


- 1 Neuropathology Unit, Hospital de Santo António, Centro Hospitalar do Porto, Porto, Portugal
- 2 Department of Neurology, Hospital de Braga, Braga, Portugal
Neurodegenerative dementias are a group of neurological disorders characterized by deterioration in several cognitive domains in which there is selective and progressive loss of specific populations of neurons. The precise neurobiological basis for the different neurodegenerative dementias remains unknown. It is expected that different pathologies reflect different mechanisms, at least early in the neurodegeneration process. The next decades promise treatments directed to causes and mechanisms, bringing an outstanding challenge to clinicians due to heterogeneous clinical presentations with the same molecular pathology. The purpose of this brief review is to describe the key neuropathological features of the most common neurodegenerative dementias (Alzheimer disease, dementia with Lewy bodies and Parkinson’s disease dementia, and frontotemporal lobar degeneration) and the relationship with the clinical syndromes described in clinico-pathological studies. We expect this overview contributes for the understanding of this broad topic integrating the two ends of the spectrum: clinical and pathological.
Introduction
Neurodegenerative dementias are a group of neurological disorders characterized by deterioration in several cognitive domains in which there is selective and progressive loss of specific populations of neurons (Dickson, 2011). The precise neurobiological basis for the selective vulnerability in the different neurodegenerative dementias remains unknown. Furthermore, recent research data showed that dementia is not only caused by “neuronal cell death”/cell loss but predominantly by dysfunction and loss of synapses in Alzheimer disease and in dementia with Lewy bodies (DLB; Jellinger, 2009). These changes cause disconnections of important nervous circuitries which can contribute to the clinical manifestations. An increasing number of hypotheses to explain the pathogenesis of Alzheimer’s disease, for instance, have been proposed but it remains a mystery a century after this dementia was first described (de la Torre, 2011). In the most common neurodegenerative disorders there are biochemical changes in a specific protein that often promotes their deposition (Dickson, 2010). Over the last decade, many researchers have investigated the neuropathological background of the phenotypic variability in neurodegenerative dementia and identified a wide spectrum of associations between clinical syndromes and molecular pathologies. The classification of neurodegenerative diseases, previously based on the anatomical systems involved, has been progressively replaced by molecular diagnosis (Jellinger and Kovacs, 2011b).
The next decades promise treatments that are directed at changing pathogenesis, increasing the importance for clinicians’ awareness of the full clinical spectrum under the umbrella of the same molecular pathology.
The purpose of this brief review is to describe the key neuropathological features of the most common neurodegenerative dementias [Alzheimer’s disease, DLB and Parkinson’s disease dementia (PDD), and frontotemporal lobar degeneration (FTLD)] and the relationship with the clinical syndromes described in clinico-pathological studies.
Alzheimer Disease
Alzheimer’s disease (AD) is the most frequent cause of dementia worldwide, and its prevalence increases steeply after age 65 years, representing a significant health-care cost in developed countries (Reitz et al., 2011). Despite significant advances have been made in the understanding of AD pathogenesis, it remains largely unknown. Monogenic causes of familial early onset AD include autosomal dominant mutations in the β-amyloid precursor protein (APP), presenilin 1, and presenilin 2 protein genes, but represent less than 5% of AD cases (Bertram, 2008). Together with identification of apolipoprotein E ε4 allele as a genetic risk factor for late onset AD (Saunders et al., 1993) and neuropathology findings (see below), this evidence supports the amyloid cascade hypothesis as an important contributor in AD pathogenesis (Hardy and Higgins, 1992), even though other converging mechanisms most certainly play important roles in non-Mendelian forms of AD. More recently, genome-wide association studies have identified multiple genetic polymorphisms which are associated with late onset, non-Mendelian AD, and suggest involvement of other molecular pathways, namely implicating immune system, synaptic, and cell membrane function (Bertram, 2011; Morgan, 2011). AD pathogenesis theories must also recognize contribution of environmental factors, since several risk factors and modifiers of disease expression such as age (Ferri et al., 2005), cognitive reserve (Roe et al., 2007), physical activity (Podewils et al., 2005), smoking (Anstey et al., 2007), obesity (Lee, 2011), diabetes (Biessels et al., 2006), and intracranial atherosclerosis (Dolan et al., 2010) have been found. Core classical clinical characteristics of AD include a gradual and progressive decline of cognitive function which affects episodic memory, involves other cognitive domains, and is not explained by other medical or psychiatric conditions. Several diagnostic criteria have been proposed, namely the National Institute of Neurological Disorders and Stroke–Alzheimer Disease and Related Disorders criteria (McKhann et al., 1984), and the Diagnostic and Statistical Manual of Mental Disorders fourth edition criteria (American Psychiatric Association, 2000), but increasing evidence concerning magnetic resonance imaging (MRI), cerebrospinal fluid (CSF), and functional neuroimaging findings has led to a new proposal of research criteria for the diagnosis of AD (Dubois et al., 2007), in which is implied a reformulation of classic concepts in AD and mild cognitive impairment (Dubois et al., 2010). In all these criteria, neuropathological findings consistent with AD are required for a definite diagnosis, since it is assumed that a clinical diagnosis, even though may have high accuracy, is probabilistic.
Pathology
Brain pathology abnormalities in AD may be classified as “positive” or accumulation lesions (Aβ peptide deposits and tau protein accumulation), “negative” lesions (neuronal loss, loss of synapses), or a third type of lesion which include dendritic and axonal changes and inflammatory reaction lesions (Gomez-Isla et al., 2008).
Aβ peptide is cleaved from APP by β-secretase and γ-secretase enzyme complexes, and it is accumulated in AD, taking the form of mature neuritic plaques (senile plaques) or different types of extracellular deposits in brain parenchyma (Figures 1A–C). Neuritic plaques, stained by Congo red, are complex lesions formed by extracellular focal deposits of Aβ, neuronal processes (axonal or dendritic), microglial cells, and astrocytic processes (Duyckaerts et al., 2009). Neuritic plaques are found evenly distributed through the isocortex, preferentially in layers II and III, with high density in associative cortices, are relatively sparse in hippocampal and parahippocampal areas and are nearly absent in striatum and presubiculum (Duyckaerts and Hauw, 1997). Diffuse Aβ deposits are weakly immunoreactive and may be found in specific regions, such as the presubiculum (Wisniewski et al., 1998) and entorhinal cortex (Thal et al., 1999). Other focal and stellate Aβ deposits were also described, with different distributions through the cortical layers (Delaère et al., 1991). In 1991, the Consortium to establish a registry for AD (CERAD) established a neuropathologic method for diagnosing “definite,” “probable,” or “possible” AD, primarily based on a semiquantitative assessment of the number of neuritic plaques and its correlation with age (Mirra et al., 1991).
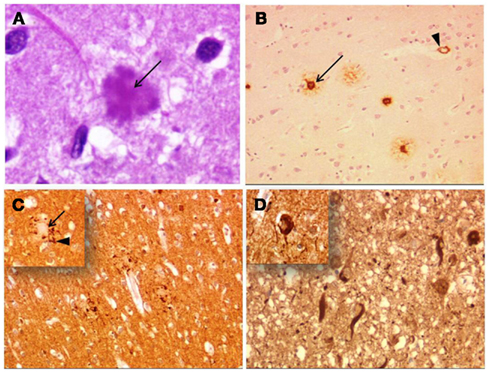
Figure 1. Alzheimer’s disease neuropathology. (A) Senile plaque (hematoxylin–eosin) with amyloid core (arrow). (B) Senile plaques with anti-Aβ antibody. The amyloid core (arrow) is surrounded by a corona of lightly labeled Aβ peptide. Arrowhead indicates vascular amyloid deposition in a capillary (Cambridge Bioscience, 4G8). (C) Neuritic plaques immunostained with tau antibody. The arrow indicate the core of the plaque and arrowhead the tau-positive processes of the neuritic crown (inset). (D) Flame-shaped neurofibrillary tangles. Inset shows a globose neurofibrillary tangle (Autogen Bioclear, AT8). Magnification, 400× (A), 100× (B,C), and 200× (D).
In AD, tau protein accumulation (three repeat and four repeat isoforms) takes the form of neurofibrillary tangles (cell body), neuropile threads (dendrites), and is also identified in the corona of neuritic plaques (Figures 1C,D). Neurofibrillary tangles, flame-, or globose-shaped silver positive intracellular inclusions, tend to accumulate in the entorhinal cortex, hippocampus, amygdala, basal nucleus of Meynert, and layers III and V of the isocortex, predominantly affecting neurons responsible for cortico-cortical projections (Arnold et al., 1991). Neuropil threads represent swollen dendrites with tau accumulation, occur in the same topography as neurofibrillary tangles and predominate in the earlier stages of the disease (Giannakopoulos et al., 2007). The neuritic plaque is where amyloid and tau pathology coincide, since the dystrophic large axonal processes in the corona at the periphery of the plaque core are tau-positive (Wang and Munoz, 1995). Despite the classical hypothesis that amyloid deposition drives the disease, neurofibrillary tangles have been shown to occur before amyloid lesions (Braak and Tredici, 2004), which highlights the poor understanding of tau–amyloid relationship, and of the role of other factors in AD pathogenesis. Braak and Braak (1991) proposed six stages for AD neuropathology, based on distribution and severity of neurofibrillary tangle pathology, and it has been demonstrated that these stages are closely related to clinical symptoms and deterioration (Riley et al., 2002). Interestingly, progression of tau pathology through a relatively predictable topography, a process which underlies progression of clinical symptoms, may result from a prion-like mechanism of neuron to neuron propagation of pathology (Braak and Tredici, 2011).
Neuronal loss is possibly the most significant microscopic correlate of gross macroscopic cerebral atrophy in patients with AD, occurring markedly in layer II of entorhinal cortex even in early clinical phases (Gomez-Isla et al., 1996), CA1, superior temporal gyrus, and supramarginal gyrus (Grignon et al., 1998).
