 Audra L. Clos1
Audra L. Clos1
- 1 Department of Dermatology, MD Anderson Cancer Center, University of Texas, Houston, TX, USA
- 2 George P. and Cynthia Woods Mitchell Center for Neurodegenerative Diseases, University of Texas Medical Branch, Galveston, TX, USA
- 3 Department of Neurology, University of Texas Medical Branch, Galveston, TX, USA
- 4 Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, TX, USA
Amyloidosis are a large group of conformational diseases characterized by abnormal protein folding and assembly which results in the accumulation of insoluble protein aggregates that may accumulate systemically or locally in certain organs or tissue. In local amyloidosis, amyloid deposits are restricted to a particular organ or tissue. Alzheimer’s, Parkinson’s disease, and amyotrophic lateral sclerosis are some examples of neurodegenerative amyloidosis. Local manifestation of protein aggregation in the skin has also been reported. Brain and skin are highly connected at a physiological and pathological level. Recently several studies demonstrated a strong connection between brain and skin in different amyloid diseases. In the present review, we discuss the relevance of the “brain–skin connection” in different neurodegenerative amyloidosis, not only at the pathological level, but also as a strategy for the treatment of these diseases.
Introduction
Amyloidosis is not a single disease, but rather a variety of different pathological processes resulting in the deposition of β-pleated sheet fibrils formed from various proteins by several different pathogenic mechanisms (Husby, 1992). Virchow introduced the term “amyloidosis” to designate an amorphous material that had a similar staining property as cellulose, a polysaccharide, not a protein at all (Kyle and Gertz, 1990; Cohen, 1993). Currently various stains have traditionally been used for the histologic detection of amyloid such as crystal violet, thioflavin S, hematoxylin–eosin, and Congo red stains. Congophilia is the main characteristic that is used to describe amyloid, and under polarized light amyloid displays apple-green birefringence due to its β-pleated sheet conformation (Kyle and Gertz, 1990; Rocken et al., 1996). Amyloidosis is an example of a disease caused by malfunctioning proteins and is thus referred to as a conformational or protein misfolding disease. The group of peptides and proteins capable of forming amyloid fibrils is very diverse (Guijarro et al., 1998; Chiti et al., 1999; West et al., 1999; Sipe and Cohen, 2000; Lopez De La Paz et al., 2002). This group consists not only of proteins involved in amyloid deposits in vivo (Sipe and Cohen, 2000), but also of non-pathogenic (von Bergen et al., 2000; Azriel and Gazit, 2001) and designed peptides and proteins (West et al., 1999; Lopez De La Paz et al., 2002). The conformational diseases or amyloid diseases include Alzheimer’s disease (AD), Parkinson diseases, amyotrophic lateral sclerosis (ALS), type 2 diabetes mellitus, and various forms of cutaneous amyloidosis (Botelho and Lupi, 2008). Hence, the question arose whether there may be evidence supporting a link between cutaneous manifestations of amyloid deposits and neurodegenerative amyloidosis. This review aims to address this question and give a novel perspective on the relation of amyloid in skin and brain.
Cutaneous and Neurodegenerative Amyloidosis
A vast range of human diseases arise from the failure of a specific peptide or protein to adopt or remain in its native functional conformational state. These pathologic conditions collectively are referred to as protein conformational diseases. The largest group of conformational diseases is associated with the conversion of specific peptides or proteins from their soluble functional state to toxic aggregates that are deposited in the tissues (Stefani and Dobson, 2003). Amyloidoses are a large group of conformational diseases in which protein aggregates accumulate either systemically or locally in certain organs or tissues (Tillement et al., 2009). In local amyloidosis, amyloid deposits are restricted to a particular organ or tissue. Local amyloidosis can be broadly grouped into neurodegenerative conditions in which aggregation occurs in the brain, including Alzheimer’s, Parkinson’s diseases (PD), and ALS (Chiti and Dobson, 2006; Meersman et al., 2006), and non-neuropathic localized amyloidosis, in which aggregation occurs in a single type of tissue other than the brain, i.e., type II diabetes (pancreas) and Cutaneous amyloidosis (skin; Chiti and Dobson, 2006). Secondary cutaneous amyloidosis is an entity of systemic amyloidosis in which amyloid can be secondarily located in the skin and others organs. In the case of the secondary cutaneous amyloidosis, the amyloid protein A (AA) is involved (Katsikas et al., 2008). Also, amyloid light chain has been involved in several cases of systemic amyloidosis in the skin (Feito-Rodriguez et al., 2008).
Primary cutaneous amyloidosis is a local amyloidosis in which amyloid deposits are restricted to the skin. This disease has been classified into three types: macular amyloidosis, lichen amyloidosis, and nodular amyloidosis (Bhat et al., 2009; Schwendiman et al., 2009). Primary cutaneous nodular amyloidosis is thought to be a plasma cell dyscrasia, where amyloid light chain is involved and diagnosis is based on the absence of clonal plasma cell proliferation in the bone marrow; however some patients thought to be afflicted with only a localized version of this disease on long-term follow-up did indeed develop systemic amyloidosis. It remains unclear if the disease was always systemic with early cutaneous manifestations, or if it was a disease with a low-grade potential to develop into a systemic disease. In the specific case of macular and lichenoid amyloidoses, it is believed that altered keratin is the main component of the amyloidogenic material (Feito-Rodriguez et al., 2008) and that epidermal cells are closely related to the deposition of amyloid (Black, 1971; Black and Jones, 1971). Work done by Hashimoto and Kumakiri (1979) more than 20 years ago demonstrated the fibrillar degeneration of epidermal cells and dropping off of the fibrillar bodies into the dermis; the authors speculated that these fibrillar bodies were modified by the surrounding histiocytes and or fibroblasts. This theory of amyloidosis has been named the fibrillar body theory (Hashimoto and Kumakiri, 1979; Kumakiri and Hashimoto, 1979; Hashimoto and Kobayashi, 1980; Kobayashi and Hashimoto, 1983; Eto et al., 1984). The second theory of deposition is the secretion theory. This theory states that in macular and lichenoid amyloidosis, the amyloid is produced in the interface of the epidermis and dermis by living basal keratinocytes, which produce the precursor of amyloid because of disturbed keratin metabolism (Yanagihara and Mori, 1982). Recent research suggests that prefibrillar aggregates, soluble oligomers, rather than fibrils per se are the most potent mediators of cytotoxicity (Herczenik and Gebbink, 2008). It has also been shown that toxic oligomers share a common structure, and that in consequence there is a generic pathogenic mechanism (Bucciantini et al., 2004; Pastor et al., 2008). However, the mechanism of amyloid formation and toxicity is still unknown; thus, there is not an effective treatment for any of the conformational diseases. Given that a common pathogenic mechanism may underlie in all of these diseases, treatments that target protein conformation could be applicable in a variety of contexts (Glabe, 2006; Glabe and Kayed, 2006). At the molecular level several models have been postulated to explain the common mechanism of amyloidogenesis in protein misfolding disease. Nucleation-dependent fibrillation is the prevalent model for assessing the self-assembly process. This model postulated that it is necessary to have a nucleus or seed to initiate the fibrillation process. Once the nucleus is formed, fibril growth is thought to proceed rapidly by further association of either monomers or oligomers with the nucleus (Jarrett and Lansbury, 1993; Harper and Lansbury, 1997; Wood et al., 1999). Recently another mechanism was proposed for amyloidogenesis: double-concerted fibrillation (Bhak et al., 2009a,b). Based on this model, the amyloid fibril formation is achieved via two consecutive, concerted associations of monomers and subsequently formed oligomeric granular species, in which the oligomeric species act as a growing unit for the fibril formation in the absences of a seed. Amyloid species can transmit conformational changes in homologous and heterologous proteins, mechanisms denominated as seeding and cross-seeding, respectively (Westermark et al., 2009). Despite a lack of similarity in the functional properties or in the primary, secondary, or tertiary structures among these proteins, the amyloids formed from them share a common ultra-structure (Rochet and Lansbury, 2000; Fu et al., 2004; Nelson et al., 2005). Several studies have described this process in vitro (Giasson et al., 2003; Lasagna-Reeves et al., 2010). However, only a few studies have described the presence of two different amyloid proteins in the same organ or tissue in human patients. For example, amyloid β-peptide (Aβ) amyloid and PrPsc were detected in a single plaque in patients with Gerstmann–Sträussler syndrome (Miyazono et al., 1992). In another study, aggregate forms of kappa-light chain and β2-microglobulin were colocalized in a patient with multiple myeloma (Takahashi et al., 1996). Aβ amyloid has also been colocalized with light chain immunoglobulin aggregates in diseased leptomeningeal and cortical vessels in patients with spontaneous intracranial hemorrhage (Galuske et al., 2004). In a recent publication, we have shown how to induce amyloid formation in the skin of wild-type mice (Clos et al., 2010). In that study we illustrated that the intradermal administration of certain amyloid can induce the aggregation of heterologous proteins (Clos et al., 2010). This study gave direct evidence of the cross-seeding mechanism in the skin. Even more, Heiko Braak and Kelly Del Tredici proposed in 2003 that PD begins in the gut, then slowly makes its way to the brain and spreads to different regions (Braak et al., 2003). The authors specifically proposed that the disease could start in the gut induced by a virus or other pathogen, and move along the vagus nerve in an upstream, or retrograde, direction toward the brain. One idea that is gaining momentum is that, instead of a virus or some other agent spreading through the nervous system to induce the pathology associated with PD, the pathology itself, in other words, the misfolded form of α-synuclein, is what spread from one neuron to the next (Braak et al., 2003; Jang et al., 2009). These studies demonstrated how the presences of amyloidogenic material outside the central nervous system can have a direct impact in neurodegenerative amyloidosis.
