 Erzsebet Kovesdi1
Erzsebet Kovesdi1 Alaa Kamnaksh2,3
Alaa Kamnaksh2,3 Daniel Wingo2 Farid Ahmed2,3
Daniel Wingo2 Farid Ahmed2,3 Neil E. Grunberg3,4
Neil E. Grunberg3,4 Joseph B. Long5 Christine E. Kasper1
Joseph B. Long5 Christine E. Kasper1 Denes V. Agoston2*
Denes V. Agoston2*- 1 U.S. Department of Veterans Affairs, Veterans Affairs Central Office, Washington, DC, USA
- 2 Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, Bethesda, MD, USA
- 3 Center for Neuroscience and Regenerative Medicine at the Uniformed Services University, Bethesda, MD, USA
- 4 Department of Medical and Clinical Psychology, School of Medicine, Uniformed Services University, Bethesda, MD, USA
- 5 Blast-Induced Neurotrauma Branch, Center for Military Psychiatry and Neuroscience, Walter Reed Army Institute of Research, Silver Spring, MD, USA
Mild traumatic brain injury (mTBI) represents a significant challenge for the civilian and military health care systems due to its high prevalence and overall complexity. Our earlier works showed evidence of neuroinflammation, a late onset of neurobehavioral changes, and lasting memory impairment in a rat model of mild blast-induced TBI (mbTBI). The aim of our present study was to determine whether acute treatment with the non-steroidal anti-inflammatory drug minocycline (Minocin®) can mitigate the neurobehavioral abnormalities associated with mbTBI, Furthermore, we aimed to assess the effects of the treatment on select inflammatory, vascular, neuronal, and glial markers in sera and in brain regions associated with anxiety and memory (amygdala, prefrontal cortex, ventral, and dorsal hippocampus) following the termination (51 days post-injury) of the experiment. Four hours after a single exposure to mild blast overpressure or sham conditions, we treated animals with a daily dose of minocycline (50 mg/kg) or physiological saline (vehicle) for four consecutive days. At 8 and 45 days post-injury, we tested animals for locomotion, anxiety, and spatial memory. Injured animals exhibited significantly impaired memory and increased anxiety especially at the later testing time point. Conversely, injured and minocycline treated rats’ performance was practically identical to control (sham) animals in the open field, elevated plus maze, and Barnes maze. Protein analyses of sera and brain regions showed significantly elevated levels of all of the measured biomarkers (except VEGF) in injured and untreated rats. Importantly, minocycline treatment normalized serum and tissue levels of the majority of the selected inflammatory, vascular, neuronal, and glial markers. In summary, acute minocycline treatment appears to prevent the development of neurobehavioral abnormalities likely through mitigating the molecular pathologies of the injury in an experimental model of mbTBI.
Introduction
Traumatic brain injury (TBI) is a prominent health concern worldwide as it is one of the major causes of death and chronic disability (Hyder et al., 2007). The mild form of traumatic brain injury (mTBI) has become an especially significant challenge for the civilian (Thurman et al., 1999) and the military healthcare systems (Hoge et al., 2008; Tanielian and Jaycox, 2008) due to its high prevalence and the absence of serious acute symptoms following injury. Blast-induced mTBI (mbTBI) was the most frequent form of mTBIs sustained during recent military conflicts (Warden, 2006; Terrio et al., 2009). There is currently no objective diagnosis for mbTBI, a minimal understanding of its underlying pathologies, and consequently a lack of specific, evidence based treatments.
Symptoms of blast-induced TBI (bTBI) include increased anxiety as well as memory impairment that may not be detectable for weeks or months after the exposure (Ryan and Warden, 2003; Okie, 2005; Nelson et al., 2009; Terrio et al., 2009; Cernak and Noble-Haeusslein, 2010; Hoffer et al., 2010). The delayed onset of neurobehavioral impairments suggests a lasting secondary injury process involving distinct brain regions (Moser and Moser, 1998). The ventral hippocampus (VHC) along with the prefrontal cortex (PFC) and the amygdala (AD) are involved in mediating anxiety, while the dorsal hippocampus (DHC) is involved in mediating spatial learning and memory (Henke, 1990; Moser and Moser, 1998; Bremner, 2005, 2007). Using a rat model of bTBI, we found that a single mild blast overpressure exposure results in increased anxiety and memory impairment (Kovesdi et al., 2011; Kwon et al., 2011). Importantly, the memory impairment was not detectable within the first week of the exposure; it became significant 2 weeks post-injury and persisted for at least 2 months after (Kovesdi et al., 2011; Kwon et al., 2011).
Our immunohistochemical and proteomics analyses of these animals showed evidence of neuronal and glial cell loss, gliosis, and neuroinflammation at 2 months post-injury. In addition to an increased presence of microglia in the DHC and the VHC of injured animals as well as increased tissue levels of interleukin-6 (IL-6) and interferon-gamma (IFNγ) in these brain regions. Neuroinflammation can adversely affect neuronal function by directly causing neuronal cell death as well as increasing neuron vulnerability to noxious factors like excitotoxins, which are also elevated after injury (Arvin et al., 1996; Morganti-Kossmann et al., 2002; Cacci et al., 2005; Floyd and Lyeth, 2007; Kochanek et al., 2008; Agoston et al., 2009; Agostinho et al., 2010; Czlonkowska and Kurkowska-Jastrzebska, 2011; Robel et al., 2011). Based on our previous evidence linking neuroinflammation to neurobehavioral abnormalities (Kovesdi et al., 2011), we hypothesized that anti-inflammatory treatment may improve the functional outcome in mbTBI.