Synaptic loss is reported to be an early event in the neurodegenerative process occurring in AD, and it is thought to be the major correlate of cognitive decline (Arendt, 2009). Distribution and degree of synapse degeneration is coincident with neurofibrillary tangle accumulation, suggesting a link between tangles and loss of synapse markers (Callahan et al., 1999).
Other neuropathology findings include local spine loss, axonal swellings, dysmorphic neurites, aberrant dendritic sprouting, inflammatory changes with activated microglia, astrocytosis, spongiosis, and Lewy bodies (LBs; Duyckaerts et al., 2009).
Recently, a new and comprehensive proposal for the neuropathological evaluation of AD was proposed by the National Institute on Aging-Alzheimer’s Association, in which there is emphasis on identification of amyloid deposits, staging of neurofibrillary tangles, scoring of neuritic plaques, and systematic search for other neurodegenerative dementia pathologies (Montine et al., 2012).
Clinico-Pathological Correlations
Classical AD clinical phenotype is characterized by: (1) decline from a previous level of function; (2) interference in daily living, work, and social interaction; (3) cognitive impairment with significant emphasis on episodic memory, accompanied by progressive and increasing involvement of other cognitive domains (visuospatial function, executive skills, attention, praxis, language); (4) progressive behavioral deterioration (depressive symptoms, disruptive behavior, apathy, anxiety, psychosis). This typical phenotype is the basis for the currently accepted diagnostic criteria. Clinical NINCDS-ADRDA criteria for AD has a reasonable sensibility and specificity for differentiating AD from normal controls, but accuracy for the differential diagnosis of neurodegenerative dementias may be very low (Ballard et al., 2011). The most recent research criteria for diagnosing AD (Dubois et al., 2007) have not been validated and there is debate on which neurocognitive tests should be used for characterization and quantification of cognitive deficits, and which are the best auxiliary biomarkers, definition of their pathological threshold and their clinical value in single patients (Oksengard et al., 2010).
Growing evidence from neuropathologic studies and new in vivo biomarkers allowed for identification and further characterization of atypical or focal presentations of AD and also an increased understanding about other neurodegenerative dementias. AD atypical presentations include posterior cortical atrophy (PCA), primary progressive aphasias (PPA), corticobasal syndrome (CBS), and frontal lobe syndrome (FLS).
Posterior cortical atrophy was first identified by Benson et al. (1998). Clinical phenotype is characterized by a progressive deterioration in complex visual functions, leading to a perceptual agnosia, Bálint syndrome, Gerstmann syndrome, and ultimately visual field defects, with no or residual impairment of other cognitive functions at least in early stages. Neuropathological studies support the hypothesis of dysfunction of the dorsal occipito-parietal visual pathway, and in the majority, AD pathology is found. Some studies have found a characteristic distribution of neuritic plaques and neurofibrillary tangles, with antero-posterior gradient in occipito-parietal regions (lower density in primary visual cortex), and relative sparing of frontal cortex (Levine et al., 1993; Hof et al., 1997). When comparing PCA and typical AD phenotype, it was found that the former had higher density of neuritic plaques and neurofibrillary tangles in visual association cortex and the later had higher density of lesions in hippocampus and subiculum, but there were no differences in other cortical areas (Tang-Wai et al., 2004). Clinical progression may disclose clues to underlying pathology: development of episodic memory impairment and involvement of other cognitive domains suggests AD; visual hallucinations, delusions, and parkinsonism may indicate DLB; asymmetric parkinsonism and ideomotor apraxia may suggest corticobasal degeneration, CBD; rapid progression of global disability, myoclonus, and cortical blindness with Anton syndrome suggest prion disease (namely the Heidenhain variant of Creutzfeldt–Jakob disease). It should be noted, however, that despite the etiological diversity of PCA, in over 75% of cases, AD pathology was shown (Renner et al., 2004; Tang-Wai et al., 2004).
Clinically, PPA may be subclassified in logopenic progressive aphasia (LPA; slow speech, impaired word retrieval, comprehension, and repetition), progressive non-fluent aphasia (PNFA; effortful speech, agrammatism, speech apraxia, dysprosody), and semantic dementia (SD; fluent, impaired confrontation naming, and word comprehension, surface dyslexia; Gorno-Tempini et al., 2011). LPA is the PPA subtype more strongly correlated with AD. Mesulam et al. (2008) found AD pathology in 7/11 patients with LPA. Even though LPA patients show atrophy in left posterior temporal and inferior parietal regions (Gorno-Tempini et al., 2008), distribution of neurofibrillary tangles did not show consistent hemispheric asymmetry in PPA/AD patients when using the CERAD protocol, while a stereological tangle quantification in four PPA/AD cases revealed higher tangle density in left hemisphere and similar entorhinal tangle density as typical AD cases (Mesulam et al., 2008). PNFA is classically associated with tau pathology but a significant proportion of patients who have come to autopsy have AD pathology (Grossman, 2010), and atypical distribution of lesions was described, with prominent involvement of left anterior perisylvian regions (Greene et al., 1996). SD is characteristically associated with TAR DNA binding protein (TDP) pathology (Seelaar et al., 2011), but 2/20 patients with SD had neuropathologic findings of AD (Alladi et al., 2007), and among 15 patients with fluent progressive aphasia, 33% had AD pathology, with striking atrophy and extensive neuritic plaque and tangle deposition in left anterior temporal and frontal lobes (Knibb et al., 2006).
Corticobasal syndrome is characterized by a slowly progressive constellation of manifestations which include asymmetric parkinsonism, asymmetric apraxia, unilateral useless limb, alien hand syndrome, cortical sensory loss, action myoclonus, and visuospatial deficits. It is now known that CBS is a neuropathological heterogeneous entity, and AD pathology was shown to be present in 24–50% in autopsy studies. Visual neglect, visual memory impairment, episodic memory deficits, and posterior extension of atrophy into precuneus and temporoparietal cortex are possible clinical indicators of CBS/AD (Alladi et al., 2007; Ling et al., 2010; Lee et al., 2011b).
Frontal lobe syndrome is dominated by deterioration of frontal functions, with dysexecutive syndrome, apathy, or disinhibition, changes in behavior, and social interaction. Initially, episodic memory impairment is absent or residual (Taylor et al., 2008), but usually progresses. Compared to typical AD, FLS/AD patients were reported to have greater impairment in Trail Making Test, phonemic fluency, and visuoconstructive skills (Johnson et al., 1999). In the previously mentioned cohort of focal dementia syndromes, among 28 patients with FLS, two had pathological findings consistent with AD (Alladi et al., 2007). In terms of neurofibrillary tangle load, entorhinal, temporal, and parietal cortex in typical AD is similar to FLS/AD, but the later have a significantly higher tangle density in frontal cortex (Johnson et al., 1999). No β-amyloid pathology distribution differences were found in this study.
In summary, AD pathology is associated principally to the classical clinical phenotype of Alzheimer’s disease with loss of episodic memory, but it should be noted that focal presentations of AD are part of the spectrum of the AD pathology (i.e., PCA, PPA, CBS, and FLS).
Parkinson’s Disease Dementia and Dementia with Lewy Bodies
Parkinson disease dementia and DLB represent two clinical phenotypes of the neurodegenerative dementia disorders diagnosed by the presence of LBs and Lewy neurites (LN).
Parkinson’s disease (PD) is one of the most frequent neurodegenerative diseases of the elderly. It is characterized clinically by bradykinesia, rigidity, resting tremor, and postural instability. The clinical diagnostic criteria require the presence of two of the four cardinal signs that are responsive to levodopa therapy (Gelb et al., 1999). The diagnosis of definitive PD requires histopathological confirmation, namely the presence of LBs in association with loss of substantia nigra neurons (Dickson et al., 2009). Clinical cohorts of patients identified with early PD are heterogeneous when referring to symptoms (resting tremor vs. akinesia and rigidity and/or postural instability and gait disorder), rates of progression (rapid vs. slow), and ages of onset (early vs. late onset), often with overlap between these phenotypes (Halliday and McCann, 2010). The prevalence of dementia in PD (i.e., PDD) is close to 30% and at least 75% of PD patients who survive for more than 10 years will develop dementia (Aarsland et al., 2005; Aarsland and Kurz, 2010). Age is an essential factor, and dementia is infrequent in patients with young onset and who are chronologically still young at the time of assessment, despite very long disease duration. The principal risk factors are older age, more severe parkinsonism (rigidity, postural instability, and gait disturbance), and mild cognitive impairment at baseline (Emre et al., 2007).
Dementia with Lewy bodies is considered to be the second most common type of degenerative dementia in the elderly, accounting for 10–15% of cases at autopsy (McKeith et al., 1996). Clinically it is characterized by prominent visuoconstructive and frontal-subcortical impairment, associated with core clinical neuropsychiatric features of fluctuating cognitive function, visual hallucinations, and spontaneous parkinsonian motor signs (McKeith et al., 2004, 2005). Both conditions have been associated to higher rates and more severe depression when compared to AD (Fritze et al., 2011).
For research purposes an arbitrary cut-off of 1 year is used to distinguish PDD from DLB. When PD develops first and dementia develops 1 year or more lately the diagnosis of PDD is made, if the cognitive impairment precedes motor symptoms or develops earlier the diagnosis is of DLB (Lippa et al., 2007).
There are no definite pathological criteria that separate the two disorders (Lippa et al., 2007) and the separation between PDD and DLB is considered by some to be artificial (Halliday et al., 2011).
