

- 1 Centre de Recherche du CHUQ, Axe Neurosciences, Université Laval, Québec, QC, Canada
- 2 Département de Psychiatrie et Neurosciences, Faculté de Médecine, Université Laval, Québec, QC, Canada
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder and is characterized by the selective loss of dopaminergic neurons and the presence of Lewy bodies. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most frequent cause of both familial and sporadic PD. One critical question is how PD-associated LRRK2 mutations cause neurodegeneration. Here, we discuss recent findings related to LRRK2-mediated regulation of gene expression and translation and provide a critical assessment of the current models that are used to address the impact of LRRK2 on the transcriptome. A better understanding of these mechanisms could provide important new clues into the function of LRRK2 during both normal and pathological conditions.
Introduction
Parkinson’s disease (PD) is the most common movement disorder and the second most common neurodegenerative disease after Alzheimer’s disease (Nussbaum and Ellis, 2003). It is estimated that more than five million people worldwide suffer from PD and related parkinsonisms. In affected individuals, motor dysfunction is caused by selective dopaminergic neuronal loss in the substantia nigra. Pathologically, PD is characterized by the presence of intraneuronal inclusions, namely Lewy bodies, which are composed mainly of alpha-synuclein protein (Spillantini et al., 1997).
Mutations in leucine-rich repeat kinase 2 (LRRK2) represent a substantial genetic component of both sporadic and familial PD (Funayama et al., 2002; Paisan-Ruiz et al., 2004). LRRK2 (also known as dardarin) is a large (approximately 280 kDa) multidomain protein harboring a central catalytic tri-domain with GTPase and kinase activities (Cookson and Bandmann, 2010). It has been shown that most, but not all, PD-associated LRRK2 mutations display increased kinase activity, which is believed to contribute to neurotoxicity (West et al., 2005; Greggio et al., 2006). Despite these advances, the underlying molecular mechanisms involved in LRRK2-mediated neurodegeneration remain poorly defined.
Accumulating evidence supports the hypothesis that abnormal regulation of gene expression and protein translation may contribute to PD development. It is well known that the duplication or triplication of the alpha-synuclein gene, which increases alpha-synuclein protein levels, can cause genetic PD (Chartier-Harlin et al., 2004; Farrer et al., 2004). Furthermore, high levels of insoluble alpha-synuclein protein in both familial and sporadic PD may be explained, at least in part, by abnormal translation of alpha-synuclein messenger RNA (mRNA). In the last 2 years, several studies have implicated LRRK2 as a potentially important player in transcriptional and translational control. Experiments in various cell and animal models have demonstrated that LRRK2 modulation is associated with significant changes in mRNA output. Moreover, LRRK2 can bind to and phosphorylate several key factors involved in posttranscriptional and translation control. However, a crucial question in the field is whether LRRK2 directly modulates these processes. Here, we discuss these findings and place them in a biological and pathological context. In addition, we provide a critical assessment of the conceptual and analytical issues presented by several studies. We hope these insights may help our colleagues to move the field forward and address these important fundamental questions about LRRK2 biology.
LRRK2 and Its Effects on the Transcriptome
It is well established that changes in gene expression networks occur during aging and that misregulation of these pathways can have serious biological consequences. Understanding the effects of LRRK2 on gene expression is of particular interest due to its role in both normal and pathological aging processes.
One of the first studies to assess the impact of LRRK2 deficiency on the transcriptome was performed by Häbig et al. (2008) using microarrays (Affymetrix U133 Plus 2.0) of human SH-SY5Y cells, a dopaminergic neuroblastoma cell line, which had been treated with small interfering RNAs (siRNAs) against LRRK2. The authors obtained approximately a 75% reduction in endogenous LRRK2 expression in siRNA-treated cells compared to control cells. They identified 187 mRNA transcripts that were significantly misregulated in the LRRK2-knockdown cells using 1.5-fold and P ≤ 0.05 (unadjusted) cut-off values. Of these transcripts, approximately half were upregulated and half were downregulated, and a subset of total genes (14 out of 16) was validated by quantitative RT-PCR (qRT-PCR). Using the ingenuity pathway analysis (IPA) software, this study identified CDC42, a protein involved in axonal guidance signaling, in five out of the nine canonical molecular pathways generated. Other differentially expressed transcripts encoded proteins involved in biological processes such as the cell cycle and differentiation, the actin cytoskeleton, nervous system development, calcium signaling, and many others.