To test our hypothesis, we selected the anti-inflammatory drug minocycline for several reasons. Minocycline hydrochloride easily crosses the blood brain barrier (BBB), is well characterized, safe, FDA approved, and has been used experimentally and clinically (Macdonald et al., 1973; Saivin and Houin, 1988). Similar to its tetracycline analogs, the side effects of minocycline treatment are mild and include discoloration of the teeth, gastrointestinal irritability, and candidiasis (Fanning et al., 1977; Gump et al., 1977). In humans, long-term treatment is generally safe and is well tolerated up to 200 mg/day. In animals, the lethal dose of minocycline is very high at 3600 mg/kg (Blum et al., 2004); the “therapeutic” dosage utilized in animal experiments ranges between 10 and 90 mg/kg with an average of 50 mg/kg for daily treatments (e.g., Wells et al., 2003; Stirling et al., 2004; Festoff et al., 2006; Li and McCullough, 2009; Abdel Baki et al., 2010; Lee et al., 2010; Siopi et al., 2011; Wixey et al., 2011; Ng et al., 2012).
Minocycline has been successfully used in various animal models of brain and spinal cord injuries as well as neurodegenerative diseases like Huntington’s (Blum et al., 2004), where it was shown to reduce tissue damage and inflammation, and improve neurological outcome (Yrjanheikki et al., 1999; Chen et al., 2000; Kriz et al., 2002; Wu et al., 2002; Wells et al., 2003; Xu et al., 2004; Zemke and Majid, 2004; Festoff et al., 2006; Marchand et al., 2009). Using a rat model of mbTBI, we report that acute treatment with minocycline mitigates the inflammatory response to injury and results in normalized neurobehavior.
Materials and Methods
Experimental Groups and Housing Conditions
Thirty-two male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA, USA) were used, weighing 245–265 g at the beginning of the experiment. All animals were kept under normal housing conditions (two rats/cage) in a reverse 12–12 h light-dark cycle and provided with food and water ad libitum for the entire length of the study. Following baseline behavioral testing (described below), animals were assigned to one of the following experimental groups: (1) sham saline treated (sham-vehicle; n = 8) and (2) sham minocycline treated (sham-mino; n = 8), which served as controls for (3) blast injured saline treated (injured-vehicle; n = 8) and (4) blast injured-minocycline treated (injured-mino; n = 8), respectively. All animals were handled according to protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Uniformed Services University (USU).
Behavioral Tests
Prior to injury, all rats underwent baseline behavioral assessments for general locomotor activity by the open field (OF) test, and for anxiety by the elevated plus maze (EPM). Rats were also trained for five consecutive days in the Barnes maze (BM) for spatial learning and memory. The baseline test results (data not shown) were used to create the aforementioned experimental groups with no statistical significance among them. Following injury or sham, two behavioral test sessions were conducted starting at 8 and 45 days. The experimental schedule is illustrated in Figure A1 in Appendix. Within each testing session, the behavioral tests were performed on separate days in the following order: OF (day 1), EPM (day 2), and BM (days 3–7). All behavioral tests were performed during animals’ dark cycle.
Open field
Tests were performed using AccuScan’s infrared light beams OF system (AccuScan Instruments, Inc.) at baseline and 1, 8, and 45 days post-injury. The OF system is a 16.5 × 16.5 × 13 (L × W × H) inches clear Plexiglas arena with a perforated lid. The system uses 16 × 16 grid light beam arrays in the X and Y axes to measure locomotor activity. The system detects beam breaks by the animal and determines the location of the rat within the cage. During the 60 min testing period, horizontal activity (number of beam breaks) and resting time (time spent with inactivity greater than or equal to 1 s) were measured. Data for each animal were recorded and analyzed automatically with Fusion 3.4 software (AccuScan Instruments, Inc.). The horizontal activity and resting time are presented as the average performance of all animals in each experimental group ±standard error of the mean (SEM) at each of the individual time points.
Elevated plus maze
The EPM is an ethologically relevant assessment of anxiety levels in rodents (Carobrez and Bertoglio, 2005; Salzberg et al., 2007; Walf and Frye, 2007). Tests were carried out prior to injury and at 9 and 46 days post-injury as described earlier in details (Kovesdi et al., 2011). Briefly, rats were placed one by one in the center of the maze facing one of the open arms. During the 5 min testing session, each animal was allowed to explore the maze freely while its movement was video-tracked. Time spent in the open and the closed arms (seconds) was recorded for each animal using ANY-maze 4.2 Software (Stoelting Company, Wood Dale, IL, USA). The maze was cleaned with a 30% ethanol solution between each trial. Data are presented as the average time (in seconds) spent in the open vs. the closed arms of the maze in each experimental group ±SEM.
