.0 ml/kg, bolus injections) unless otherwise indicated (Table 1).
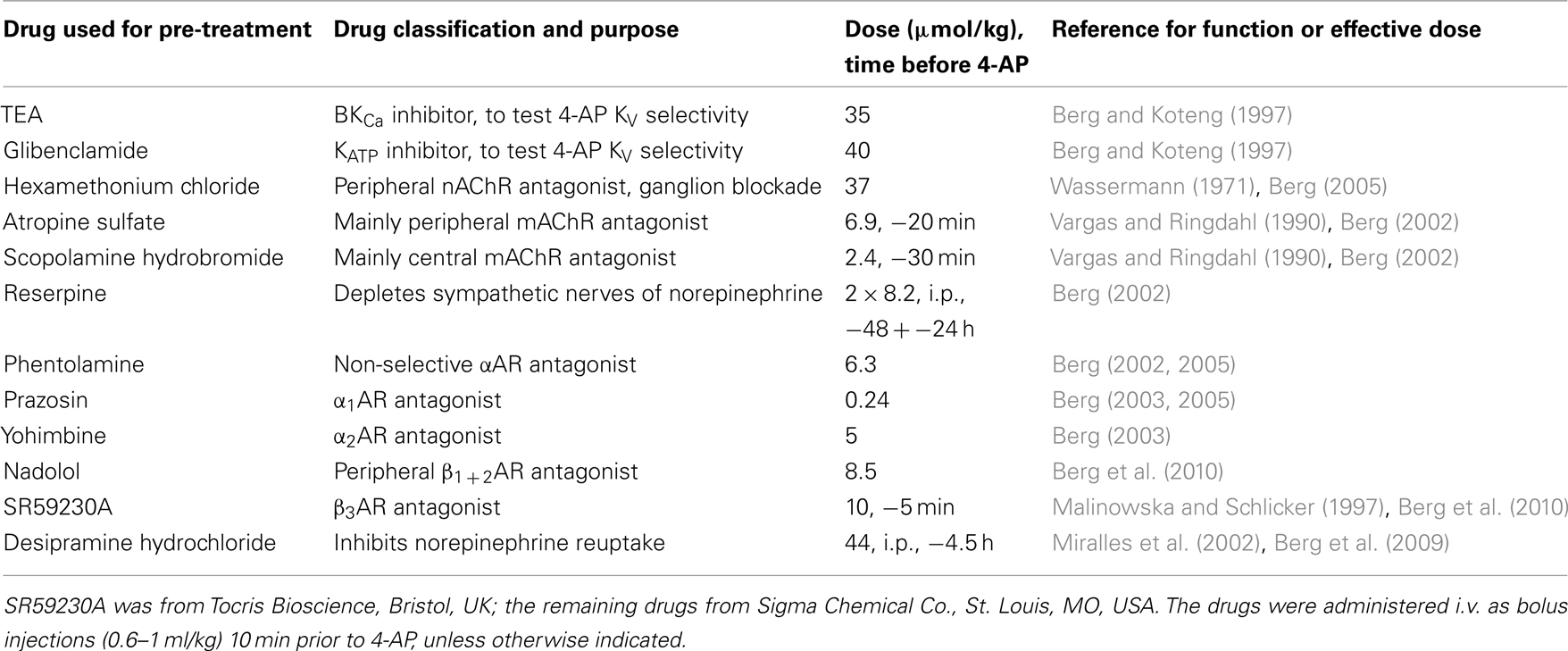
Table 1. Details on the drugs used for pre-treatment.
Experimental Protocols
Experimental details and function of drugs included in the experiments are outlined in Table 1. All rats received an intravenous, bolus injection containing a sub-maximal dose (Berg and Koteng, 1997) of the KV inhibitor 4-AP (34.5 μmol/kg, 1.3 ml/kg), and were monitored for 25 min. Control rats were pre-treated with PBS. To control 4-AP KV channel selectivity, the PBS-sham pre-treatment was substituted with other K+ channel inhibitors, i.e., tetraethyl ammonium chloride (TEA, high conductance Ca2+-activated K+ channels, BKCa, inhibitor) or glibenclamide (ATP-sensitive K+ channels, KATP, inhibitor). To identify autonomic nervous system components responsible for the cardiovascular response to 4-AP, rats were pre-treated with various autonomic nervous system interventions. Involvement of ganglion transmission in the response to 4-AP was investigated by pre-treatment with the ganglion blocker hexamethonium. To identify the involvement of mAChR activation in the cardiovascular response to 4-AP, rats were pre-treated with atropine sulfate or scopolamine hydrobromide, which have a central:peripheral action of 1:10 and 1:1, respectively. Scopolamine would therefore identify a role of CNS mACh-dependant sympathetic output, and atropine predominantly an involvement of peripheral parasympathetic nerve activation. The ability of the vagal nerve to influence the cardiovascular response was verified by electrical stimulation of the right vagal nerve, which primarily influences HR by inhibiting sympathetic activation of the sinus node. The vagal nerve was accessed through a cervical section, dissected free from the cervical sympathetic chain and cut. The distal end of the nerve was stimulated electrically using a suction electrode (2 ms duration, 7.5 V, 10 Hz) from 5 min before injection of 4-AP to the end of the 25-min 4-AP-observation-period. The role of activation of the adrenergic system in the cardiovascular response to 4-AP was studied by pre-treatment with sympathetic nerve transmitter depletion using reserpine, the αAR antagonists phentolamine, prazosin, and yohimbine, the βAR antagonists nadolol and SR59230A, or bilateral adrenalectomy to identify an involvement of adrenal catecholamine secretion (AdrX, performed 30 min before the first injection, (Berg, 2002, 2003; Berg et al., 2010). To augment the effect of adrenergic stimulation, we inhibited norepinephrine reuptake with desipramine. The number of rats per group is given in Table 2.
