

- Department of Neurology, Loyola University Medical Center, Stritch School of Medicine, Maywood, IL, USA
Tumors or chronic inflammatory lesions of the occipital condyle may cause occipital pain associated with an ipsilateral hypoglossal nerve injury (occipital condyle syndrome). We describe a young woman with recurrent otitis media and occipital condyle syndrome associated with a limited form of Wegener’s disease.
A 47-year-old woman had a 2-week history of “troubles handling food inside her mouth” resulting in a 7-pound weight loss. Few days prior to this complaint, the patient experienced continuous, “ice-pick-like” pains on the right occipital area radiating to the right temporal area. The pain improved by turning her head to the right and worsened by head turning to the left. Past medical history was remarkable for a 6-month history of recurrent bilateral otitis media requiring bilateral myringotomies, and one episode of transient right lid drooping. Pertinent findings on neurological examination included dysarthric speech, right nasolabial fold flattening, and right hemilingual atrophy with fasciculations.
Initial complete blood count (CBC), complete metabolic profile (CMP), activated partial thromboplastin time (aPTT), international normalized ratio (INR), and prothrombin time (PT) were unremarkable. Erythrocyte sedimentation rate (ESR) was 29 mm/h and C-reactive protein (CRP) was 0.4 mg/dl. MRI of the brain with gadolinium showed diffuse enhancement of the nasopharynx, petrous apices, and prevertebral soft tissues with extension to the medial margins of the carotid artery spaces, medial pterygoid muscles, and dura at the level of the pre-medullary cistern (Figure 1). CT of the neck with contrast failed to identify any lesion in the nasopharynx. CT of the chest, abdomen, and pelvis showed bilateral pulmonary nodules with irregular margins (Figure 2). A percutaneous transthoracic biopsy of the dominant lung nodule, showed changes consistent with a non-caseating granulomatous reaction (Figure 3). Cerebrospinal fluid (CSF) analysis showed a protein of 70 mg/dl, no red blood cells (RBCs), 1 white blood cell (WBC), and a glucose of 53 mg/dl (55% of serum glucose). Cytology was negative for malignancy. CSF acetylcholinesterase (ACE), PCR for Epstein Barr virus, Varicella Zoster virus, Herpes Simplex virus type 1 and 2, JC virus as well as antibodies against Borrelia Burgdorferi, Arbovirus, and Coccidioides were unremarkable. HIV 1 and 2 antibodies, serum rheumatoid factor, ACE, rapid plasma regain (RPR), carcinoembryonic antigen (CEA), Lyme antibodies, beta 2 glycoprotein antibodies (IgG and IgM), cardiolipin antibodies (IgG and IgM), Aspergillus, Coccidioides, and perinuclear-Anti neutrophil cytoplasmic antibody (P-ANCA) were negative. Serum sample for classical-antineutrophil cytoplasmic antibody (C-ANCA) was positive. Urinalysis and urine Histoplasma antigen were negative. AFB staining in the CSF, blood, and lung tissue was unremarkable. A flexible endoscopic naso-pharyngo-laryngoscopy showed hypomotility of the right true vocal cord.
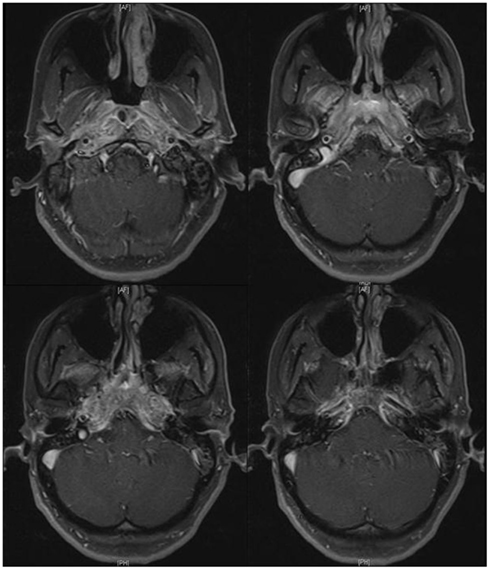
Figure 1. MRI (T1W with contrast) shows diffuse enhancement in the nasopharynx, petrous apices, prevertebral soft tissues with extension to the medial margins of the carotid spaces, medial pterygoid muscles and dura mater at the pre-medullary cistern.