Barnes maze
Barnes maze represents a widely used and less stressful alternative to the Morris water maze for assessing spatial memory in rodents (Barnes, 1979; Maegele et al., 2005; Doll et al., 2009; Harrison et al., 2009). Tests were carried out prior to injury (training session), and at 10 and 47 days post-injury (Test Session I and II, respectively; Kovesdi et al., 2011). The maze is a circular platform (1.2 m in diameter) that contains 18 evenly spaced holes around the periphery. One of the holes is the entrance to a darkened escape box that is not visible from the surface of the board. The position of the escape chamber relative to the other holes and the testing room remains fixed during all BM trials. On the first day of the training session, each rat was placed in the escape box and covered for 30 s. The escape box was then removed with the animal inside and moved to the center of the maze. The rat was allowed to explore the maze for a few seconds after which it was returned to its home cage. In the second and third trial (only day 1 of the BM training session has three trials), the same rat was placed under a start box in the center of the maze for 30 s. The start box was removed and the rat was allowed to explore freely to find the escape box. Training sessions ended after the animal had entered the escape box or when a pre-determined time (240 s) had elapsed. If the animal had not found the escape box during the given time period, it was placed in the escape box for 1 min at the end of the trial. During the baseline BM session, animals were trained until their daily latency time averaged 10 s. The two post-injury BM test sessions were run for five consecutive days; every rat was tested twice per day as described above. In each trial, the latency to enter the escape box was measured and recorded using ANY-maze 4.2 Software (Stoelting Company, Wood Dale, IL, USA). The escape box and the maze were cleaned with a 30% ethanol solution between each trial and animal. Data are presented as the average latency times of two daily trials per animal per experimental group ±SEM.
Mild Blast Injury
On the day of the injury all rats (average weight ∼300 g) were transferred to Walter Reed Army Institute of Research (Silver Spring, MD, USA) as described in detail (Kamnaksh et al., 2011). Sixteen rats were exposed to whole body mbTBI as described earlier (Long et al., 2009; Kovesdi et al., 2011; Kwon et al., 2011). Briefly, rats were anesthetized with 4% Isoflurane for 6 min in an induction chamber (Forane, Baxter Healthcare Corporation, Deerfield, IL, USA), placed in an animal holder within the shock tube in a transverse prone position, and exposed to whole body blast overpressure (20.6 ± 3 psi) while wearing chest protection. The other 16 rats were similarly anesthetized, placed in the shock tube, but were not exposed to blast overpressure (sham). Following blast injury or sham, rats were moved back to their home cages and transported back to the USU animal facility.
Pharmacological Treatment
Four hours after injury or sham, rats received a total volume of 0.25 ml/100 g body weight of either physiological saline alone (vehicle) or 50 mg/kg of clinical grade minocycline (Minocin®, Triax Pharmaceuticals, Italy) dissolved in saline (mino) intraperitoneally (i.p.). Animals received minocycline or saline for four consecutive days at identical times each day. Our minocycline dosage and treatment paradigm was based on previous studies using rodent models of various neurological conditions where minocycline was administered i.p. at an average dose of 50 mg/kg (see Table A1 in Appendix).
Tissue Collection and Processing
At the completion of the last behavioral test session (51 days post-injury or sham), animals were placed inside an induction chamber saturated with Isoflurane and deeply anesthetized until a tail pinch produced no reflex movement. Anesthesia was maintained using a mask/nose cone attached to the anesthetic vaporizer and blood was collected (1.5 ml) from a tail vein; serum was prepared as described earlier (Kwon et al., 2011). For measuring tissue levels of protein markers, rats were decapitated and brains were immediately removed and placed on ice. The amygdala (AD), PFC, VHC, and DHC were dissected, frozen, and stored at −80°C until use as described earlier (Kwon et al., 2011).
Protein measurements
Sample preparation, printing, scanning, and data analysis of serum and brain regions were performed using Reverse Phase Protein Microarray (RPPM) as described earlier (Kovesdi et al., 2011; Kwon et al., 2011). Briefly, frozen brain tissues were pulverized in liquid nitrogen, the powder was transferred into a lysis buffer (Thermo Fisher, Waltham, MA, USA) with protease and phosphatase inhibitors (Thermo Fisher), sonicated, centrifuged, and the supernatants aliquoted and stored at −80°C. Protein concentrations were measured by BCA assay (Thermo Fisher). Blood samples were centrifuged at 10,000 × g for 15 min at 4°C; supernatants were aliquoted, flash-frozen, and stored at −80°C.
Tissue samples were diluted in print buffer and then subjected to an 11-point serial 1:2 dilution and transferred into Genetix 384-well plates (X7022, Fisher Scientific, Pittsburg, PA, USA) using a JANUS Varispan Integrator and Expanded Platform Workstation (PerkinElmer, Waltham, MA, USA). Plates were transferred into an Aushon 2470 Arrayer (Aushon Biosystem, Billerica, MA, USA) to be printed on ONCYTE Avid (brain samples) or ONCYTE Nova (serum samples) single-pad nitrocellulose coated glass slides (Grace Bio-Labs, Bend, OR, USA; Gyorgy et al., 2010).
Primary antibodies (Table A2 in Appendix) were diluted to 10× the optimal Western analysis concentration in antibody incubation buffer as described earlier (Gyorgy et al., 2010). The primary antibody solution was incubated overnight at 4°C with a cover slip. The following day slides were washed and then incubated with an Alexa Fluor® 635 goat anti-mouse (Cat# A-31574), goat anti-rabbit (Cat# A-31576), or rabbit anti-goat IgG (H + L; Cat# A-21086) secondary antibodies from Invitrogen at 1:6000 dilution in antibody incubation buffer for 1 h at room temperature. After washing and drying, fluorescent signals were measured by a Scan Array Express HT microarray scanner (Perkin Elmer, Waltham, MA, USA) using a 633 nm wavelength laser and a 647 nm filter.
