


- 1 Departamento de Psicobiologia, Universidade Federal de São Paulo, Sao Paulo, Brazil
- 2 Centro de Matemática, Computação e Cognição, Universidade Federal do ABC, Sao Paulo, Brazil
Stress and sleep are related to each other in a bidirectional way. If on one hand poor or inadequate sleep exacerbates emotional, behavioral, and stress-related responses, on the other hand acute stress induces sleep rebound, most likely as a way to cope with the adverse stimuli. Chronic, as opposed to acute, stress impairs sleep and has been claimed to be one of the triggering factors of emotional-related sleep disorders, such as insomnia, depressive- and anxiety-disorders. These outcomes are dependent on individual psychobiological characteristics, conferring even more complexity to the stress-sleep relationship. Its neurobiology has only recently begun to be explored, through animal models, which are also valuable for the development of potential therapeutic agents and preventive actions. This review seeks to present data on the effects of stress on sleep and the different approaches used to study this relationship as well as possible neurobiological underpinnings and mechanisms involved. The results of numerous studies in humans and animals indicate that increased sleep, especially the rapid eye movement phase, following a stressful situation is an important adaptive behavior for recovery. However, this endogenous advantage appears to be impaired in human beings and rodent strains that exhibit high levels of anxiety and anxiety-like behavior.
Introduction
The impact of stress on sleep has been recognized for millennia since Hippocrates stated that sleep loss is a signal of pain and suffering that may lead to mental disorders, whereas excessive diurnal somnolence indicates the existence of disease. He also emphasized the importance of sleep as a medicine for physical and psychological stress (Vgontzas et al., 2000). However, there are more subtleties to this relation than previously thought. For instance, the nature and length of the stressor, and, most importantly, the individual capacity to cope with stressful situations determine the sleep outcome. Moreover, not only stress alters the sleep pattern but also inadequate sleep influences the activity of stress response systems, i.e., whether one is sleep-deprived or not, determines the ability to respond to stressors (Spiegel et al., 1999; Suchecki et al., 2002). This bidirectional association can, thus, represent a vicious circle with detrimental consequences to physical and mental health.
The bidirectional influence between stress and sleep can be attested by evidence showing that, on the one hand, daily stressful events reduce sleep time and trigger insomnia (Beary et al., 1984), although that effect also depends on individual features (Morin et al., 2003). On the other hand, insomnia (Vgontzas et al., 1998, 2001) and sleep apnea patients (Carneiro et al., 2008), who are chronically sleep-deprived, exhibit dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic nervous system, the main physiological stress response systems (Joels and Baram, 2009). Chronic psychosocial stressors represent powerful sleep disrupting stimuli as can be seen in studies with divorcees (Cartwright and Wood, 1991), with people who experience lack of social support in the work environment (Gadinger et al., 2009; Nomura et al., 2009), with children and adolescents exposed to traumatic events (Charuvastra and Cloitre, 2009) and in burnout patients (Armon et al., 2008). Interestingly, women who suffer from chronic burnout and report being more anxious and having more problems to sleep also exhibit lower prolactin levels (Grossi et al., 2003), a hormone that, later in this review, will be discussed as a sleep-inducing factor under stressful situations.
It is noteworthy that sleep disorders and dysregulation of the systems involved in stress response are hallmarks of many psychiatric disorders, such as depression, generalized anxiety, and post-traumatic stress disorder (PTSD; Papadimitriou and Linkowski, 2005). In major depression, hypercortisolemia, reduced time of delta sleep and of rapid eye movement sleep (REMS) latency, and increased percentage of REMS and sleep fragmentation are usually reported (for review, see Holsboer-Trachsler and Seifritz, 2000; Shaffery et al., 2003; Armitage, 2007). Although there is some controversy as to whether PTSD patients display impaired sleep (Klein et al., 2002), several studies do report increased time waking after sleep onset (Raboni et al., 2006; Habukawa et al., 2007), more stage 1 of non-REM sleep (NREMS; Kobayashi et al., 2007), more transitions from REMS to stage 1 and to waking (Breslau et al., 2004; Habukawa et al., 2007), indicating more sleep fragmentation, as part of the characteristic hyperarousal, and increased sympathetic activity observed in these patients (Woodward et al., 2000).
Recently it has become more evident that sleep, and specially REMS, play a fundamental role on the emotional and mental recovery from adverse situations. It has been proposed that one of the functions of REMS is to weaken undesirable and persistent memories, by a reverse learning, or remodeling, process (Crick and Mitchison, 1983), a process that, supposedly, could help individuals to cope with aversive and stressful situations. Indeed, victims of motor vehicle accidents who slept longer and more consolidated episodes of REMS in the aftermath period following the traumatic event did not develop PTSD (Mellman et al., 2002, 2007; Mellman and Hipolito, 2006). Moreover, the individuals who did not develop PTSD displayed more relative beta frequency during REMS, interpreted by the authors to be an indication of high cognitive activation during this sleep phase as this frequency is negatively correlated with the occurrence of nightmares and with symptoms of PTSD (Mellman et al., 2007). From the neurophysiological point of view, REMS represents a period of noradrenergic quiescence (Pace-Schott and Hobson, 2002), suggesting that this phase of sleep is an ideal period for reframing negative experiences and restoring favorable emotional reactivity (Walker and van der Helm, 2009). This was demonstrated in a study carried out with healthy volunteers who exhibited reduced amygdala and behavioral reactivity to emotionally loaded pictures after one night of sleep, as opposed to volunteers kept awake during the day. These findings were correlated with diminished gamma EEG activity during REMS (van der Helm et al., 2011), a marker of adrenergic activity (Cape and Jones, 1998; Hajos et al., 2003; Walling et al., 2011). Based on these data REMS can be viewed as a period involving the reduction of emotional salience associated with prior waking (van der Helm et al., 2011). This is exactly what Gujar et al. (2011) observed in individuals who had REMS periods during a 90-min nap and displayed less reaction to fearful facial expressions and more positive reactions to happy faces. Further evidence that sleep protects the organism against stress-induced psychiatric disorders stems from epidemiological studies indicating that insomnia and difficulty to sleep during stressful periods are risk factors for depression (Breslau et al., 1996; Chang et al., 1997; Riemann et al., 2001).
Animal studies are essential to disclose the mechanisms involved in this protective effect of REMS on emotionality and how differences in anxiety-like and coping behaviors, and their neurobiological underpinnings, determine the sleep response of animals to numerous stressful situations. In the present review, we will briefly describe the main physiological systems responsible for the adaptive response to acute stress and will describe the importance of considering the nature of the stressors and the individual characteristics of the subjects on the physiological and sleep outcomes as a way to determine how stress-induced REMS rebound may protect the subject against development of psychiatric disorders.
Systems Involved in Stress Response
Stress, as much as sleep, is a global phenomenon that involves the entire central nervous system (CNS), with numerous neurotransmitter systems playing a role in their regulation. On the one hand, noradrenaline (NA), acetylcholine (ACh), and serotonin (5-HT) stimulate, whereas gamma aminobutyric acid (GABA) inhibits the activity of the paraventricular nucleus of the hypothalamus (PVN), where neurons that synthesize corticotrophin releasing hormone (CRH) are located. On the other hand, these neurotransmitters operate in an orchestrated manner to regulate sleep, so much so that NA induces waking and GABA, sleep; administration of ACh into specific brain regions in the pons induces REM sleep and 5-HT is involved in the regulation of delta sleep (Pace-Schott and Hobson, 2002).
The stress response entails two main systems, the locus coeruleus/medulla adrenal and the HPA axis, although other hormones are important for the optimal stress response, as is the case of Prolactin (PRL), Oxytocin, and Vasopressin (AVP). When facing either a physical or psychological threatening stimulus, the organism responds by activating these two systems, which major function is to promote behavioral and metabolic adaptations to the situation so as to guarantee survival. These systems are briefly described below (for review, see Sapolsky et al., 2000; Morilak et al., 2005; Gunnar and Quevedo, 2007).
Locus Coeruleus/Adrenal Medulla
The locus coeruleus–adrenal medulla is also known as the sympathetic-adrenomedullary system and, together with the parasympathetic system, is part of the autonomic nervous system. The activation of preganglionic sympathetic neurons located in the intermediolateral cell column of the thoracolumbar spinal cord. These neurons exit the spinal cord via the ventral root to form cholinergic synapses on the chromaffin cells of the adrenal medulla. When these cells are stimulated, adrenaline (80%) and NA (20%) secretion ensues and these catecholamines affect organs and tissues by binding to specific alpha and beta adrenergic receptors. The net result of activation of these receptors involves vasodilatation of muscle and vasoconstriction of skin blood vessels, increased blood pressure and heart rate, increased oxygen and glucose supply to skeletal muscles and brain, so as to assure the best conditions for the fight or flight reaction (Ulrich-Lai and Herman, 2009).
Although neither Adrenaline nor NA cross the blood–brain barrier, activation of the locus coeruleus parallels the peripheral actions of these catecholamines (Svensson, 1987) and vigilance, arousal and increased attention to the source of the stressful stimulus is ensured. In addition, this system also stimulates the neuroendocrine limb of the stress response, represented by the HPA axis (Morilak et al., 2005).