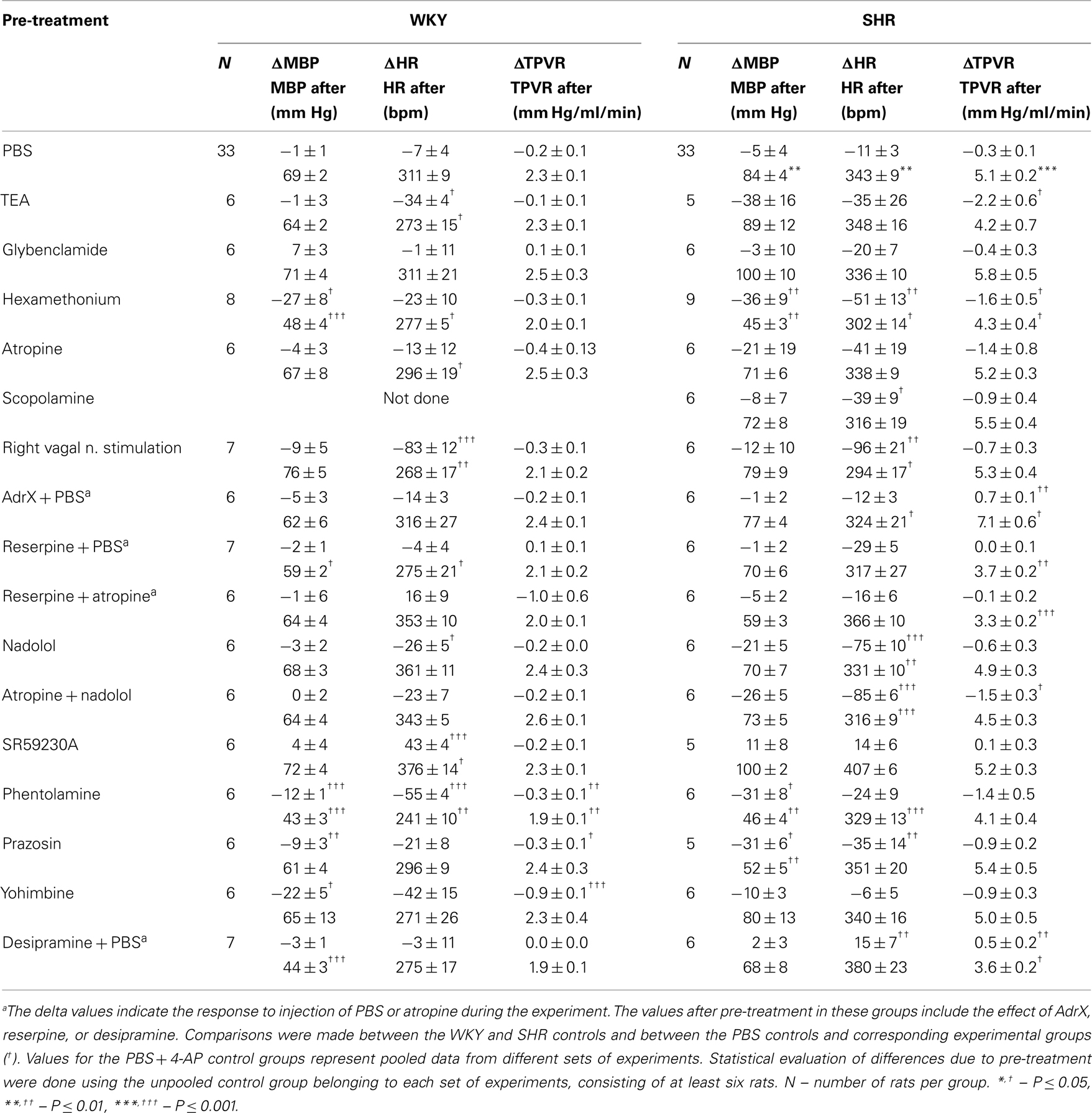
Table 2. The response to pre-treatment and the resulting MBP, HR, and TPVR after pre-treatment, i.e., baselines prior to 4-AP.
Plasma Catecholamines
In some of the experiments, 1.5 ml blood was sampled from the femoral artery after the 25-min observation-period into tubes containing 40 μl 0.2 M glutathione with 0.2 M ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA; 4°C). Plasma was stored at −80°C, and the concentration of epinephrine and norepinephrine determined using an HPLC-electrochemical detection method as previously described (Jensen et al., 2005).
Saliva Measurements
Salivation is not present in anesthetized rats, but can be activated by parasympathetic or adrenergic stimulation; here by 4-AP. Whole saliva was collected from the oral cavity with a pipette, and saliva volume was determined by weight. The saliva was stored at −20°C. Salivary kallikrein, in the rat primarily a submandibular salivary gland component, which is stimulated predominantly by α-adrenoceptor activation (Ørstavik and Gautvik, 1977), was measured by S2266 amidolytic activity (Berg et al., 1992).
Statistical Analyses
Results are presented as mean values ± SEM. The HR-response to 4-AP are shown as real change, since ΔHR in response to 4-AP will appear exaggerated when pre-treatment greatly reduced baseline HR. The-TPVR-response is expressed in percent of baseline to take into account that resistance ∼1/r4, where r represents resistance vessel radius. Recorded data were averaged every minute throughout the experiment. During the initial response (1–2 min), data were averaged every seventh heart beat to accurately determine HRnadir and TPVRmax. Significant effects of pre-treatment compared to that of PBS in the controls, i.e., from before pre-treatment to before 4-AP, were determined by one-way ANOVA, followed by two-sample Student’s t-tests. The 4-AP-response-curves were evaluated with Repeated Measures Analyses of Variance and Covariance, first as over-all tests, then for one or between two group(s). One- and two-sample Student’s t-tests were used to locate significant responses and differences between groups, respectively, at the initial response and after 25 min. The P-value was for all tests and each step adjusted according to Bonferroni, proceeding only when the presence of significant responses or differences was indicated. One-way ANOVA and two-sample Student’s t-tests were used to evaluate differences in saliva volume, and saliva kallikrein and plasma catecholamine concentrations (P ≤ 0.05). In the presence of outliers, Kruskal Wallis tests substituted the two-sample Student’s t-tests. Correlations were determined with the Pearson Correlation test (P ≤ 0.05).
Results
The Response to 4-AP
The bolus injection of 4-AP-induced an immediate and sustained increase in MBP in both strains (Figure 1). This response comprised an initial bradycardia in WKY with a subsequent, sustained tachycardia in both strains, and a sustained increase in CO in WKY (Figure 1). There was also an immediate rise in TPVR, which was transient in WKY but sustained in SHR (Figure 1). 4-AP-induced an increase in the plasma concentration of norepinephrine, which was greater in SHR than in WKY (P < 0.001), but had no effect on the concentration of epinephrine (Table 3). Since our purpose was to study autonomic nerve activation and influence, we focused on the changes elicited in HR and TPVR.