These findings reveal that there may be a connection of cutaneous forms of amyloidoses and neurodegenerative amyloidoses. Furthermore, skin physiology is widely altered in these types of diseases.
Functional Amyloids
An increasing number of examples suggest that amyloid fibers, or amyloidogenic intermediates, can be used to facilitate a particular physiological task (Fowler et al., 2007; Greenwald and Riek, 2010). These functional amyloids have been found in many organisms, including bacteria, fungi, and mammals (Chiti and Dobson, 2006; Fowler et al., 2007). Well-studied functional amyloid in Homo sapiens includes Pmel17, which serves as a structural scaffold for covalent polymerization of molecules during melanin assembly. It has recently been shown that peptide and protein hormones in secretory granules of the mammalian endocrine system are stored in an amyloid-like cross-β-sheet-rich conformation (Maji et al., 2009). The natural storage of hormones in amyloid-like structures does not produce any toxic effect to neuronal cells. Hydrophobins are key components of fungal coats (e.g., Aspergillus fumigatus), and in bacteria curlin subunits (csgA) is necessary for biofilm formation and host invasion. In yeast several functional amyloidogenic proteins have been identified, such as Ure2p, Sup35p, Rnq1p, and adhesins which is involved in cellular aggregation (Chiti and Dobson, 2006; Fowler et al., 2007; Wang et al., 2007; Epstein and Chapman, 2008; Maurer-Stroh et al., 2010; Ramsook et al., 2010). Other examples of functional amyloids in nature includes silkmoth chorion protein, fish chorion proteins, and type I antifreeze protein (AFP, ice-structuring protein preventing ice growth) of winter flounder (Iconomidou and Hamodrakas, 2008). Many of these functional amyloids are involved in protection from environmental influences, for example Pmel17 in human melanin assembly to prevent UV-damage, AFP in winter flounder to protect from ice crystals. Again, this shows that we are far from really understanding the underlying processes. However, as shown in this paragraph, there are functional amyloids in both skin and brain (Fowler et al., 2007; Greenwald and Riek, 2010).
Skin–Brain Connection
As of late there is increasing evidence for the “brain–skin connection.” The skin is a highly innervated organ and has been coined a diffuse brain (Urpe et al., 2005). Many neuropeptides originate in the skin, including catecholamines, substance P, calcitonin gene related peptide (CGRP), vasoactive intestinal peptide (VIP), and nerve growth factor (NGF). In psoriasis it has been shown that activation of the autonomic nervous system increases neuropeptides and is implicated in the development of psoriatic lesions. It has also been found that psoriatic plaques have increased nerve fiber density, and that high expression of NGF mediates T cell and keratinocyte proliferation, mast cell migration and degranulation, and memory T cell migration; all important in the pathogenesis of psoriasis (Halevy and Livni, 1993; Steinkraus et al., 1993; Raychaudhuri et al., 2000). Also important in psoriasis, is the impact of negative psychosocial consequences, including stress, depression, and mood disorders, which in some cases have been shown to precede the diagnosis of psoriasis as well as appear prior to the onset of disease flares (Fortune et al., 2002, 2005). In atopic dermatitis interactions between mast cells, the nervous system, and neuropeptides have also been implicated in the disease process and in murine models of sound-produced stress lends support to a NGF-Substance P mast cell communication in neurogenic skin inflammation (Arck et al., 2003; Jarvikallio et al., 2003; Peters et al., 2004).
Many other dermatologic conditions and diseases are linked to neuroimmunological stress such as pruritus, hyperhidrosis, flushing, seborrhoeic eczema, prurigo nodularis, lichen planus, and alopecia areata (Biro et al., 2005) In fact the hair follicle is one of the most hormone sensitive tissues in human physiology. It is very sensitive to stress associated hormones such, as cortisol releasing hormone (CRH), cortisol, prolactin, catecholamines, among others. The hair follicle is not only an endpoint for neuropeptides, but also a source (Slominski et al., 2000). It is not surprising that stress has been implicated as a major aggravating factor in hair loss (Arck et al., 2003).
As discussed previously pruritus is linked to neuroimmunological stress; endpoints of pruritus leads to scratching (itch-scratch cycle), scratching leads to prolonged inflammation and inflammation may induce conformational changes of proteins. Certain proteins, in the skin, are more susceptible to conformational changes, an example of this is cytokeratin; implicated in the pathogenesis of macular and lichenoid amyloidosis. Primary localized cutaneous amyloidosis (PLCA) is a notoriously itchy skin condition associated with amyloid deposition in the superficial dermis (Brownstein and Helwig, 1970). Of note it is reported that often pruritus is the first symptom reported in both familial and sporadic forms of PLCA, implicating the importance of pruritus and the nervous system in the pathogenesis of this disease (Brownstein and Helwig, 1970).
Alzheimer’s Disease and Skin
Alzheimer’s disease, is the most common cause of dementia in the elderly, is marked by a progressive degeneration of limbic and cortical brain structures, mainly in the temporal lobe. By 2050 the number of people with AD is expected to triple (Hebert et al., 2001; Editorial, 2010), placing an enormous burden on health care and social care system. AD is a neurodegenerative disorder that is characterized clinically by progressive loss of memory, language problems, social withdrawal, deterioration of executive functions, and eventually culminates in death (Citron, 2002). Most AD cases are sporadic, with multiple risk factors, such as aging, environmental stress, and diet, suggested to play critical pathogenic roles. The remaining AD cases, which account for 5–10% of total AD cases, are rare, but inherited from one generation to the next and are referred to as familial AD (FAD). The major neuropathological features of AD are synaptic and neuronal degeneration and the presence of amyloid plaques and neurofibrillary tangles (NFTs). The major protein component of the plaques is the Aβ, which is a 39–43 amino acid peptide that is found by a much larger transmembrane protein, the amyloid precursor protein (APP; Selkoe, 1994), whereas NFTs are composed of hyperphosphorylated forms of the microtubule-binding protein tau. These two insoluble protein aggregates are believed to play critical roles in the neurodegenerative process.