In a similar study, Devine et al. (2011) performed global mRNA microarray analyses (Illumina) using two cellular models: LRRK2-inducible HEK293 cells (wild-type and R1441C) and fibroblasts isolated from LRRK2 mutation (G2019S and Y1699C) carriers or healthy non-mutant controls. Upon LRRK2 induction in HEK cells, the exogenous levels of the wild-type LRRK2 protein or the LRRK2 R1441C mutant protein increased by approximately 1.5- and 2.5-fold, respectively, above the endogenous LRRK2 levels. In both cell lines and in all experimental conditions tested, the authors did not identify any significant changes in mRNA transcript levels when 1.5-fold cut-off values and P values ≤ 0.05 (after correction for multiple testing) were applied. As no independent validation was provided in this study, it remains unclear whether high-ranking (e.g., P ≤ 0.05, without correction for multiple testing) and/or lowly expressed genes (e.g., ≤1.5-fold) were indeed misregulated in these models, and no further functional assays were performed in this study.
Using embryonic stem (ES) cell-derived neurons, Schulz et al. (2011) analyzed gene expression patterns via microarray (GeneChip Mouse Genome 430 v2.0) during the neuronal differentiation of wild-type and haploinsufficient LRRK2+/− cells. Interestingly, only 23 mRNA transcripts were significantly misregulated in undifferentiated LRRK2+/− ES cells when compared to controls after 1.5-fold cut-off values and P values ≤ 0.01 (adjusted) were applied. On the other hand, almost 3000 genes were affected in LRRK2+/− differentiated neurons using similar cut-off criteria. The majority (approximately 80%) was downregulated, and some showed greater than 20-fold differences. Several neurotransmitter receptors as well as neurotransmitter release (GABA and glutamate) were increased in the LRRK2+/− cultures, suggesting that LRRK2 not only actively participates in neuronal differentiation but also accelerates this process. Interestingly, other misregulated genes included alpha-synuclein and the eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1). By western blot, the authors validated that three genes, Nanog, 4E-BP1, as well as ezrin, radixin, and moesin (ERM), were specifically downregulated in the LRRK2+/− neurons.
Very recently, Nikonova et al. (2012) performed global mRNA profiling experiments in LRRK2 knockout (KO) mice as well as in PD-associated LRRK2 G2019S transgenic (Tg) mice. In the Tg model, mutant LRRK2 is driven from the neuronal-specific promoter CMVE–PDGF-β and is expressed three- to four-fold over endogenous LRRK2. Using microarrays (Affymetrix GeneChip), the authors identified approximately 600 mRNA transcripts that were significantly misregulated (P ≤ 0.01, adjusted) in some tissues, including the cortex and striatum. In these assays, the authors compared three biological replicates (n = 2 per genotype, thus 12 animals in total) per group (LRRK2 KO vs. WT). For the LRRK2 G2019S mice, they used six biological replicates (n = 1 per genotype, thus 12 animals in total) per group (G2019S Tg vs. non-Tg). Interestingly, a significant portion of mRNA transcripts showed opposite patterns of fold changes in KO vs. Tg mice (after background correction using WT/non-Tg mice), suggesting that increased LRRK2 kinase activity associated with the most frequent LRRK2 G2019S mutation causes a gain of function with regard to gene expression regulation. Biological pathway analysis demonstrated that LRRK2 was significantly enriched for mRNA processing and ribosomal/translation functions. It is noteworthy that the overall false discovery rates used in this study varied significantly between the different mouse models and tissues tested and, in some cases, were above 80%. Thus, although a subset of genes (between 5 and 30%, depending on the model) was validated by qRT-PCR, the physiological relevance of these findings requires further evaluation. Also, the effects of LRRK2 WT overexpression remain to be established. Finally, the age of the mice used in this study was not provided, which makes it difficult to place these results within the context of aging. Nevertheless, this was the first study linking LRRK2 modulation and function with gene expression misregulation in vivo in mammals.