Hypothalamic–Pituitary–Adrenal Axis
The PVN convey information from ascending pathways originating in the brainstem and midbrain, more specifically from the nucleus of the solitary tract, locus coeruleus, and raphe nucleus, and from descending pathways originating in the limbic system (hippocampus and amygdala) and prefrontal cortex, which signal, respectively, physical and psychological threats to homeostasis. Stimulation of the PVN triggers a cascade of neuroendocrine events, which involves the release of CRH – the major stimulator of the synthesis and release of adrenocorticotropin (ACTH) from the anterior pituitary and arginine vasopressin (AVP) – known to increase the CRH signal at the corticotropes. ACTH reaches the adrenal cortex and induces the synthesis and secretion of glucocorticoids (GC) – cortisol in human beings and primates and corticosterone (CORT) in rodents. GC regulate the activity of the HPA axis by means of a feedback signal at the prefrontal cortex, hippocampus, hypothalamus, and pituitary by binding to the low affinity glucocorticoid receptor (GR or type II), widely distributed throughout the body (Figure 1). These receptors mediate the suppressive effects of GC, including immunosuppression, increased waking (suppression of sleep), and the negative feedback regulatory function (suppression of HPA axis activity). In addition to these receptors, high affinity mineralocorticoid receptors (MR or type I) mediate basal and permissive effects of GC, including the induction of digestive enzymes, stimulation of fibrinogen production (in case of tissue injury), regulation of memory consolidation and regulation of REMS. Binding of GC to their cytoplasmic receptors leads to changes in gene transcription explaining the long time lag for initiation of their effects.
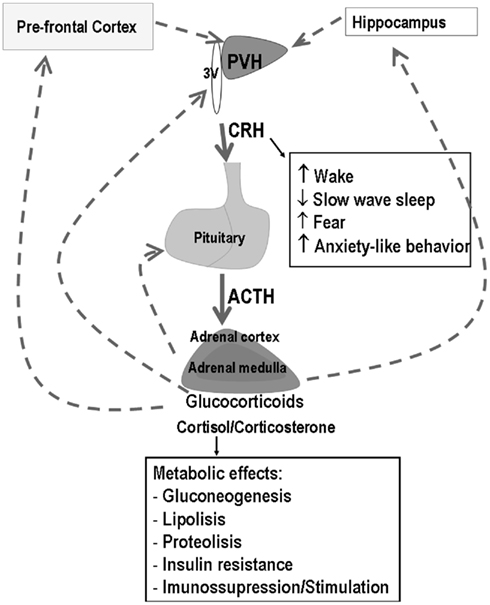
Figure 1. Schematic representation of the hypothalamic–pituitary–adrenal (HPA) axis and main effects of corticotropin releasing hormone (CRH) and glucocorticoids (GC). Filled arrows represent stimulatory effects, whereas dotted arrows represent inhibition. The main sites of GC negative feedback are the prefrontal cortex, hippocampus, hypothalamus, and pituitary.
Effects of Stress Hormones on Sleep
Exogenous administration of the hormones of HPA axis induces waking, as should be expected to happen under stressful or threatening situations. Classically, the release of these hormones under stressful situations is to increase stimulus directed-attention and to promote metabolic changes required to provide energy for appropriate “fight or flight” response.
The evidence indicates that CRH is one of the main humoral mediators of waking and intracerebroventricularly (i.c.v.) infusion reduces slow wave sleep in rats, and induces a typical waking EEG pattern (Ehlers et al., 1986). Even in REMS-deprived rats that display greater homeostatic pressure to engage REMS rebound, CRH administration impairs this compensatory phenomenon (Machado et al., 2010). Blockade of CRH receptors, on the contrary, induces NREMS, an effect that is likely mediated, at least in part, by interleukin-1 (IL-1), because administration of CRH antagonist increases mRNA for IL-1α and IL-1β (Chang and Opp, 1998). An additional evidence of the waking-promoting effect of this neuropeptide comes from a study with Lewis rats, which are deficient in CRH production and that display less waking time and more NREMS during the active phase than the stress hyperresponsive Fisher rats (Opp, 1997). ACTH, in turn, increases waking by reducing REMS both during the light and dark periods of the circadian rhythm (Chastrette et al., 1990).
CORT influence on sleep is more of a regulatory fashion. On the one hand, adrenalectomy, i.e., removal of the adrenal gland and hence of the CORT source, results in shorter sleep episodes, less NREMS and more waking during the resting phase in the rat, with an opposite pattern during the active phase. Replacement with basal levels of CORT (approximately 5 μg/dl), restores normal sleep pattern, whereas higher doses (resulting in twice as much plasma levels) inhibits NREMS (Bradbury et al., 1998) and increases waking time (Vazquez-Palacios et al., 2001). Addison’s Disease patients (who are characterized by adrenal insufficiency) also display impairment of sleep continuity, increased REMS latency and reduced percentage of REMS, problems that can be normalized by hydrocortisone replacement at bed time (Garcia-Borreguero et al., 2000). Conversely, Cushing’s syndrome patients (who present very high levels of plasma cortisol) exhibit sleep fragmentation, reduced REMS latency and augmented REM density in the first episode (Shipley et al., 1992a,b). Additional evidence is given by a study with control and REM sleep-deprived rats submitted to different regimens of stress; the group that exhibit intermediate plasma levels of CORT was the same that displayed the greater REMS rebound, whereas the groups that secreted the lowest or the highest levels also showed the smallest rebound (Machado et al., 2008). Therefore, it appears as though a narrow range of GC concentrations is necessary to maintain normal sleep pattern. This effect is likely mediated by the balance of MR and GR occupation that results from the distinct affinity of these receptors for CORT. Maximum performance on a number of function, such as memory, sleep, activity of the immune system, is obtained by GC concentrations capable to saturate MR and occupy approximately 50% of GR; very low or very high GC concentrations impair the performance of these functions, in an inverted U-shape (de Kloet et al., 1991, 1999; De Kloet et al., 1998; Marinesco et al., 1999; Lupien et al., 2007; Machado et al., 2008).
Possible mechanistic explanation for the suppressive effects of GC on sleep involves the modulatory effect of these hormones on GABAA receptors located in the thalamus. Muscimol binding to GABAA receptors is greatly diminished by adrenalectomy and can be restored by corticosterone (that binds both MR and GR), but not by dexamethasone (that binds only GR). Therefore, absence of GC or exclusive stimulation of GR reduces muscimol affinity for GABAA receptors (Majewska et al., 1985). GABAergic inhibition of the thalamocortical pathway, especially in the reticular nucleus of the thalamus, is one of the main factors of NREM generation resulting in electroencephalogram synchronized activity, including spikes and slow wave activity during NREM sleep (Steriade et al., 1993; von Krosigk et al., 1993; McCormick and Bal, 1997; Huntsman et al., 1999). Therefore, impairment of GABAergic action (by high levels of GC, for instance) could hinder the generation and maintenance of NREMS.
Peculiarities of Stress-Induced Sleep Changes
The Length of the Stressor
Initial studies on the relationship between stress and sleep were prompted by the fact that REMS deprivation induced by the flower pot method, while being stressful and causing activation the HPA axis, induces REMS rebound. Moreover, ACTH derivatives – desacetyl-α-MSH and corticotropin-like intermediate lobe peptide (CLIP) induce, respectively, NREMS and REMS rebound after exogenous administration (Chastrette et al., 1990). The authors then questioned whether other types of stressful situations would also lead to sleep rebound and demonstrated that indeed, exposure to a 2-h period of immobilization stressor before the active phase increases REMS time (Rampin et al., 1991). There is a clear time course for this phenomenon to be manifested and for a few hours after the stressor exposure, sleep is inhibited by the obvious mechanisms discussed above (activation of the HPA axis and of LC/adrenal medulla). This inhibition appears to be quite independent of the length of the stressor (Marinesco et al., 1999; Tiba et al., 2004), suggesting a lack of habituation. Following this initial inhibitory period, there is an increase in the time spent sleeping, with the highest levels being reached during the dark part of the light–dark cycle, possibly due to the lowest expression of baseline sleep during this period (Koehl et al., 2002; Tiba et al., 2003, 2004, 2008; Dewasmes et al., 2004; Machado et al., 2008).
The length of stressor exposure is also an important variable that determines the magnitude of sleep rebound, inasmuch as 1 h of immobilization stress leads to a large increase in REMS (63% above baseline sleep) and a moderate increase of NREMS (16% above baseline; Cespuglio et al., 1995), whereas 2 h of immobilization results in 32% increase in REMS and no change in NREMS (Rampin et al., 1991). By extending the period of stressor exposure to 4 h, the expression of sleep rebound is prevented, and the increase in REMS or in NREMS is no longer observed (Marinesco et al., 1999). One possible mediator of this wake-promoting effect of stress seems to be CORT, given the progressive increase of plasma levels of this hormone from 1 to 4 h of stressor exposure. These findings also indicate that inhibition of NREMS by stress is a linear inverse function (the more intense the stressor, reflected by CORT levels, the less NREMS rebound is obtained), whereas stress modulation of REMS follows an inverted U-shape curve, with very low or very high levels producing a smaller expression of REMS rebound, and intermediate levels inducing a high expression (Figure 2). Interestingly, in adrenalectomized rats, i.e., removal of adrenal glands, and consequently, of the source of corticosterone production, REMS rebound is impaired. Administration of dexamethasone, a synthetic glucocorticoid that binds GR, also inhibits immobilization-induced REMS rebound (Marinesco et al., 1999), evidencing once again, the inverted U-shape curve effect of stress on sleep.
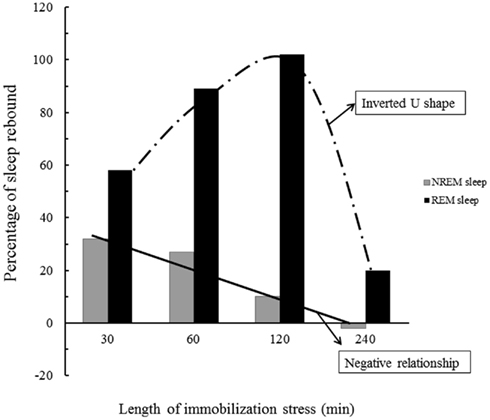
Figure 2. Schematic representation the effects of stress on NREMS and REMS rebound. The longer the stress period the higher the corticosterone (CORT) secretion and as a consequence, there is a gradual inhibition of NREMS rebound. REMS, however, is regulated by optimum levels of CORT, so either very low or very high levels (induced by very short or very long periods of stress) result in meager rebound. Data is adapted from Marinesco et al. (1999).