Pathology
There is no “gold standard” for the pathological diagnosis of DLB or PDD. The hallmark pathology is α-synuclein (αSyn) in form of LBs (both classical and cortical types) and LN (Halliday et al., 2011; Ince, 2011). Classical LBs (Figure 2A) are easily recognizable by standard histological methods as large, spherical, highly eosinophilic intracytoplasmatic inclusions with a clear halo in the dopaminergic neurons of substantia nigra and the locus coeruleus (McKeith et al., 1996; Kövari et al., 2009). Cortical LBs (Figures 2B,C) are seen in limbic and neocortical regions, predominantly in the small neurons of deep layers of the cortex. Because of their small size they are easily identified using immunohistochemistry with αSyn antibodies (Figures 2E,F; Kövari et al., 2009). LN are curvilinear or dot-like processes (Figures 2D–F) that are found in regions with the highest density of LBs, such as limbic cortex and amygdala (Saito et al., 2003; Dickson, 2010).
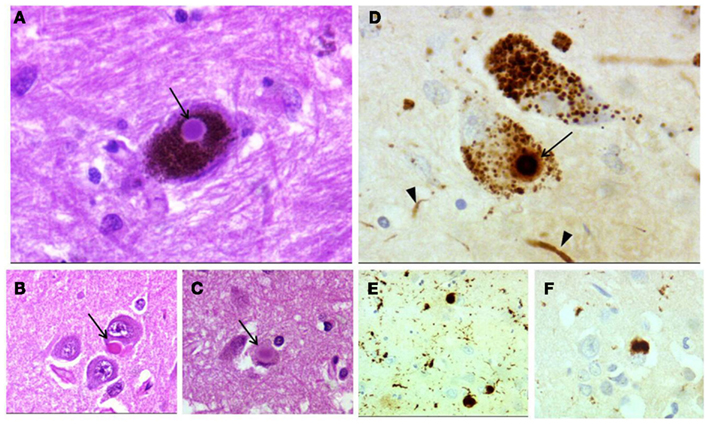
Figure 2. Dementia with Lewy bodies neuropathology. Lewy body in a neuron of the substantia nigra (A), in a pyramidal cell of CA1 area of the hippocampus (B), and in cingulated cortex (C) (arrows). Lewy body (arrow) and Lewy neurites (arrowheads) in the substantia nigra (D). Cortical Lewy bodies (E,F). (A–C) hematoxylin–eosin; (D–F) anti-α-synuclein immunostaining. Magnification, 400× (A–C,F) and 200× (E).
In spite of being the hallmark of DLB and PD, LBs they can be detected in the amygdala in up to 50% of patients with clinically and pathologically confirmed AD (Hamilton, 2000) and in up to 10% of neurologically normal elderly individuals over age of 60 years (Gibb and Lees, 1998). α-synuclein is a small, presynaptic protein without a well-defined function. Some data implicate the misfolding or aggregation of αSyn in the disease pathogenesis, but the mechanisms that underlie the aberrant functions of α-synuclein and how these impacts on disease pathogenesis remain poorly understood (Forman et al., 2004; Vekrellis et al., 2011).
Clinico-Pathological Correlations
A staging system, based on the number and location of LBs, with a caudal to rostral six-stage progression has been proposed for sporadic PD (Braak et al., 2003). The first two stages, with LB pathology involving medulla oblongata and pontine tegmentum, are considered asymptomatic or presymptomatic and may explain the early non-motor symptoms (autonomic and olfactory). Stages 3 and 4, with extension of LB pathology to midbrain and basal prosencephalon and mesocortex, have been correlated to clinical symptomatic stages. The terminal stages 5 and 6, characterized by widespread neocortical LB degeneration, are correlated with significant cognitive decline associated with severe parkinsonism (Hurtig et al., 2000). Although there is an acceptable correlation between pathological findings and clinical data in this staging system, mainly in a subgroup with early onset and prolonged duration (Halliday et al., 2008), recent studies revealed exceptions to the general order of progression suggested by Braak and colleagues (Jellinger, 2008; Parkkinen et al., 2008; Dickson et al., 2009; Kalaitzakis et al., 2009). Another interesting observation from a number of recent clinico-pathological studies that assessed the progression of pathology in subtypes of PD is that in patients with non-tremor-dominant and postural instability and gait dominant clinical pictures there are significantly more cortical LBs and amyloid β plaques compared with tremor dominant or younger onset patients (Selikhova et al., 2009; Halliday et al., 2011). Furthermore, PD patients with dementia have higher amounts of cortical αSyn pathology as compared to those without dementia and a correlation between its severity and AD pathology is also present in such patients (Halliday et al., 2011).
According to the consensus pathologic guidelines for DLB, LBs are scored semiquantitatively according to the severity and anatomical distribution, separating brainstem-predominant, limbic (or transitional), and neocortical types, depending on the anatomical distribution of the αSyn pathology (McKeith et al., 1996, 2005). More recently, a new protocol for assessing αSyn pathology and currently recommended by the DLB Consortium, showed higher inter-observer agreement for both the assignment to brainstem, limbic, neocortical and amygdala-predominant categories of synucleinopathy, and Braak stages (1–6; Alafuzoff et al., 2009). Alzheimer’s disease pathology is the most common co-occurring pathology that accompanies Lewy body pathology (PDD or DLB; Dickson et al., 2009) and most cases with cortical LBs show in some degree concomitant AD pathology (i.e., NFTs and neuritic plaques; Kövari et al., 2009). The methods proposed by the third Consortium for DLB (McKeith et al., 2005) recommend the description of Alzheimer disease-type pathology using the National Institute on Aging-Reagan Institute criteria (Hyman and Trojanowski, 1997). It is proposed that the DLB clinical syndrome is directly related to the severity of Lewy-related pathology, and inversely related to the severity of concurrent AD-type pathology (McKeith et al., 2005). In cases of “pure” DLB (i.e., without excessive tau neuritic pathology) clinical picture appears more similar to the dementia phenotype of PD than to AD (Emre et al., 2007). There is evidence suggesting that the increase of neocortical αSyn is associated with cognitive decline in DLB and PD (Hurtig et al., 2000; Kövari et al., 2003) and some clinico-pathological studies demonstrated that visual hallucinations are strongly related to the αSyn burden in the amygdala in both (Casanova et al., 2011).
Within the DLB phenotype spectrum, some clinico-pathological studies and case reports disclose a subset of DLB patients with rapid symptoms progression to death within 1–2 years (Armstrong et al., 1991; Haik et al., 2000). These patients can fall within the differential diagnosis of rapid progressive dementias such as Creutzfeldt–Jakob disease, and more recent data show that 7–10% of autopsy confirmed diffuse Lewy body disorder cases can have a rapid PD and dementia progression (Gaig et al., 2011; Jellinger and Attems, 2011a). The neuropathology of these cases did not show particular features that could differentiate rapidly progressing from classical forms.
In summary, PDD and DLB refers to a form of dementia that has the pathological signature of abnormal aggregates of α-synuclein in the form of LBs and LN. There is a considerable clinical heterogeneity explained, at least partially, by topographic distribution of αSyn aggregates and the presence of additional neuropathologies such as AD pathology. In fact, large autopsy series show that although the specificity of ante-mortem diagnosis of DLB when correlated to pathological diagnosis was over 95%, the sensitivity of the clinical diagnoses was quite low (32%). Furthermore, these studies show that that in late-stage cognitive impairment, specifically documented signs and symptoms associated with DLB (visual hallucinations, extrapyramidal signs, and fluctuating cognition) do not contribute for predicting the presence of neocortical LBs at autopsy. Consequently, while these clinical symptoms may be useful in milder cases of dementia, caution should be used when providing a diagnosis of LBD in patients with more advanced dementia (Nelson et al., 2010).
Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration refers to a clinical, genetic, and pathological heterogeneous group of disorders that constitute a common cause of dementia with onset usually before 65 years of age (Cairns et al., 2007; Pickering-Brown et al., 2008). FTLD is a macro-anatomical descriptive term reflecting the relatively selective involvement of frontal and temporal lobes that characterizes most cases (Rohrer et al., 2011). Epidemiological studies suggest that FTLD is the second most common cause of young onset dementia after AD (Ratnavalli et al., 2002; Rosso et al., 2003). The clinical spectrum of FTLD encompasses three canonical syndromes that are distinguished by the presenting symptoms and regional pattern of atrophy: the behavioral variant frontotemporal dementia (bvFTD), with predominant behavioral symptoms; PNFA, a disorder of expressive language; and SD, a disorder of conceptual knowledge (Neary et al., 1998; Kertesz et al., 1999, 2005). There is also overlap of FTLD with motor neuron disease (FTD–MND), as well as the parkinsonian syndromes progressive supranuclear palsy and CBD (Litvan et al., 1996; Neary et al., 1998; Boeve et al., 2003).
A positive family history is common in FTLD, with up to 40% cases showing a pattern of inheritance consistent with autosomal dominant transmission of disease (Neary et al., 2005; Seelaar et al., 2011). Genetic heterogeneity of FTLD is reflected by the identification of seven different genes that are associated with FTLD. Mutations in genes encoding for microtubule-associated protein tau (MAPT) and progranulin (GRN) are responsible for approximately 50% of the familial cases, where other genes associated with FTLD pathology are extremely rare and include mutations in the valosin-containing protein (VCP) gene, the charged multivesicular body protein 2B (CHMP2B) gene, the TAR DNA binding protein 43 (TARDBP) gene and the fused in sarcoma (FUS) gene (Josephs et al., 2011; Seelaar et al., 2011). More recently, an expansion in chromosome 9 (C9ORF72 gene) was identified as cause of Chromosome 9p21-Linked FTD–MND (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
Pathology
The pathology of the group of disorders under the FTLD umbrella term overlaps in gross and histological features. All share the findings of selective atrophy of the frontal and temporal cortex (Figure 3), with neuronal loss, gliosis and spongiosis of the superficial layers, especially of layer II. In some patients there is asymmetry of atrophy, typically reflected in perisylvian loss on one side of the brain. Specific diagnosis of the neurodegenerative disease within the FTLD group is established by the identification of the protein that constitutes the cellular inclusions (Cairns et al., 2007). Three proteins have been identified as important players in the mechanism of neurodegeneration of the FTLD: MAPT, the transactive response DNA binding protein of 43 kD (TDP-43), and the tumor associated protein FUS (Josephs et al., 2011). Therefore, the majority of FTLDs can be subclassified at molecular level as FTLD–tau, FTLD–TDP, and FTLD–FUS (Mackenzie et al., 2010a). However, the exact mechanisms by which cell death occurs are not known. We will use this subclassification to characterize further the pathology features within each subgroup and, in the next section, to serve as basis for the clinico-pathological correlates reported in the literature.