To our knowledge, only two groups have analyzed gene expression profiles in post-mortem brain tissues of patients with LRRK2 mutations. Häbig et al. (2008) compared mRNA transcript levels between idiopathic PD (IPD) patients (n = 5) and LRRK2 mutation (G2019S) carriers (n = 5). In these assays, RNA was extracted from the occipital cortex, where LRRK2 is expressed but where little or no pathology (e.g., Lewy bodies or cell loss) was detected. As in the previously mentioned cell lines (see above), the authors observed no major changes in gene expression patterns between LRRK2 carriers and either neurologically normal controls or IPD patients. In an independent study, Botta-Orfila et al. (2012) performed microarrays (Affymetrix GeneChip Human Exon 1.0 ST) using RNA extracted from the putamen. In this study, the authors compared groups of IPD (n = 5) and LRRK2 mutation (G2019S) carriers (n = 3) to neurologically healthy controls (n = 5) and a single asymptomatic G2019S-carrier. A total of 2874 genes were significantly altered (P ≤ 0.05, unadjusted) in the IPD group when compared to controls, whereas only 38 genes were misregulated in the LRRK2 G2019S group. However, only 227 and 5 genes in the IPD and LRRK2 G2019S groups, respectively, were significantly altered after applying a twofold change cut-off. Within the IPD group, 8 out of 10 genes were validated by qRT-PCR. Altered gene expression related to biological processes involved diverse cell signaling cascades and synaptic plasticity for both the sporadic PD cases as well as the asymptomatic LRRK2 mutation carrier. However, no gene ontology categories were reported in the LRRK2 mutant group, even after lower thresholds were used in the analyses. Unfortunately, the RNA integrity number (RIN) values or other biochemical assessments of RNA quality were not included in these studies. This is an important issue, as most tissues had high (>24 h) post-mortem intervals. Thus, the clinical and physiological relevance of these results remain uncertain, and larger cohorts, who will increase the statistical power of these findings, as well as better tissue quality, are needed to draw definitive conclusions.
LRRK2 and Protein Translation Regulation: Focus on 4E-BP1 and the eIF4 Complex
The misregulation of translation can lead to misfolding and an accumulation of aggregation-prone proteins, which are common features of many age-related diseases including PD (Hindle, 2010). Protein translation is a highly energetic process, and its stimulation can perturb cellular energy homeostasis. Below, we focus on LRRK2-mediated regulation of protein synthesis and particularly on the formation of the eIF4 translation initiation complex.
In Drosophila, it has been shown that LRRK2 interacts genetically with the TOR/4E-BP1 pathway (Imai et al., 2008), which is involved in cell growth via protein synthesis (Wang and Proud, 2006). At the post-synapse, LRRK2 binds to and phosphorylates 4E-BP1, a binding inhibitor of the eukaryotic translation initiation factor 4E (eIF4E; Figure 1). The phosphorylation and consequent release of 4E-BP1 enables eIF4E to engage in protein translation (Imai et al., 2008). Notably, pathogenic LRRK2 mutations can further deregulate protein synthesis through hyperphosphorylation of 4E-BP1, which can cause an age-dependent loss of dopaminergic neurons (Imai et al., 2008). However, whether LRRK2 PD-associated mutations that do not display increased kinase activity have similar effects on 4E-BP1 and eIF4E function remains to be determined. In another study, Pons et al. (2012) showed that LRRK2 downregulation using siRNAs in various cancer cell lines caused a significant decrease in 4E-BP1 protein (but not mRNA) levels, which is consistent with the results of previous studies (Schulz et al., 2011). They further found that decreased LRRK2 (and 4E-BP1) expression was associated with increased cellular proliferation. Thus, LRRK2 seems equally required for the regulation of 4E-BP1 phosphorylation and protein stability, potentially in a regulatory feedback loop.