There are several findings of altered skin physiology in patients with AD. In AD presenilin 1 (PS-1) is an enzyme that is essential for proteolytic processing of APP and has been implicated in skin cancer in animals (Xia et al., 2001). PS-1 is also involved in epidermal growth factor receptor (EGRF) regeneration and a partial loss of presenilin-1 may lead to the development of seborrhea keratoses and inflammatory skin conditions (Tournoy et al., 2004; Repetto et al., 2007). Antioxidant defenses in skin fibroblasts from patients with familial AD were shown to be lower when compared to control patients (Cecchi et al., 2002). In patients with AD, it is also been discovered that cutaneous vascular function is altered, just as in the brain (Kalman et al., 2002; Khalil et al., 2007), as well as mastocyte expression of β amyloid and tau protein (Soininen et al., 1992; Okada et al., 1994; Wen et al., 1994; Kvetnoi et al., 2003).
In addition to the connection between AD and skin pathophysiology, the skin is also being used as a vector to deliver medication in AD patients. The Rivastigmine patch is the first transdermal treatment to be approved for AD. Rivastigmine is an established cholinesterase inhibitor indicated for the treatment of mild to moderate dementia of AD. Two drug classes have been approved for the treatment of AD in the US: cholinesterase inhibitors and N-methyl-D-aspartate-receptor antagonists. Several of rivastigmine’s chemical characteristics make it appropriate for transdermal patch delivery; the drug can pass through the skin with ease, entering the blood stream and crossing the blood brain barrier (Nitti et al., 2006). Compared to rivastigmine capsules the patch shows less fluctuation in plasma concentration and fewer GI side effects, making this drug more tolerable for patients and lowering the discontinuation rate (Rosler et al., 1999; Mercier et al., 2007; Winblad et al., 2007a). The patch and oral form of this medication show similar efficacy, other than less gastrointestinal side effects than the oral form, there is also a caregiver preference for the patch form of Rivastigmine (Blesa et al., 2007; Winblad et al., 2007b).
Several experimental immunizations, in animal models, are also utilizing the skin and nasal mucosa to deliver β amyloid. Nikolic et al. (2007) demonstrated that transcutaneous immunization with aggregated β amyloid peptide with adjuvant cholera toxin, lead to high titers of Aβ antibodies and lead to significant decreases in cerebral amyloid β levels. Other experiments show that Aβ derived peptides, with an adjuvant, delivered intranasally to mucosal epithelial tissues results in effective clearance of Aβ plaques and improvement of cognitive function in animal models of AD (Maier et al., 2005, 2006). The skin is a well known effective route for vaccination of peptide based vaccines; strong humoral and cellular immune activity has been summoned secondary to trans cutaneous vaccinations. This can be attributed to the diverse populations of resident antigen presenting cells and other immune cells in the dermis (Beignon et al., 2005; Itoh and Celis, 2005; Dell et al., 2006; Giudice and Campbell, 2006).
Parkinson’s Disease and Skin
Parkinson’s disease is a progressive and devastating neurodegenerative disorder, affecting 1% of individuals over 60 years old (Samii et al., 2004). The three cardinal clinical features of PD are rigidity, resting tremor and bradykinesia, and these occur when approximately 50% of dopaminergic neurons projecting from the substantia nigra pars compacta (SNc) to the striatum are lost (Samii et al., 2004). The neuropathological hallmark of the disease is the presence in surviving SNc neurons of intracellular inclusions known as Lewy bodies (LBs), which are composed mainly of fibrillar form of the protein α-synuclein (Spillantini et al., 1997; Samii et al., 2004). The pathology of PD is not solely confined to the nigrostriatal pathway because LBs are also found in the cortex, amygdala, locus coeruleus, and peripheral autonomic system (Dickson et al., 2009; Wolters, 2009). Dysfunction of these extranigral neuronal populations and the presence of LBs correlate with the non-motor manifestations of PD, including autonomic, sleep, and olfactory dysfunctions, and these can precede the appearance of motor symptoms (Dickson et al., 2009; Wolters, 2009).
Interestingly there is a higher than expected frequency of the development of melanoma, a malignant tumor of melanin manufacturing cells in the skin, in patients with PD, and even more, melanoma patients are more likely to have PD. Malfunctions in melanin correlate with various skin cancers, such as melanoma, whereas abnormalities in neuromelanin correlate with PD, this proposes that melanin has a function in predisposition to development of PD and melanoma. Pigmentation genes determine the amount and type of melanin, such genes that are shared in these two diseases, include those that encode tyrosinase and hydroxylase (Halaban et al., 1997). Melanocytes and dopaminergic neurons contain different substances, but they both share a precursor of tyrosine and make 3,4 dihydroxyphenylalanine (DOPA). Thus genes involved in the synthesis of dopamine and melanin appear to be a likely source of the relationship between PD and melanoma (Gudbjartsson et al., 2008; Duffy et al., 2010).
As we already mentioned, α-synuclein plays a large role in the pathogenesis of PD; it accumulates within SN neurons (Spillantini et al., 1997). It has been shown that α-synuclein is increased in the cutaneous peripheral nerves of PD patients when compared to other neurodegenerative diseases (Paisan-Ruiz et al., 2009). α-Synuclein is widely expressed in melanoma cells. Even more melanoma cells that do express high levels of α-synuclein do not produce much melanin pigment, and it is thought that melanin pigment is protective against malignant melanomas. This could be a plausible explanation for why patients with PD have an increased risk for melanoma (Paris et al., 2001).
As in AD, in PD, the skin is also being utilized to deliver Rivastigmine, an inhibitor of acetylcholinesterase and butyrylcholinesterase, transdermally. Cholinergic deficits are pronounced in patients who have dementia related with PD. In a study performed by Emre et al. (2007) oral Rivastigmine was associated with moderate improvements in dementia associated with PD. Rivastigmine patch is also approved for treatment in PD and has been shown to have similar efficacy and improved tolerability as compared to the oral form of the drug (Mercier et al., 2007; Winblad et al., 2007a).
It has also been shown that the transdermal delivery of dopamine agonist, Rotigotine, is well tolerated and is beneficial in the treatment of early and late stage PD. Rotigotine has been also shown to improve sleep as well as early morning motor symptoms (Trenkwalder et al., 2011).
Amyotrophic Lateral Sclerosis and Skin
Amyotrophic lateral sclerosis is a neurologic disease with an annual incidence rate of approximately 1 in 100,000 (Morrison and Morrison, 1999). There are both sporadic and familial forms of the disease, with familial ALS accounting for approximately 5% of cases. The symptoms and pathology of patients with the familial form of ALS are identical to those of patients with the sporadic form, suggesting that the mechanisms of neurodegeneration for sporadic and familial ALS share common components. The early symptoms of ALS are muscle atrophy, weakness, and fasciculations. The disease progresses over an average of 5 years, leading to paralysis and premature death (Morrison and Morrison, 1999). Pathologically, ALS is characterized by extensive loss of lower motor neurons in the spinal cord and brainstem, atrophy of ventral roots, degeneration of upper motor neurons in the motor cortex, corticospinal tract degeneration, somatic and axonal inclusions of aberrant neurofilament proteins (NFP), and reactive astrocytosis (Hirano, 1991; Leigh and Swash, 1991).
Superoxide dismutase 1 (SOD1) is an antioxidant protein expressed abundantly and ubiquitously (Zelko et al., 2002) and since the discovery in 1993 that mutations in SOD1 cause ALS (Deng et al., 1993; Rosen, 1993) a further 140 mutations have since been found scattered throughout the 153 amino acid Cu/Zn-homodimer. Mutations are mostly substitutions of single amino acid residues although some deletions, insertions, and C-terminal truncations have been identified. SOD1 mutations are known to be causative in 10–20% fALS cases, and have been observed in 2% sporadic cases (Dion et al., 2009). The presence of inclusion bodies or aggresomes is one of the neuropathological hallmarks of ALS. Spinal cord sections from SOD1-fALS patients have been examined by immunohistochemistry and found to contain aggregates that are reactive to anti-SOD1 antibodies (Kato, 2008). More recent evidence has shown that in non-SOD1 ALS (fALS and sALS), aggregates also contain SOD1 (Shibata et al., 1994; Matsumoto et al., 1996), and there is no marked difference in disease presentation and progression, either clinically or neuropathologically, between fALS and sALS (Gros-Louis et al., 2006).