Data from the scanned images were imported into a Microsoft Excel-based bioinformatics program developed in-house for analysis (Gyorgy et al., 2010). The linear regression of the log–log data was calculated after the removal of flagged data, which include signal to noise ratios of less than 2, spot intensities in the saturation range or noise range, or high variability between duplicate spots (>10–15%). The total amount of antigen is determined by the y-axis intercept (Y-cept; Gyorgy et al., 2010). Data is reported as the mean Y-cept ±SEM.
Corticosterone assay
Serum corticosterone (CORT) levels were measured with Cayman’s Corticosterone EIA Kit according to the manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI, USA). Each sample was diluted 1:500 and measured in triplicate (Kwon et al., 2011). Data is reported as the mean concentration (in pg/mg) ±SEM.
Statistical Analysis
All data were analyzed using Graph Pad Instat software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was verified by one-way analysis of variance (ANOVA), followed by Tukey post hoc test for multiple comparison. Differences with a p value of <0.05 were considered significant.
Results
Behavioral Tests
One day following blast exposure, injured rats showed reduced horizontal activity and slightly increased resting time in the OF compared to sham animals, but the differences were not statistically significant (Figure 1A). At 8 days post-injury, the horizontal activity of injured-vehicle animals further decreased. On the other hand, injured-mino rats had a similar horizontal activity to animals in the two sham groups. The horizontal activity of animals in all groups was the lowest at 45 days after injury. Similarly, animals in all experimental groups spent more time resting with injured-vehicle animals spending significantly more time resting than animals in the other three groups (Figure 1B).
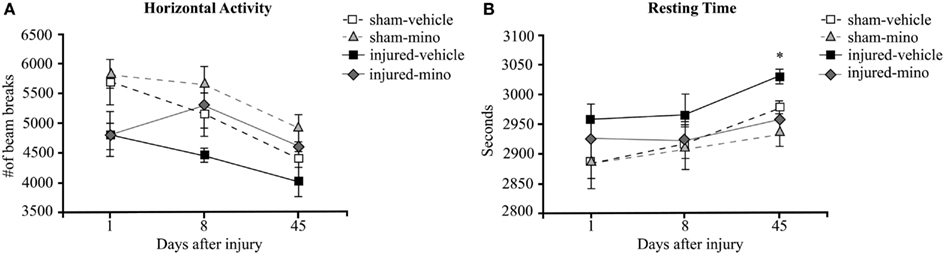
Figure 1. The effect of injury and minocycline treatment on basic locomotor activities at different time points after mbTBI. (A) Horizontal activity (number of beam breaks), and (B) Resting time (seconds) were measured in Open field. Data are presented as mean ± SEM. *p < 0.05 for injured-vehicle vs. sham-mino rats.
During the first EPM testing performed 9 days after exposure, injured-vehicle animals spent less time in the open arms and more time in the closed arms of the maze than animals in the other three groups (Figures 2A,B). However, the difference at this time point was not statistically significant. At 46 days after injury, the differences in the time spent in the open and closed arms of the maze became significant between injured-vehicle and injured-mino animals. At this later time point, injured-vehicle animals barely spent any time in the open arms of the maze and practically spent all of their time in the closed arms of the maze (Figures 2A,B). By contrast, injured-mino animals spent a comparable amount of time to animals in the two other groups did in the open and closed arms of the maze.
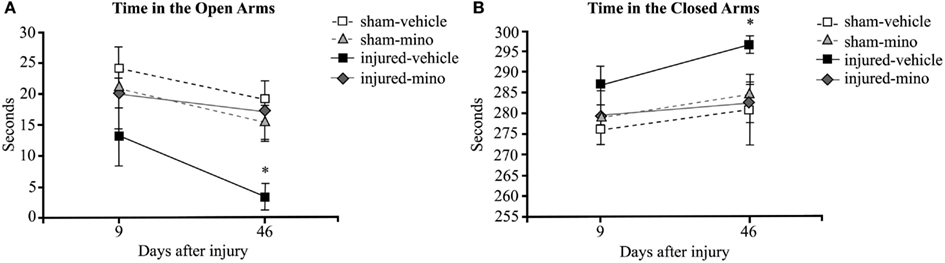
Figure 2. The effect of injury and minocycline treatment on anxiety levels at different time points after mbTBI. (A) Time spent in the open arms (seconds), and (B) time spent in the closed arms (seconds) were measured for all animals in the elevated plus maze. Data are presented as mean ± SEM. *p < 0.05 for injured-vehicle vs. sham-vehicle rats.
In order to assess time-dependent changes in spatial memory, we performed two tests in the BM at two different time points. Test Session I started at 10 days after injury and lasted for 5 days. Injured-vehicle animals performed poorly during the first 2 days of the test (Figure 3A). They required approximately twice as much time as animals in the other experimental groups to find the escape box. While their performance improved slightly on the second day of testing, injured-vehicle animals still required significantly more time to find the escape box compared to their sham group. On the third day of testing, their performance became roughly similar to animals in the other experimental groups. By contrast, the performance of injured-mino animals was very similar to uninjured (sham) animals; their measured latency times to locate and enter the escape box were almost identical on days 11 through 14. They found the escape box with slightly improved efficiency every day.