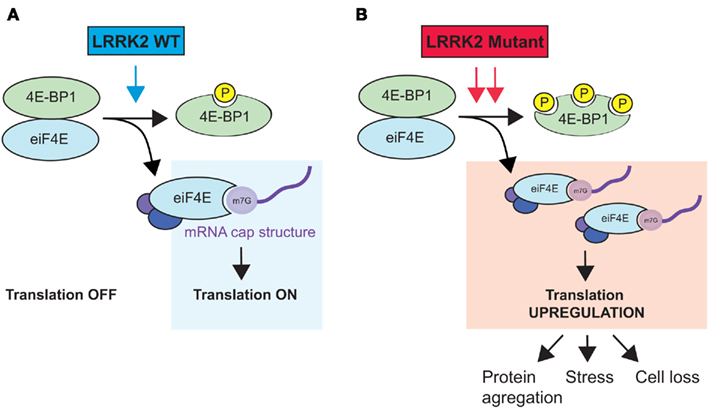
Figure 1. Hypothetical model of LRRK2-mediated regulation of protein translation. (A) Wild-type LRRK2 binds to and phosphorylates the eiF4E inhibitor 4E-BP1. Free eiF4E interacts with other initiation complex factors (eiF4A and eiF4G) to bind to the mRNA cap structure and initiate protein translation. (B) PD-associated LRRK2 mutants with increased kinase activity cause 4E-BP1 hyperphosphorylation, which over-stimulates protein synthesis. The upregulation of protein translation can lead to various consequences, such as protein aggregation, cell stress, and ultimately, cell death. Note that binding of LRRK2 to 4E-BP1 is not depicted here.
Previous studies have shown that mutations in eIF4E are involved in carcinogenesis and that mutant eIF4E is overexpressed in various tumors (Wendel et al., 2007), which likely allows for the translation of oncogenes and promotes cancer progression. Interestingly, Chartier-Harlin et al. (2011) recently identified mutations in eIF4G1, a binding partner of eIF4E and an essential component of the eIF4F translation initiation complex (composed of eIF4A, eIF4E, and eIF4G), in families with genetic PD. Two specific mutations impaired the formation of the complex, and functional analyses demonstrated a disruption in the binding of the eIF4G1 mutant A1205H to eIF3E as well as a defect in the binding of the eIF4G1 A502V mutant to eIF4E. These mutants were also associated with impaired mitochondrial dysfunction in response to oxidative stress. This study proposes the exciting new viewpoint that mutations in eIF4G1, and perhaps other components of the protein synthesis machinery, are directly implicated in abnormal mRNA translation in PD pathogenesis.