The Nature of the Stressor
It would, however, be naive to believe that all kinds of stressors would lead to similar outcomes. Different stressors recruit distinct brain areas and neurotransmitter systems, as has been shown by Pácak and Palkovits (2001) by c-fos activation. Thus, the comparative study of sleep changes induced by 1 h of physical (cold stress), psychological (restraint stress), or mixed (2 mA inescapable footshocks) stimuli showed that these stressors result in increased NREMS, REMS, and waking times, respectively (Palma et al., 2000). For instance, several studies show that footshock, a fairly intense stressor, retards the onset of REMS and prolongs the wake time following its application, sometimes even preventing the expression of sleep rebound during the night time (Palma et al., 2000; Vazquez-Palacios and Velazquez-Moctezuma, 2000; Tang et al., 2007). However, previous manual restraint of the rat, a routine laboratory procedure, prevents the impairment of REMS rebound in the light period following the stressor (Tang et al., 2007).
There are specific aspects regarding footshock that determines the sleep outcome. Rodents can learn to associate a neutral stimulus (sound, light, context) with an aversive stimulus, such a footshock. After this association, the light, the sound, or the context are no longer neutral and the animal reacts to them, almost as if it were exposed to the footshock. Single or multiple sound-shock pairings lead to shorter periods of NREMS and REMS although these reductions are more evident on the day following multiple pairings. Even 27 days after the training, presentation of tone results in reduction of REMS, indicating that the strength of the pairing has a long-lasting effect on sleep (Sanford et al., 2003a).
Recently Sanford’s group showed that by giving mice the opportunity to control the footshock in a paradigm of fear conditioning the sleep pattern is modified. In this study mice were trained to avoid a shock by moving to a safe compartment of a shuttle-box (escapable shock – ES); when they did so, their action also terminated the shock of the yoked group submitted to inescapable shock (IS). Sleep recordings were performed after two training sessions and after context re-exposure (without delivery of footshock) and in all three occasions, ES mice exhibited REMS rebound, whereas IS mice displayed less REMS. Interestingly, both groups displayed similar amount of freezing behavior and, presumably, learning, when exposed to the aversive context (Sanford et al., 2010), indicating that REMS rebound is a likely consequence of predictable, controlled stressful situations. The group also demonstrated that these shock regimens result in differential activation (production of Fos) of selected brain regions involved with stress response and wake regulation. Both ES and IS induce Fos in the PVN (indicating augmented stress response), but the magnitude is greater in IS than ES mice. Interestingly, Fos immunoreactivity is greater in the LC, dorsal raphe nucleus (DRN), laterodorsal tegmental nucleus (LDT) and amygdala of IS than ES and control mice (Liu et al., 2009a), indicating that, to a certain extent, stress controllability lessens the activation of structures that are involved with stress and arousal.
In rats, fear conditioning has also been employed to study the neurobiology of the relationship between associative fear learning and sleep (Jha et al., 2005; Pawlyk et al., 2005, 2008; Tang et al., 2005b; Wellman et al., 2008; Liu et al., 2009a). This paradigm has proven particularly useful as a model of PTSD-induced sleep impairment because, not only the footshock can induce sleep changes, but also reminders of the aversive situation. With this idea in mind, Jha et al. (2005) evaluated the sleep pattern of rats trained in tone-shock fear conditioning and reported that rats re-exposed only to the tones display a clear reduction of REMS percentage compared to their respective baseline sleep. Similar results are observed when rats are re-exposed to the aversive context, e.g., they display less REMS during the light period. However, if re-exposure to the aversive context (by virtue of pairing it to the footshocks) is sufficiently long to allow extinction of the conditioned fear, the time spent in REMS is restored to baseline levels (Wellman et al., 2008). In addition, when rats sleep in an aversive environment, they take longer to initiate NREMS and REMS and exhibit reduced percentage of REMS; however, when allowed to sleep in a neutral environment, these rats exhibit rebound of NREMS and REMS indicating that discrimination between neutral and aversive contexts influences sleep (Pawlyk et al., 2005).
In human beings, the most potent stressors are those of a social nature, including social hierarchy, family and/or work-related problems, inter-personal relationships. One example of social stressor in rodents is the social defeat paradigm, in which an intruder is attacked almost immediately upon being introduced in the resident’s cage (Koolhaas et al., 1997). Physiological changes seen in the defeated animal include increased catecholamine, corticosterone, prolactin and testosterone levels, increased heart rate, blood pressure, and body temperature (Bohus et al., 1987). The consequences of social defeat on sleep include increased slow wave activity (SWA) during NREMS, reflecting the augmented sleep intensity. Based on this result, it has been proposed that the function of SWA is to restore the inner balance following a traumatic event (Meerlo et al., 1997, 2001a; Kinn et al., 2008). Predation also results in changes in macro- and microstructure of sleep with an initial reduction of SWA, followed by an increase approximately 90 min after the stimulus. Regarding REMS the reduction takes place for the first 3 h that follows the encounter and increases thereafter reaching statistical significance in the last 3 h of sleep recording – from 15:00 to 18:00 h (Lesku et al., 2008). Predictably, under the stressful situation of being chased or threatened of being chased by a predator, REMS is reduced because this phase of sleep is characterized by elevated arousal threshold, when compared to NREM sleep (Dillon and Webb, 1965; Van Twyver and Garrett, 1972; Piallat and Gottesmann, 1995; Lima et al., 2005) and loss of muscle tone obligating the animal to lay down (Tobler, 1992), likely increasing vulnerability to predation. In this respect the same authors also reported that mammals living in riskier areas or those at the lower end of the food chain spend less time in REMS than carnivores living in secure areas (Lesku et al., 2006). It is worth mentioning that in human beings the waking threshold is similar between tonic REM sleep stage 2 NREM sleep; however the highest behavioral arousal threshold occurs during phasic REM sleep and stage 4 sleep (Price and Kremen, 1980; Ermis et al., 2010). In rats, tonic and phasic components of REM sleep are present, although not as clearly as in humans (Sanchez-Lopez and Escudero, 2011).
Individual Characteristics Influence the Response to Stress
The modulatory role that genetic background and previous stress history exert on the sleep responses to stress is quite remarkable. Such characteristics are so determinant that even after REMS deprivation that always results in a compensatory phenomenon, REMS rebound is greater in young volunteers characterized by being more extroverted, optimistic, and having more friends, than in those with opposite personality features (Nakazawa et al., 1975). Even in young children, poor sleep quality seems to be related to negative emotionality and higher cortisol levels (Scher et al., 2010). Therefore, it is only natural to suspect that the sleep response to stress also depends on how the subject faces and interprets stressful stimuli. In this section we will describe the studies that explored the environmental and genetic influences on stress-induced sleep rebound. The gene-environment diathesis hypothesis has permeated numerous studies on the impact of stress on behavior, both in humans and in animal models. According to this hypothesis, the resulting resilience or vulnerability to stress-induced disorders are the consequence of the interplay between the genetic background and the environmental influence during specific time-windows, including infancy and adolescence. Adversity during these periods may result in augmented vulnerability, whereas social support throughout life leads to resilience (Plotsky et al., 1998).
Genetic Influences
The genetic background is one of the most important determining variables of behavioral expression, so much so that the same stimulus may result in different outcomes, depending on how the animal reacts to it. For instance, Fisher rats, in addition to being stress hyperresponsive are also more anxious in the open-field test than Wistar and Sprague-Dawley rats. This strain also exhibits less baseline NREMS and REMS and in response to a 30-min exposure to an open-field, Fisher rats exhibit increased REMS in the light period, whereas this rebound occurs in Wistar rats in the dark period (Tang et al., 2005a). In the training of contextual fear conditioning, Fisher rats are also more reactive than Wistar, presenting greater fear response to both training (in the presence of footshock) and re-exposure to the context (in the absence of the noxious stimulus), but no impairment in REMS rebound (Tang et al., 2005b). Although surprising, these results may be explained by the fact that Fisher rats secrete more corticosterone and prolactin in response to stressful stimuli (Sarrieau et al., 1998), and stress-induced prolactin secretion is closely related to REMS (Bodosi et al., 2000). This possibility will be discussed in greater detail in the section devoted to the mechanisms of stress-induced sleep rebound.
In mice, the level of anxiety-like behavior and reactivity to stress predicts the sleep outcome in response to exposure to noxious and non-noxious stimuli. Shock delivery in fear conditioning protocols produces a more intense reduction of REMS in the “anxious” BALB/cJ (BALB) mice, compared to the less “anxious” C57BL/6J (C57). When exposed to either sound or context that had been paired to the shock, the latter shows REMS rebound, whereas BALB mice do not (Sanford et al., 2003b,c). A similar pattern of sleep reaction follows non-noxious manipulations, such as exposure to an open-field (Tang et al., 2004) or restraint stress (Meerlo et al., 2001b): C57BL/6J mice display the characteristic REMS rebound, whereas BALB/cJ strain displays sleep impairment. These data are in line with human studies that indicate that anxious individuals have difficulty to conciliate sleep after a stressful day or highly emotional situation (Morin et al., 2003; Drake et al., 2004).