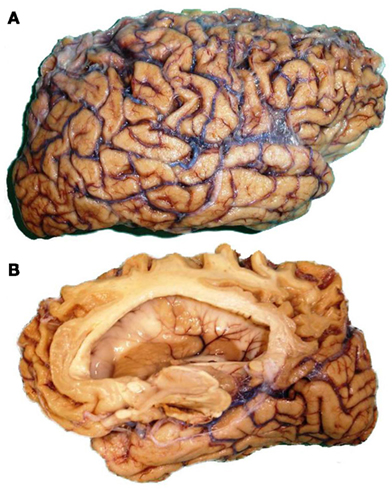
Figure 3. Macroscopical findings of a FTLD–TDP brain. Lateral (A) and medial view (B) showing cortical atrophy that involves frontal, temporal, and parietal lobes, with better preserved sensorimotor cortex and occipital lobe.
FTLD–tau
In this group the major abnormal protein identified by immunohistochemistry is the tau protein. This group includes Pick’s disease (Dickson, 2001) and the pathological entities CBD and progressive nuclear palsy, which can fall under the FTLD clinical presentation (Litvan et al., 1996; Boeve et al., 2003; Kertesz, 2003; Scaravilli et al., 2005; Josephs et al., 2006). Tau is a phosphoprotein that promotes microtubule polymerization and stabilization. The discovery of multiple mutations in the tau gene that lead to the abnormal aggregation of tau and cause FTLD demonstrates that tau dysfunction is sufficient to produce neurodegenerative disease, but the precise mechanisms remain to be completely elucidated (Lee et al., 2001). The microtubule binding domain of the tau protein contains three of four repeat regions (tau 3R and 4R) depending on the splicing of the RNA. There is preferential accumulation of 3R or 4R tau in the different tauopathies, allowing a biochemical subclassification within this group.
In Pick’s disease (PiD), the most characteristic neuropathological feature is the presence of Pick bodies. Pick bodies are spherical cytoplasmatic neuronal inclusions, that are well demarcated, amorphous, and faintly basophilic on hematoxylin–eosin staining (Figures 4A,B). They are strongly argyrophilic but do not stain with Gallyas (Dickson et al., 2011b). They are abundant in the dentate gyrus of the hippocampus and also common on the cerebral cortex, particularly in layers II and III (Munoz et al., 2011). Pick bodies contain deposition of tau protein (Figures 4C,D) that is abnormally hyperphosphorylated and biochemistry analysis showed that most of the tau consists of 3R tau (Delacorte et al., 1998; Bronner et al., 2005). Mutations in the tau gene (MAPT), most commonly associated to bvFTD-like phenotype in which extrapyramidal features may also be present (Josephs et al., 2011), account for the most pathologically confirmed cases of familial PiD (Dickson et al., 2011b). The neuropathological characteristics associated to MAPT gene mutations vary substantially, but the hallmark is the presence of tau protein deposits in neurons and/or glia (Ghetti et al., 2011). The pathology can resemble other tauopathies as PiD, PSP, or CBD and, for instances, neuropathological criteria for CBD states that for differentiate it from MAPT gene mutation cases additional clinical or molecular genetic information is required to make an accurate diagnosis (Dickson et al., 2002).
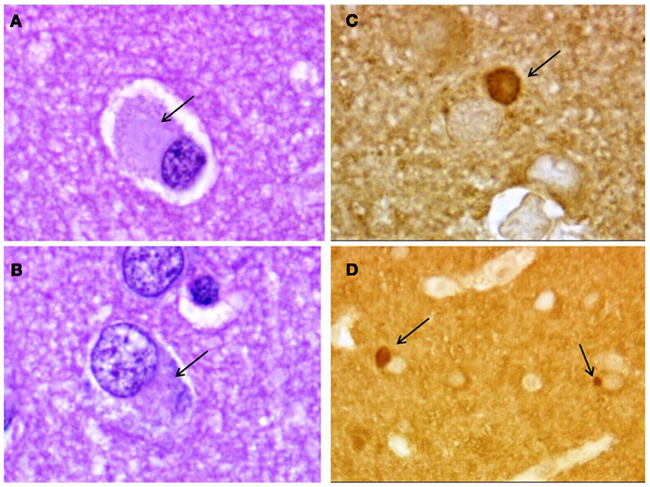
Figure 4. Neuropathology of Pick’s disease. Pick bodies (arrows) in frontal cortex. (A,B) Hematoxylin–eosin; (C,D) tau immunostaining. Magnification, 1000× (A–C), 400× (D).
Corticobasal degeneration, as a pathological entity, is associated to a wide range of clinical presentations (see below). CBD is a 4R tauopathy (Dickson et al., 2011b). The characteristic pathology in CBD is tau immunoreactive inclusions in the cell processes of neurons and glia in the cortex, basal ganglia, thalamus, and brainstem. Tau-reactive thin, thread-like processes of glial and neuronal origin are also seen throughout the gray and white matter and are an important feature of the pathology of CBD. The most specific histopathological lesion in CBD is the astrocytic plaque, a distinctive annular cluster of thick, short tau immunoreactive deposits within the distal processes of astrocytes (Dickson et al., 2002). Ballooned neurons, described since the first report of the disease under the name “corticodentatonigral degeneration with neuronal achromasia” (Rebeiz et al., 1967), are swollen cortical neurons, eosinophilic in hematoxylin–eosin staining, most often found in the third, fifth, and sixth cortical layers. They are immunoreactive to phosphorylated neurofilaments and alpha–beta-crystallin (Dickson et al., 2002). Despite of being one of the histological hallmarks of CBD, this type of neuronal degeneration itself is known to be a non-specific change and can be seen in other pathological conditions (Ikeda, 1997).
Progressive supranuclear palsy is also a 4R tauopathy. The most characteristic neuronal lesion on histopathology is the globose neurofibrillary tangle, while the most significant astrocytic lesion is the tufted astrocyte (Nishimura et al., 1992; Yamada et al., 1992), characterized by a tuft like arrangement of cell processes around the astrocyte cell body. They are both best appreciated with Gallyas silver stain or tau immunohistochemistry. The core neuroanatomical regions affected include basal ganglia, subthalamic nucleus, and substantia nigra, with cortical involvement more pronounced in motor and premotor cortices (Dickson et al., 2011b). Neuropathological criteria for PSP are based on the distribution of tau pathology and the exclusion of other neurodegenerative disorders associated to parkinsonism and dementia (Hauw et al., 1994). However, these criteria did not take into account the atypical clinical PSP presentations that can present under the FTLD spectrum.
FTLD–TDP
In this group the pathological changes signature consists in the presence of immunoreactive TDP-43 neuronal cytoplasmatic inclusions (NCI), dystrophic neurites (DN), and in some cases neuronal intranuclear inclusions (NII) in the frontotemporal neocortex and dentate granule cells of the hippocampus (Figure 5; Neumann et al., 2006; Davidson et al., 2007). TDP-43 is a ubiquitously expressed, highly conserved nuclear protein that regulates RNA in a variety of ways. Converging lines of research suggest that TDP-43 is mechanistically linked to neurodegeneration, with many pathways probably involved, including gain of toxic functions and loss of normal functions (Lee et al., 2011a). Four subtypes of FTLD–TDP are currently recognized based on morphology and anatomical distribution of TDP-43 lesions (Mackenzie et al., 2011a). Type A is characterized by numerous short DN and crescentic or oval NCI, as well as moderate numbers of NII (Figure 5), type B consists of moderate numbers of NCI and minimal or absent DN and Type C have a predominance of elongated and minimal to absent NCI. Finally, type D refers to the pathology associated to inclusion body myopathy with Paget’s disease of bone and frontotemporal dementia caused by CHMP2B VCP gene mutations, and is characterized by numerous short DN and frequent lentiform NII. There is good association between FTLD–TDP types and clinical syndromes (see below).
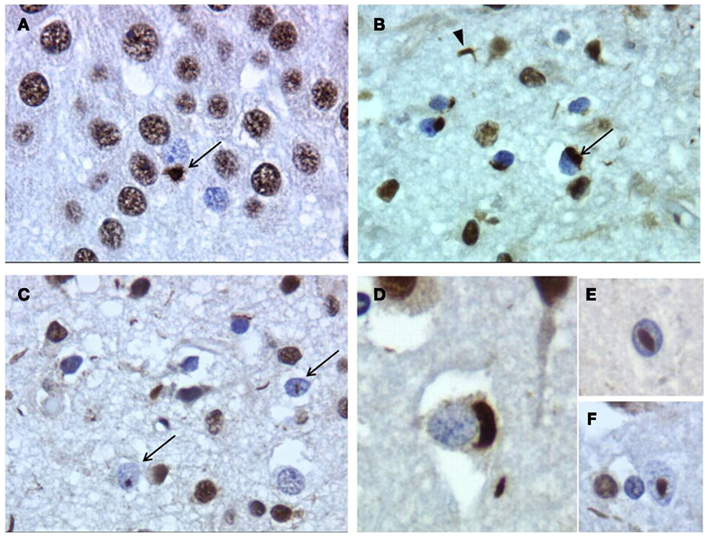
Figure 5. Frontotemporal lobar degeneration–TDP neuropathology of patient with progranulin gene mutation. (A) Neuronal cytoplasmatic inclusion (NCI) in the hippocampus (arrow). (B) Superficial frontal neocortex showing NCIs (arrow) and dystrophic neurites (DN; arrowhead). (C) Superficial parietal neocortex showing NCIs, DN, and neuronal intranuclear inclusions (NII; arrow). (D) NCI in frontal cortex. (E,F) Lentiform and round NII in frontal cortex. Magnification, 400×.