LRRK2 and Non-Coding RNAs: A Link between Transcription and Translation
As mentioned above, LRRK2 harbors several protein–protein interaction domains that are potentially involved in the scaffolding of protein complexes and the binding of substrates (Li et al., 2011). To date, two independent studies have demonstrated an interaction between LRRK2 and the microRNA (miRNA) pathway. These small (approximately 21–23 nucleotides) non-protein-coding RNAs are essential for brain development and neuronal survival and control precise neuronal functions (Hebert and De Strooper, 2009; Bagetta et al., 2010). While the exact mode of action of miRNAs remains under scrutiny, it has been shown that they function to promote either the inhibition of translation or mRNA degradation (Huntzinger and Izaurralde, 2011). In one study, Dachsel et al. (2007) used HEK293 cells overexpressing wild-type human LRRK2 and co-immunoprecipitation followed by mass spectrometry to identify 14 proteins that interacted specifically with exogenous LRRK2, including eukaryotic translation initiation factor 2C 1 (eIF2c1/Ago1) and eukaryotic translation initiation factor 2C 2 (eIF2c2/Ago2). While not discussed in this study, argonaute (Ago) proteins are essential components of the miRNA machinery (Peters and Meister, 2007). In a second study, Gehrke et al. (2010) validated the interaction between LRRK2 and Ago by co-immunoprecipitation in transfected HEK293 cells as well as in brain extracts of transgenic Drosophila. By immunofluorescence, the authors further showed that ectopic LRRK2 partially co-localized with Ago1 in dopaminergic neurons in vivo. Notably, 4E-BP1 also interacted with Ago1 in an age-dependent manner. Moreover, the LRRK2 PD-associated mutant G2019S, which displayed increased kinase activity, stimulated this interaction, suggesting a greater affinity between Ago and phosphorylated 4E-BP1 (Figure 2). The authors identified two downstream effector miRNAs (let-7 and miR-184*) as well as target genes (DP1 and E2F1) involved in LRRK2-mediated neurodegeneration. Interestingly, earlier studies have shown that the central domain of Ago proteins exhibits sequence similarities to that of eIF4E and that human Ago2 can compete with eIF4E to bind the cap structure of mRNAs (Kiriakidou et al., 2007; Eulalio et al., 2008). Finally, it has been reported that Ago2 can be phosphorylated by unidentified kinases, which reduces its binding affinity for small RNAs (Rudel et al., 2011). Taken together, these results highlight a potential cross talk between LRRK2, the miRNA pathway, and eukaryotic transcription factors for the modulation of protein synthesis and neuronal survival. Clearly, these concepts need to be validated experimentally as well as investigated using other biological models, including the mammalian brain.
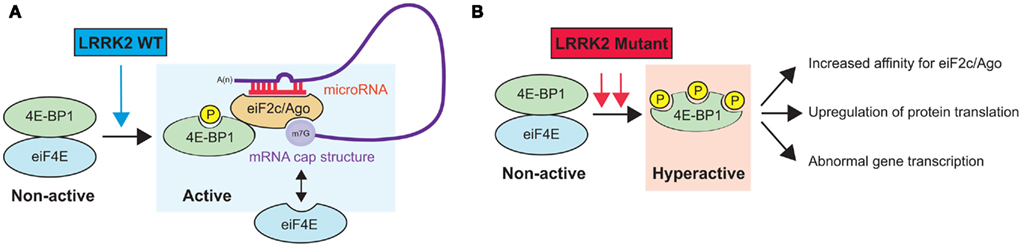
Figure 2. Hypothetical model of LRRK2-mediated regulation of the miRNA pathway. (A) Wild-type LRRK2 binds to and phosphorylates 4E-BP1. Phosphorylated 4E-BP1 then binds to eiF2c/Ago, which can potentially modulate miRNA binding to its mRNA targets and affect gene expression. Interestingly, eiF2c/Ago can also compete with free eiF4E to bind the mRNA cap structure, possibly affecting protein translation in addition to gene transcription via miRNAs. Note that the interaction between LRRK2 and eiF2c/Ago is not shown here. (B) PD-associated LRRK2 mutants could affect the miRNA pathway by increasing miRNA stability and function, potentially through the increase in the affinity between Ago and phospho-4E-BP1. Obviously, various changes in translation and/or transcription regulation are conceivable in this model and need to be investigated further. Misregulation of these pathways could have important biological consequences.