The Wistar-Kyoto rat strain, originated from Wistar rats, was initially used as a normotensive control for the spontaneously hypertensive strain. Later, it was shown that these animals also exhibited physiological, behavioral, and hormonal alterations compatible with symptoms of depression (Will et al., 2003), including stress hyperresponsiveness (Redei et al., 1994), increased stress-induced ulcer formation and shortened latency to acquire an immobility posture in the forced swimming test (Pare and Redei, 1993). Regarding the sleep architecture, these rats show sleep pattern that emulate those typically displayed by depressive patients, e.g., reduced latency to REMS, increased REMS percentage and increased sleep fragmentation (Dugovic et al., 2000). When re-exposed to a tone that was paired with footshocks, Wistar-Kyoto rats exhibit more freezing behavior and bouts of sequential REMS (less than 3-min inter-REM intervals) than Wistar rats (Dasilva et al., 2011), whereas Sprague-Dawley rats exhibit increased single REMS bouts (more than 3-min inter-REM interval; Madan et al., 2008). This means that in Wistar-Kyoto rats REMS rebound induced by stressful situations is fragmented, whilst that of Sprague-Dawley rats is consolidated. Such a pattern of REMS has been attributed to protect automobile accident victims from developing PTSD (Mellman et al., 2002).
Early Life Stress
Epidemiological studies show that adversity during infancy and/or adolescence represent a risk factor for psychiatric disorders in humans, including anxiety, depression, and schizophrenia (Agid et al., 1999). In rats, disruptions of mother–infant relationship, a form of early life stress, also precipitate psychopathologic-like behaviors in adulthood, such as increased preference for ethanol intake (Huot et al., 2001), increased novelty-induced fear (Caldji et al., 2000), increased anxiety-like behavior (Faturi et al., 2010; Barbosa Neto et al., 2012) and hyper-responsiveness of the HPA axis to stress (Plotsky and Meaney, 1993). Given the waking-promoting effect of CRH (Chang and Opp, 2001) and CORT (Vazquez-Palacios et al., 2001) and the fact that anxious individuals are at greater risk of developing chronic insomnia after a stressful situation (Waters et al., 1993; Morin et al., 2003; Larsson et al., 2008), we hypothesized that early life stress would impair the REMS rebound induced by acute stress. The paradigm used to disrupt mother–infant relationship was long maternal separation that consists of removing the litter from the home-cage for 3 h/day, for the first 2 weeks of life. To our surprise, we found that male rats submitted to long maternal separation during infancy displayed more baseline REMS in the light period than control rats, without changes in the amount of NREMS or in REMS latency (Tiba et al., 2004). The female offspring of both groups exhibited stress-induced sleep rebound in the dark period, but females submitted to maternal separation exhibited the largest REMS rebound (Tiba et al., 2008). A possible explanation for these findings is the fact that Wistar mothers are very attentive to their pups’ needs and compensate the offspring demands upon reunion (Pryce et al., 2001; Tiba et al., 2004).
Mechanisms and Mediators of Stress-Induced Sleep Changes
The first hypothesis put forward to explain the mechanisms of how acute stressors induce sleep rebound involves the serotonergic system, based on a classical study showing that blockade of serotonin synthesis or lesions of the Raphe nuclei induce insomnia (Jouvet and Renault, 1966). Indeed several subsequent studies confirmed that dysregulation of the serotonergic system hinders the capacity of the animal to cope with stress, e.g., impairment of REMS rebound, as reported for 5-HT1A (Boutrel et al., 2002; Popa et al., 2006) and 5-HT1B knockout mice (Boutrel et al., 1999) and rats treated with para-chlorophenylalanine, a serotonin synthesis inhibitor (Sinha, 2006). A recent study shows that in wild-type mice, the short-term effects of restraint stress (up to 2 h) encompass inhibition of REMS, increased release of serotonin in the hypothalamus and increased pre-pro-orexin immunoreactivity in the lateral hypothalamic area. Following this initial REMS inhibition (subsequent 8 h), there is a major REMS rebound in wild-type, but not in mutant mice lacking the serotonin transporter that fails to engage in the compensatory phenomenon (Rachalski et al., 2009).
Classical studies show that serotonin is differentially released, depending on the phase of the light–dark cycle so that during waking, serotonin is released from the axon, whereas during sleep, it is released from the dendrites (Cespuglio et al., 1992). The axonal release prepares the brain to sleep, due to increased and cumulative synthesis of hypnogenic substances in the arcuate nucleus and pre-optic area (Jouvet, 1999), initiating a cascade of genomic events (Garcia de Yebenes et al., 1994; Churruca et al., 2008). In the arcuate nucleus, serotonin induces the synthesis of corticotropin-like intermediate lobe peptide (CLIP or ACTH18–39), a pro-opiomelanocortin (POMC) derivative (Cespuglio et al., 1992), claimed to be a major mediator of stress-induced sleep rebound, due to its well-known REMS inducing properties (Wetzel et al., 1994, 1997, 2003; Cespuglio et al., 1995; Bonnet et al., 1997). Thus, increased serotonin transmission induces POMC mRNA in the arcuate nucleus, which, in turn, is cleaved originating several derivatives, including ACTH, α-melanocyte stimulating hormone (α-MSH) and CLIP (Figure 3; Cespuglio et al., 1995; Bonnet et al., 1997). Although ACTH is a well-known waking promoter, α-MSH and CLIP induce sleep (Wetzel et al., 1994). CLIP content and its phosphorylated form (ph-CLIP) increase in the dorsal Raphe nucleus immediately after immobilization stress, whereas such an increase in the arcuate nucleus takes place 4 h after the end of the stress, when REMS rebound reaches its maximum (Bonnet et al., 1997). Importantly, in adrenalectomized rats, 1 h of immobilization stress leads to a delayed and smaller REMS rebound than in intact rats and, in both cases, the sleep rebound is preceded by increased production of ph-CLIP in the dorsal Raphe nucleus (Bonnet et al., 2000). These data indicate that corticosterone seems to be important for the full expression of REMS rebound following stress. In fact, the expected increase of ph-CLIP in the arcuate nucleus is absent in adrenalectomized rats, which might explain the smaller REMS rebound seen under this condition (Bonnet et al., 2000). Finally, exogenous administration of CLIP or its N-terminal fragments ACTH20–24 and ACTH18–24 induces a significant increase of REMS, by increasing episode length (Wetzel et al., 1994, 1997).
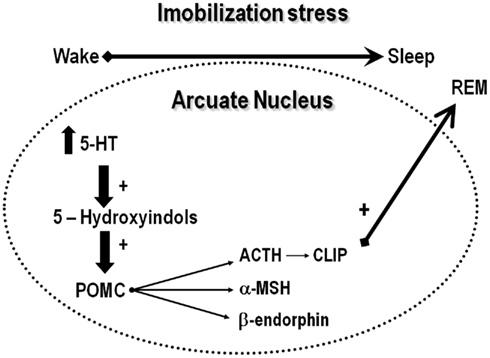
Figure 3. The serotonergic hypothesis of acute stress-induced sleep rebound. According to this hypothesis, stress induces the release of serotonin, which is metabolized to 5-hydroxyindole substances that stimulates the cleavage of pro-opiomelanocortin, originating adrenocorticotropic hormone (ACTH), alpha-melanocyte stimulating hormone (α-MSH) and β-endorphin. ACTH is metabolized originating corticotropin-like intermediate lobe peptide (CLIP), which is a major inducer of REMS.
Interestingly, serotonin is a major stimulator of PRL secretion (Balsa et al., 1998); PRL, in turn, increases the activity of cholinergic neurons in the mesopontine tegmental area (Takahashi et al., 2000), involved in the induction of REMS. Numerous studies indicate that PRL is involved with induction of REMS. For instance, in the absence of stressful stimuli, hyperprolactinemic rats spend more time in REMS during the light period (Obal et al., 1997), whereas administration of PRL anti-serum reduces REMS, without effecting NREMS (Obal et al., 1992). Subcutaneous (s.c.), i.c.v. administration (Roky et al., 1993) or intra-dorsolateral hypothalamus infusion (Roky et al., 1994) of PRL during the dark phase of the light–dark cycle inhibits REMS, whereas the opposite is seen when s.c. administration takes place during the light phase. Moreover, PRL deficient mice and rats display less REMS than normal animals, which is reversed by PRL replacement (Obal et al., 2005).
Prolactin seems to be a protective factor against harmful effects of stress, as indicated by a recent study showing that it protects hippocampal neurons from chronic stress-induced neuronal death, facilitating neurogenesis in the dentate gyrus (Torner et al., 2009). The stress-protective feature could also be, at least partly, attributed to its REMS regulatory role (Roky et al., 1995), since total sleep and REMS deprivation induced by various methods impairs hippocampal neurogenesis (Guzman-Marin et al., 2008; Meerlo et al., 2009; Sportiche et al., 2010), with controversial findings on the participation of corticosterone on this effect (Mirescu et al., 2006; Mueller et al., 2008). PRL involvement in stress-induced REMS rebound is attested by several findings, such that both ether and restraint stress induce PRL secretions, which is followed by increased REMS (Bodosi et al., 2000; Meerlo et al., 2001b). Moreover, the comparative hormonal response and REMS rebound of C57 and BALB mice to restraint stress strengthens PRL regulatory role on stress-induced REMS, because in response to restraint stress both mice strains secrete similar levels of corticosterone; however, only C57 mice exhibit augmented PRL secretion and REMS rebound (Meerlo et al., 2001b). A recent evidence of the importance of PRL on REMS induction after stress stems from a study in which REMS-deprived rats, exposed to a high intensity stressor, displayed very long REMS episodes during the recovery period, concomitant to high plasma levels of PRL, in the presence of intermediate corticosterone levels (Machado et al., 2008). The precise mechanism for such an effect still needs full clarification, but PRL infusion in the DRN results in delayed and long REMS sleep episodes, of about 5–6 min (unpublished data). A proposed mechanism put forward by our group to explain this result involves a probable stimulant effect of PRL on 5-HT release from the DRN, which in turn, leads to activation of other REMS inducing factors, such as hypothalamic hypnogenic peptides (Figure 4).