Sporadic ALS is a degenerative disorder of the upper and lower motor neurons. There is no determined cause for this disease. Interestingly it was reported in 1996 by Kolde et al. (1996) that there was deposition of amyloid β protein in the skin of patients with ALS, but not in control matched patients. Findings from Tamaoka and colleagues (Tamaoka et al., 2000) suggest that changes in the extracellular matrix in the skin of ALS patients may facilitate aggregation and deposition of Amyloid β in the skin. This conclusion was extracellular matrix components including type 4 collagen and glycosaminoglycans have been found and these components affect processing and accumulation of Amyloid β in the human brain (Asami-Odaka et al., 1995; Ono et al., 1998a,b).
In ALS skin-like and spherical inclusions within the somatodendritic compartment of vulnerable neurons is a hallmark of the disease process. A large constituent of the inclusions is a phosphorylated, aggregated, and ubiquitinated intranuclear protein, Transactivation-responsive DNA-binding protein-43 (TDP-43; Arai et al., 2006; Davidson et al., 2007; Geser et al., 2009). TDP-43 is a transcription-related protein that is localized within the cell nucleus; it may act as a transcription repressor of nuclear bodies through communication with survival motor neuron protein (Neumann et al., 2006). It has also been shown that missense mutations in the TDP-43 gene have been discovered in familial and sporadic cases of ALS, lending credence to the role that TDP-43 plays in the degeneration and cell death of motor neurons (Gitcho et al., 2008; Sreedharan et al., 2008; Daoud et al., 2009). Suzuki et al. (2010) investigated the skin from Sporadic ALS patients, and found that there was a high frequency of TDP-43 positive cells. Furthermore they reported that the skin from patients with longer standing ALS was more immunoreactive for TDP-43 than patients who had not been afflicted with the disease for as long a period of time. Based on their discovery, it may be postulated that a common disease process may be occurring in the skin and in the motor neurons, with regards to accumulation of TDP-43 (Suzuki et al., 2010).
Matrix metalloproteinase (MMP) are a family of Zn2+ endopeptides that have the ability to digest elements of the extracellular matrix such as collagen (Kolde et al., 1996). In ALS there is an increased activity of MMPs in the CNS (Beach et al., 1986). Reports from experiments in mutant SOD1 transgenic mice have implicated a role of MMPs in the early stage of the disease and that inhibition of certain MMPs may serve a protective role in ALS (Vincenti and Brinckerhoff, 2007). It has been observed that patients with ALS do not develop many bedsores (Furukawa and Toyokura, 1976). Reports from skin biopsies of patients with ALS show altered skin ultra-structure with decreased collagen crosslinking and thinner collagen fiber diameter; this could possibly be due to increased MMP activity in the skin (Bonnefont-Rousselot et al., 2000; Lorenzl et al., 2006; Fang et al., 2009). Fang et al. (2010) discovered increased MMP-9 activity in the skin of ALS patients, as well as in the CNS, thus linking the pathophysiology of neurodegeneration and skin alterations in ALS.
Several systemic treatments, utilizing subcutaneous injections, have been proposed in ALS, although currently there is no curative treatment to date. Ciliary neurotrophic growth factor (CNTF) is a neurotrophic factor which has been proven to promote motor neuron survival in cell cultures and in murine ALS models. Two randomized clinical trials with ALS patients utilized this growth factor, but no significant efficacy was observed (Miller et al., 1996; Bongioanni et al., 2004).
Insulin like growth factor I (IGF-I) is a peptide with neurotrophic effects; the recombinant form of this peptide has also been injected subcutaneously into patients with ALS is several clinical trials with varying results (Lai et al., 1997; Borasio et al., 1998; Mitchell et al., 2007). In neuropathy models using transgenic animals, beneficial outcomes have been reported in a number of studies (Mitchell et al., 2007).
Conclusion
There is now solid evidence for the existence of a defined “brain–skin connection,” which includes the over-activation of neuropeptides in psoriasis and atopic dermatitis and several dermatologic conditions linked to neuroimmunological stress (Fortune et al., 2002; Biro et al., 2005). Several studies discussed in the present review, have suggested that cutaneous amyloidosis shares a common amyloidogenesis and pattern of deposition similar to other well characterized amyloid-related diseases and provide the first evidence showing that all these diseases share common amyloidogenic structures associated with a generic pathogenic mechanism (Bucciantini et al., 2004; Pastor et al., 2008; Clos et al., 2011). Even more, if we take in consideration the newly published data which demonstrates that amyloidogenic proteins can spread in a seeding mechanism (Westermark et al., 2009) and may possibly start outside the central nervous system (Braak et al., 2003). In light of the evidence discussed in this review, is not illogical to think that the presence of amyloid in the skin can induce protein aggregation in the brain either directly of indirectly. As indicated in Figure 1, the “brain–skin connection” may have a physiological relevance in terms of amyloid diseases, their pathogenesis, and progression. This clear and direct axis can been explored as a safe and simple way to deliver different medications to treat neurodegenerative amyloidosis via cutaneous administration.
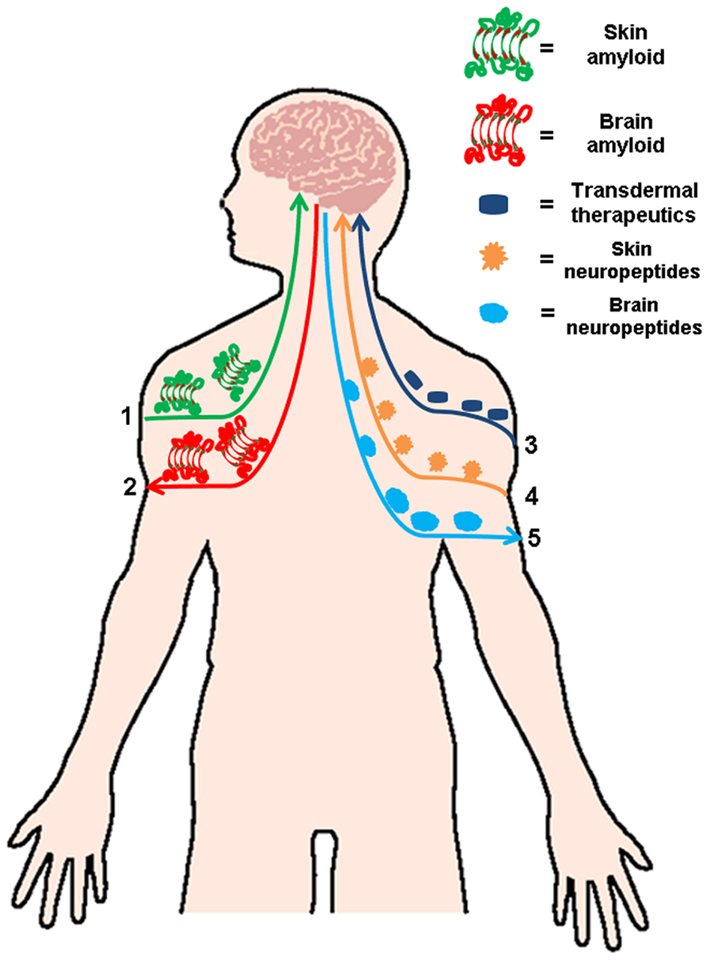
Figure 1. Linking skin and brain amyloidosis. Skin and brain are connected in several ways. Based on the published data describing the spread amyloid deposits, it is feasible that cutaneous amyloid spread to other organs including the brain (1) or vice versa (2). This strong connection between both organs makes it a viable and attractive option for the treatment several neurodegenerative amyloidosis via transdermal administration of therapeutics (3). The skin–brain connection not only involved amyloid treatment and formation, but also has an important relevance in several dermatologic conditions linked to neuroimmunological stress, in which an increase of neuropeptides in the skin (4) or the brain (5) has been reported.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y., and Oda, T. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611.