Another interesting study provided a link between LRRK2 and transcriptional control via non-coding RNAs. Indeed, Liu et al. (2011) recently demonstrated that in mice, LRRK2 binds the potent negative transcriptional regulator NFAT, which functions in immune responses. LRRK2 can also interact with the NFAT repressor complex that includes the large (800–3700 nucleotides) non-coding RNA NRON. Knockdown or overexpression of NRON abrogated or increased, respectively, the binding of exogenous LRRK2 with the NFAT complex. Without affecting NFAT phosphorylation, LRRK2 sequestered the NFAT complex in the cytosol and, as expected, prevented its functional translocation into the nucleus. As most experiments were conducted in peripheral cells or tissues, it would be interesting to study this pathway in the brain using LRRK2 animal models. Whether NFAT and/or NRON mutations are found in familial or sporadic PD patients remains to be investigated.
Conclusion
Over the past 20 years, many advances have been made regarding the identification of genes and pathways associated with PD. However, the exact contribution and interaction between these genes for the etiology of the disease remain unclear. In this review, we discussed recent findings related to the influence of LRRK2 in the regulation of gene transcription and protein translation, two ubiquitous and fundamental biological processes. The current literature provides ample evidence that LRRK2 participates actively in translation initiation through the phosphorylation of 4E-BP1 and likely other factors as well. Furthermore, both LRRK2 and 4E-BP1 interact with core components of the miRNA pathway to potentially act as a “molecular hub” for gene expression and protein synthesis. Mutations in genes related to protein synthesis in genetic PD highlight the significance of protein translation dysfunction in this disease.
To date, most researchers have relied on microarrays to assess the effects of LRRK2 on the genome. As discussed herein, the choice of platform, the number of replicates, the power of the statistical test, and the cut-off criteria can considerably impact the overall conclusions, especially when the effects are subtle. These factors, combined with the high false-negative and false-positive rates associated with microarrays in general, necessitate the independent validation of selected genes to make definitive conclusions. We believe that it is safe to conclude that LRRK2 does influence mRNA levels. However, whether these effects are direct and whether they reflect a global or specific loss (or gain) of function remains open to debate. As of now, the consequences of LRRK2 modulation on protein output remain largely unexplored, such that additional studies are required to fully appreciate its role in the cell. Obviously, the choice of biological model, ranging from cells to humans, adds to the complexity of these analyses. Similarly, the selection of LRRK2 mutations may also have an impact on the experimental outcome. As noted previously, not all PD-associated LRRK2 mutants (e.g., R1441C/G/H) display increased kinase activity. In these specific cases, it is thought that the GTPase domain contributes to LRRK2-mediated toxicity (Xiong et al., 2010), possibly independently of binding to GTP/GDP nucleotides (Taymans et al., 2011). Moreover, the function (or dysfunction) of LRRK2 may be cell and/or tissue-specific. In agreement with this hypothesis, Tong et al. (2010) showed that genetic deletion through germ-line transmission of targeted LRRK2 in mice had no measurable effect on dopaminergic neurons, whereas it was highly toxic to the kidneys. Finally, it remains possible that LRRK2 may function mainly under stress conditions. Undoubtedly, the results discussed herein provide a strong foundation for further validation and functional studies with regard to the role of LRRK2 in the regulation of transcription and translation as well as in the pathogenesis of PD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Scottish Rite Charitable Foundation of Canada and the Fonds de recherche du Québec – Santé.
References
Bagetta, V., Ghiglieri, V., Sgobio, C., Calabresi, P., and Picconi, B. (2010). Synaptic dysfunction in Parkinson’s disease. Biochem. Soc. Trans. 38, 493–497.
Botta-Orfila, T., Tolosa, E., Gelpi, E., Sanchez-Pla, A., Marti, M. J., Valldeoriola, F., Fernandez, M., Carmona, F., and Ezquerra, M. (2012). Microarray expression analysis in idiopathic and LRRK2-associated Parkinson’s disease. Neurobiol. Dis. 45, 462–468.