FTLD–FUS
Fused in sarcoma is a ubiquitously expressed protein that binds to RNA and DNA and is involved in diverse cellular processes (Neumann et al., 2009). Given the fact that Both TDP-43 and FUS are ubiquitously expressed DNA/RNA-binding proteins involved in multiple aspects of gene expression, transcription regulation, RNA splicing, transport and translation, although its precise function is poorly characterized. The understanding of the mechanisms underlying the pathophysiology of FUS accumulation and FUS-mediated neurodegeneration is still limited. As for TDP-43 proteinopathies, a toxic gain-of-function mechanism, a loss-of-function mechanism by depletion of physiological FUS and maybe co-sequestration of other vital factors, or both, is possible (Mackenzie et al., 2010b). This recently described FTLD category is pathological characterized by the presence of NCI and NII that are strongly immunoreactive for FUS protein and negative for the other proteins associated to neurodegenerative dementias (Neumann et al., 2009). Another consistent and striking feature of this group is the severe atrophy of the head of caudate nucleus (Roeber et al., 2008) that can be a useful clinical predictor of this pathology when detected by neuroimaging (Josephs et al., 2010). The true incidence and prevalence of FTLD–FUS is unknown. Based on brain bank studies has an estimate frequency of approximately 5% of all FTLD patients (Mackenzie et al., 2011b).
Clinico-Pathological Correlations
The recent discoveries on FTLD pathologies and their classification according to the major protein deposited in brain allowed to established associations between the FTLD pathologies and the clinical syndromes. In this section we will described the most important clinico-pathological associations taking as starting point the clinical syndrome and genetic variability.
The syndrome bvFTD, the most common clinical syndrome in FTLD spectrum, is histopathologically heterogeneous (Josephs et al., 2011; Rohrer et al., 2011), with half of the patients having tau pathology and the other 50% have tau-negative FTLD with ubiquitin immunoreactive inclusions (FTLD-U), which in the majority of cases are TPD-43 positive (Hodges et al., 2004; Snowden et al., 2007). In the FTLD-tau group presenting with bvFTD, Pick’s disease account for the majority of the cases (∼70%) followed by CBD and a minority of cases have PSP pathology (Wadia and Lang, 2007; Ling et al., 2010; Josephs et al., 2011). It should keep in mind that classical PSP clinical presentation, recently referred as Richardson syndrome, permit accurate ante-mortem diagnosis in most cases (Josephs and Dickson, 2003). The atypical clinical presentations, which can fall under the umbrella of FTLD, reflect varying anatomical distribution of tau pathology. In the FTLD–TDP group, bvFTD is not strongly associated to any TDP particular type (Josephs et al., 2011). A subtype of bvFTD characterized by a very young onset patient (∼40 years), with a clinical syndrome dominated by hypersexual and hyperphagic behavior, prominent stereotypy, and obsessionality, together with striatal atrophy was tightly associated to FTLD–FUS (Roeber et al., 2008; Urwin et al., 2010; Snowden et al., 2011).
The phenotype FTD–MND is highly specific of FTLD–TDP pathology (Josephs et al., 2011; Rohrer et al., 2011), with type B being the most common (Mackenzie et al., 2011a). The associated neuropathology of patients with mutations on C9ORF72 gene is also a FTLD–TDP type B (Stewart et al., 2012). Interestingly, this subset of patients (MND/ALS and FTLD–TDP with C9ORF72 repeat expansion) have also p62 positive, TDP-43 negative, neuronal cytoplasmic, and intranuclear inclusions in the cerebellum and hippocampus that seems to be specific for this condition (Al-Sarraj et al., 2011).
Progressive non-fluent aphasia is associated predominantly with FTLD–tau (70%; Josephs et al., 2011) and no particular association with specific tau pathology can be made (i.e., PiD, CBD, or PSP).
Semantic dementia is predominantly associated to FTLD–TDP (83%), particularly type C (Josephs et al., 2011; Rohrer et al., 2011).
Although clinically heterogeneous, even among family members carrying the same mutation, certain characteristics have been linked more frequently to FTLD patients with progranulin mutations, such extrapyramidal features and parietal lobe deficits (Rohrer et al., 2008; van Swieten and Heutink, 2008; Taipa et al., 2012).
In summary, FTLD is the umbrella term for a heterogeneous group of clinical (bvFTD, PNFA, SD, FTD–MND) and pathological disorders (FTLD–tau, FTLD–TDP, and FTLD–FUS), with strong clinico-pathological associations in certain groups (i.e., FTD–MND/FTLD–TDP, SD/FTLD–TDP, PNFA–FTLD–tau, young onset bvFTD/FTLD–FUS).
Conclusion
The precise mechanisms involved in neurodegeneration remain largely unknown, but some proteins have emerged as important players in the mechanism of neurodegeneration. This suggests, at least partially and probably early in the process, specific pathophysiological characteristics in the different neurodegenerative dementias. Consequently, the rational use of disease modifying treatments will almost certainly imply a specific diagnosis at a molecular level. This brings an outstanding challenge to clinicians due to heterogeneous clinical presentations with the same molecular pathology. Clinico-pathological studies helped in refining diagnosis and continue to be essential in order to pursuit in vivo biomarkers to achieve higher diagnostic specificity. Adding to the clinical overlap of distinct neuropathological diagnosis, it must be taken into account that while evaluating post-mortem brains, pathologists have to assess numerous pathologies, keeping in mind the clinical presentation, but also to be aware of the frequent findings of comorbidity or unexpected pathologies which characterize the aging brain (Alafuzoff et al., 2009).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Isabel Pires, Aurora Rodriges, and Carlos Gouveia for histological and immunohistochemistry technical assistance.
References
Aarsland, D., and Kurz, M. W. (2010). The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 20, 633–639.
Aarsland, D., Zaccai, J., and Brayne, C. (2005). A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov. Disord. 20, 1255–1263.
Alafuzoff, I., Ince, P. G., Arzberger, T., Al-Sarraj, S., Bell, J., Bodi, I., Bogdanovic, N., Bugiani, O., Ferrer, I., Gelpi, E., Gentleman, S., Giaccone, G., Ironside, J. W., Kavantzas, N., King, A., Korkolopoulou, P., Kovács, G. C., Meyronet, D., Monoranu, C., Parchi, P., Parkkinen, L., Patsouris, E., Roggendorf, W., Rozemuller, A., Stadelmann-Nessler, C., Streichenberger, N., Thal, D. R., and Kretzschmar, H. (2009). Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol. 117, 635–652.
Alladi, S., Xuereb, J., Bak, T., Nestor, P., Knibb, J., Patterson, K., and Hodges, J. R. (2007). Focal cortical presentations of Alzheimer’s disease. Brain 130, 2636–2645.
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., Rogelj, B., Al-Chalabi, A., Hortobágyi, T., and Shaw, C. E. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702.
American Psychiatric Association. (2000). Diagnosis and Statistic Manual of Mental Disorders (IV-TR), 4th Edn. Washington, DC: Amer Psychiatric Pub.
Anstey, K. J., von Sanden, C., Salim, A., and O’Kearney, R. (2007). Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. Am. J. Epidemiol. 166, 367–378.
Armstrong, T. P., Hansen, L. A., Salomon, D. P., Masliah, E., Pay, M., Kunin, J. M., and Katzman, R. (1991). Rapidly progressive dementia in a patient with Lewy body variant of Alzheimer’s disease. Neurology 41, 1178–1180.
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R., and Van Hoesen, G. W. (1991). The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1, 103–116.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Benson, F., Davis, J., and Snyder, B. D. (1998). Posterior cortical atrophy. Arch. Neurol. 45, 789–793.
Bertram, L. (2008). “Dementias,” in Handbook of Clinical Neurology, Vol. 89, 3rd series, eds C. Duyckaerts and I. Litvan (Amsterdam: Elsevier), 223–232.
Bertram, L. (2011). Alzheimer’s genetics in the GWAS era: a continuing story of ‘replications and refutations.’ Curr. Neurol. Neurosci. Rep. 11, 246–253.
Biessels, J. G., Staekenborg, S., Brunner, E., Brayne, C., and Scheltens, P. (2006). Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 5, 64–74.
Boeve, B. F., Lang, A. E., and Litvan, I. (2003). Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann. Neurol. 54(Suppl. 5), S15–S19.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Braak, H., and Tredici, K. D. (2004). Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-β formation. Neurobiol. Aging 25, 713–718.
Braak, H., and Tredici, K. D. (2011). Alzheimer’s pathogenesis: is there neuron to neuron propagation? Acta Neuropathol. 121, 589–595.
Bronner, I. F., ter Meulen, B. C., Azmani, A., Severijnen, L. A., Willemsen, R., Kamphorst, W., Ravid, R., Heutink, P., and van Swieten, J. C. (2005). Hereditary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain 128, 2645–2653.
Cairns, N. J., Bigio, E. H., Mackenzie, I. R., Neumann, M., Lee, V. M., Hatanpaa, K. J., White, C. L. III, Scheinder, J. A., Grinberg, L. T., Halliday, G., Duyckaerts, C., Lowe, J. S., Holm, I. E., Tolnay, M., Okamoto, K., Yokoo, H., Murayama, S., Woulfe, J., Munoz, D. G., Dickson, D. W., Ince, P. G., Trojanowski, J. Q., Mann, D. M., and Consortium for Frontotemporal Lobar Degeneration. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 114, 5–22.
Callahan, L. M., Vaules, W. A., and Coleman, P. D. (1999). Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 275–287.