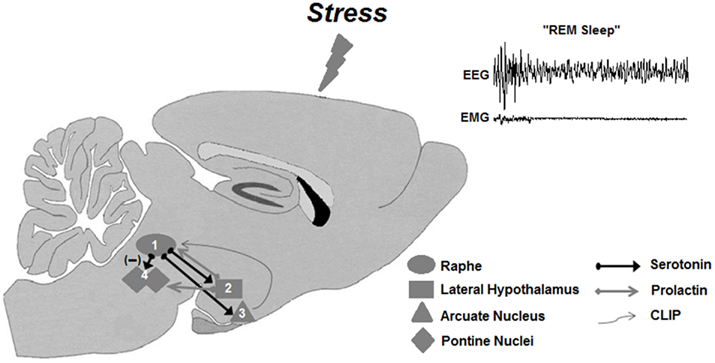
Figure 4. Working model of the prolactinergic hypothesis of stress-induced REMS rebound. Stressors (including REMS deprivation) increase serotonin release from the Raphe nuclei (1), which, in turn, could stimulate the release of PRL from the lateral hypothalamic area – LHA (2). Descending PRL projections from the LHA to the Raphe nuclei would close the self-stimulatory circuit, producing a further release of serotonin. In parallel, serotonergic projection to the arcuate nucleus – Arc (3) stimulates POMC cleavage resulting in production and secretion of CLIP in this region. Descending projections from the Arc to the Raphe nuclei would lead to accumulation of CLIP in this area, inhibiting serotonin secretion, thus releasing the cholinergic activity in pontine nuclei (4), leading to REMS. PRL would also produce excitation of pontine cholinergic neurons, contributing to the expression of REMS rebound.
Although a major role for the amygdala on regulation of REMS and stress-induced sleep has also been proposed, this does not appear to be a locus for PRL-induced REMS, since infusion in the central nucleus of the amygdala (CNA) reduces NREMS and increases waking (Sanford et al., 1998). Trains of electrical pulses in the CNA inhibit the activity of LC (Bouret et al., 2003), probably by means of CRH projections that synapse on the peri-LC (Van Bockstaele et al., 1998). The peri-LC contains GABAergic neurons that inhibit the LC and, as a consequence of their stimulation, result in the onset of REMS (Luppi et al., 2006). The involvement of this circuit in stress-induced REMS was confirmed in a series of studies employing infusion of GABAergic agonist, muscimol, or antagonist, bicuculline, into the CNA. Muscimol temporarily inactivates the CNA, leading to reduction of REMS, whereas bicuculline increases this phase of sleep (Sanford et al., 2002). In response to IS, bicuculline reverses the expected suppression of REMS, whereas muscimol maintains this impairment. In agreement with the behavioral outcomes, Fos staining in LC is increased after IS and partially reversed by infusion of bicuculline in rats exposed to IS. Moreover, a negative correlation between Fos in LC and REMS was observed (Liu et al., 2009b).
Final Remarks
Stress-induced sleep rebound is an adaptive strategy that represents a major evolutionary acquisition, and inability to engage in it may have disastrous consequences. A good example of the importance of REMS rebound after a stressful situation is attested by the sleep assessment of individuals who experienced a traumatic event; those who exhibited long episodes of REMS did not develop PTSD, whereas those who had several, but very short, episodes of REMS developed the disorder (Mellman et al., 2002). These findings suggest that long consolidated REMS episodes are essential for elaboration and integration of traumatic memories into the auto-biography of the patients (Stickgold, 2007), so they can resume their daily life.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by Associação Fundo de Incentivo à Pesquisa (AFIP) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP – CEPID, grant # 98/14303-3). Deborah Suchecki is the recipient of a research fellowship from the National Research Council (CNPq). Ricardo Borges Machado is the recipient of a post-doctoral fellowship from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 2010/09087-3).
References
Agid, O., Shapira, B., Zislin, J., Ritsner, M., Hanin, B., Murad, H., Troudart, T., Bloch, M., Heresco-Levy, U., and Lerer, B. (1999). Environment and vulnerability to major psychiatric illness: a case control study of early parental loss in major depression, bipolar disorder and schizophrenia. Mol. Psychiatry 4, 163–172.
Armitage, R. (2007). Sleep and circadian rhythms in mood disorders. Acta. Psychiatr. Scand. Suppl. 115, 104–115.
Armon, G., Shirom, A., Shapira, I., and Melamed, S. (2008). On the nature of burnout-insomnia relationships: a prospective study of employed adults. J. Psychosom. Res. 65, 5–12.
Balsa, J. A., Sanchez-Franco, F., Pazos, F., Lara, J. I., Lorenzo, M. J., Maldonado, G., and Cacicedo, L. (1998). Direct action of serotonin on prolactin, growth hormone, corticotropin and luteinizing hormone release in cocultures of anterior and posterior pituitary lobes: autocrine and/or paracrine action of vasoactive intestinal peptide. Neuroendocrinology 68, 326–333.
Barbosa Neto, J. B., Tiba, P. A., Faturi, C. B., de Castro-Neto, E. F., da Graca Naffah-Mazacoratti, M., de Jesus Mari, J., de Mello, M. F., and Suchecki, D. (2012). Stress during development alters anxiety-like behavior and hippocampal neurotransmission in male and female rats. Neuropharmacology 62, 518–526.
Beary, M. D., Lacey, J. H., Crutchfield, M. B., and Bhat, A. V. (1984). Psycho-social stress, insomnia and temazepam: a sleep laboratory evaluation in a “general practice” sample. Psychopharmacology (Berl.) 83, 17–19.
Bodosi, B., Obal, F. Jr., Gardi, J., Komlodi, J., Fang, J., and Krueger, J. M. (2000). An ether stressor increases REM sleep in rats: possible role of prolactin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1590–R1598.
Bohus, B., Benus, R. F., Fokkema, D. S., Koolhaas, J. M., Nyakas, C., van Oortmerssen, G. A., Prins, A. J., de Ruiter, A. J., Scheurink, A. J., and Steffens, A. B. (1987). Neuroendocrine states and behavioral and physiological stress responses. Prog. Brain Res. 72, 57–70.
Bonnet, C., Leger, L., Baubet, V., Debilly, G., and Cespuglio, R. (1997). Influence of a 1 h immobilization stress on sleep states and corticotropin-like intermediate lobe peptide (CLIP or ACTH18-39, Ph-ACTH18-39) brain contents in the rat. Brain Res. 751, 54–63.
Bonnet, C., Marinesco, S., Debilly, G., Kovalzon, V., and Cespuglio, R. (2000). Influence of a 1-h immobilization stress on sleep and CLIP (ACTH(18-39)) brain contents in adrenalectomized rats. Brain Res. 853, 323–329.
Bouret, S., Duvel, A., Onat, S., and Sara, S. J. (2003). Phasic activation of locus ceruleus neurons by the central nucleus of the amygdala. J. Neurosci. 23, 3491–3497.
Boutrel, B., Franc, B., Hen, R., Hamon, M., and Adrien, J. (1999). Key role of 5-HT1B receptors in the regulation of paradoxical sleep as evidenced in 5-HT1B knock-out mice. J. Neurosci. 19, 3204–3212.
Boutrel, B., Monaca, C., Hen, R., Hamon, M., and Adrien, J. (2002). Involvement of 5-HT1A receptors in homeostatic and stress-induced adaptive regulations of paradoxical sleep: studies in 5-HT1A knock-out mice. J. Neurosci. 22, 4686–4692.
Bradbury, M. J., Dement, W. C., and Edgar, D. M. (1998). Effects of adrenalectomy and subsequent corticosterone replacement on rat sleep state and EEG power spectra. Am. J. Physiol. 275, R555–R565.
Breslau, N., Roth, T., Burduvali, E., Kapke, A., Schultz, L., and Roehrs, T. (2004). Sleep in lifetime posttraumatic stress disorder: a community-based polysomnographic study. Arch. Gen. Psychiatry 61, 508–516.
Breslau, N., Roth, T., Rosenthal, L., and Andreski, P. (1996). Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol. Psychiatry 39, 411–418.
Caldji, C., Francis, D., Sharma, S., Plotsky, P. M., and Meaney, M. J. (2000). The effects of early rearing environment on the development of GABAA and central benzodiazepine receptor levels and novelty-induced fearfulness in the rat. Neuropsychopharmacology 22, 219–229.
Cape, E. G., and Jones, B. E. (1998). Differential modulation of high-frequency gamma-electroencephalogram activity and sleep-wake state by noradrenaline and serotonin microinjections into the region of cholinergic basalis neurons. J. Neurosci. 18, 2653–2666.
Carneiro, G., Togeiro, S. M., Hayashi, L. F., Ribeiro-Filho, F. F., Ribeiro, A. B., Tufik, S., and Zanella, M. T. (2008). Effect of continuous positive airway pressure therapy on hypothalamic-pituitary-adrenal axis function and 24-h blood pressure profile in obese men with obstructive sleep apnea syndrome. Am. J. Physiol. Endocrinol. Metab. 295, E380–E384.
Cartwright, R. D., and Wood, E. (1991). Adjustment disorders of sleep: the sleep effects of a major stressful event and its resolution. Psychiatry Res. 39, 199–209.
Cespuglio, R., Houdouin, F., Oulerich, M., El Mansari, M., and Jouvet, M. (1992). Axonal and somato-dendritic modalities of serotonin release: their involvement in sleep preparation, triggering and maintenance. J. Sleep Res. 1, 150–156.
Cespuglio, R., Marinesco, S., Baubet, V., Bonnet, C., and el Kafi, B. (1995). Evidence for a sleep-promoting influence of stress. Adv. Neuroimmunol. 5, 145–154.
Chang, F. C., and Opp, M. R. (1998). Blockade of corticotropin-releasing hormone receptors reduces spontaneous waking in the rat. Am. J. Physiol. 275, R793–R802.