Arck, P. C., Handjiski, B., Peters, E. M., Peter, A. S., Hagen, E., Fischer, A., Klapp, B. F., and Paus, R. (2003). Stress inhibits hair growth in mice by induction of premature catagen development and deleterious perifollicular inflammatory events via neuropeptide substance P-dependent pathways. Am. J. Pathol. 162, 803–814.
Asami-Odaka, A., Ishibashi, Y., Kikuchi, T., Kitada, C., and Suzuki, N. (1995). Long amyloid beta-protein secreted from wild-type human neuroblastoma IMR-32 cells. Biochemistry 34, 10272–10278.
Azriel, R., and Gazit, E. (2001). Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. An experimental support for the key role of the phenylalanine residue in amyloid formation. J. Biol. Chem. 276, 34156–34161.
Beach, R. L., Rao, J. S., Festoff, B. W., Reyes, E. T., Yanagihara, R., and Gajdusek, D. C. (1986). Collagenase activity in skin fibroblasts of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 72, 49–60.
Beignon, A. S., Brown, F., Eftekhari, P., Kramer, E., Briand, J. P., Muller, S., and Partidos, C. D. (2005). A peptide vaccine administered transcutaneously together with cholera toxin elicits potent neutralising anti-FMDV antibody responses. Vet. Immunol. Immunopathol. 104, 273–280.
Bhak, G., Choe, Y. J., and Paik, S. R. (2009a). Mechanism of amyloidogenesis: nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep. 42, 541–551.
Bhak, G., Lee, J. H., Hahn, J. S., and Paik, S. R. (2009b). Granular assembly of alpha-synuclein leading to the accelerated amyloid fibril formation with shear stress. PLoS ONE 4, e4177. doi:10.1371/journal.pone.0004177
Bhat, A., Selmi, C., Naguwa, S. M., Cheema, G. S., and Gershwin, M. E. (2009). Currents concepts on the immunopathology of amyloidosis. Clin. Rev. Allergy Immunol. 38, 97–106.
Biro, T., Ko, M. C., Bromm, B., Wei, E. T., Bigliardi, P., Siebenhaar, F., Hashizume, H., Misery, L., Bergasa, N. V., Kamei, C., Schouenborg, J., Roostermann, D., Szabó, T., Maurer, M., Bigliardi-Qi, M., Meingassner, J. G., Hossen, M. A., Schmelz, M., and Steinhoff, M. (2005). How best to fight that nasty itch – from new insights into the neuroimmunological, neuroendocrine, and neurophysiological bases of pruritus to novel therapeutic approaches. Exp. Dermatol. 14, 225–240.
Black, M. M. (1971). The role of the epidermis in the histopathogenesis of lichen amyloidosis. Histochemical correlations. Br. J. Dermatol. 85, 524–530.
Black, M. M., and Jones, E. W. (1971). Macular amyloidosis. A study of 21 cases with special reference to the role of the epidermis in its histogenesis. Br. J. Dermatol. 84, 199–209.
Blesa, R., Ballard, C., Orgogozo, J. M., Lane, R., and Thomas, S. K. (2007). Caregiver preference for rivastigmine patches versus capsules for the treatment of Alzheimer disease. Neurology 69, S23–S28.
Bongioanni, P., Reali, C., and Sogos, V. (2004). Ciliary neurotrophic factor (CNTF) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. CD004302.
Bonnefont-Rousselot, D., Lehmann, E., Jaudon, M. C., Delattre, J., Perrone, B., and Rechke, J. P. (2000). Blood oxidative stress and lipoprotein oxidizability in haemodialysis patients: effect of the use of a vitamin E-coated dialysis membrane. Nephrol. Dial. Transplant. 15, 2020–2028.
Borasio, G. D., Robberecht, W., Leigh, P. N., Emile, J., Guiloff, R. J., Jerusalem, F., Silani, V., Vos, P. E., Wokke, J. H., and Dobbins, T. (1998). A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 51, 583–586.
Botelho, M. G., and Lupi, O. (2008). Protein folding and cutaneous diseases. Int. J. Dermatol. 47, 1225–1233.
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Brownstein, M. H., and Helwig, E. B. (1970). The cutaneous amyloidosis. I. Localized forms. Arch. Dermatol. 102, 8–19.
Bucciantini, M., Calloni, G., Chiti, F., Formigli, L., Nosi, D., Dobson, C. M., and Stefani, M. (2004). Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 279, 31374–31382.
Cecchi, C., Fiorillo, C., Sorbi, S., Latorraca, S., Nacmias, B., Bagnoli, S., Nassi, P., and Liguri, G. (2002). Oxidative stress and reduced antioxidant defenses in peripheral cells from familial Alzheimer’s patients. Free Radic. Biol. Med. 33, 1372–1379.
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366.
Chiti, F., Webster, P., Taddei, N., Clark, A., Stefani, M., Ramponi, G., and Dobson, C. M. (1999). Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. U.S.A. 96, 3590–3594.
Citron, M. (2002). Alzheimer’s disease: treatments in discovery and development. Nat. Neurosci. 5(Suppl.), 1055–1057.
Clos, A. L., Role of oligomers Lasagna-Reeves, C. A., Kelly, B., Wagner, R., Wilkerson, M., Jackson, G. R., and Kayed, R. (2011). Role of oligomers in the amyloidogenesis of primary cutaneous amyloidosis. J. Am. Acad. Dermatol. 65, 1023–1031.
Clos, A. L., Lasagna-Reeves, C. A., Wagner, R., Kelly, B., Jackson, G. R., and Kayed, R. (2010). Therapeutic removal of amyloid deposits in cutaneous amyloidosis by localised intra-lesional injections of anti-amyloid antibodies. Exp. Dermatol. 19, 904–911.
Daoud, H., Valdmanis, P. N., Kabashi, E., Dion, P., Dupré, N., Camu, W., Meininger, V., and Rouleau, G. A. (2009). Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 46, 112–114.
Davidson, Y., Kelley, T., Mackenzie, I. R., Pickering-Brown, S., Du Plessis, D., Neary, D., Snowden, J. S., and Mann, D. M. (2007). Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533.
Dell, K., Koesters, R., Linnebacher, M., Klein, C., and Gissmann, L. (2006). Intranasal immunization with human papillomavirus type 16 capsomeres in the presence of non-toxic cholera toxin-based adjuvants elicits increased vaginal immunoglobulin levels. Vaccine 24, 2238–2247.
Deng, H. X., Hentati, A., Tainer, J. A., Iqbal, Z., Cayabyab, A., Hung, W. Y., Getzoff, E. D., Hu, P., Herzfeldt, B., Roos, R. P., et al.. (1993). Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 261, 1047–1051.
Dickson, D. W., Fujishiro, H., Orr, C., DelleDonne, A., Josephs, K. A., Frigerio, R., Burnett, M., Parisi, J. E., Klos, K. J., and Ahlskog, J. E. (2009). Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat. Disord. 15(Suppl. 3), S1–S5.
Dion, P. A., Daoud, H., and Rouleau, G. A. (2009). Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat. Rev. Genet. 10, 769–782.
Duffy, D. L., Zhao, Z. Z., Sturm, R. A., Hayward, N. K., Martin, N. G., and Montgomery, G. W. (2010). Multiple pigmentation gene polymorphisms account for a substantial proportion of risk of cutaneous malignant melanoma. J. Invest. Dermatol. 130, 520–528.
Emre, M., Cummings, J. L., and Lane, R. M. (2007). Rivastigmine in dementia associated with Parkinson’s disease and Alzheimer’s disease: similarities and differences. J. Alzheimers Dis. 11, 509–519.
Epstein, E. A., and Chapman, M. R. (2008). Polymerizing the fibre between bacteria and host cells: the biogenesis of functional amyloid fibres. Cell. Microbiol. 10, 1413–1420.
Eto, H., Hashimoto, K., Kobayashi, H., Fukaya, T., Matsumoto, M., and Sun, T. T. (1984). Differential staining of cytoid bodies and skin-limited amyloids with monoclonal anti-keratin antibodies. Am. J. Pathol. 116, 473–481.