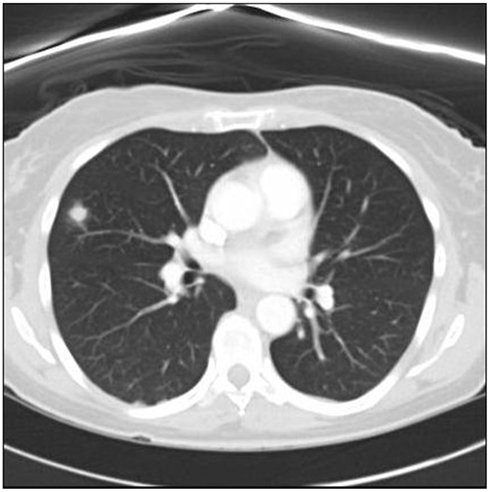
Figure 2. Chest CT shows the dominant nodule located on the right superior lobe.

Figure 3. Lung fine needle aspiration shows granulomatous reaction with epithelioid histiocytes, acute and chronic inflammation (Diff-Quik stain, 400 ×).
The patient was started on cefepime and voriconazole. Non-steroidal anti-inflammatory medications failed to relieve her headaches. Antibiotics were discontinued after all smears and cultures from the lung lesion, blood, urine, and CSF returned negative. The patient received 5 days of intravenous methylprednisolone followed by daily 60 mg of oral prednisone. Subsequent serum sample for anti-proteinase 3 (APR3) was positive. Methotrexate (MTX) and folic acid were started.
After 4 weeks of therapy with prednisone and MTX, a repeat C-ANCA, CRP, ESR, and APR3 were found to be normal. The patient was slowly tapered from steroids with almost immediate recurrence of headaches, hemilingual paresis, and dysphagia. Repeat MRI showed a similar pattern of enhancement. Multiple biopsies of the right nasopharynx mucosa, right periclival area, right descending palatine artery, skeletal muscle of the right infratemporal fossa, bone, and cartilage fragments of the maxillary, ethmoid, and sphenoid sinus showing geographic suppurative necrosis, focal vasculitis, and granulomatous inflammation (Figures 4 and 5). These findings supported the diagnosis of Wegener’s disease (WD). Immunostains (CD20, CD3, CD5, CD56, kappa, lambda) and Epstein–Barr virus-encoded ribonucleic acid study (EBER) showed no evidence of sinonasal lymphoma. AFB, bacterial, and fungal cultures were negative.
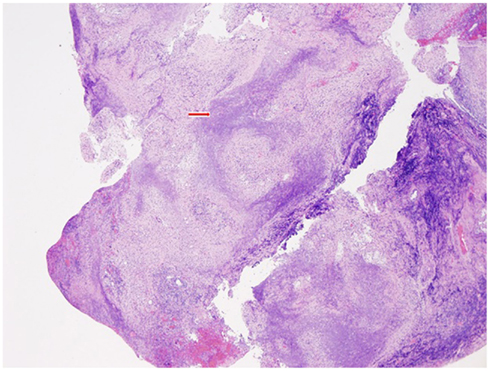
Figure 4. At low magnification, the histologic features include ”geographic” type necrosis with a basophilic appearance in right nasal pharynx (H&E stain, 40×). Red arrow shows basophilic, “geographic” type necrosis.
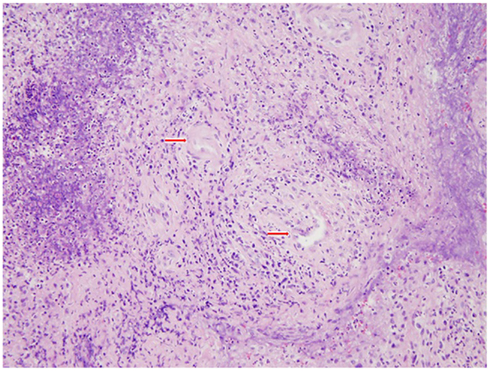
Figure 5. Right nasal pharynx biopsy shows the inflammatory infiltrate surrounding the blood vessels (vasculitis) and showing angioinvasion (H&E stain, 200×). Red arrows mark the vessels surrounded by inflammatory infiltrates.
MTX was discontinued, due to protracted nausea and vomiting, and the patient was given on intravenous rituximab 325 mg/m2. A repeat pulse of methylprednisolone controlled the recurrent symptoms. The patient is currently free of symptoms on 20 mg of prednisone daily and 160/800 mg of trimethoprim–sulfamethoxazole twice daily.