Chang, F. C., and Opp, M. R. (2001). Corticotropin-releasing hormone (CRH) as a regulator of waking. Neurosci. Biobehav. Rev. 25, 445–453.
Chang, P. P., Ford, D. E., Mead, L. A., Cooper-Patrick, L., and Klag, M. J. (1997). Insomnia in young men and subsequent depression. The Johns Hopkins Precursors Study. Am. J. Epidemiol. 146, 105–114.
Charuvastra, A., and Cloitre, M. (2009). Safe enough to sleep: sleep disruptions associated with trauma, posttraumatic stress, and anxiety in children and adolescents. Child Adolesc. Psychiatr. Clin. N. Am. 18, 877–891.
Chastrette, N., Cespuglio, R., and Jouvet, M. (1990). Proopiomelanocortin (POMC)-derived peptides and sleep in the rat. Part 1 – hypnogenic properties of ACTH derivatives. Neuropeptides 15, 61–74.
Churruca, I., Portillo, M. P., Casis, L., Gutierrez, A., Macarulla, M. T., and Echevarria, E. (2008). Effects of fluoxetine administration on hypothalamic melanocortin system in obese Zucker rats. Neuropeptides 42, 293–299.
Dasilva, J. K., Lei, Y., Madan, V., Mann, G. L., Ross, R. J., Tejani-Butt, S., and Morrison, A. R. (2011). Fear conditioning fragments REM sleep in stress-sensitive Wistar-Kyoto, but not Wistar, rats. Prog. Neuropsychopharmacol. Biol. Psychiatry. 35, 67–73.
de Kloet, E. R., Joels, M., Oitzl, M., and Sutanto, W. (1991). Implication of brain corticosteroid receptor diversity for the adaptation syndrome concept. Methods Achiev. Exp. Pathol. 14, 104–132.
de Kloet, E. R., Oitzl, M. S., and Joels, M. (1999). Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 22, 422–426.
De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S., and Joels, M. (1998). Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 19, 269–301.
Dewasmes, G., Loos, N., Delanaud, S., Dewasmes, D., and Ramadan, W. (2004). Pattern of rapid-eye movement sleep episode occurrence after an immobilization stress in the rat. Neurosci. Lett. 355, 17–20.
Dillon, R. F., and Webb, W. B. (1965). Threshold of arousal from “activated” sleep in the rat. J. Comp. Physiol. Psychol. 59, 446–447.
Drake, C., Richardson, G., Roehrs, T., Scofield, H., and Roth, T. (2004). Vulnerability to stress-related sleep disturbance and hyperarousal. Sleep 27, 285–291.
Dugovic, C., Solberg, L. C., Redei, E., Van Reeth, O., and Turek, F. W. (2000). Sleep in the Wistar-Kyoto rat, a putative genetic animal model for depression. Neuroreport 11, 627–631.
Ehlers, C. L., Reed, T. K., and Henriksen, S. J. (1986). Effects of corticotropin-releasing factor and growth hormone-releasing factor on sleep and activity in rats. Neuroendocrinology 42, 467–474.
Ermis, U., Krakow, K., and Voss, U. (2010). Arousal thresholds during human tonic and phasic REM sleep. J. Sleep Res. 19, 400–406.
Faturi, C. B., Tiba, P. A., Kawakami, S. E., Catallani, B., Kerstens, M., and Suchecki, D. (2010). Disruptions of the mother-infant relationship and stress-related behaviours: altered corticosterone secretion does not explain everything. Neurosci. Biobehav. Rev. 34, 821–834.
Gadinger, M. C., Fischer, J. E., Schneider, S., Fischer, G. C., Frank, G., and Kromm, W. (2009). Female executives are particularly prone to the sleep-disturbing effect of isolated high-strain jobs: a cross-sectional study in German-speaking executives. J. Sleep Res. 18, 229–237.
Garcia de Yebenes, E., Li, S., and Pelletier, G. (1994). Role of serotonin in the regulation of prolactin gene expression in the male rat as evaluated by in situ hybridization. Brain Res. Bull. 34, 437–440.
Garcia-Borreguero, D., Wehr, T. A., Larrosa, O., Granizo, J. J., Hardwick, D., Chrousos, G. P., and Friedman, T. C. (2000). Glucocorticoid replacement is permissive for rapid eye movement sleep and sleep consolidation in patients with adrenal insufficiency. J. Clin. Endocrinol. Metab. 85, 4201–4206.
Grossi, G., Perski, A., Evengard, B., Blomkvist, V., and Orth-Gomer, K. (2003). Physiological correlates of burnout among women. J. Psychosom. Res. 55, 309–316.
Gujar, N., McDonald, S. A., Nishida, M., and Walker, M. P. (2011). A role for rem sleep in recalibrating the sensitivity of the human brain to specific emotions. Cereb. Cortex 21, 115–123.
Gunnar, M., and Quevedo, K. (2007). The neurobiology of stress and development. Annu. Rev. Psychol. 58, 145–173.
Guzman-Marin, R., Suntsova, N., Bashir, T., Nienhuis, R., Szymusiak, R., and McGinty, D. (2008). Rapid eye movement sleep deprivation contributes to reduction of neurogenesis in the hippocampal dentate gyrus of the adult rat. Sleep 31, 167–175.
Habukawa, M., Uchimura, N., Maeda, M., Kotorii, N., and Maeda, H. (2007). Sleep findings in young adult patients with posttraumatic stress disorder. Biol. Psychiatry 62, 1179–1182.
Hajos, M., Hoffmann, W. E., Robinson, D. D., Yu, J. H., and Hajos-Korcsok, E. (2003). Norepinephrine but not serotonin reuptake inhibitors enhance theta and gamma activity of the septo-hippocampal system. Neuropsychopharmacology 28, 857–864.
Holsboer-Trachsler, E., and Seifritz, E. (2000). Sleep in depression and sleep deprivation: a brief conceptual review. World J. Biol. Psychiatry 1, 180–186.
Huntsman, M. M., Porcello, D. M., Homanics, G. E., DeLorey, T. M., and Huguenard, J. R. (1999). Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283, 541–543.
Huot, R. L., Thrivikraman, K. V., Meaney, M. J., and Plotsky, P. M. (2001). Development of adult ethanol preference and anxiety as a consequence of neonatal maternal separation in Long Evans rats and reversal with antidepressant treatment. Psychopharmacology (Berl.) 158, 366–373.
Jha, S. K., Brennan, F. X., Pawlyk, A. C., Ross, R. J., and Morrison, A. R. (2005). REM sleep: a sensitive index of fear conditioning in rats. Eur. J. Neurosci. 21, 1077–1080.
Jouvet, M., and Renault, J. (1966). Persistence of insomnia after lesions of the nuclei of the raphe in the cat. C. R. Seances Soc. Biol. Fil. 160, 1461–1465.
Kinn, A. M., Gronli, J., Fiske, E., Kuipers, S., Ursin, R., Murison, R., and Portas, C. M. (2008). A double exposure to social defeat induces sub-chronic effects on sleep and open field behaviour in rats. Physiol. Behav. 95, 553–561.
Klein, E., Koren, D., Arnon, I., and Lavie, P. (2002). No evidence of sleep disturbance in post-traumatic stress disorder: a polysomnographic study in injured victims of traffic accidents. Isr. J. Psychiatry Relat. Sci. 39, 3–10.
Kobayashi, I., Boarts, J. M., and Delahanty, D. L. (2007). Polysomnographically measured sleep abnormalities in PTSD: a meta-analytic review. Psychophysiology 44, 660–669.
Koehl, M., Bouyer, J. J., Darnaudery, M., Le Moal, M., and Mayo, W. (2002). The effect of restraint stress on paradoxical sleep is influenced by the circadian cycle. Brain Res. 937, 45–50.
Koolhaas, J. M., De Boer, S. F., De Rutter, A. J., Meerlo, P., and Sgoifo, A. (1997). Social stress in rats and mice. Acta Physiol. Scand. Suppl. 640, 69–72.
Larsson, M. R., Backstrom, M., and Johanson, A. (2008). The interaction between baseline trait anxiety and trauma exposure as predictor of post-trauma symptoms of anxiety and insomnia. Scand. J. Psychol. 49, 447–450.
Lesku, J. A., Roth, T. C. II, Amlaner, C. J., and Lima, S. L. (2006). A phylogenetic analysis of sleep architecture in mammals: the integration of anatomy, physiology, and ecology. Am. Nat. 168, 441–453.
Lesku, J. A., Roth, T. C., Rattenborg, N. C., Amlaner, C. J., and Lima, S. L. (2008). Phylogenetics and the correlates of mammalian sleep: a reappraisal. Sleep Med. Rev. 12, 229–244.
Lima, S. L., Rattenborg, N. C., Lesku, J. A., and Amlaner, C. J. (2005). Sleeping under the risk of predation. Anim. Behav. 70, 723–736.
Liu, X., Tang, X., and Sanford, L. D. (2009a). Stressor controllability and Fos expression in stress regulatory regions in mice. Physiol. Behav. 97, 321–326.
Liu, X., Yang, L., Wellman, L. L., Tang, X., and Sanford, L. D. (2009b). GABAergic antagonism of the central nucleus of the amygdala attenuates reductions in rapid eye movement sleep after inescapable footshock stress. Sleep 32, 888–896.
Lupien, S. J., Maheu, F., Tu, M., Fiocco, A., and Schramek, T. E. (2007). The effects of stress and stress hormones on human cognition: implications for the field of brain and cognition. Brain Cogn. 65, 209–237.
Luppi, P. H., Gervasoni, D., Verret, L., Goutagny, R., Peyron, C., Salvert, D., Leger, L., and Fort, P. (2006). Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. J. Physiol. Paris 100, 271–283.
Machado, R. B., Tufik, S., and Suchecki, D. (2008). Chronic stress during paradoxical sleep deprivation increases paradoxical sleep rebound: association with prolactin plasma levels and brain serotonin content. Psychoneuroendocrinology 33, 1211–1224.