Fang, L., Huber-Abel, F., Teuchert, M., Hendrich, C., Dorst, J., Schattauer, D., Zettlmeissel, H., Wlaschek, M., Scharffetter-Kochanek, K., Tumani, H., Ludolph, A. C., and Brettschneider, J. (2009). Linking neuron and skin: matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 285, 62–66.
Fang, L., Teuchert, M., Huber-Abel, F., Schattauer, D., Hendrich, C., Dorst, J., Zettlmeissel, H., Wlaschek, M., Scharffetter-Kochanek, K., Kapfer, T., Tumani, H., Ludolph, A. C., and Brettschneider, J. (2010). MMP-2 and MMP-9 are elevated in spinal cord and skin in a mouse model of ALS. J. Neurol. Sci. 294, 51–56.
Feito-Rodriguez, M., Garcia-Macarron, J., Pagán-Muñoz, B., Mariño-Enríquez, A., Vidaurrázaga-Díaz, Y., Arcaya, C., Díaz-Díaz, R. M., and Casado-Jiménez, M. (2008). Disseminated nodular primary localized cutaneous amyloidosis. Actas Dermosifiliogr. 99, 648–652.
Fortune, D. G., Richards, H. L., and Griffiths, C. E. (2005). Psychological factors in psoriasis: consequences, mechanisms, and interventions. Dermatol. Clin. 23, 681–694.
Fortune, D. G., Richards, H. L., Griffiths, C. E., and Main, C. J. (2002). Psychological stress, distress and disability in patients with psoriasis: consensus and variation in the contribution of illness perceptions, coping and alexithymia. Br. J. Clin. Psychol. 41(Pt 2), 157–174.
Fowler, D. M., Koulov, A. V., Balch, W. E., and Kelly, J. W. (2007). Functional amyloid – from bacteria to humans. Trends Biochem. Sci. 32, 217–224.
Fu, X., Korenaga, T., Fu, L., Xing, Y., Guo, Z., Matsushita, T., Hosokawa, M., Naiki, H., Baba, S., Kawata, Y., Ikeda, S., Ishihara, T., Mori, M., and Higuchi, K. (2004). Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils. FEBS Lett. 563, 179–184.
Furukawa, T., and Toyokura, Y. (1976). Letter: amyotrophic lateral sclerosis and bedsores. Lancet 1, 862.
Galuske, R. A., Drach, L. M., Nichtweiss, M., Marquardt, G., Franz, K., Bohl, J., and Schlote, W. (2004). Colocalization of different types of amyloid in the walls of cerebral blood vessels of patients suffering from cerebral amyloid angiopathy and spontaneous intracranial hemorrhage: a report of 5 cases. Clin. Neuropathol. 23, 113–119.
Geser, F., Martinez-Lage, M., Kwong, L. K., Lee, V. M., and Trojanowski, J. Q. (2009). Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J. Neurol. 256, 1205–1214.
Giasson, B. I., Lee, V. M., and Trojanowski, J. Q. (2003). Interactions of amyloidogenic proteins. Neuromolecular Med. 4, 49–58.
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., Hatanpaa, K. J., White, C. L. III, Bigio, E. H., Caselli, R., Baker, M., Al-Lozi, M. T., Morris, J. C., Pestronk, A., Rademakers, R., Goate, A. M., and Cairns, N. J. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538.
Giudice, E. L., and Campbell, J. D. (2006). Needle-free vaccine delivery. Adv. Drug Deliv. Rev. 58, 68–89.
Glabe, C. G. (2006). Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575.
Glabe, C. G., and Kayed, R. (2006). Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66(Suppl. 1), S74–S78.
Greenwald, J., and Riek, R. (2010). Biology of amyloid: structure, function, and regulation. Structure 18, 1244–1260.
Gros-Louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972.
Gudbjartsson, D. F., Sulem, P., Stacey, S. N., Goldstein, A. M., Rafnar, T., Sigurgeirsson, B., Benediktsdottir, K. R., Thorisdottir, K., Ragnarsson, R., Sveinsdottir, S. G., Magnusson, V., Lindblom, A., Kostulas, K., Botella-Estrada, R., Soriano, V., Juberías, P., Grasa, M., Saez, B., Andres, R., Scherer, D., Rudnai, P., Gurzau, E., Koppova, K., Kiemeney, L. A., Jakobsdottir, M., Steinberg, S., Helgason, A., Gretarsdottir, S., Tucker, M. A., Mayordomo, J. I., Nagore, E., Kumar, R., Hansson, J., Olafsson, J. H., Gulcher, J., Kong, A., Thorsteinsdottir, U., and Stefansson, K. (2008). ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat. Genet. 40, 886–891.
Guijarro, J. I., Sunde, M., Jones, J. A., Campbell, I. D., and Dobson, C. M. (1998). Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. U.S.A. 95, 4224–4228.
Halaban, R., Cheng, E., Zhang, Y., Moellmann, G., Hanlon, D., Michalak, M., Setaluri, V., and Hebert, D. N. (1997). Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells. Proc. Natl. Acad. Sci. U.S.A. 94, 6210–6215.
Halevy, S., and Livni, E. (1993). Beta-adrenergic blocking drugs and psoriasis: the role of an immunologic mechanism. J. Am. Acad. Dermatol. 29, 504–505.
Harper, J. D., and Lansbury, P. T. Jr. (1997). Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407.
Hashimoto, K., and Kobayashi, H. (1980). Histogenesis of amyloid in the skin. Am. J. Dermatopathol. 2, 165–171.
Hashimoto, K., and Kumakiri, M. (1979). Colloid-amyloid bodies in PUVA-treated human psoriatic patients. J. Invest. Dermatol. 72, 70–80.
Hebert, L. E., Beckett, L. A., Scherr, P. A., and Evans, D. A. (2001). Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord. 15, 169–173.
Herczenik, E., and Gebbink, M. F. (2008). Molecular and cellular aspects of protein misfolding and disease. FASEB J. 22, 2115–2133.
Husby, G. (1992). Nomenclature and classification of amyloid and amyloidoses. J. Intern. Med. 232, 511–512.
Iconomidou, V. A., and Hamodrakas, S. J. (2008). Natural protective amyloids. Curr. Protein Pept. Sci. 9, 291–309.
Itoh, T., and Celis, E. (2005). Transcutaneous immunization with cytotoxic T-cell peptide epitopes provides effective antitumor immunity in mice. J. Immunother. 28, 430–437.
Jang, H., Boltz, D., Sturm-Ramirez, K., Shepherd, K. R., Jiao, Y., Webster, R., and Smeyne, R. J. (2009). Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 14063–14068.
Jarrett, J. T., and Lansbury, P. T. Jr. (1993). Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73, 1055–1058.
Jarvikallio, A., Harvima, I. T., and Naukkarinen, A. (2003). Mast cells, nerves and neuropeptides in atopic dermatitis and nummular eczema. Arch. Dermatol. Res. 295, 2–7.
Kalman, J., Szakacs, R., Török, T., Rózsa, Z., Barzó, P., Rudas, L., Papp, J. G., and Janka, Z. (2002). Decreased cutaneous vasodilatation to isometric handgrip exercise in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 17, 371–374.
Kato, S. (2008). Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol. 115, 97–114.
Katsikas, G. A., Maragou, M., Rontogianni, D., Gouma, P., Koutsouvelis, I., and Kappou-Rigatou, I. (2008). Secondary cutaneous nodular AA amyloidosis in a patient with primary Sjogren syndrome and celiac disease. J. Clin. Rheumatol. 14, 27–29.
Khalil, Z., LoGiudice, D., Khodr, B., Maruff, P., and Masters, C. (2007). Impaired peripheral endothelial microvascular responsiveness in Alzheimer’s disease. J. Alzheimers Dis. 11, 25–32.