Chartier-Harlin, M. C., Dachsel, J. C., Vilarino-Guell, C., Lincoln, S. J., Lepretre, F., Hulihan, M. M., Kachergus, J., Milnerwood, A. J., Tapia, L., Song, M. S., Le Rhun, E., Mutez, E., Larvor, L., Duflot, A., Vanbesien-Mailliot, C., Kreisler, A., Ross, O. A., Nishioka, K., Soto-Ortolaza, A. I., Cobb, S. A., Melrose, H. L., Behrouz, B., Keeling, B. H., Bacon, J. A., Hentati, E., Williams, L., Yanagiya, A., Sonenberg, N., Lockhart, P. J., Zubair, A. C., Uitti, R. J., Aasly, J. O., Krygowska-Wajs, A., Opala, G., Wszolek, Z. K., Frigerio, R., Maraganore, D. M., Gosal, D., Lynch, T., Hutchinson, M., Bentivoglio, A. R., Valente, E. M., Nichols, W. C., Pankratz, N., Foroud, T., Gibson, R. A., Hentati, F., Dickson, D. W., Destee, A., and Farrer, M. J. (2011). Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 89, 398–406.
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., Levecque, C., Larvor, L., Andrieux, J., Hulihan, M., Waucquier, N., Defebvre, L., Amouyel, P., Farrer, M., and Destee, A. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169.
Cookson, M. R., and Bandmann, O. (2010). Parkinson’s disease: insights from pathways. Hum. Mol. Genet. 19, R21–R27.
Dachsel, J. C., Taylor, J. P., Mok, S. S., Ross, O. A., Hinkle, K. M., Bailey, R. M., Hines, J. H., Szutu, J., Madden, B., Petrucelli, L., and Farrer, M. J. (2007). Identification of potential protein interactors of Lrrk2. Parkinsonism Relat. Disord. 13, 382–385.
Devine, M. J., Kaganovich, A., Ryten, M., Mamais, A., Trabzuni, D., Manzoni, C., McGoldrick, P., Chan, D., Dillman, A., Zerle, J., Horan, S., Taanman, J. W., Hardy, J., Marti-Masso, J. F., Healey, D., Schapira, A. H., Wolozin, B., Bandopadhyay, R., Cookson, M. R., van der Brug, M. P., and Lewis, P. A. (2011). Pathogenic LRRK2 mutations do not alter gene expression in cell model systems or human brain tissue. PLoS ONE 6, e22489. doi:10.1371/journal.pone.0022489
Eulalio, A., Huntzinger, E., and Izaurralde, E. (2008). GW182 interaction with argonaute is essential for miRNA-mediated translational repression and mRNA decay. Nat. Struct. Mol. Biol. 15, 346–353.
Farrer, M., Kachergus, J., Forno, L., Lincoln, S., Wang, D. S., Hulihan, M., Maraganore, D., Gwinn-Hardy, K., Wszolek, Z., Dickson, D., and Langston, J. W. (2004). Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 55, 174–179.
Funayama, M., Hasegawa, K., Kowa, H., Saito, M., Tsuji, S., and Obata, F. (2002). A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 51, 296–301.
Gehrke, S., Imai, Y., Sokol, N., and Lu, B. (2010). Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 466, 637–641.
Greggio, E., Jain, S., Kingsbury, A., Bandopadhyay, R., Lewis, P., Kaganovich, A., van der Brug, M. P., Beilina, A., Blackinton, J., Thomas, K. J., Ahmad, R., Miller, D. W., Kesavapany, S., Singleton, A., Lees, A., Harvey, R. J., Harvey, K., and Cookson, M. R. (2006). Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341.
Häbig, K., Walter, M., Poths, S., Riess, O., and Bonin, M. (2008). RNA interference of LRRK2-microarray expression analysis of a Parkinson’s disease key player. Neurogenetics 9, 83–94.
Hebert, S. S., and De Strooper, B. (2009). Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 32, 199–206.
Huntzinger, E., and Izaurralde, E. (2011). Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12, 99–110.
Imai, Y., Gehrke, S., Wang, H. Q., Takahashi, R., Hasegawa, K., Oota, E., and Lu, B. (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 27, 2432–2443.