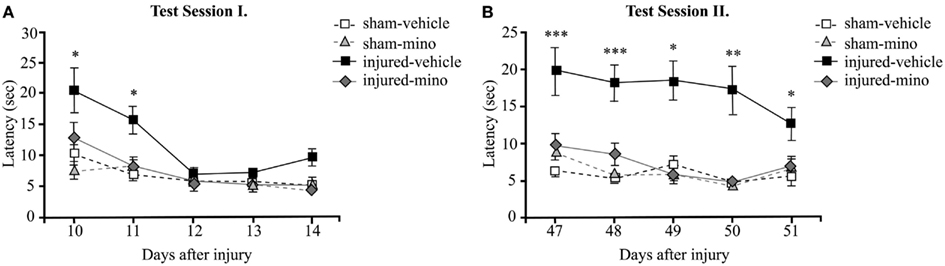
Figure 3. The effect of injury and minocycline treatment on spatial memory at different time points after mbTBI. Latency (seconds) to find and enter the escape box was measured for five consecutive days in the Barnes maze starting at (A) 10 days, and (B) 47 days after injury or sham. Data are presented as the average of the 2 daily trials per animal in each experimental group ±SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 for injured-vehicle vs. sham-vehicle rats.
During Test Session II (beginning at 47 days post-injury), the performance of injured-vehicle animals was significantly worse than sham-vehicle animals on all five testing days (Figure 3B). While their performance slightly improved on each subsequent testing day, injured-vehicle rats still needed significantly more time to find the escape box, even on the last day of testing. Conversely, injured-mino animals performed similar to animals in the two control groups (sham-vehicle and sham-mino). Their performance during Test Session II was similar to that in Test Session I; they required about the same time to find the escape box on each testing day.
Protein Analyses
Select protein marker levels were measured in the serum and dissected brain regions of animals in all four experimental groups. Injury without minocycline treatment caused a significant increase in the serum levels of all biomarkers measured (Figure 4). Both inflammatory markers, CRP and MCP-1, were significantly elevated in injured-vehicle animals; minocycline treatment resulted in normal or near normal (i.e., sham) sera levels in the injured-mino group. Claudin 5 levels were also elevated following blast injury in the vehicle-treated group, but were reduced to sham levels in injured-mino animals. Similarly, neuronal and glial loss and/or damage markers like NSE, NF-H, Tau, S100β, and GFAP were all significantly elevated in the sera of injured-vehicle animals. Minocycline treatment resulted in a significant reduction in serum levels of all of the markers except for GFAP. Lastly, serum CORT levels were also significantly increased in injured-vehicle rats, but minocycline treatment resulted in significantly lower serum CORT levels in injured-mino animals.
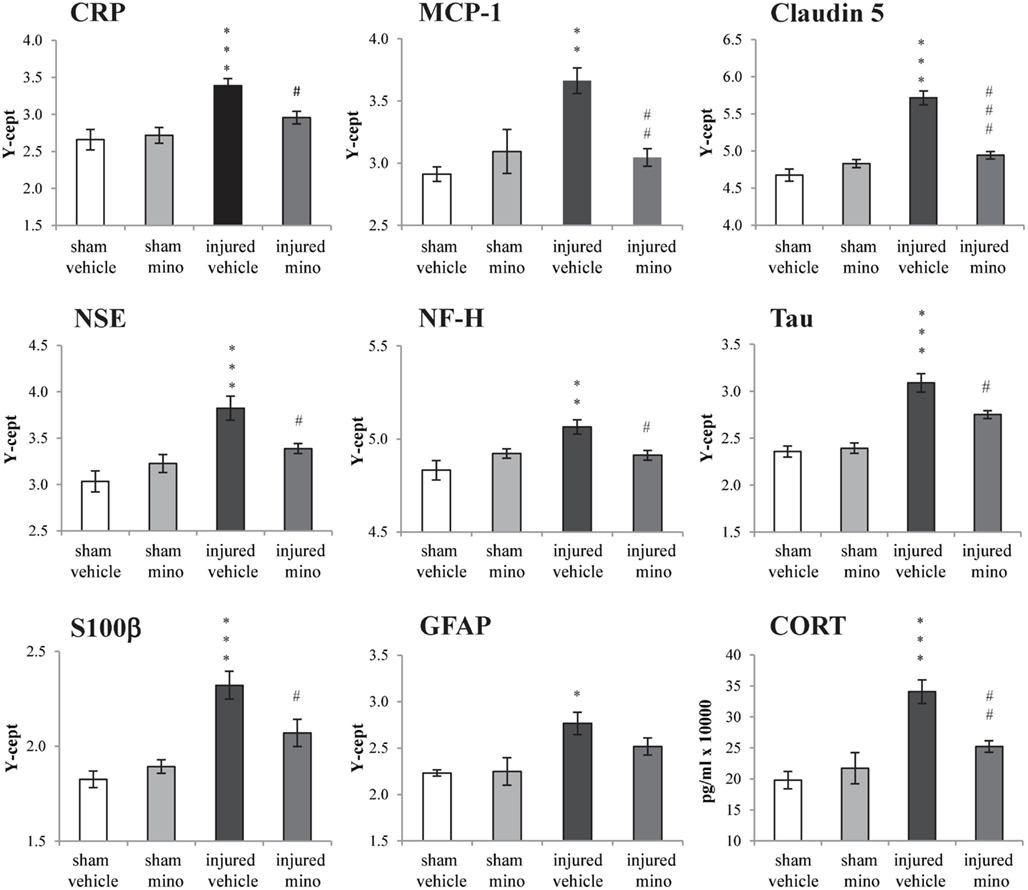
Figure 4. The effect of injury and minocycline treatment on serum levels of selected markers in the different experimental groups. Serum levels of 8 protein markers were assayed by RPPM; CORT levels were assayed by ELISA. Protein values are expressed as y-axis intercept (Y-cept) and CORT values are expressed as pg/ml. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 for injured-vehicle vs. sham-vehicle rats. #p < 0.05, ##p < 0.01, and ###p < 0.001 for injured-vehicle vs. injured-mino rats.