Casanova, M. F., Starkstein, S. E., and Jellinger, K. A. (2011). Clinicopathological correlates of behavioral and psychological symptoms of dementia. Acta Neuropathol. 122, 117–135.
Davidson, Y., Kelley, T., Mackenzie, I. R., Pickering-Brown, S., Du Plessis, D., Neary, D., Snowden, J. S., and Mann, D. M. (2007). Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533.
de la Torre, J. C. (2011). Three postulates to help identify the cause of Alzheimer’s disease. J. Alzheimers Dis. 24, 657–668.
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., Nicholson, A. M., Finch, N. A., Flynn, H., Adamson, J., Kouri, N., Wojtas, A., Sengdy, P., Hsiung, G. Y., Karydas, A., Seeley, W. W., Josephs, K. A., Coppola, G., Geschwind, D. W., Wszolek, Z. K., Feldman, H., Knopman, D. S., Petersen, R. C., Miller, B. L., Dickson, D. W., Boylan, K. B., Graff-Radford, N. R., and Rademakers, R. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256.
Delacorte, A., Sergeant, N., Wattez, A., Gauvreau, D., and Robittaile, Y. (1998). Vulnerable neuronal subsets in Alzheimer’s disease and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann. Neurol. 43, 193–204.
Delaère, P., Duyckaerts, C., He, Y., Piette, F., and Hauw, J. J. (1991). Subtypes and differential laminar distribution of βA4 deposits in Alzheimer’s disease: relationship with the intellectual status of 26 cases. Acta Neuropathol. 81, 328–335.
Dickson, D. W. (2010). Neuropathology of non-Alzheimer degenerative disorders. Int. J. Clin. Exp. Pathol. 3, 1–23.
Dickson, D. W. (2011). “Introduction to neurodegeneration: the molecular pathology of dementia and movement disorders,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, eds D. W. Dickson and R. O. Weller (Chichester: Wiley-Blackwell), 3–5.
Dickson, D. W., Bergeron, C., Chin, S. S., Duyckaerts, C., Horoupian, D., Ikeda, K., Jellinger, K., Lantos, P. L., Lippa, C. F., Mirra, S. S., Tabaton, M., Vonsattel, J. P., Wakabayashi, K., and Litvan, I. (2002). Office of rare diseases neuropathologic criteria for corticobasal degeneration. J. Neuropathol. Exp. Neurol. 61, 935–946.
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., Hardy, J., Leverenz, J. B., Del Tredici, K., Wszolek, Z. K., and Litvan, I. (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157.
Dickson, D. W., Kouri, N., Murray, M. E., and Josephs, K. A. (2011b). Neuropathology of frontotemporal lobar degeneration-tau (FTLD-Tau). J. Mol. Neurosci. 45, 384–489.
Dolan, H., Crain, B., Troncoso, J., Resnick, S. M., Zonderman, A. B., and O’Brien, R. J. (2010). Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann. Neurol. 68, 231–240.
Dubois, B., Feldman, H. H., Jacova, C., Cummings, J. L., DeKosky, S. T., Barbeger-Gateau, P., Delacourte, A., Frisoni, G., Fox, N. C., Galasko, D., Gauthier, S., Hampel, H., Jicha, G. A., Meguro, K., O’Brien, J., Pasquier, F., Robert, P., Rossor, M., Salloway, S., Sarazin, M., de Sousa, L. C., Stern, Y., Visser, P. J., and Scheltens, P. (2010). Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 9, 1118–1127.
Dubois, B., Feldman, H. H., Jacova, C., DeKosky, S. T., Barbeger-Gateau, P., Cummings, J., Delacourte, A., Galasko, D., Gauthier, S., Jicha, G., Meguro, K., O’Brien, J., Pasquier, F., Robert, P., Rossor, M., Salloway, S., Stern, Y., Visser, P. J., and Scheltens, P. (2007). Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 6, 734–746.
Duyckaerts, C., Delatour, B., and Potier, M. C. (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36.
Duyckaerts, C., and Hauw, J. J. (1997). Diagnosis and staging of Alzheimer disease. Neurobiol. Aging 18, S33–S42.
Emre, M., Arsland, D., Brown, R., Burn, D. J., Duyckaerts, C., Mizuno, Y., Broe, G. A., Cummings, J., Dickson, D. W., Gauthier, S., Goldman, J., Goetz, C., Korczyn, A., Lees, A., Levy, R., Litvan, I., McKeith, I., Olanow, W., Poewe, W., Quinn, N., Sampaio, C., Tolosa, E., and Dubois, B. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 22, 1689–1707.
Ferri, C. P., Prince, M., Brayne, C., Brodaty, H., Fratiglioni, L., Ganguli, M., Hall, K., Hasegawa, K., Hendrie, H., Huang, Y., Jorm, A., Mathers, C., Menezes, P. R., Rimmer, E., and Scazufca, M. (2005). Global prevelance of dementia: a Delphi consensus study. Lancet 366, 2112–2117.
Forman, M. S., Trojanowski, J. Q., and Lee, V. M. (2004). Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 10, 1055–1063.
Fritze, F., Ehrt, U., Sønnesyn, H., Kurz, M., Hortobágyi, T., Nore, S. P., Ballard, C., and Aarsland, D. (2011). Depression in mild dementia: associations with diagnosis, APOE genotype and clinical features. Int. J. Geriatr. Psychiatry 26, 1054–1061.
Gaig, C., Valldeoriola, F., Gelpi, E., Ezquerra, M., Llufriu, S., Buongiorno, M., Rey, M. J., Martí, M. J., Graus, F., and Tolosa, E. (2011). Rapidly progressive diffuse Lewy body disease. Mov. Disord. 26, 1316–1323.
Gelb, D. J., Oliver, E., and Gilman, S. (1999). Diagnostic criteria for Parkinson’s disease. Arch. Neurol. 56, 33–39.
Ghetti, B., Wszolek, Z. K., Boeve, B. F., Spina, S., and Goedert, M. (2011). “Frontotemporal dementia and parkinsonism linked to chromosome 17,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, eds D. W. Dickson and R. O. Weller (Chichester: Wiley-Blackwell), 110–134.
Giannakopoulos, G., von Gunten, A., Kövari, E., Gold, G., Herrmann, F. R., Hof, P. R., and Bouras, C. (2007). Stereological analysis of neuropil threads in the hippocampal formation: relationships with Alzheimer’s disease neuronal pathology and cognition. Neuropathol. Appl. Neurobiol. 33, 334–343.
Gibb, W. R., and Lees, A. J. (1998). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 51, 745–752.
Gomez-Isla, T., Price, J. L., McKeel, D. W., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occur in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500.
Gomez-Isla, T., Spires, T., Calignon, A., and Hyman, B. T. (2008). “Dementias,” in Handbook of Clinical Neurology, Vol. 89, 3rd series, eds C. Duyckaerts and I. Litvan (Amsterdam: Elsevier), 233–243.
Gorno-Tempini, M. L., Brambati, S. M., Ginex, V., Ogar, J., Dronker, N. F., Marcone, A., Perani, D., Garibotto, V., Cappa, S. F., and Miller, B. L. (2008). The logopenic/phonologic variant of primary progressive aphasia. Neurology 71, 1227–1234.
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., Ogar, J. M., Rohrer, J. D., Black, S., Boeve, B. F., Manes, F., Dronkers, N. F., Vandenberghe, R., Rascovsky, K., Patterson, K., Miller, B. L., Knopman, D. S., Hodges, J. R., Mesulam, M. M., and Grossman, M. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014.
Greene, J. D., Patterson, K., Xuereb, J., and Hodges, J. R. (1996). Alzheimer disease an nonfluent progressive aphasia. Arch. Neurol. 53, 1072–1078.
Grignon, Y., Duyckaerts, C., Bennecib, M., and Hauw, J. J. (1998). Cytoarchitectonic alterations in the supramarginal gyrus of late onset Alzheimer’s disease. Acta Neuropathol. 95, 395–406.
Grossman, M. (2010). Primary progressive aphasia: clinicopathological correlations. Nat. Rev. Neurol. 6, 88–97.
Haik, S., Brandel, J. P., Sazdovitch, V., Delasnerie-Lauprêtre, N., Peoc’h, K., Laplanche, J. L., Privat, N., Duyckaerts, C., Kemeny, J. L., Kopp, N., Laquerrière, A., Mohr, M., Deslys, J. P., Dormont, D., and Hauw, J. J. (2000). Dementia with Lewy bodies in a neuropathologic series of suspected Creutzfeldt-Jakob disease. Neurology 55, 1401–1404.
Halliday, G., Hely, M., Reid, W., and Morris, J. (2008). The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 115, 409–415.
Halliday, G. M., Holton, J. L., Revesz, T., and Dickson, D. W. (2011). Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 122, 187–204.
Halliday, G. M., and McCann, H. (2010). The progression of pathology in Parkinson’s disease. Ann. N. Y. Acad. Sci. 1184, 188–195.
Hamilton, R. L. (2000). Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohisto-chemistry. Brain Pathol. 10, 378–384.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Hauw, J. J., Daniel, S. E., Dickson, D., Horoupian, D. S., Jellinger, K., Lantos, P. L., McKee, A., Tabaton, M., and Litvan, I. (1994). Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44, 2015–2019.
Hodges, J. R., Davies, R. R., Xuereb, J. H., Casey, B., Broe, M., Bak, T. H., Kril, J. J., and Halliday, G. M. (2004). Clinicopathological correlates in frontotemporal dementia. Ann. Neurol. 56, 399–406.
Hof, P. R., Vogt, B. A., Bouras, C., and Morrison, J. H. (1997). Atypical form of Alzheimer’s disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res. 37, 3609–3625.
Hurtig, H. I., Trojanowski, J. Q., Galvin, J., Ewbank, D., Schmidt, M. L., Lee, V. M., Clark, C. M., Glosser, G., Stern, M. B., Gollomp, S. M., and Arnold, S. E. (2000). Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921.