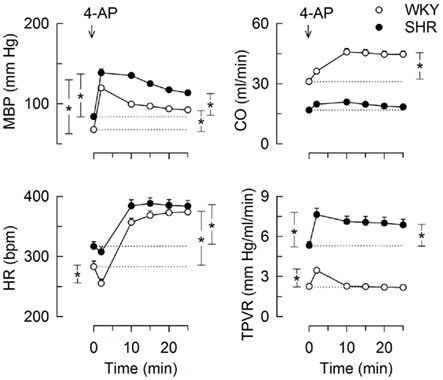
Figure 1. The cardiovascular response to 4-AP in WKY and SHR. 4-AP was injected as a bolus injection (arrow). The SBP- and DBP-responses paralleled, MBP was therefore used to demonstrate the BP-response. Curve evaluation was done by Repeated Measures Analyses of Variance and Covariance with significant responses located as described in Materials and Methods, at HR nadir (brackets left of curves) and at 25 min (brackets right of curves). Please see Table 2 for cardiovascular baselines prior to 4-AP. * – P < 0.01.
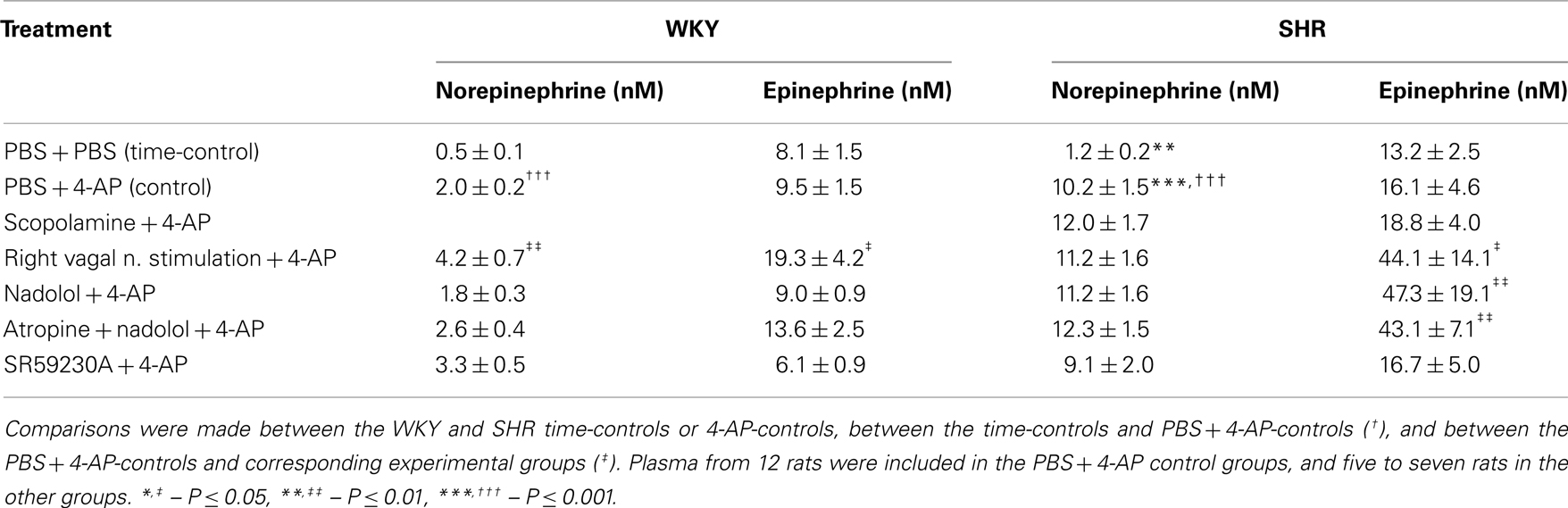
Table 3. The effect of 4-AP on plasma catecholamine concentrations.
The HR- and TPVR-response to 4-AP was not significantly different after prior inhibition of BKCa channels with TEA or KATP channels with glibenclamide (Figure 2), indicating that the cardiovascular response to 4-AP selectively involved KV.
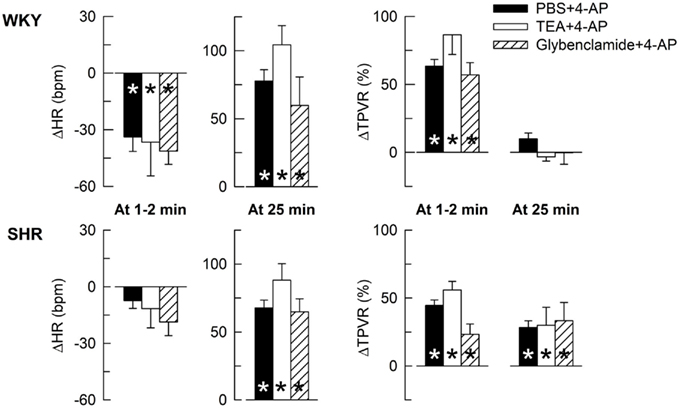
Figure 2. The HR- and TPVR-response to 4-AP in WKY and SHR after pre-treatment with other K+ channel inhibitors. Rats were pre-treated with the BKCa inhibitor TEA or the KATP channel inhibitor glibenclamide as indicated by symbol legend. Significant responses during the initial response and at 25 min were subsequently located as indicated (* within symbol). Significant group differences were not detected. The effects of TEA and glibenclamide on baselines are shown in Table 2. * – P ≤ 0.025.
Autonomic Involvement in the Initial 4-AP-Induced Bradycardia, i.e., the Parasympathetic Component
The 4-AP-induced, initial bradycardia in WKY was abolished by the mAChR antagonist atropine (Figure 3) and hexamethonium (Figure 4; P = NS one-sample Student’s t-tests, and P = 0.023 and 0.009, respectively, compared to the controls). AdrX, reserpine, nadolol, or SR59230A, did not influence the bradycardia (Figure 5). These observations showed that 4-AP activated ACh release and negative chronotropic mAChR, and that this response also relied on nAChR, possibly within the ganglion. The mAChR bradycardia was not further enhanced by electrical stimulation of the right vagal nerve (Figure 3) or influenced by the norepinephrine reuptake inhibitor desipramine (Figure 6).
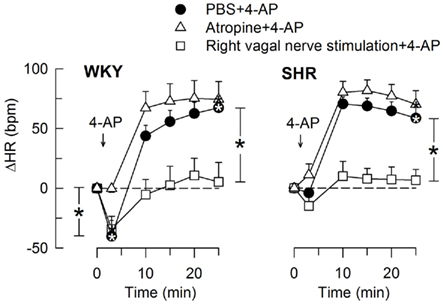
Figure 3. The HR-response to 4-AP after pre-treatment with the mAChR antagonist atropine or electrical stimulation of the right, efferent vagal nerve. After curve evaluation, significant responses (* within symbol) and group differences at HR nadir (brackets left of curves) and at 25 min (brackets right of curves) were located as indicated. Atropine had little effect on the HR baseline, whereas vagal stimulation greatly reduced HR in both strains (Table 2). * – P < 0.025.