Machado, R. B., Tufik, S., and Suchecki, D. (2010). Modulation of sleep homeostasis by corticotropin releasing hormone in rem sleep-deprived rats. Int. J. Endocrinol. 2010, 326151.
Madan, V., Brennan, F. X., Mann, G. L., Horbal, A. A., Dunn, G. A., Ross, R. J., and Morrison, A. R. (2008). Long-term effect of cued fear conditioning on REM sleep microarchitecture in rats. Sleep 31, 497–503.
Majewska, M. D., Bisserbe, J. C., and Eskay, R. L. (1985). Glucocorticoids are modulators of GABAA receptors in brain. Brain Res. 339, 178–182.
Marinesco, S., Bonnet, C., and Cespuglio, R. (1999). Influence of stress duration on the sleep rebound induced by immobilization in the rat: a possible role for corticosterone. Neuroscience 92, 921–933.
McCormick, D. A., and Bal, T. (1997). Sleep and arousal: thalamocortical mechanisms. Annu. Rev. Neurosci. 20, 185–215.
Meerlo, P., de Bruin, E. A., Strijkstra, A. M., and Daan, S. (2001a). A social conflict increases EEG slow-wave activity during subsequent sleep. Physiol. Behav. 73, 331–335.
Meerlo, P., Easton, A., Bergmann, B. M., and Turek, F. W. (2001b). Restraint increases prolactin and REM sleep in C57BL/6J mice but not in BALB/cJ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R846–R854.
Meerlo, P., Mistlberger, R. E., Jacobs, B. L., Heller, H. C., and McGinty, D. (2009). New neurons in the adult brain: the role of sleep and consequences of sleep loss. Sleep Med. Rev. 13, 187–194.
Meerlo, P., Pragt, B. J., and Daan, S. (1997). Social stress induces high intensity sleep in rats. Neurosci. Lett. 225, 41–44.
Mellman, T. A., Bustamante, V., Fins, A. I., Pigeon, W. R., and Nolan, B. (2002). REM sleep and the early development of posttraumatic stress disorder. Am. J. Psychiatry 159, 1696–1701.
Mellman, T. A., and Hipolito, M. M. (2006). Sleep disturbances in the aftermath of trauma and posttraumatic stress disorder. CNS Spectr. 11, 611–615.
Mellman, T. A., Pigeon, W. R., Nowell, P. D., and Nolan, B. (2007). Relationships between REM sleep findings and PTSD symptoms during the early aftermath of trauma. J. Trauma Stress 20, 893–901.
Mirescu, C., Peters, J. D., Noiman, L., and Gould, E. (2006). Sleep deprivation inhibits adult neurogenesis in the hippocampus by elevating glucocorticoids. Proc. Natl. Acad. Sci. U.S.A. 103, 19170–19175.
Morilak, D. A., Barrera, G., Echevarria, D. J., Garcia, A. S., Hernandez, A., Ma, S., and Petre, C. O. (2005). Role of brain norepinephrine in the behavioral response to stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 1214–1224.
Morin, C. M., Rodrigue, S., and Ivers, H. (2003). Role of stress, arousal, and coping skills in primary insomnia. Psychosom. Med. 65, 259–267.
Mueller, A. D., Pollock, M. S., Lieblich, S. E., Epp, J. R., Galea, L. A., and Mistlberger, R. E. (2008). Sleep deprivation can inhibit adult hippocampal neurogenesis independent of adrenal stress hormones. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1693–R1703.
Nakazawa, Y., Kotorii, M., Kotorii, T., Tachibana, H., and Nakano, T. (1975). Individual differences in compensatory rebound of REM sleep, with particular reference to their relationship to personality and behavioral characteristics. J. Nerv. Ment. Dis. 161, 18–25.
Nomura, K., Nakao, M., Takeuchi, T., and Yano, E. (2009). Associations of insomnia with job strain, control, and support among male Japanese workers. Sleep Med. 10, 626–629.
Obal, F. Jr., Garcia-Garcia, F., Kacsoh, B., Taishi, P., Bohnet, S., Horseman, N. D., and Krueger, J. M. (2005). Rapid eye movement sleep is reduced in prolactin-deficient mice. J. Neurosci. 25, 10282–10289.
Obal, F. Jr., Kacsoh, B., Alfoldi, P., Payne, L., Markovic, O., Grosvenor, C., and Krueger, J. M. (1992). Antiserum to prolactin decreases rapid eye movement sleep (REM sleep) in the male rat. Physiol. Behav. 52, 1063–1068.
Obal, F. Jr., Kacsoh, B., Bredow, S., Guha-Thakurta, N., and Krueger, J. M. (1997). Sleep in rats rendered chronically hyperprolactinemic with anterior pituitary grafts. Brain Res. 755, 130–136.
Opp, M. R. (1997). Rat strain differences suggest a role for corticotropin-releasing hormone in modulating sleep. Physiol. Behav. 63, 67–74.
Pacak, K., and Palkovits, M. (2001). Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr. Rev. 22, 502–548.
Pace-Schott, E. F., and Hobson, J. A. (2002). The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat. Rev. Neurosci. 3, 591–605.
Palma, B. D., Suchecki, D., and Tufik, S. (2000). Differential effects of acute cold and footshock on the sleep of rats. Brain Res. 861, 97–104.
Papadimitriou, G. N., and Linkowski, P. (2005). Sleep disturbance in anxiety disorders. Int. Rev. Psychiatry 17, 229–236.
Pare, W. P., and Redei, E. (1993). Depressive behavior and stress ulcer in Wistar Kyoto rats. J. Physiol. Paris 87, 229–238.
Pawlyk, A. C., Jha, S. K., Brennan, F. X., Morrison, A. R., and Ross, R. J. (2005). A rodent model of sleep disturbances in posttraumatic stress disorder: the role of context after fear conditioning. Biol. Psychiatry 57, 268–277.
Pawlyk, A. C., Morrison, A. R., Ross, R. J., and Brennan, F. X. (2008). Stress-induced changes in sleep in rodents: models and mechanisms. Neurosci. Biobehav. Rev. 32, 99–117.
Piallat, B., and Gottesmann, C. (1995). The reticular arousal threshold during the transition from slow wave sleep to paradoxical sleep in the rat. Physiol. Behav. 58, 199–202.
Plotsky, P. M., and Meaney, M. J. (1993). Early, postnatal experience alters hypothalamic corticotropin-releasing factor (CRF) mRNA, median eminence CRF content and stress-induced release in adult rats. Brain Res. Mol. Brain Res. 18, 195–200.
Plotsky, P. M., Owens, M. J., and Nemeroff, C. B. (1998). Psychoneuroendocrinology of depression. Hypothalamic-pituitary-adrenal axis. Psychiatr. Clin. North Am. 21, 293–307.
Popa, D., El Yacoubi, M., Vaugeois, J. M., Hamon, M., and Adrien, J. (2006). Homeostatic regulation of sleep in a genetic model of depression in the mouse: effects of muscarinic and 5-HT1A receptor activation. Neuropsychopharmacology 31, 1637–1646.
Price, L. J., and Kremen, I. (1980). Variations in behavioral response threshold within the REM period of human sleep. Psychophysiology 17, 133–140.
Pryce, C. R., Bettschen, D., and Feldon, J. (2001). Comparison of the effects of early handling and early deprivation on maternal care in the rat. Dev. Psychobiol. 38, 239–251.
Raboni, M. R., Tufik, S., and Suchecki, D. (2006). Treatment of PTSD by eye movement desensitization reprocessing (EMDR) improves sleep quality, quality of life, and perception of stress. Ann. N. Y. Acad. Sci. 1071, 508–513.
Rachalski, A., Alexandre, C., Bernard, J. F., Saurini, F., Lesch, K. P., Hamon, M., Adrien, J., and Fabre, V. (2009). Altered sleep homeostasis after restraint stress in 5-HTT knock-out male mice: a role for hypocretins. J. Neurosci. 29, 15575–15585.
Rampin, C., Cespuglio, R., Chastrette, N., and Jouvet, M. (1991). Immobilisation stress induces a paradoxical sleep rebound in rat. Neurosci. Lett. 126, 113–118.
Redei, E., Pare, W. P., Aird, F., and Kluczynski, J. (1994). Strain differences in hypothalamic-pituitary-adrenal activity and stress ulcer. Am. J. Physiol. 266, R353–R360.
Riemann, D., Berger, M., and Voderholzer, U. (2001). Sleep and depression–results from psychobiological studies: an overview. Biol. Psychol. 57, 67–103.
Roky, R., Obal, F. Jr., Valatx, J. L., Bredow, S., Fang, J., Pagano, L. P., and Krueger, J. M. (1995). Prolactin and rapid eye movement sleep regulation. Sleep 18, 536–542.
Roky, R., Valatx, J. L., and Jouvet, M. (1993). Effect of prolactin on the sleep-wake cycle in the rat. Neurosci. Lett. 156, 117–120.
Roky, R., Valatx, J. L., Paut-Pagano, L., and Jouvet, M. (1994). Hypothalamic injection of prolactin or its antibody alters the rat sleep-wake cycle. Physiol. Behav. 55, 1015–1019.
Sanchez-Lopez, A., and Escudero, M. (2011). Tonic and phasic components of eye movements during REM sleep in the rat. Eur. J. Neurosci. 33, 2129–2138.
Sanford, L. D., Fang, J., and Tang, X. (2003a). Sleep after differing amounts of conditioned fear training in BALB/cJ mice. Behav. Brain Res. 147, 193–202.
Sanford, L. D., Tang, X., Ross, R. J., and Morrison, A. R. (2003b). Influence of shock training and explicit fear-conditioned cues on sleep architecture in mice: strain comparison. Behav. Genet. 33, 43–58.