Discussion
The association of pain in the occipital region and ipsilateral cranial nerve XII (CN XII) paresis was consistent with the occipital condyle syndrome, first identified by Greenberg et al. (1981). The occipital condyle syndrome has been associated with metastases to the base of the skull and primary head and neck tumors (Greenberg et al., 1981; Moris et al., 1998; Capobianco et al., 2002). The largest case series ever reported about CN XII palsy indicated that almost half of these cases are due to malignancies (Keane, 1996). Our patient had many of the typical features of the occipital condyle syndrome (Capobianco et al., 2002) including radiation of pain to the ipsilateral temporal area, improvement of symptoms by turning the head toward the painful side with worsening of pain by contralateral neck rotation. Our patient had some atypical features including absence of neck stiffness or worsening pain by neck flexion. Probably the most important syndromatic difference in this case is related to the etiology. To our knowledge, the occipital condyle syndrome has not been previously associated to non-neoplastic etiologies such as WD.
Wegener’s disease is a clinicopathological syndrome characterized by extravascular granulomatous inflammation, granulomatous vasculitis of small size vessels, necrosis of upper and lower respiratory tracts, and pauci-immune glomerulonephritis (DeRemee et al., 1976; Leavitt et al., 1990). Recurrent middle ear involvement, as in our patient, has been well documented (McCaffrey et al., 1980; Ridley et al., 1988). Neurological manifestations in WD include cerebral and subarachnoid hemorrhage, cerebral arterial and venous thrombosis, cranial and peripheral neuropathies (primarily mononeuritis multiplex), diabetes insipidus, retinal vascular and optic nerve lesions, ocular myositis, orbital pseudotumor, and myopathy (Drachman, 1963; Biller and Grau, 2004; Holle and Gross, 2011). A localized granulomatous involvement of the meninges, described as chronic granulomatous pachymeningitis, presents a very broad clinical spectrum including seizures, encephalopathy and, as in the case of our patient, headaches that only resolve with corticosteroids, meningismus, and cranial neuropathies (Anderson et al., 1975; Katrib et al., 1998). In this setting, the most common cranial nerves affected are the optic nerve (CN II) followed by the olfactory (CN I; Nishino et al., 1993). A presentation with multiple cranial neuropathies, although uncommon, has been reported (Parker and Sobel, 1988; Fujikawa et al., 2008). CSF analyses in these patients show lymphocytic pleocytosis with high protein concentration and normal glucose level (Reinhold-Keller et al., 2001) C-ANCA and anti PR3 are of great diagnostic value but do not reflect disease activity and should not be used to assess therapeutic response (Nowack et al., 2009). Untreated patients with WD have a mean survival of 5 months and a 1-year mortality of 82% (Walton, 1958). In 1993, cyclophosphamide was the first immunosuppressant shown to reverse the meningeal disease (Weinberger et al., 1993). Since then, corticosteroids and cyclophosphamide have been documented to achieve excellent control of disease activity (de Groot et al., 2001). Modern immunosuppressants including infliximab and rituximab have been reported to successfully manage refractory cases of this condition (Mukhtyar and Luqmani, 2005; Sharma et al., 2010). The use of trimethoprim–sulfamethoxazole has been described to reduce the incidence of relapses on patients with WD. These effect is maximum on patient in remission who presented with limited disease (Reinhold-Keller et al., 1996; Stegeman et al., 1996).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, J. M., Jamieson, D. G., and Jefferson, J. M. (1975). Non-healing granuloma and the nervous system. Q. J. Med. 44, 309–323.
Biller, J., and Grau, R. G. (2004). “Cerebral vasculitis,” in Handbook of Cerebrovascular Diseases, ed. H. P. Adams Jr, 2nd edn. (New York: Marcel Dekker), 653–680.
Capobianco, D. J., Brazis, P. W., Rubino, F. A., and Dalton, J. N. (2002). Occipital condyle syndrome. Headache 42, 142–146.
de Groot, K., Schmit, D. K., Arlt, A. C., Gross, W. L., and Reinhold-Keller, E. (2001). Standardized neurologic evaluations of 128 patients with Wegener’s granulomatosis. Arch. Neurol. 58, 1215–1221.
DeRemee, R. A., McDonald, T. J., Harrison, E. G. Jr., and Coles, D. T. (1976). Wegener’s granulomatosis: anatomic correlates, a proposed classification. Mayo Clin. Proc. 51, 777–781.
Drachman, D. A. (1963). Neurological complications of Wegener’s granulomatosis. Arch. Neurol. 8, 145–155.
Fujikawa, K., Kawakami, A., and Eguchi, K. (2008). Recovery from multiple cranial nerve palsy of Wegener’s granulomatosis with infliximab. J. Rheumatol. 35, 1471–1472.
Greenberg, H. S., Deck, M. D., Vkram, B., Chu, F. C., and Posner, J. B. (1981). Metastasis to the base of the skull: clinical findings in 43 patients. Neurology 31, 530–537.