Tissue levels of 13 selected protein biomarkers (Figure 5; Table A3 in Appendix) were determined in the AD, PFC, VHC, and DHC of animals in the various experimental groups. We found significantly elevated levels of all three inflammatory markers (CRP, MCP-1, and TLR9) in the brains of injured-vehicle animals (Figure 5). Importantly, minocycline treatment of injured animals resulted in normal or near normal levels of these inflammatory markers; tissue levels of these markers in all four brain regions of injured-mino rats were not statistically different from those of sham-vehicle or sham-mino animals. NSE, S100β, and GFAP similarly showed injury-induced increases in all four brain regions. Minocycline treatment normalized their tissue levels with the exception of GFAP in the PFC, where GFAP levels of injured-vehicle and injured-mino animals were practically the same.
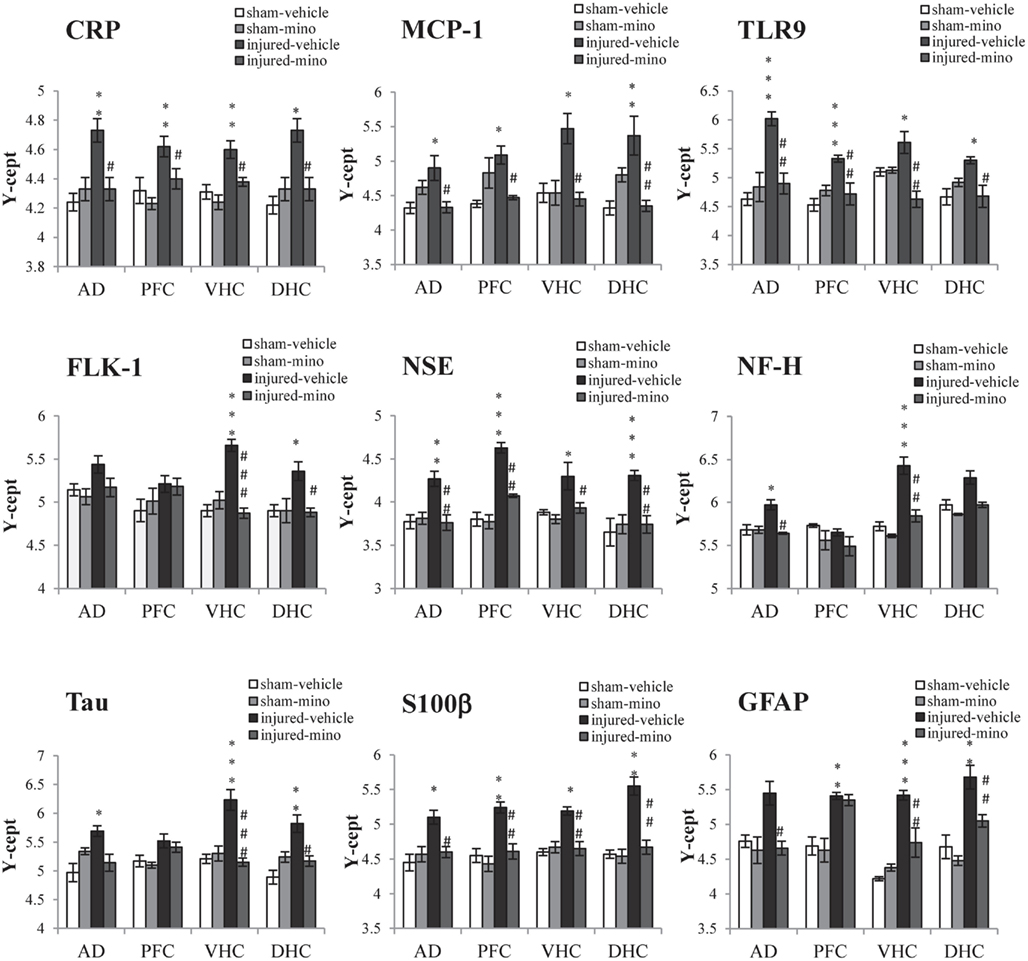
Figure 5. The effect of injury and minocycline treatment on the levels of protein markers in various brain regions in the different experimental groups. Tissue levels of 9 protein markers were measured in the AD, PFC, VHC, and DHC of rats by RPPM. Protein values are expressed as y-axis intercept (Y-cept) and data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 for injured-vehicle vs. sham-vehicle rats. #p < 0.05 and ##p < 0.01 for injured-vehicle vs. injured-mino rats.
Some of the protein biomarkers that were analyzed showed brain region-dependent increases in response to injury. Of the vascular markers, tissue levels of FLK-1 (Figure 5), Claudin 5 and AQP4 (Table A3 in Appendix) were significantly elevated in the VHC following injury; FLK-1 and AQP4 levels were also elevated in the DHC and the AD, respectively. Similarly, neuronal and glial markers showed brain region-specific increases to injury. For instance, all three markers (NF-H, Tau, and MBP) showed injury-induced increases in the VHC but not in the PFC. Minocycline treatment of injured animals significantly reduced the tissue levels of all of the markers with the exception of Tau, which was not significantly reduced in the AD. Interestingly, VEGF did not show any significant changes in response to injury in any of the analyzed brain regions.