Hyman, B. T., and Trojanowski, J. Q. (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 1095–1097.
Ince, P. G. (2011). “Dementia with Lewy bodies and Parkinson’s disease dementia,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, eds D. W. Dickson and R. O. Weller (Chichester: Wiley-Blackwell), 224–241.
Jellinger, K. A. (2008). A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 116, 1–16.
Jellinger, K. A. (2009). Criteria for the neuropathological diagnosis of dementing disorders: routes out of the swamp? Acta Neuropathol. 117, 101–110.
Jellinger, K. A., and Attems, J. (2011a). Rapidly progressing diffuse Lewy body disease. Mov. Disord. 26, 2584–2585.
Jellinger, K. A., and Kovacs, G. G. (2011b). Clinico-pathological correlations in neurodegeneration. Acta Neuropathol. 122, 115–116.
Johnson, J. K., Head, E., Kim, R., Starr, A., and Cotman, C. W. (1999). Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch. Neurol. 56, 1233–1239.
Josephs, K. A., and Dickson, D. W. (2003). Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov. Disord. 18, 1018–1026.
Josephs, K. A., Hodges, J. R., Snowden, J. S., Mackenzie, I. R., Neumann, M., Mann, D. M., and Dickson, D. W. (2011). Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 122, 137–153.
Josephs, K. A., Petersen, R. C., Knopman, D. S., Boeve, B. F., Whitwell, J. L., Duffy, J. R., Parisi, J. E., and Dickson, D. W. (2006). Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66, 41–48.
Josephs, K. A., Whitwell, J. L., Parisi, J. E., Petersen, R. C., Boeve, B. F., Jack, C. R. Jr., and Dickson, D. W. (2010). Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur. J. Neurol. 17, 969–975.
Kalaitzakis, M. E., Graeber, M. B., Gentleman, S. M., and Pearce, R. K. (2009). Evidence against a reliable staging system of alpha-synuclein pathology in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 35, 125–126.
Kertesz, A. (2003). Pick complex: an integrative approach to frontotemporal dementia: primary progressive aphasia, corticobasal degeneration and progressive supranuclear palsy. Neurologist 9, 311–317.
Kertesz, A., Davidson, W., and Munoz, D. G. (1999). Clinical and pathological overlap between frontotemporal dementia, primary progressive aphasia and corticobasal degeneration: the Pick complex. Dement. Geriatr. Cogn. Disord. 10(Suppl. 1), 46–49.
Kertesz, A., McMonagle, P., Blair, M., Davidson, W., and Munoz, D. G. (2005). The evolution and pathology of frontotemporal dementia. Brain 128, 1996–2005.
Knibb, J. A., Xuereb, J. H., Patterson, K., and Hodges, J. R. (2006). Clinical and pathological characterization of progressive aphasia. Ann. Neurol. 59, 156–165.
Kövari, E., Gold, G., Herrmann, F. R., Canuto, A., Hof, P. R., Bouras, C., and Giannakopoulos, P. (2003). Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 106, 83–88.
Kövari, E., Horvath, J., and Bouras, C. (2009). Neuropathology of Lewy body disorders. Brain Res. Bull. 80, 203–210.
Lee, E. B., Lee, V. M., and Trojanowski, J. Q. (2011a). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50.
Lee, S. E., Rabinovici, G. D., Mayo, M. C., Wilson, S. M., Seeley, W. W., DeArmond, S. J., Huang, E. J., Trojanowski, J. Q., Growdon, M. E., Jang, J. Y., Sidhu, M., See, T. M., Karydas, A. M., Gorno-Tempini, M. L., Boxer, A. L., Weiner, M. W., Geschwind, M. D., Rankin, K. P., and Miller, B. L. (2011b). Clinicopathological correlations in corticobasal degeneration. Ann. Neurol. 70, 327–340.
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159.
Levine, D. N., Lee, J. M., and Fisher, C. M. (1993). The visual variant of Alzheimer’s disease: clinicopathologic case study. Neurology 43, 305–313.
Ling, H., O’Sullivan, S. S., Holton, J. L., Revesz, T., Massey, L. A., Williams, D. R., Paviour, D. C., and Lees, A. J. (2010). Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 133, 2045–2057.
Lippa, C. F., Duda, J. E., Grossman, M., Hurtig, H. I., Aarsland, D., Boeve, B. F., Brooks, D. J., Dickson, D. W., Dubois, B., Emre, M., Fahn, S., Farmer, J. M., Galasko, D., Galvin, J. E., Goetz, C. G., Growdon, J. H., Gwinn-Hardy, K. A., Hardy, J., Heutink, P., Iwatsubo, T., Kosaka, K., Lee, V. M., Leverenz, J. B., Masliah, E., McKeith, I. G., Nussbaum, R. L., Olanow, C. W., Ravina, B. M., Singleton, A. B., Tanner, C. M., Trojanowski, J. Q., and Wszolek, Z. K. (2007). DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68, 812–819.
Litvan, I., Agid, Y., Calne, D., Campbell, G., Dubois, B., Duvoisin, R. C., Goetz, C. G., Golbe, L. I., Grafman, J., Growdon, J. H., Hallett, M., Jankovic, J., Quinn, N. P., Tolosa, E., and Zee, D. S. (1996). Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47, 1–9.
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., Perry, R. H., Trojanowski, J. Q., Mann, D. M., and Lee, V. M. (2011a). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113.
Mackenzie, I. R., Neumann, M., Cairns, N. J., Munoz, D. G., and Isaacs, A. M. (2011b). Novel types of frontotemporal lobar degeneration: beyond tau and TDP-43. J. Mol. Neurosci. 45, 402–408.
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., Kovacs, G. C., Ghetti, B., Halliday, G., Holm, I. E., Ince, P. G., Kamphorst, W., Revesz, T., Rozemuller, A. J., Kumar-Singh, S., Akiyama, H., Baborie, A., Spina, S., Dickson, D. W., Trojanowski, J. Q., and Mann, D. M. (2010a). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4.
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010b). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007.
McKhann, G., Drachman, D. A., Folstein, M., Katzman, R., Price, D. L., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease – report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34, 939–944.
McKeith, I., Mintzer, J., Aarsland, D., Burn, D., Chiu, H., Cohen-Mansfield, J., Dickson, D., Dubois, B., Duda, J. E., Feldman, H., Gauthier, S., Halliday, G., Lawlor, B., Lippa, C., Lopez, O. L., Carlos Machado, J., O’Brien, J., Playfer, J., and Reid, W. (2004). Dementia with Lewy bodies. Lancet Neurol. 3, 19–28.
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., Cummings, J., Duda, J. E., Lippa, C., Perry, E. K., Aarsland, D., Arai, H., Ballard, C. G., Boeve, B., Burn, D. J., Costa, D., Del Ser, T., Dubois, B., Galasko, D., Gauthier, S., Goetz, C. G., Gomez-Tortosa, E., Halliday, G., Hansen, L. A., Hardy, J., Iwatsubo, T., Kalaria, R. N., Kaufer, D., Kenny, R. A., Korczyn, A., Kosaka, K., Lee, V. M., Lees, A., Litvan, I., Londos, E., Lopez, O. L., Minoshima, S., Mizuno, Y., Molina, J. A., Mukaetova-Ladinska, E. B., Pasquier, F., Perry, R. H., Schulz, J. B., Trojanowski, J. Q., and Yamada, M. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65, 1863–1872.
McKeith, I. G., Galasko, D., Kosaka, K., Perry, E. K., Dickson, D. W., Hansen, L. A., Salmon, D. P., Lowe, J., Mirra, S. S., Byrne, E. J., Lennox, G., Quinn, N. P., Edwardson, J. A., Ince, P. G., Bergeron, C., Burns, A., Miller, B. L., Lovestone, S., Collerton, D., Jansen, E. N., Ballard, C., de Vos, R. A., Wilcock, G. K., Jellinger, K. A., and Perry, R. H. (1996). Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47, 1113–1124.
Mesulam, M., Wicklund, A., Johnson, N., Rogalski, E., Léger, G. C., Rademaker, A., Weintraub, S., and Bigio, E. H. (2008). Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann. Neurol. 63, 709–719.
Mirra, S. S., Heyman, A., McKeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., Vogel, F. S., Hughes, J. P., van Belle, G., and Berg, L. (1991). The Consortium to Establish a Registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486.
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., and Dickson, D. W. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11.
Morgan, K. (2011). Three new pathways leading to Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 37, 353–357.
Munoz, D. G., Morris, H. R., and Rossor, M. (2011). “Picks disease,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, eds D. W. Dickson and R. O. Weller (Chichester: Wiley-Blackwell), 156–164.
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., Freedman, M., Kertesz, A., Robert, P. H., Albert, M., Boone, K., Miller, B. L., Cummings, J., and Benson, D. F. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554.
Neary, D., Snowden, J. S., and Mann, D. M. (2005). Frontotemporal dementia. Lancet Neurol. 4, 771–780.
Nelson, P. T., Jicha, G. A., Kryscio, R. J., Abner, E. L., Schmitt, F. A., Cooper, G., Xu, L. O., Smith, C. D., and Markesbery, W. R. (2010). Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J. Neurol. 257, 359–366.
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., McCluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H., Trojanowski, J. Q., and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Nishimura, M., Namba, Y., Ikeda, K., and Oda, M. (1992). Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci. Lett. 143, 35–38.
Oksengard, A. R., Cavallin, L., Axelsson, R., Andersson, C., Nägga, K., Winblad, B., Eriksdotter-Jönhagen, M., and Wahlund, L. O. (2010). Lack of accuracy for the proposed “Dubois criteria” in Alzheimer’s disease: a validation study from the Swedish brain power initiative. Dement. Geriatr. Cogn. Disord. 30, 374–380.