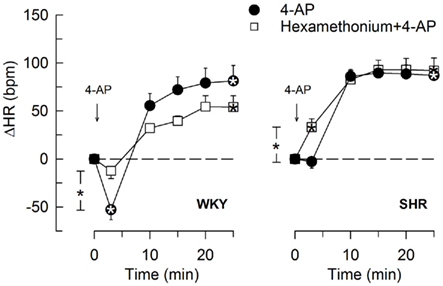
Figure 4. The HR-response to 4-AP after pre-treatment with nAChR antagonist, i.e., the ganglion blocker hexamethonium. Significant responses (* within symbol) and group differences at the initial HRnadir (brackets left of curves) and after 25 min (none detected) were as indicated. Hexamethonium reduced baseline HR in both strains (Table 2). * – P ≤ 0.025.
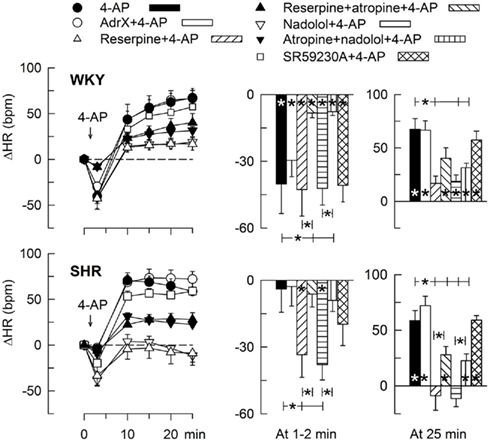
Figure 5. HR-response to 4-AP after removal of components of the adrenergic system. The rats were pre-treated to eliminate adrenal (AdrX) or neuronal (reserpine) catecholamines, peripheral β1 + 2AR (peripherally restricted antagonist nadolol), or β3AR-activity (β3AR antagonist SR59230A). Reserpine and nadolol were also combined with mAChR antagonist (atropine), as indicated by symbol legends. Column graphs to the right shows HRnadir during the initial response (1–2 min) and ΔHR at 25 min. Significant responses (* within column) and group differences (brackets) were located as indicated. Baseline HR was slightly reduced after reserpine and nadolol in WKY and clearly after nadolol in SHR (Table 2). * – P ≤ 0.025.
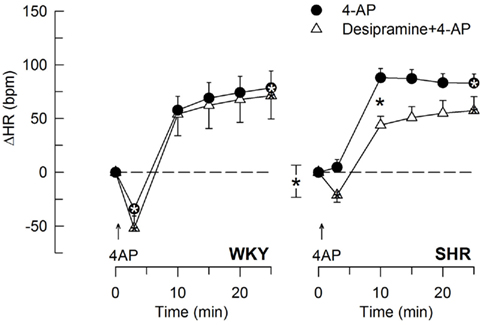
Figure 6. The effect of the norepinephrine reuptake inhibitor desipramine on the HR-response to 4-AP. Significant responses (* within symbol) at HR nadir and at 25 min were located as indicated. A difference between the SHR control and the desipramine-treated group was detected at 10 min (* between curves). Baseline HR was not different after desipramine (Table 2). * – P ≤ 0.025 and 0.0167 for significant responses and group differences, respectively.
In SHR, 4-AP-induced bradycardia only after pre-treatment with reserpine or nadolol (P ≤ 0.01), and this response was abolished by additional pre-treatment with atropine (Figure 5). Instead of bradycardia, 4-AP-induced an initial tachycardia in hexamethonium- (Figure 4) and scopolamine-treated (Figure 7) SHR (P ≤ 0.001). These results demonstrated that in SHR, 4-AP activated a sympathetic nerve-dependent, β1/2AR-mediated tachycardia, which shielded the initial parasympathetic response. This initial balance was not altered by vagal nerve stimulation (Figure 3).
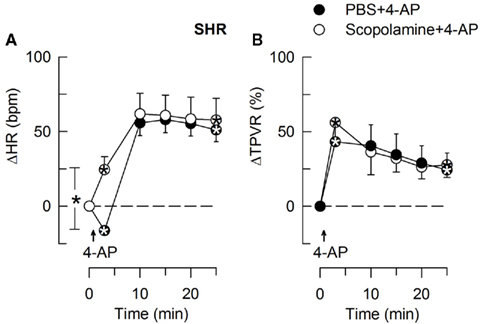
Figure 7. The effect of scopolamine on the HR- (A) and TPVR- (B) response to 4-AP in SHR. Scopolamine had no effect on the MBP-response to 4-AP (not shown). We have previously shown that scopolamine did not significantly alter the response in WKY (Berg, 2002). Significant responses (* within symbol) during the initial response and after 25 min, as well as group differences at these times (brackets) were detected as indicated. Scopolamine caused a minor reduction in baseline HR (Table 2). * – P ≤ 0.025.
There was no significant correlation between the 4-AP-induced, initial bradycardia, and the simultaneous increase in MBP within each control group or within all groups in each strain.
Autonomic Involvement in the Subsequent 4-AP-Induced Tachycardia, i.e., the Sympathetic Component
The subsequent 4-AP-induced tachycardia was abolished as previously described (Berg, 2002, 2003) by reserpine in both strains (Figure 5). The tachycardia was also eliminated by the peripherally restricted β1 + 2AR antagonist nadolol, but was not significantly different after acute AdrX or the β3AR antagonist SR59230A (Figure 5). Like previously shown in WKY (Berg, 2002), the 4-AP-induced adrenergic tachycardia in SHR was not different after the centrally active mAChR antagonist scopolamine (Figure 7) or the ganglion blocker hexamethonium (Figure 4). In SHR, the tachycardia was delayed by desipramine (P ≤ 0.003), which also allowed a minor initial bradycardia (Figure 6).
Stimulation of the right vagal nerve abolished the 4-AP-induced tachycardia in both strains, whereas atropine had no effect (Figure 3). However, when the sympathetic component had been eliminated by reserpine or nadolol, atropine increased the HR-response to 4-AP in SHR (P ≤ 0.014 and 0.006, respectively) but not in WKY (Figure 5). These results demonstrated that in SHR, 4-AP-induced a sustained, parasympathetic mAChR activation, the effect of which was totally shielded by the sympathetic nerve response.
Autonomic Involvement in the 4-AP-Induced TPVR-Response
In agreement with previously published results (Berg, 2002, 2003), the initial rise in TPVR in response to 4-AP was not reduced by reserpine (Figure 8) or α1AR antagonist in either strain (P = NS; Figure 9), compatible with vasoconstriction being due to inhibition of VSMC KV channels. In addition to our previous results, we found that this vasoconstriction was not significantly different after SR59230A (Figure 8), hexamethonium (Figure 10), or vagal nerve stimulation (Figure 11) in either strain, but was reduced after reserpine + atropine in SHR (Figure 8).