Kobayashi, H., and Hashimoto, K. (1983). Amyloidogenesis in organ-limited cutaneous amyloidosis: an antigenic identity between epidermal keratin and skin amyloid. J. Invest. Dermatol. 80, 66–72.
Kolde, G., Bachus, R., and Ludolph, A. C. (1996). Skin involvement in amyotrophic lateral sclerosis. Lancet 347, 1226–1227.
Kumakiri, M., and Hashimoto, K. (1979). Histogenesis of primary localized cutaneous amyloidosis: sequential change of epidermal keratinocytes to amyloid via filamentous degeneration. J. Invest. Dermatol. 73, 150–162.
Kvetnoi, I. M., Kvetnaia, T. V., Riadnova, I. I. U., Fursov, B. B., Ernandes-Jago, H., and Blesa, J. R. (2003). Expression of beta-amyloid and tau-protein in mastocytes in Alzheimer disease. Arkh. Patol. 65, 36–39.
Lai, E. C., Felice, K. J., Festoff, B. W., Gawel, M. J., Gelinas, D. F., Kratz, R., Murphy, M. F., Natter, H. M., Norris, F. H., and Rudnicki, S. A. (1997). Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 49, 1621–1630.
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Guerrero-Muoz, M. J., Jackson, G. R., and Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041.
Leigh, P. N., and Swash, M. (1991). Cytoskeletal pathology in motor neuron diseases. Adv. Neurol. 56, 115–124.
Lopez De La Paz, M., Goldie, K., Zurdo, J., Lacroix, E., Dobson, C. M., Hoenger, A., and Serrano, L. (2002). De novo designed peptide-based amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 16052–16057.
Lorenzl, S., Narr, S., Angele, B., Krell, H. W., Gregorio, J., Kiaei, M., Pfister, H. W., and Beal, M. F. (2006). The matrix metalloproteinases inhibitor Ro 28-2653 [correction of Ro 26-2853] extends survival in transgenic ALS mice. Exp. Neurol. 200, 166–171.
Maier, M., Seabrook, T. J., Lazo, N. D., Jiang, L., Das, P., Janus, C., and Lemere, C. A. (2006). Short amyloid-beta (Abeta) immunogens reduce cerebral Abeta load and learning deficits in an Alzheimer’s disease mouse model in the absence of an Abeta-specific cellular immune response. J. Neurosci. 26, 4717–4728.
Maier, M., Seabrook, T. J., and Lemere, C. A. (2005). Modulation of the humoral and cellular immune response in Abeta immunotherapy by the adjuvants monophosphoryl lipid A (MPL), cholera toxin B subunit (CTB) and E. coli enterotoxin LT(R192G). Vaccine 23, 5149–5159.
Maji, S. K., Perrin, M. H., Sawaya, M. R., Jessberger, S., Vadodaria, K., Rissman, R. A., Singru, P. S., Nilsson, K. P., Simon, R., Schubert, D., Eisenberg, D., Rivier, J., Sawchenko, P., Vale, W., and Riek, R. (2009). Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325, 328–332.
Matsumoto, S., Kusaka, H., Ito, H., Shibata, N., Asayama, T., and Imai, T. (1996). Sporadic amyotrophic lateral sclerosis with dementia and Cu/Zn superoxide dismutase-positive Lewy body-like inclusions. Clin. Neuropathol. 15, 41–46.
Maurer-Stroh, S., Debulpaep, M., Kuemmerer, N., Lopez de la Paz, M., Martins, I. C., Reumers, J., Morris, K. L., Copland, A., Serpell, L., Serrano, L., Schymkowitz, J. W., and Rousseau, F. (2010). Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 7, 237–242.
Meersman, F., Dobson, C. M., and Heremans, K. (2006). Protein unfolding, amyloid fibril formation and configurational energy landscapes under high pressure conditions. Chem. Soc. Rev. 35, 908–917.
Mercier, F., Lefevre, G., Huang, H. L., Schmidli, H., Amzal, B., and Appel-Dingemanse, S. (2007). Rivastigmine exposure provided by a transdermal patch versus capsules. Curr. Med. Res. Opin. 23, 3199–3204.
Miller, R. G., Petajan, J. H., Bryan, W. W., Armon, C., Barohn, R. J., Goodpasture, J. C., Hoagland, R. J., Parry, G. J., Ross, M. A., and Stromatt, S. C. (1996). A placebo-controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Ann. Neurol. 39, 256–260.
Mitchell, J. D., Wokke, J. H., and Borasio, G. D. (2007). Recombinant human insulin-like growth factor I (rhIGF-I) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. CD002064.
Miyazono, M., Kitamoto, T., Iwaki, T., and Tateishi, J. (1992). Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Straussler syndrome. Acta Neuropathol. 83, 333–339.
Morrison, B. M., and Morrison, J. H. (1999). Amyotrophic lateral sclerosis associated with mutations in superoxide dismutase: a putative mechanism of degeneration. Brain Res. Brain Res. Rev. 29, 121–135.
Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. Ø., Riekel, C., Grothe, R., and Eisenberg, D. (2005). Structure of the cross-beta spine of amyloid-like fibrils. Nature 435, 773–778.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., McCluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H. A., Trojanowski, J. Q., and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Nikolic, W. V., Bai, Y., Obregon, D., Hou, H., Mori, T., Zeng, J., Ehrhart, J., Shytle, R. D., Giunta, B., Morgan, D., Town, T., and Tan, J. (2007). Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc. Natl. Acad. Sci. U.S.A. 104, 2507–2512.
Nitti, V. W., Sanders, S., Staskin, D. R., Dmochowski, R. R., Sand, P. K., MacDiarmid, S., and Maibach, H. I. (2006). Transdermal delivery of drugs for urologic applications: basic principles and applications. Urology 67, 657–664.
Okada, A., Urakami, K., Takahashi, K., Ohno, K., Sato, K., and Endo, H. (1994). Expression of amyloid beta-protein precursor mRNAs in cultured skin fibroblasts taken from patients with dementia of the Alzheimer type. Dementia 5, 55–56.
Ono, S., Imai, T., Aso, A., Yamano, T., Shimizu, N., and Nagao, K. (1998a). Alterations of skin glycosaminoglycans in patients with ALS. Neurology 51, 399–404.
Ono, S., Imai, T., Takahashi, K., Jinnai, K., Yamano, T., Nagao, K., Shimizu, N., and Yamauchi, M. (1998b). Decreased type IV collagen of skin and serum in patients with amyotrophic lateral sclerosis. Neurology 51, 114–120.
Paisan-Ruiz, C., Bhatia, K. P., Li, A., Hernandez, D., Davis, M., Wood, N. W., Hardy, J., Houlden, H., Singleton, A., and Schneider, S. A. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 65, 19–23.
Paris, I., Dagnino-Subiabre, A., Marcelain, K., Bennett, L. B., Caviedes, P., Caviedes, R., Azar, C. O., and Segura-Aguilar, J. (2001). Copper neurotoxicity is dependent on dopamine-mediated copper uptake and one-electron reduction of aminochrome in a rat substantia nigra neuronal cell line. J. Neurochem. 77, 519–529.
Pastor, M. T., Kummerer, N., Schubert, V., Esteras-Chopo, A., Dotti, C. G., López de la Paz, M., and Serrano, L. (2008). Amyloid toxicity is independent of polypeptide sequence, length and chirality. J. Mol. Biol. 375, 695–707.
Peters, E. M., Handjiski, B., Kuhlmei, A., Hagen, E., Bielas, H., Braun, A., Klapp, B. F., Paus, R., and Arck, P. C. (2004). Neurogenic inflammation in stress-induced termination of murine hair growth is promoted by nerve growth factor. Am. J. Pathol. 165, 259–271.
Ramsook, C. B., Tan, C., Garcia, M. C., Fung, R., Soybelman, G., Henry, R., Litewka, A., O’Meally, S., Otoo, H. N., Khalaf, R. A., Dranginis, A. M., Gaur, N. K., Klotz, S. A., Rauceo, J. M., Jue, C. K., and Lipke, P. N. (2010). Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryotic Cell 9, 393–404.