Discussion
Minocycline is an FDA approved, semisynthetic, second-generation tetracycline drug that exhibits anti-inflammatory and/or neuroprotective effects in various experimental models of CNS disorders. These include focal and cerebral ischemia (Yrjanheikki et al., 1998; Xu et al., 2004), TBI (Sanchez Mejia et al., 2001), amyotrophic lateral sclerosis (Zhu et al., 2002), Parkinson’s disease (Wu et al., 2002), kainic acid treatment (Heo et al., 2006), Huntington’ disease (Chen et al., 2000; Du et al., 2001; Wu et al., 2002; Wang et al., 2003), multiple sclerosis (Brundula et al., 2002; Popovic et al., 2002), Alzheimer’s disease (Choi et al., 2007), and spinal cord injury (Wells et al., 2003; Stirling et al., 2004; Festoff et al., 2006; Table A1 in Appendix). Minocycline’s ability to improve outcome in distinct types of CNS disease models may stem from its ability to find multiple targets in different biochemical cascades that play a role in the development of the abovementioned diseases. Previous studies indicated that minocycline acts as a pleiotropic molecule; it can reduce the release of various chemokines and cytokines (Sanchez Mejia et al., 2001; Bye et al., 2007), lipid mediators of inflammation, matrix metalloproteinases (MMPs), and nitric oxide (NO; Stirling et al., 2005). Minocycline can also inhibit microglia activation (Yrjanheikki et al., 1998, 1999; Tikka and Koistinaho, 2001). The inhibition of microglial inflammatory responses has been reported in various neurodegenerative diseases (Yrjanheikki et al., 1999) including Huntington’s (Chen et al., 2000; Popovic et al., 2002; Wu et al., 2002); additional anti-inflammatory actions may be through the impediment of molecules like cyclooxigenase-2 (Patel et al., 1999; Yrjanheikki et al., 1999). Minocycline exerts its neuroprotective effects (Kriz et al., 2002; Wells et al., 2003; Stirling et al., 2004; Zemke and Majid, 2004; Marchand et al., 2009) through the repression of poly (ADP-ribose) polymerase-1 activity (Alano et al., 2006), which plays a central role in caspase-independent apoptosis (Susin et al., 1999; Zhang et al., 2002; Cao et al., 2003; Du et al., 2003), and the suppression of caspase-1 and caspase-3 expression (Chen et al., 2000) and cytochrome c release from the mitochondria (Zhu et al., 2002). Moreover, minocycline has been shown to sequester excess Ca2+ released after injury (Antonenko et al., 2010), and block the injury-induced decrease of soluble alpha amyloid precursor protein in the attenuation of diffuse axonal injury (Siopi et al., 2011). Based on all of these findings, we were compelled to test the effects of minocycline in our rat model of mbTBI.
During our pilot studies we followed a reported treatment schedule of 90 mg/kg of minocycline administered i.p. twice on the first day, 50 mg/kg twice per day for 2 subsequent days, and 50 mg/kg once per day for three additional days (Lee et al., 2003; Teng et al., 2004; Festoff et al., 2006; Yune et al., 2007). However, we found that this treatment caused substantial weight loss likely due to gastrointestinal problems (i.e., diarrhea). Based on these preliminary findings, we decided to modify the treatment paradigm by lowering the dose to 50 mg/kg once per day for four consecutive days. Our conservative treatment schedule caused light and transient diarrhea, and animals recovered and gained weight normally from the third day post-injury until the termination of the experiment on day 51 (data not shown).
Consistent with our previous findings, injured rats had reduced horizontal activity and a somewhat higher resting time than sham animals in the OF 1 day after injury (Kwon et al., 2011). Interestingly, all of the rats, independent of injury and treatment, showed gradually decreasing horizontal activities during the two subsequent OF sessions. There are two plausible explanations for this behavior. Rodents actively explore new areas, but inadvertently become less active on subsequent exposures to the same environment, a process called habituation (Pitkänen et al., 2006). We also observed on numerous occasions in other experiments that the horizontal activity of naïve rats in the OF at baseline is higher than it is 24 h later. We believe that since the OF represents a novel environment for the rats, they actively explore it (Bolivar et al., 2000; Daenen et al., 2001). However, repeated testing may cause the animals to habituate to the OF and in turn spend less time exploring and more time resting. Another possible explanation may be aging, especially during the last OF session, as young rodents have higher motor activity levels than more mature rodents (Sprott and Eleftheriou, 1974; Ingram et al., 1981; Gage et al., 1984; Lamberty and Gower, 1993). The effects of aging have also been observed as decreases in distance traveled in the EPM over time in both, sham and blast injured animals (Kovesdi et al., 2011).
Epidemiological studies have indicated that soldiers frequently develop neurobehavioral abnormalities like increased anxiety and memory impairments in mbTBI (Belanger et al., 2007; Brenner et al., 2009). Anxiety affects rehabilitation, psychosocial adjustment, and cognition in humans (Kersel et al., 2001; Rapoport et al., 2005). The EPM is a simple behavioral assay for evaluating the anxiety responses of rodents (Pellow et al., 1985) and studying the brain sites (limbic regions, hippocampus, amygdala; Silveira et al., 1993; Gonzalez and File, 1997) and the mechanisms underlying anxiolytic behavior (GABA, glutamate, serotonin, hypothalamic–pituitary–adrenal axis neuromodulators; Handley and Mithani, 1984; Pellow et al., 1985; Rodgers et al., 1992; Silva and Brandao, 2000; Korte and De Boer, 2003; Overstreet et al., 2003; Cortese and Phan, 2005). Rodents naturally prefer dark, enclosed spaces, and demonstrate an aversion to open spaces and a fear of heights (Barnett, 1975). Despite these natural inclinations, non-anxious rodents possess exploratory behaviors that cause them to investigate the open arms of the maze while more anxious rats remain in the closed arms of the maze for longer periods of time.