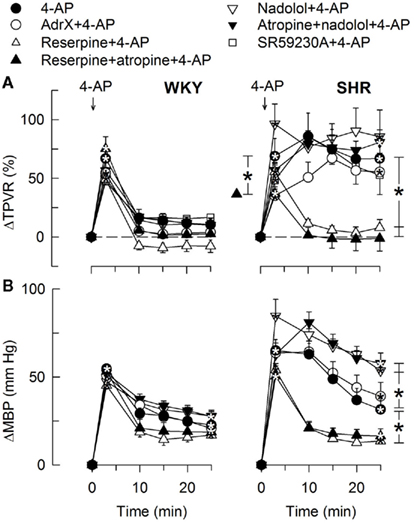
Figure 8. The TPVR- (A) and MBP- (B) response to 4-AP after blockade of components of the adrenergic system. Adrenal catecholamines were eliminated by AdrX, neuronal norepinephrine by reserpine, β1 + 2AR activity with the peripherally restricted antagonist nadolol, and β3AR-activity with the β3AR antagonist SR59230A. Reserpine and nadolol were given alone or combined with mAChR antagonist (atropine), as indicated by symbol legends. Significant responses (* within symbol) and group differences (brackets) were located as indicated. Baseline TPVR was higher in AdrX SHR, and reduced after reserpine, reserpine + atropine, and nadolol + atropine in SHR. MBP was lower after reserpine in WKY (Table 2). * – P ≤ 0.025.
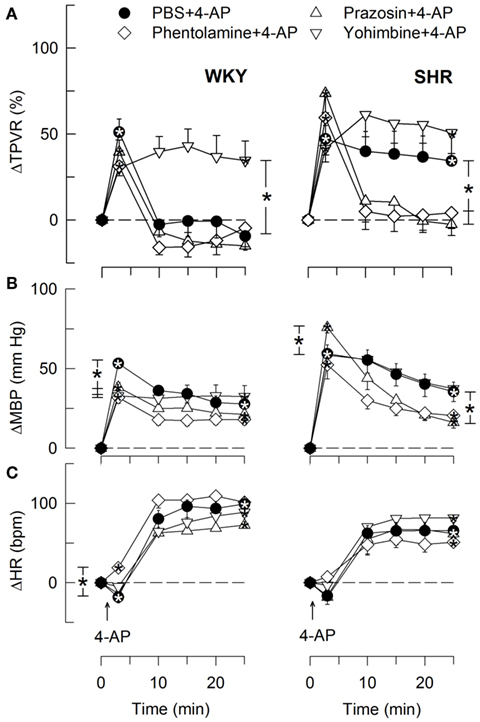
Figure 9. The TPVR- (A), MBP- (B), and HR- (C) response to 4-AP in WKY and SHR after pre-treatment with αAR antagonists. Significant responses (* within symbol) and group differences during the immediate TPVRmax and at 25 min (brackets) were located as indicated. All antagonists reduced baseline TPVR in WKY, but not significantly in SHR (Table 2). Baseline MBP was lower after all three antagonists in WKY and after phentolamine and prazosin in SHR. Baseline HR was lower after phentolamine in WKY and after phentolamine and prazosin in SHR (Table 2). * – P ≤ 0.025.
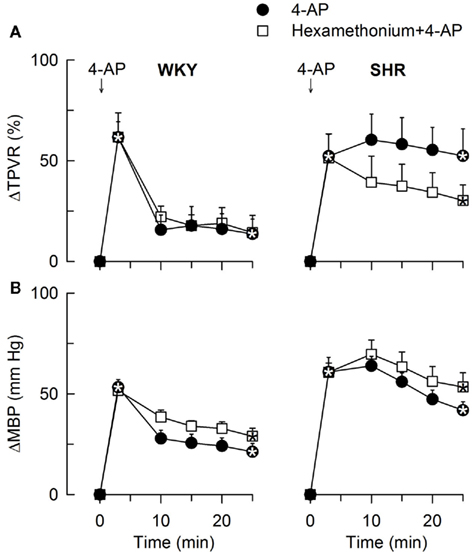
Figure 10. The TPVR- (A) and MBP- (B) response to 4-AP after pre-treatment with the peripherally restricted nAChR antagonist, ganglion blocker hexamethonium. Significant responses (* within symbol) during the initial response and after 25 min, were detected as indicated. Significant group differences were not detected. Hexamethonium reduced baseline MBP in both strains and also TPVR in SHR (Table 2). * – P ≤ 0.025.
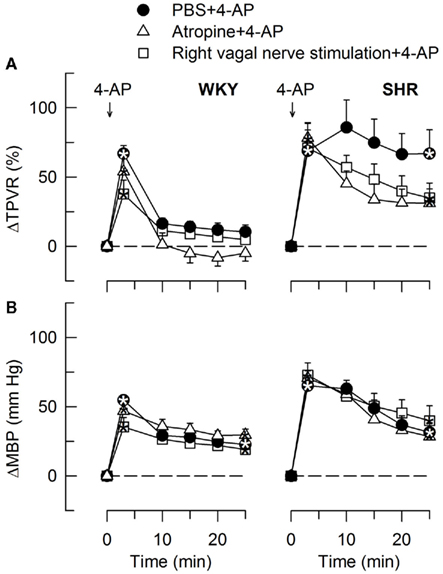
Figure 11. The TPVR- (A) and MBP- (B) response to 4-AP after atropine and vagal nerve stimulation. Significant responses (* within symbols) at the initial peak response and after 25 min were detected as indicated. Significant group differences were not detected. Atropine and vagal nerve stimulation did not alter MBP and TPVR baselines (Table 2). * – P ≤ 0.025.
The subsequent down-regulation of TPVR in WKY was not influenced by reserpine (Figure 8), phentolamine, or prazosin, but was prevented by the α2AR antagonist yohimbine (Figure 9). The return to TPVR baseline in this strain was also prevented in part by the reuptake inhibitor desipramine (Figure 12). The sustained late TPVR-response in SHR was due to neuronal norepinephrine release and activation of a α1AR-mediated vasoconstriction since it was eliminated by reserpine (Figure 8), phentolamine, and prazosin but not by yohimbine (Figure 9). The late TPVR-response to 4-AP was not different after scopolamine in SHR (Figure 7), and in both strains not altered by hexamethonium (Figure 10), vagal nerve stimulation, atropine, alone (Figure 11) or combined with nadolol, nadolol alone, AdrX, or SR59230A (Figure 8).