Raychaudhuri, S. P., Farber, E. M., and Raychaudhuri, S. K. (2000). Role of nerve growth factor in RANTES expression by keratinocytes. Acta Derm. Venereol. 80, 247–250.
Repetto, E., Yoon, I. S., Zheng, H., and Kang, D. E. (2007). Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J. Biol. Chem. 282, 31504–31516.
Rochet, J. C., and Lansbury, P. T. Jr. (2000). Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 10, 60–68.
Rocken, C., Schwotzer, E. B., Linke, R. P., and Saeger, W. (1996). The classification of amyloid deposits in clinicopathological practice. Histopathology 29, 325–335.
Rosen, D. R. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362.
Rosler, M., Anand, R., Cicin-Sain, A., Gauthier, S., Agid, Y., Dal-Bianco, P., Stähelin, H. B., Hartman, R., and Gharabawi, M. (1999). Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ 318, 633–638.
Schwendiman, M. N., Beachkofsky, T. M., Wisco, O. J., Owens, N. M., and Hodson, D. S. (2009). Primary cutaneous nodular amyloidosis: case report and review of the literature. Cutis 84, 87–92.
Selkoe, D. J. (1994). Alzheimer’s disease: a central role for amyloid. J. Neuropathol. Exp. Neurol. 53, 438–447.
Shibata, N., Hirano, A., Kobayashi, M., Sasaki, S., Kato, T., Matsumoto, S., Shiozawa, Z., Komori, T., Ikemoto, A., and Umahara, T. (1994). Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 179, 149–152.
Sipe, J. D., and Cohen, A. S. (2000). Review: history of the amyloid fibril. J. Struct. Biol. 130, 88–98.
Slominski, A., Wortsman, J., Luger, T., Paus, R., and Solomon, S. (2000). Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 80, 979–1020.
Soininen, H., Syrjanen, S., Heinonen, O., Neittaanmäki, H., Miettinen, R., Paljärvi, L., Syrjänen, K., Beyreuther, K., and Riekkinen, P. (1992). Amyloid beta-protein deposition in skin of patients with dementia. Lancet 339, 245.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., Ackerley, S., Durnall, J. C., Williams, K. L., Buratti, E., Baralle, F., de Belleroche, J., Mitchell, J. D., Leigh, P. N., Al-Chalabi, A., Miller, C. C., Nicholson, G., and Shaw, C. E. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672.
Stefani, M., and Dobson, C. M. (2003). Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699.
Steinkraus, V., Steinfath, M., Stöve, L., Körner, C., Abeck, D., and Mensing, H. (1993). Beta-adrenergic receptors in psoriasis: evidence for down-regulation in lesional skin. Arch. Dermatol. Res. 285, 300–304.
Suzuki, M., Mikami, H., Watanabe, T., Yamano, T., Yamazaki, T., Nomura, M., Yasui, K., Ishikawa, H., and Ono, S. (2010). Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 122, 367–372.
Takahashi, M., Hoshii, Y., Kawano, H., Gondo, T., Ishihara, T., and Isobe, T. (1996). Ultrastructural evidence for colocalization of kappa light chain- and beta 2-microglobulin-derived amyloids using double labelling immunogold electron microscopy. Virchows Arch. 429, 383–388.
Tamaoka, A., Matsumoto, S., Ono, S., Shimizu, N., and Shogi, S. (2000). Increased amyloid beta protein in the skin of patients with amyotrophic lateral sclerosis. J. Neurol. 247, 633–635.
Tillement, J. P., Lecanu, L., and Papadopoulos, V. (2009). Amyloidosis and neurodegenerative diseases: current treatments and new pharmacological options. Pharmacology 85, 1–17.
Tournoy, J., Bossuyt, X., Snellinx, A., Regent, M., Garmyn, M., Serneels, L., Saftig, P., Craessaerts, K., De Strooper, B., and Hartmann, D. (2004). Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum. Mol. Genet. 13, 1321–1331.
Trenkwalder, C., Kies, B., Rudzinska, M., Fine, J., Nikl, J., Honczarenko, K., Dioszeghy, P., Hill, D., Anderson, T., Myllyla, V., Kassubek, J., Steiger, M., Zucconi, M., Tolosa, E., Poewe, W., Surmann, E., Whitesides, J., Boroojerdi, B., Chaudhuri, K. R., Recover Study Group. (2011). Rotigotine effects on early morning motor function and sleep in Parkinson’s disease: a double-blind, randomized, placebo-controlled study (RECOVER). Mov. Disord. 26, 90–99.
Urpe, M., Buggiani, G., and Lotti, T. (2005). Stress and psychoneuroimmunologic factors in dermatology. Dermatol. Clin. 23, 609–617.
Vincenti, M. P., and Brinckerhoff, C. E. (2007). Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you? J. Cell. Physiol. 213, 355–364.
von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E. M., and Mandelkow, E. (2000). Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134.
Wang, X., Smith, D. R., Jones, J. W., and Chapman, M. R. (2007). In vitro polymerization of a functional Escherichia coli amyloid protein. J. Biol. Chem. 282, 3713–3719.
Wen, G. Y., Wisniewski, H. M., Blondal, H., Benedikz, E., Frey, H., Pirttila, T., Rudelli, R., and Kim, K. S. (1994). Presence of non-fibrillar amyloid beta protein in skin biopsies of Alzheimer’s disease (AD), Down’s syndrome and non-AD normal persons. Acta Neuropathol. 88, 201–206.
West, M. W., Wang, W., Patterson, J., Mancias, J. D., Beasley, J. R., and Hecht, M. H. (1999). De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. U.S.A. 96, 11211–11216.
Westermark, P., Lundmark, K., and Westermark, G. T. (2009). Fibrils from designed non-amyloid-related synthetic peptides induce AA-amyloidosis during inflammation in an animal model. PLoS ONE 4, e6041. doi:10.1371/journal.pone.0006041
Winblad, B., Cummings, J., Andreasen, N., Grossberg, G., Onofrj, M., Sadowsky, C., Zechner, S., Nagel, J., and Lane, R. (2007a). A six-month double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease – rivastigmine patch versus capsule. Int. J. Geriatr. Psychiatry 22, 456–467.
Winblad, B., Kawata, A. K., Beusterien, K. M., Thomas, S. K., Wimo, A., Lane, R., Fillit, H., and Blesa, R. (2007b). Caregiver preference for rivastigmine patch relative to capsules for treatment of probable Alzheimer’s disease. Int. J. Geriatr. Psychiatry 22, 485–491.
Wolters, E. (2009). Non-motor extranigral signs and symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 15(Suppl. 3), S6–S12.
Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M., and Biere, A. L. (1999). Alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 274, 19509–19512.
Xia, X., Qian, S., Soriano, S., Wu, Y., Fletcher, A. M., Wang, X. J., Koo, E. H., Wu, X., and Zheng, H. (2001). Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 98, 10863–10868.
Yanagihara, M., and Mori, S. (1982). Histogenesis of macular amyloidosis. Nippon Hifuka Gakkai Zasshi 92, 851–859.
Keywords: amyloid, skin, brain and neurodegenerative amyloidosis
Citation: Clos AL, Kayed R and Lasagna-Reeves CA (2012) Association of skin with the pathogenesis and treatment of neurodegenerative amyloidosis. Front. Neur. 3:5. doi: 10.3389/fneur.2012.00005
Received: 06 October 2011; Accepted: 04 January 2012;
Published online: 20 January 2012.
Edited by:
Philip P. Foster, University of Texas Medical Branch, USAReviewed by:
Daniel Michaelson, Tel-Aviv University, IsraelSalahadin Abdi, Harvard Medical School, USA
Philip P. Foster, University of Texas Medical Branch, USA
Copyright: © 2012 Clos, Kayed and Lasagna-Reeves. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Cristian A. Lasagna-Reeves, University of Texas Medical Branch, 301 University Blvd, Medical Research Building, Room 10.130, Galveston, TX 77555, USA. e-mail:Y2FsYXNhZ25AdXRtYi5lZHU=