Holle, J. U., and Gross, W. L. (2011). Neurological involvement in Wegener’s granulomatosis. Curr. Opin. Rheumatol. 23, 7–11.
Katrib, A., Portek, I., Corbett, A. J., Hersch, M. I., and Zagami, A. S. (1998). Meningeal involvement in Wegener’s granulomatosis. J. Rheumatol. 25, 1009–1011.
Leavitt, R. Y., Fauci, A. S., Bloch, D. A., Michel, B. A., Hunder, G. G., Arend, W. P., Calabrese, L. H., Fries, J. F., Lie, J. T., Lightfoot, R. W. Jr., Masi, A. T., McShane, D. J., Mills, J. A., Stevens, M. B., Wallace, S. L., and Zvaifler, N. J. (1990). The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 33, 1101–1107.
McCaffrey, T. V., McDonald, T. J., Facer, G. W., and DeRemee, R. A. (1980). Otologic manifestations of Wegener’s granulomatosis. Otolaryngol. Head Neck Surg. 88, 586–593.
Moris, G., Roig, C., Misiego, M., Alvarez, A., Berciano, J., and Pascual, J. (1998). The distinctive headache of the occipital condyle syndrome: a report of 4 cases. Headache 38, 308–311.
Mukhtyar, C., and Luqmani, R. (2005). Current state of TNF alpha blockade in Wegener’s granulomatosis. Ann. Rheum. Dis. 64, 31–36.
Nishino, H., Rubino, F. A., DeRemee, R. A., Swanson, J. W., and Parisi, J. E. (1993). Neurological involvement in Wegener’s granulomatosis: an analysis of 324 consecutive patients at the Mayo clinic. Ann. Neurol. 33, 4–9.
Nowack, R., Wachtler, P., Kunz, J., and Rasmussen, N. (2009). Cranial nerve palsy in Wegener’s granulomatosis – lessons from clinical cases. Neurology 256, 299–304.
Parker, S. W., and Sobel, R. A. (1988). Case records of the Massachusetts General Hospital (Case 12-1988). N. Engl. J. Med. 318, 760–768.
Reinhold-Keller, E., de Groot, K., Holl-Ulrich, K., Arlt, A. C., Heller, M., Feller, A. C., and Gross, W. L. (2001). Severe CNS manifestations as the clinical hallmark in generalized Wegener’s granulomatosis consistently negative fr ANCA: a report of 3 cases and review of the literature. Clin. Exp. Rheumatol. 19, 541–549.
Reinhold-Keller, E., De Groot, K., Rudert, H., Nolle, B., Heller, M., and Grose, W. L. (1996). Response to trimethoprim/sulfamethoxazole in Wegener’s granulomatosis depends on the phase of disease. QJM 89, 15–23.
Ridley, M. G., Wolfe, C. S., Asherson, R. A., and Hughes, G. R. V. (1988). Recurrent otitis media and pulmonary infiltrates as presenting features in Wegener’s granulomatosis. BMJ 297, 352–353.
Sharma, A., Kunar, S., Wanchu, A., Lal, V., Singh, R., Gupta, V., Singh, S., and Gupta, A. (2010). Successful treatment of hypertrophic pachymeningitis in refractory Wegener’s granulomatosis with rituximab. Clin. Rheumatol. 29, 107–110.
Stegeman, C. A., Tervaert, J. W. C., de Jong, P. E., and Kallenberg, C. G. M. (1996) Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N. Engl. J. Med. 335, 16–20.
Walton, E. W. (1958). Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br. Med. J. 2, 265–270.
Keywords: Wegener’s disease, occipital condyle syndrome, hypoglossal nerve paralysis
Citation: Hornik A, Rodriguez-Porcel F, Ersahin CH, Kadanoff R and Biller J (2012) Wegener’s disease presenting with occipital condyle syndrome. Front. Neur. 3:53. doi: 10.3389/fneur.2012.00053
Received: 07 February 2012; Paper pending published: 28 February 2012;
Accepted: 23 March 2012; Published online: 12 April 2012.
Edited by:
Richard Mayeux, Columbia University, USAReviewed by:
Kevin N. Sheth, University of Maryland School of Medicine, USAValentina Garibotto, Geneva University and Geneva University Hospital, Switzerland
Copyright: © 2012 Hornik, Rodriguez-Porcel, Ersahin, Kadanoff and Biller. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: José Biller, Department of Neurology, Loyola University Medical Center, Stritch School of Medicine, 2160 South, 1st Avenue, Building 105, Room 2700, Maywood, IL 60153, USA. e-mail:amJpbGxlckBsdW1jLmVkdQ==