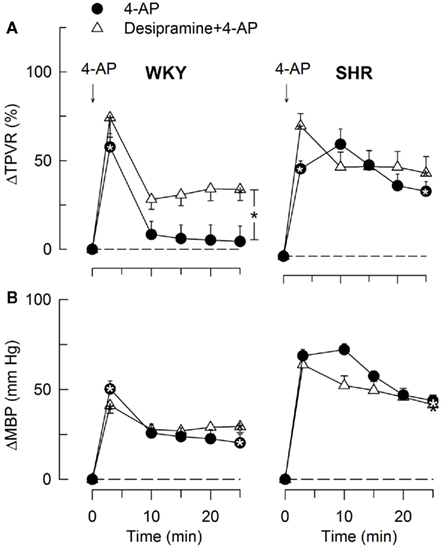
Figure 12. The TPVR- (A) and MBP- (B) response to 4-AP after inhibition of norepinephrine reuptake with desipramine. Significant responses (* within symbol) and group differences during the immediate TPVRmax (none detected) and after 25 min (brackets right of curve) were located as indicated. Desipramine reduced baseline TPVR in both strains and also MBP in WKY (Table 2). * – P ≤ 0.025.
Autonomic Involvement in the 4-AP-Induced Changes in Plasma Catecholamine Concentrations
Norepinephrine and epinephrine overflow to plasma in 4-AP-treated WKY was increased by vagal nerve stimulation (Table 3). In SHR, we detected no differences in the plasma concentration of norepinephrine, but the concentration of epinephrine was increased after vagal nerve stimulation, nadolol, and atropine + nadolol, but not after scopolamine or SR59230A (Table 3).
4-AP-Induced Salivation
4-AP activated salivary secretion with a greater efficacy in WKY than in SHR (40 ± 8 and 13 ± 4 μl whole saliva, respectively, P = 0.003), starting 4–6 min after the injection of 4-AP. Salivary kallikrein (S2266-activity) concentration and total secretion were higher in SHR (348 ± 82 U/ml and 15.6 ± 5.7 U, respectively) than in WKY (112 ± 35 U/ml and 3.4 ± 0.2 U; P ≤ 0.009). 4-AP-induced salivation was absent in all groups pre-treated with atropine, scopolamine, or hexamethonium, but was not much influenced by other pre-treatments (data not shown).
Discussion
The present results demonstrated that 4-AP activated both the sympathetic and the parasympathetic branch of the autonomic nervous system in anesthetized rats. The HR-response elicited by 4-AP, substituted an initial parasympathetic bradycardia followed by a sympathetic tachycardia. The HR-response in SHR reflected the shifted autonomic control of HR in hypertension, i.e., sympathetic hyperactivity with low parasympathetic influence, in spite of that parasympathetic activation was actually greater in SHR than in WKY. Norepinephrine release, elicited by 4-AP, elevated TPVR in SHR but not WKY, due to the absence of an α2AR vasodilatory component, which down-regulated tension in WKY. We also detected a βAR-mediated inhibition of adrenal epinephrine secretion in SHR.
The 4-AP-induced parasympathetic activation was demonstrated through the ability of the mAChR antagonist atropine to eliminate the initial 4-AP-induced bradycardia in WKY. In addition, 4-AP activated salivary secretion, which was blocked by pre-treatment with atropine. The initial bradycardia was also prevented by the nAChR antagonist, ganglion blocker hexamethonium, suggesting a baroreflex activation. However, there was no correlation between the initial ΔMBP and ΔHR, and, under similar conditions, a rise in BP due to selective stimulation of sympathetic nerve norepinephrine release by tyramine, elicited tachycardia only (Berg et al., 2010). Moreover, hexamethonium also abolished the atropine-sensitive salivation, which is not part of the baroreflex. We therefore concluded that 4-AP activated presynaptic ACh release and, through that, postsynaptic nAChR in parasympathetic ganglions, parallel to that observed in isolated rabbit superior cervical ganglions (Simmons and Dun, 1984). Subsequent release of ACh from the postsynaptic parasympathetic neuron was responsible for activation of the mAChR, which caused the initial bradycardia.
The tachycardia, which followed the initial bradycardia, was caused in both strains by 4-AP-activated norepinephrine release. This conclusion was based on the fact that reserpine, like previously described (Berg, 2002, 2003), but not AdrX, eliminated this response. The same was true for the late rise in TPVR in SHR. The tachycardia was also eliminated by the peripherally restricted β1 + 2AR antagonist nadolol, and the late TPVR-response in SHR also by the α1AR antagonist prazosin. In addition, 4-AP increased the plasma concentration of norepinephrine but not epinephrine. 4-AP evidently crossed the blood brain barrier, evidenced by the occurrence of muscular twitches due to cortical stimulation (Paskov et al., 1986). However, scopolamine and hexamethonium did not alter the 4-AP-induced tachycardia or the late TPVR-response. Thus, these 4-AP-induced adrenergic responses did not rely on CNS ACh release and increased sympathetic output, or on ganglion transmission. We therefore concluded that 4-AP activated release of norepinephrine from peripheral sympathetic nerve terminals.
In WKY, the parasympathetic and sympathetic activation was separated in time, since the initial bradycardia was not influenced by reserpine or nadolol and the subsequent tachycardia was not altered by atropine, even in the presence of sympatho-inhibition with reserpine or nadolol. In SHR, the sympathetic nerve-dependent, positive chronotropic response totally dominated the HR-response to 4-AP, in spite of that ACh release did occur even throughout the 4-AP-observation-period. This conclusion was made because an initial, atropine-sensitive bradycardia was observed in SHR only after pre-treatment with the sympatholytics reserpine or nadolol, and after such pre-treatment, atropine increased also the late HR-response to 4-AP in this strain. Furthermore, after inhibition of the parasympathetic component with hexamethonium or scopolamine, 4-AP-induced an initial tachycardia in SHR. The functionality of vagal mAChR inhibition of sympathetic nerve-induced stimulation of the sinus node was also verified by that the 4-AP-induced tachycardia was eliminated by electrical stimulation of the right vagal nerve in SHR, as in WKY. The HR-response to 4-AP in SHR therefore fully reflected the shifted autonomic balance in hypertension, with a sympathetic component dominating over parasympathetic HR control (Palatini and Julius, 2009; Tjugen et al., 2010), in spite of an augmented parasympathetic activation in SHR.
A sympathetic dominance was also detected for the 4-AP-induced salivation. Saliva flow, a 4-AP-induced parasympathetic response since it was inhibited by atropine, was lower in SHR than in WKY. In addition, the saliva concentration and total secretion of kallikrein, in the rat primarily an αAR-activated salivary component (Ørstavik and Gautvik, 1977), was higher in SHR than in WKY.