Kiriakidou, M., Tan, G. S., Lamprinaki, S., De Planell-Saguer, M., Nelson, P. T., and Mourelatos, Z. (2007). An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell 129, 1141–1151.
Li, T., Yang, D., Sushchky, S., Liu, Z., and Smith, W. W. (2011). Models for LRRK2-linked parkinsonism. Parkinsons Dis. 2011, 942412.
Liu, Z., Lee, J., Krummey, S., Lu, W., Cai, H., and Lenardo, M. J. (2011). The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 12, 1063–1070.
Nikonova, E. V., Xiong, Y., Tanis, K. Q., Dawson, V. L., Vogel, R. L., Finney, E. M., Stone, D. J., Reynolds, I. J., Kern, J. T., and Dawson, T. M. (2012). Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum. Mol. Genet. 21, 163–174.
Nussbaum, R. L., and Ellis, C. E. (2003). Alzheimer’s disease and Parkinson’s disease. N. Engl. J. Med. 348, 1356–1364.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W., and Singleton, A. B. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600.
Peters, L., and Meister, G. (2007). Argonaute proteins: mediators of RNA silencing. Mol. Cell 26, 611–623.
Pons, B., Armengol, G., Livingstone, M., Lopez, L., Coch, L., Sonenberg, N., and Ramon, Y. C. S. (2012). Association between LRRK2 and 4E-BP1 protein levels in normal and malignant cells. Oncol. Rep. 27, 225–231.
Rudel, S., Wang, Y., Lenobel, R., Korner, R., Hsiao, H. H., Urlaub, H., Patel, D., and Meister, G. (2011). Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res. 39, 2330–2343.
Schulz, C., Paus, M., Frey, K., Schmid, R., Kohl, Z., Mennerich, D., Winkler, J., and Gillardon, F. (2011). Leucine-rich repeat kinase 2 modulates retinoic acid-induced neuronal differentiation of murine embryonic stem cells. PLoS ONE 6, e20820. doi:10.1371/journal.pone.0020820
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Taymans, J. M., Vancraenenbroeck, R., Ollikainen, P., Beilina, A., Lobbestael, E., De Maeyer, M., Baekelandt, V., and Cookson, M. R. (2011). LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE 6, e23207. doi:10.1371/journal.pone.0023207
Tong, Y., Yamaguchi, H., Giaime, E., Boyle, S., Kopan, R., Kelleher, R. J. III, and Shen, J. (2010). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884.
Wang, X., and Proud, C. G. (2006). The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 21, 362–369.
Wendel, H. G., Silva, R. L., Malina, A., Mills, J. R., Zhu, H., Ueda, T., Watanabe-Fukunaga, R., Fukunaga, R., Teruya-Feldstein, J., Pelletier, J., and Lowe, S. W. (2007). Dissecting eIF4E action in tumorigenesis. Genes Dev. 21, 3232–3237.
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., Dawson, V. L., and Dawson, T. M. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847.
Keywords: LRRK2, 4E-BP1, eIF4, transcription, translation, Parkinson’s disease
Citation: Dorval V and Hébert SS (2012) LRRK2 in transcription and translation regulation: relevance for Parkinson’s disease. Front. Neur. 3:12. doi: 10.3389/fneur.2012.00012
Received: 12 December 2011;
Accepted: 17 January 2012;
Published online: 10 February 2012.
Edited by:
Emmanuel Planel, Centre Hospitalier de l’Université Laval, CanadaReviewed by:
Alejandra Alvarez, Pontificia Universidad Católica de Chile, ChileKimberly Scearce-Levie, Genentech, USA
Copyright: © 2012 Dorval and Hébert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sébastien S. Hébert, Centre de Recherche du CHUQ, Axe Neurosciences, Université Laval, 2705 boul. Laurier, RC-9800, Québec, QC, Canada G1V4G2. e-mail:c2ViYXN0aWVuLmhlYmVydEBjcmNodWwudWxhdmFsLmNh