Sanford, L. D., Yang, L., and Tang, X. (2003c). Influence of contextual fear on sleep in mice: a strain comparison. Sleep 26, 527–540.
Sanford, L. D., Nassar, P., Ross, R. J., Schulkin, J., and Morrison, A. R. (1998). Prolactin microinjections into the amygdalar central nucleus lead to decreased NREM sleep. Sleep Res. Online 1, 109–113.
Sanford, L. D., Parris, B., and Tang, X. (2002). GABAergic regulation of the central nucleus of the amygdala: implications for sleep control. Brain Res. 956, 276–284.
Sanford, L. D., Yang, L., Wellman, L. L., Liu, X., and Tang, X. (2010). Differential effects of controllable and uncontrollable footshock stress on sleep in mice. Sleep 33, 621–630.
Sapolsky, R. M., Romero, L. M., and Munck, A. U. (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 21, 55–89.
Sarrieau, A., Chaouloff, F., Lemaire, V., and Mormede, P. (1998). Comparison of the neuroendocrine responses to stress in outbred, inbred and F1 hybrid rats. Life Sci. 63, 87–96.
Scher, A., Hall, W. A., Zaidman-Zait, A., and Weinberg, J. (2010). Sleep quality, cortisol levels, and behavioral regulation in toddlers. Dev. Psychobiol. 52, 44–53.
Shaffery, J., Hoffmann, R., and Armitage, R. (2003). The neurobiology of depression: perspectives from animal and human sleep studies. Neuroscientist 9, 82–98.
Shipley, J. E., Schteingart, D. E., Tandon, R., Pande, A. C., Grunhaus, L., Haskett, R. F., and Starkman, M. N. (1992a). EEG sleep in Cushing’s disease and Cushing’s syndrome: comparison with patients with major depressive disorder. Biol. Psychiatry 32, 146–155.
Shipley, J. E., Schteingart, D. E., Tandon, R., and Starkman, M. N. (1992b). Sleep architecture and sleep apnea in patients with Cushing’s disease. Sleep 15, 514–518.
Sinha, R. K. (2006). P-CPA pretreatment reverses the changes in sleep and behavior following acute immobilization stress rats. J Physiol Sci 56, 123–129.
Spiegel, K., Leproult, R., and Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet 354, 1435–1439.
Sportiche, N., Suntsova, N., Methippara, M., Bashir, T., Mitrani, B., Szymusiak, R., and McGinty, D. (2010). Sustained sleep fragmentation results in delayed changes in hippocampal-dependent cognitive function associated with reduced dentate gyrus neurogenesis. Neuroscience 170, 247–258.
Steriade, M., McCormick, D. A., and Sejnowski, T. J. (1993). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685.
Suchecki, D., Tiba, P. A., and Tufik, S. (2002). Hormonal and behavioural responses of paradoxical sleep-deprived rats to the elevated plus maze. J. Neuroendocrinol. 14, 549–554.
Svensson, T. H. (1987). Stress, central neurotransmitters, and the mechanism of action of alpha 2-adrenoceptor agonists. J. Cardiovasc. Pharmacol. 10(Suppl. 12), S88–S92.
Takahashi, K., Koyama, Y., Kayama, Y., and Yamamoto, M. (2000). The effects of prolactin on the mesopontine tegmental neurons. Psychiatry Clin. Neurosci. 54, 257–258.
Tang, X., Liu, X., Yang, L., and Sanford, L. D. (2005a). Rat strain differences in sleep after acute mild stressors and short-term sleep loss. Behav. Brain Res. 160, 60–71.
Tang, X., Yang, L., and Sanford, L. D. (2005b). Rat strain differences in freezing and sleep alterations associated with contextual fear. Sleep 28, 1235–1244.
Tang, X., Xiao, J., Liu, X., and Sanford, L. D. (2004). Strain differences in the influence of open field exposure on sleep in mice. Behav. Brain Res. 154, 137–147.
Tang, X., Yang, L., and Sanford, L. D. (2007). Interactions between brief restraint, novelty and footshock stress on subsequent sleep and EEG power in rats. Brain Res. 1142, 110–118.
Tiba, P. A., Palma, B. D., Tufik, S., and Suchecki, D. (2003). Effects of early handling on basal and stress-induced sleep parameters in rats. Brain Res. 975, 158–166.
Tiba, P. A., Tufik, S., and Suchecki, D. (2004). Effects of maternal separation on baseline sleep and cold stress-induced sleep rebound in adult Wistar rats. Sleep 27, 1146–1153.
Tiba, P. A., Tufik, S., and Suchecki, D. (2008). Long lasting alteration in REM sleep of female rats submitted to long maternal separation. Physiol. Behav. 93, 444–452.
Torner, L., Karg, S., Blume, A., Kandasamy, M., Kuhn, H. G., Winkler, J., Aigner, L., and Neumann, I. D. (2009). Prolactin prevents chronic stress-induced decrease of adult hippocampal neurogenesis and promotes neuronal fate. J. Neurosci. 29, 1826–1833.
Ulrich-Lai, Y. M., and Herman, J. P. (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 10, 397–409.
Van Bockstaele, E. J., Colago, E. E., and Valentino, R. J. (1998). Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J. Neuroendocrinol. 10, 743–757.
van der Helm, E., Yao, J., Dutt, S., Rao, V., Saletin, J. M., and Walker, M. P. (2011). REM sleep depotentiates amygdala activity to previous emotional experiences. Curr. Biol. 21, 2029–2032.
Van Twyver, H., and Garrett, W. (1972). Arousal threshold in the rat determined by “meaningful” stimuli. Behav. Biol. 7, 205–215.
Vazquez-Palacios, G., Retana-Marquez, S., Bonilla-Jaime, H., and Velazquez-Moctezuma, J. (2001). Further definition of the effect of corticosterone on the sleep-wake pattern in the male rat. Pharmacol. Biochem. Behav. 70, 305–310.
Vazquez-Palacios, G., and Velazquez-Moctezuma, J. (2000). Effect of electric foot shocks, immobilization, and corticosterone administration on the sleep-wake pattern in the rat. Physiol. Behav. 71, 23–28.
Vgontzas, A. N., Bixler, E. O., and Kales, A. (2000). “Sleep, sleep disorders, and stress,” in Encyclopedia of Stress, Vol. 3, ed. G. Fink (San Diego: Academic Press).
Vgontzas, A. N., Bixler, E. O., Lin, H. M., Prolo, P., Mastorakos, G., Vela-Bueno, A., Kales, A., and Chrousos, G. P. (2001). Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: clinical implications. J. Clin. Endocrinol. Metab. 86, 3787–3794.
Vgontzas, A. N., Tsigos, C., Bixler, E. O., Stratakis, C. A., Zachman, K., Kales, A., Vela-Bueno, A., and Chrousos, G. P. (1998). Chronic insomnia and activity of the stress system: a preliminary study. J. Psychosom. Res. 45, 21–31.
von Krosigk, M., Bal, T., and McCormick, D. A. (1993). Cellular mechanisms of a synchronized oscillation in the thalamus. Science 261, 361–364.
Walker, M. P., and van der Helm, E. (2009). Overnight therapy? The role of sleep in emotional brain processing. Psychol. Bull. 135, 731–748.
Walling, S. G., Brown, R. A., Milway, J. S., Earle, A. G., and Harley, C. W. (2011). Selective tuning of hippocampal oscillations by phasic locus coeruleus activation in awake male rats. Hippocampus 21, 1250–1262.
Waters, W. F., Adams, S. G. Jr., Binks, P., and Varnado, P. (1993). Attention, stress and negative emotion in persistent sleep-onset and sleep-maintenance insomnia. Sleep 16, 128–136.
Wellman, L. L., Yang, L., Tang, X., and Sanford, L. D. (2008). Contextual fear extinction ameliorates sleep disturbances found following fear conditioning in rats. Sleep 31, 1035–1042.
Wetzel, K., Baltatu, O., Nafz, B., Persson, P. B., Haase, H., and Morano, I. (2003). Expression of smooth muscle MyHC B in blood vessels of hypertrophied heart in experimentally hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R607–R610.
Wetzel, W., Balschun, D., Janke, S., Vogel, D., and Wagner, T. (1994). Effects of CLIP (corticotropin-like intermediate lobe peptide) and CLIP fragments on paradoxical sleep in rats. Peptides 15, 237–241.
Wetzel, W., Wagner, T., Vogel, D., Demuth, H. U., and Balschun, D. (1997). Effects of the CLIP fragment ACTH 20-24 on the duration of REM sleep episodes. Neuropeptides 31, 41–45.
Will, C. C., Aird, F., and Redei, E. E. (2003). Selectively bred Wistar-Kyoto rats: an animal model of depression and hyper-responsiveness to antidepressants. Mol. Psychiatry 8, 925–932.
Keywords: sleep, REM sleep, stress, HPA axis, prolactin, anxiety-related behavior, animal models
Citation: Suchecki D, Tiba PA and Machado RB (2012) REM sleep rebound as an adaptive response to stressful situations. Front. Neur. 3:41. doi: 10.3389/fneur.2012.00041
Received: 02 November 2011; Paper pending published: 21 November 2011;
Accepted: 02 March 2012; Published online: 02 April 2012.
Edited by:
Larry Sanford, Eastern Virginia Medical School, USAReviewed by:
Laurie Wellman, Eastern Virginia Medical School, USARichard Ross, Philadelphia VA Medical Center, USA
Copyright: © 2012 Suchecki, Tiba and Machado. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Deborah Suchecki, Departamento de Psicobiologia, Universidade Federal de São Paulo, Rua Botucatu, 862, Vila Clementino, 04023062 Sao Paulo, Brazil. e-mail:c3VjaGVja2lAcHNpY29iaW8uZXBtLmJy