An initial bradycardia and a delayed development of the tachycardia were seen in desipramine-treated SHR. This observation suggested that increased βAR activation hampered the positive chronotropic response to norepinephrine release, in agreement with that excessive signaling may hamper cardiac function (Noor et al., 2010). A negative chronotropic effect due to β3AR-activity did not influence the HR-response to 4-AP in either strain, although the β3AR antagonist SR59230A, as in previous studies (Berg et al., 2010), increased resting HR in WKY but not SHR.
In addition to inhibiting VSMC and autonomic nerve KV, 4-AP also inhibits K+ channels mediating cardiac repolarization currents, such as the transient outward current and the delayed rectifier currents (Ridley et al., 2003; Tamargo et al., 2004). Inhibition of these currents results in action potential prolongation, which, in the absence of other significant electrophysiological changes, is a hallmark of failing ventricular myocardium and also produces arrhythmias (Tomaselli and Marban, 1999). Studies on the HR-response to 4-AP may therefore be of particular interest for understanding mechanisms responsible for arrhythmia and cardiac events in hypertensive disease.
Also the 4-AP-induced rise in TPVR was mediated through peripheral actions, first, in both strains, by inhibition of VSMC KV (Berg, 2002, 2003), and subsequently, in SHR, through activation of peripheral neuronal norepinephrine release. This was concluded since scopolamine and the ganglion blocker hexamethonium did not influence the immediate or the late TPVR-response to 4-AP. In addition, reserpine did not influence the initial but hampered the late TPVR-response in SHR. Still, there was a decrease in the initial TPVR-response in SHR pre-treated with reserpine + atropine, suggesting that the VSMC KV activity relied on ACh and mAChR. In isolated aortic rings, the 4-AP-induced vasoconstriction was sustained (Berg, 2002, 2003), whereas a down-regulation of tension was observed in WKY in vivo. This down-regulation in WKY involved an α2AR vasodilatory component since the non-selective α2AR antagonist yohimbine prevented the return to baseline. This elevated late TPVR-response in WKY after α2AR inhibition, represented a α1AR-mediated vasoconstriction, since TPVR was down-regulated after the α1 + 2AR antagonist phentolamine. The α2AR vasodilatory component opposing the α1AR vasoconstriction in WKY may possibly represent endothelial α2AR, which may activate nitric oxide synthesis (NO; Shafaroudi et al., 2005). When shifting this balance by augmenting the sympathetic component by desipramine, α2AR-mediated vasodilatation failed to fully down-regulate TPVR also in WKY.
We previously observed that down-regulation of TPVR in response to tyramine-stimulated, selective norepinephrine release, depended in SHR exclusively on adrenal catecholamine(s) and activation of β2 + 3AR-mediated vasodilatation, with no influence of the nerve-dependent activation of postsynaptic β1AR, which down-regulated tension in WKY (Berg et al., 2010). Since AdrX, nadolol, and SR59230A did not influence the late TPVR-response to 4-AP in either strain, 4-AP apparently blocked βAR-mediated vasodilatation. This conclusion was supported by that 4-AP halved the fall in TPVR in response to the βAR agonist isoprenaline (data not shown). Therefore, when βAR-mediated vasodilatation was prevented by 4-AP, α2AR-mediated vasodilatation was revealed, but in WKY only, in agreement with the presence of α2AR disorders in SHR (Feres et al., 1998; Zugck et al., 2003).
Although 4-AP itself did not influence the plasma epinephrine concentration, efferent vagal stimulation increased circulating epinephrine in both strains, demonstrating a positive vagal influence on adrenal epinephrine secretion. In SHR, plasma epinephrine also increased after pre-treatment with nadolol, alone or combined with atropine, but not SR59230A, and without simultaneous changes in norepinephrine overflow. The latter observation was likely to be explained by a negative feedback loop, where adrenal catecholamine(s) activated β1/2AR on vagal, afferent nerves, which through a CNS relay has been shown to hamper adrenal catecholamine secretion (Mravec, 2011). This mechanism apparently involved 4-AP-induced ACh release, since nadolol did not alter plasma epinephrine concentration in SHR infused with tyramine (Berg, unpublished observations). This feedback mechanism was not detected in WKY, possibly due to a lower sympathetic tone in this strain, although an elevated norepinephrine overflow to plasma was observed in vagal-stimulated WKY. This latter increase may result from activation of presynaptic release-stimulating mAChR (Boehm and Kubista, 2002).
Conclusion
4-AP activated both branches of the autonomic nervous system, and their influence on the cardiovascular system could be separated and identified. In SHR, a sympathetic nerve-dependent norepinephrine release dominated and prevented parasympathetic influence on HR, even though the parasympathetic component actually was augmented in SHR. In addition, in the presence of 4-AP, norepinephrine release activated an α2AR-mediated vasodilatory component, which opposed and prevented an α1AR-mediated increase in TPVR in WKY. This α2AR-mediated vasodilatory mechanism was not functional in SHR. We also detected a β1/2AR-dependent inhibition of adrenal catecholamine release in SHR, possibly due to activation of a negative feedback mechanism mediated by β1/2AR on afferent vagal fibers. 4-AP therefore exposed mechanisms involved in autonomic nervous system disorders in SHR, and may allow identification of factors responsible for these important changes in hypertension.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by The Norwegian Council on Cardiovascular diseases (Torill Berg), Anders Jahres fond (Torill Berg), and Novo Nordisk Foundation, Denmark (Jørgen Jensen).
References
Berg, T. (2002). Analysis of the pressor response to the K+ channel inhibitor 4-aminopyridine. Eur. J. Pharmacol. 452, 325–337.
Berg, T. (2003). The vascular response to the K+ channel inhibitor 4-aminopyridine in hypertensive rats. Eur. J. Pharmacol. 466, 301–310.
Berg, T. (2005). Increased counteracting effect of eNOS and nNOS on an alpha(1)-adrenergic rise in total peripheral vascular resistance in spontaneous hypertensive rats. Cardiovasc. Res. 67, 736–744.
Berg, T., Degerman, E., and Tasken, K. (2009). Increased cAMP signaling can ameliorate the hypertensive condition in spontaneously hypertensive rats. J. Vasc. Res. 46, 25–35.
Berg, T., and Koteng, O. (1997). Signalling pathways in bradykinin- and nitric oxide-induced hypotension in the normotensive rat; role of K+-channels. Br. J. Pharmacol. 121, 1113–1120.
Berg, T., Piercey, B. W., and Jensen, J. (2010). Role of beta 1-3-adrenoceptors in blood pressure control at rest and during tyramine-induced norepinephrine release in spontaneously hypertensive rats. Hypertension 55, 1224–1230.