Parkkinen, L., Pirttilä, T., and Alafuzoff, I. (2008). Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 115, 399–407.
Pickering-Brown, S. M., Rollinson, S., Du Plessis, D., Morrison, K. E., Varma, A., Richardson, A. M., Neary, D., Snowden, J. S., and Mann, D. M. (2008). Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131, 721–731.
Podewils, L. J., Guallar, E., Kuller, L. H., Fried, L. P., Lopez, O. L., Carlson, M., and Lyketsos, C. G. (2005). Physical activity, APOE genotype, and dementia risk: findings from the Cardiovascular Health Cognition Study. Am. J. Epidemiol. 161, 639–651.
Ratnavalli, E., Brayne, C., Dawson, K., and Hodges, J. R. (2002). The prevalence of frontotemporal dementia. Neurology 58, 1615–1621.
Rebeiz, J. J., Kolodny, E. H., and Richardson, E. P. Jr. (1967). Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans. Am. Neurol. Assoc. 92, 23–26.
Reitz, C., Brayne, C., and Mayeux, R. (2011). Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152.
Renner, J. A., Burns, J. M., Hou, C. E., McKeel, D. W., Storandt, M., and Morris, J. C. (2004). Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 63, 1175–1180.
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., Schymick, J. C., Laaksovirta, H., van Swieten, J. C., Myllykangas, L., Kalimo, H., Paetau, A., Abramzon, Y., Remes, A. M., Kaganovich, A., Scholz, S. W., Duckworth, J., Ding, J., Harmer, D. W., Hernandez, D. G., Johnson, J. O., Mok, K., Ryten, M., Trabzuni, D., Guerreiro, R. J., Orrell, R. W., Neal, J., Murray, A., Pearson, J., Jansen, I. E., Sondervan, D., Seelaar, H., Blake, D., Young, K., Halliwell, N., Callister, J. B., Toulson, G., Richardson, A., Gerhard, A., Snowden, J., Mann, D., Neary, D., Nalls, M. A., Peuralinna, T., Jansson, L., Isoviita, V. M., Kaivorinne, A. L., Hölttä-Vuori, M., Ikonen, E., Sulkava, R., Benatar, M., Wuu, J., Chiò, A., Restagno, G., Borghero, G., Sabatelli, M., Italsgen Consortium, Heckerman, D., Rogaeva, E., Zinman, L., Rothstein, J. D., Sendtner, M., Drepper, C., Eichler, E. E., Alkan, C., Abdullaev, Z., Pack, S. D., Dutra, A., Pak, E., Hardy, J., Singleton, A., Williams, N. M., Heutink, P., Pickering-Brown, S., Morris, H. R., Tienari, P. J., and Traynor, B. J. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268.
Riley, K. P., Snowdon, D. A., and Markesbery, W. R. (2002). Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann. Neurol. 51, 567–577.
Roe, C. M., Xiong, C., Miller, J. P., and Morris, J. C. (2007). Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology 68, 223–228.
Roeber, S., Mackenzie, I. R., Kretzschmar, H. A., and Neumann, M. (2008). TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 116, 147–157.
Rohrer, J. D., Lashley, T., Schott, J. M., Warren, J. E., Mead, S., Isaacs, A. M., Beck, J., Hardy, J., de Silva, R., Warrington, E., Troakes, C., Al-Sarraj, S., King, A., Borroni, B., Clarkson, M. J., Ourselin, S., Holton, J. L., Fox, N. C., Revesz, T., Rossor, M. N., and Warren, J. D. (2011). Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134, 2565–2581.
Rohrer, J. D., Warren, J. D., Omar, R., Mead, S., Beck, J., Revesz, T., Holton, J., Stevens, J. M., Al-Sarraj, S., Pickering-Brown, S. M., Hardy, J., Fox, N. C., Collinge, J., Warrington, E. K., and Rossor, M. N. (2008). Parietal lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch. Neurol. 65, 506–513.
Rosso, S. M., Donker Kaat, L., Baks, T., Joosse, M., de Koning, I., Pijnenburg, Y., de Jong, D., Dooijes, D., Kamphorst, W., Ravid, R., Niermeijer, M. F., Verheij, F., Kremer, H. P., Scheltens, P., van Duijn, C. M., Heutink, P., and van Swieten, J. C. (2003). Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 126, 2016–2022.
Saito, Y., Kawashima, A., Ruberu, N. N., Fujiwara, H., Koyama, S., Sawabe, M., Arai, T., Nagura, H., Yamanouchi, H., Hasegawa, M., Iwatsubo, T., and Murayama, S. (2003). Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 62, 644–654.
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., Rosi, B. L., Gusella, J. F., Crapper-MacLachlan, D. R., Alberts, M. J., Hulette, C., Crain, B., Goldgaber, D., and Roses, A. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472.
Scaravilli, T., Tolosa, E., and Ferrer, I. (2005). Progressive supranuclear palsy and corticobasal degeneration: lumping versus splitting. Mov. Disord. 20, S21–S28.
Seelaar, H., Rohrer, J. D., Pijnenburg, Y. A., Fox, N. C., and van Swieten, J. C. (2011). Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J. Neurol. Neurosurg. Psychiatr. 82, 476–486.
Selikhova, M., Williams, D. R., Kempster, P. A., Holton, J. L., Revesz, T., and Less, A. J. (2009). A clinicopathological study of subtypes in Parkinson’s disease. Brain 132, 2947–2957.
Snowden, J., Neary, D., and Mann, D. (2007). Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 114, 31–38.
Snowden, J. S., Hu, Q., Rollinson, S., Halliwell, N., Robinson, A., Davidson, Y. S., Momeni, P., Baborie, A., Griffiths, T. D., Jaros, E., Perry, R. H., Richardson, A., Pickering-Brown, S. M., Neary, D., and Mann, D. M. (2011). The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 122, 99–110.
Stewart, H., Rutherford, N. J., Briemberg, H., Krieger, C., Cashman, N., Fabros, M., Baker, M., Fok, A., Dejesus-Hernandez, M., Eisen, A., Rademakers, R., and Mackenzie, I. R. (2012). Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 123, 409–417.
Taipa, R., Tuna, A., Damásio, J., Pinto, P. S., Cavaco, S., Pereira, S., Milterberger-Miltenyi, G., Galimberti, D., and Melo-Pires, M. (2012). Clinical, neuropathological and genetic characteristics of the novel IVS9+1delG GRN mutation in a patient with frontotemporal dementia. J. Alzheimers Dis. [Epub ahead of print].
Tang-Wai, D. F., Graff-Radford, N. R., Boeve, B. F., Dickson, D. W., Parisi, J. E., Crook, R., Caselli, R. J., Knopman, D. S., and Petersen, R. C. (2004). Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 63, 1168–1174.
Taylor, K. I., Probst, A., Miserez, A. R., Monsch, A. U., and Tolnay, M. (2008). Clinical course of neuropathologically confirmed frontal-variant Alzheimer’s disease. Nat. Clin. Pract. Neurol. 4, 226–232.
Thal, D. R., Sassing, I., Schultz, C., Haass, C., Braak, E., and Braak, H. (1999). Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Abeta. J. Neuropathol. Exp. Neurol. 58, 210–216.
Urwin, H., Josephs, K. A., Rohrer, J. D., Mackenzie, I. R., Neumann, M., Authier, A., Seelaar, H., Van Swieten, J. C., Brown, J. M., Johannsen, P., Nielsen, J. E., Holm, I. E., FReJA Consortium, Dickson, D. W., Rademakers, R., Graff-Radford, N. R., Parisi, J. E., Petersen, R. C., Hatanpaa, K. J., White, C. L. III, Weiner, M. F., Geser, F., Van Deerlin, V. M., Trojanowski, J. Q., Miller, B. L., Seeley, W. W., van der Zee, J., Kumar-Singh, S., Engelborghs, S., De Deyn, P. P., Van Broeckhoven, C., Bigio, E. H., Deng, H. X., Halliday, G. M., Kril, J. J., Munoz, D. G., Mann, D. M., Pickering-Brown, S. M., Doodeman, V., Adamson, G., Ghazi-Noori, S., Fisher, E. M., Holton, J. L., Revesz, T., Rossor, M. N., Collinge, J., Mead, S., and Isaacs, A. M. (2010). FUS pathology defines the majority of tau and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 120, 33–41.
van Swieten, J. C., and Heutink, P. (2008). Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 7, 965–974.
Vekrellis, K., Xilouri, M., Emmanouilidou, E., Rideout, H. J., and Stefanis, L. (2011). Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 10, 1015–1025.
Wadia, P. M., and Lang, A. E. (2007). The many faces of corticobasal degeneration. Parkinsonism Relat. Disord. 13, S336–S340.
Wang, D., and Munoz, D. G. (1995). Qualitative and quantitative differences in senile plaque dystrophic neuritis of Alzheimer’s disease and normal aged brain. J. Neuropathol. Exp. Neurol. 54, 548–556.
Keywords: neurodegenerative dementia, neuropathology, clinical syndromes
Citation: Taipa R, Pinho J and Melo-Pires M (2012) Clinico-pathological correlations of the most common neurodegenerative dementias. Front. Neur. 3:68. doi: 10.3389/fneur.2012.00068
Received: 06 February 2012; Paper pending published: 22 February 2012;
Accepted: 10 April 2012; Published online: 01 May 2012.
Edited by:
João Massano, University of Porto, PortugalReviewed by:
Tibor Hortobágyi, University of Debrecen, HungaryDouglas Watt, Quincy Medical Center/Cambridge Health Alliance, USA
Copyright: © 2012 Taipa, Pinho and Melo-Pires. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ricardo Taipa, Neuropathology Unit, Hospital de Santo António, Centro Hospitalar do Porto, Largo Prof. Abel Salazar – 4099-001, Porto, Portugal. e-mail:cnRhaXBhLm5ldXJvcGF0QGNocG9ydG8ubWluLXNhdWRlLnB0