Berg, T., Schøyen, H., Wassdal, I., Hull, R., Gerskowitch, V. P., and Toft, K. (1992). Characterization of a new kallikrein-like enzyme (KLP-S3) of the rat submandibular gland. Biochem. J. 281, 819–828.
Bjørnstad-Østensen, A., and Berg, T. (1994). The role of nitric oxide, adrenergic activation and kinin-degradation in blood pressure homeostasis following an acute kinin-induced hypotension. Br. J. Pharmacol. 113, 1567–1573.
Boehm, S., and Kubista, H. (2002). Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol. Rev. 54, 43–99.
Borovikova, L. V., Ivanova, S., Nardi, D., Zhang, M., Yang, H., Ombrellino, M., and Tracey, K. J. (2000). Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton. Neurosci. 85, 141–147.
Esler, M., Rumantir, M., Kaye, D., Jennings, G., Hastings, J., Socratous, F., and Lambert, G. (2001). Sympathetic nerve biology in essential hypertension. Clin. Exp. Pharmacol. Physiol. 28, 986–989.
Feres, T., Borges, A. C., Silva, E. G., Paiva, A. C., and Paiva, T. B. (1998). Impaired function of alpha-2 adrenoceptors in smooth muscle of mesenteric arteries from spontaneously hypertensive rats. Br. J. Pharmacol. 125, 1144–1149.
Freeman, R. (2006). Assessment of cardiovascular autonomic function. Clin. Neurophysiol. 117, 716–730.
Jensen, J., Ruzzin, J., Jebens, E., Brennesvik, E. O., and Knardahl, S. (2005). Improved insulin-stimulated glucose uptake and glycogen synthase activation in rat skeletal muscles after adrenaline infusion: role of glycogen content and PKB phosphorylation. Acta Physiol. Scand. 184, 121–130.
Malinowska, B., and Schlicker, E. (1997). Further evidence for differences between cardiac atypical beta-adrenoceptors and brown adipose tissue beta3-adrenoceptors in the pithed rat. Br. J. Pharmacol. 122, 1307–1314.
Miralles, A., Asensio, V. J., and Garcia-Sevilla, J. A. (2002). Acute treatment with the cyclic antidepressant desipramine, but not fluoxetine, increases membrane-associated G protein-coupled receptor kinases 2/3 in rat brain. Neuropharmacology 43, 1249–1257.
Mravec, B. (2011). Role of catecholamine-induced activation of vagal afferent pathways in regulation of sympathoadrenal system activity: negative feedback loop of stress response. Endocr. Regul. 45, 37–41.
Noor, N., Patel, C. B., and Rockman, H. A. (2010). beta-Arrestin: a signaling molecule and potential therapeutic target for heart failure. J. Mol. Cell Cardiol. 51, 534–541.
*Ørstavik, T. B., and Gautvik, K. M. (1977). Regulation of salivary kallikrein secretion in the rat submandibular gland. Acta Physiol. Scand. 100, 33–44.
Palatini, P., and Julius, S. (1997). Association of tachycardia with morbidity and mortality: pathophysiological considerations. J. Hum. Hypertens. 11(Suppl. 1), S19–S27.
Palatini, P., and Julius, S. (2009). The role of cardiac autonomic function in hypertension and cardiovascular disease. Curr. Hypertens. Rep. 11, 199–205.
Paskov, D. S., Agoston, S., and Bowman, W. C. (1986). “4-Aminopyridine hydrochloride (pymadin),” in Handbook of Experimental Pharmacology, ed. D. A. Karckevich (Springer: Berlin), 679–717.
Ridley, J. M., Milnes, J. T., Zhang, Y. H., Witchel, H. J., and Hancox, J. C. (2003). Inhibition of HERG K+ current and prolongation of the guinea-pig ventricular action potential by 4-aminopyridine. J. Physiol. 549, 667–672.
Shafaroudi, M. M., McBride, M., Deighan, C., Wokoma, A., Macmillan, J., Daly, C. J., and McGrath, J. C. (2005). Two “knockout” mouse models demonstrate that aortic vasodilatation is mediated via alpha2a-adrenoceptors located on the endothelium. J. Pharmacol. Exp. Ther. 314, 804–810.
Simmons, M. A., and Dun, N. J. (1984). Actions of 4-aminopyridine on mammalian ganglion cells. Brain Res. 298, 149–153.
Tamargo, J., Caballero, R., Gomez, R., Valenzuela, C., and Delpon, E. (2004). Pharmacology of cardiac potassium channels. Cardiovasc. Res. 62, 9–33.
Thrasher, T. N. (2005). Baroreceptors, baroreceptor unloading, and the long-term control of blood pressure. Am. J. Physiol Regul. Integr. Comp. Physiol. 288, R819–R827.
Tjugen, T. B., Flaa, A., and Kjeldsen, S. E. (2010). The prognostic significance of heart rate for cardiovascular disease and hypertension. Curr. Hypertens. Rep. 12, 162–169.
Tomaselli, G. F., and Marban, E. (1999). Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 42, 270–283.
Vargas, H. M., and Ringdahl, B. (1990). Centrally active antimuscarinic analogs of oxotremorine selectively block physostigmine-induced hypertension, but not peripheral muscarinic vasodepression. J. Pharmacol. Exp. Ther. 253, 165–170.
Wassermann, O. (1971). Studies on the pharmacokinetics of bis-quaternary ammonium compounds. II. Autoradiographic studies on the distribution of 3 H-hexamethonium in mice. Naunyn Schmiedebergs Arch. Pharmacol. 270, 419–427.
Keywords: total peripheral vascular resistance, heart rate, norepinephrine, epinephrine, parasympathetic nerves, sympathetic nerves, adrenal, 4-aminopyridine
Citation: Berg T and Jensen J (2011) Simultaneous parasympathetic and sympathetic activation reveals altered autonomic control of heart rate, vascular tension, and epinephrine release in anesthetized hypertensive rats. Front. Neur. 2:71. doi: 10.3389/fneur.2011.00071
Received: 12 September 2011; Accepted: 29 October 2011;
Published online: 23 November 2011.
Edited by:
Vaughan G. Macefield, University of Western Sydney, AustraliaReviewed by:
Anna P. Malykhina, University of Pennsylvania, USAChangfeng Tai, University of Pittsburgh, USA
Copyright: © 2011 Berg and Jensen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Torill Berg, Department of Physiology, Institute of Basic Medical Sciences, P.O. BOX 1103, Blindern, 0317 Oslo, Norway. e-mail:dG9yaWxsLmJlcmdAbWVkaXNpbi51aW8ubm8=