We previously found increased anxiety in our rodent model of mbTBI (Kovesdi et al., 2011). As our current EPM data illustrates, acute minocycline treatment prevented the increase in anxiety following blast overpressure. The time spent in the closed arms of the maze by injured-mino rats was indistinguishable from that of the two sham groups at both testing time points. Conversely, injured-vehicle animals showed signs of increased anxiety early on; they spent less time on the open arms of the maze than animals in the other three experimental groups. While the difference was not statistically significant at this early time point, injured-vehicle animals barely spent any time outside of the closed arms of the maze 46 days after the injury. Even though there is very little information available about the effects of minocycline on anxiety, especially in brain injury, minocycline treatment reduced anxiety in the EPM in models of cardiac arrest/cardiopulmonary resuscitation and fragile X syndrome (Bilousova et al., 2009; Neigh et al., 2009).
Current treatments of increased anxiety are mostly symptomatic (Tenovuo, 2006; Silver et al., 2009), and patients frequently experience side effects from the use of drugs like benzodiazepines (Rickels et al., 1991; Baldwin et al., 2005). Acute minocycline treatment may provide an alternative to the use of these drugs. Interestingly, injured-mino animals also had lower serum CORT levels than injured-vehicle animals at 51 days after the injury. While serum CORT levels have been used as indicators of stress (Dunn et al., 2004), the correlation between serum CORT levels and anxiety is rather complex and likely involve multiple regulatory pathways.
Consistent with available epidemiological data and our previous studies, the memory impairment associated with mbTBI develops over several weeks after the insult (Kovesdi et al., 2011; Kwon et al., 2011). Importantly, the deficit persists for at least 2 months post-injury (Kovesdi et al.). Given that 2 months in the lifespan of a rat roughly translates into several human years (Quinn, 2005), the observed memory impairment mirrors the chronic condition that manifests in humans reasonably well. The BM has been extensively used to study spatial learning and memory in rats (Barnes, 1979), and is considered a less anxiogenic alternative to the Morris water maze since it does not involve swimming (Pompl et al., 1999; Miyakawa et al., 2001; Deacon and Rawlins, 2002; Holmes et al., 2002). BM has been applied to studies of TBI; rodents with hippocampal damage show impaired performance in the maze, supporting the spatial nature of the task (Fox et al., 1998; Paylor et al., 2001; Deacon and Rawlins, 2002; Raber et al., 2004). In BM animals are presumed to learn the location of an escape hole using spatial reference points that are either fixed in relation to the maze (extra-maze cues) or are fixed on the maze itself in relation to the escape hole (proximal cues). It is important to note that during our acclimation and baseline behavioral testing, all animals were exposed to the maze and were trained to “learn” the task of locating and entering the escape box.
Early signs of the memory deficit were detected in the first testing session. Injured-vehicle animals required approximately twice as long to locate the escape box on the first day of testing, while injured-mino animals performed similar to the uninjured shams. On the second day of testing, injured-vehicle rats still needed more time than the other groups. During the last 3 days of testing, injured-vehicle rats relearned and remembered the task, requiring about the same amount of time as the other groups. However, during the second testing session, injured-vehicle rats performed poorly on all five testing days with only minor improvements in their speed from day to day. Conversely, injured-mino rats performed as well as sham animals did throughout. A similar effect was found in a study by Siopi et al. (2011) where acute treatment with minocycline significantly improved recognition memory; the effects lasted for up to 13 weeks in a mouse closed head injury model. There are currently no effective treatments in clinical use for memory impairment. Existing therapies predominantly target symptoms associated with mood disorders (e.g., depression) that can also improve memory performance (Tenovuo, 2006; Silver et al., 2009). Therefore, acute minocycline treatment has the potential to offer a potentially effective alternative.
The observed neurobehavioral impairments implicate the AD, PFC, VHC, and DHC due to their involvement in mediating anxiety and memory (Henke, 1990; Moser and Moser, 1998). In our earlier works we found indications of inflammation, axonal, glial, and neuronal damage in these brain regions (Kovesdi et al., 2011; Kwon et al., 2011). The neuroinflammatory response to various brain insults has been suggested as a potential link between injury and altered behavior, including increased anxiety. As reported earlier, blast can trigger a systemic inflammatory process even when the body is fully protected and only the head is exposed (Cernak et al., 2011). It is crucial to note that the similarities and the dissimilarities between mbTBI and other better-characterized forms of closed head injuries are currently not known with regards to their primary and secondary injury mechanisms. Nevertheless, it has been hypothesized that the different types of TBIs may share pathological components like neuroinflammation, neuronal and glial cell loss, and axonal injuries (Agoston et al., 2009).
In our current study, we found that minocycline treatment normalized significantly elevated sera levels of the inflammatory markers CRP and MCP-1 following exposure to mild blast. CRP and MCP-1 levels are routinely monitored in clinical settings and are used as an indicator of inflammation (Berman et al., 1996; Glabinski et al., 1996; Du Clos, 2000; Lobo et al., 2003). CRP is a component of the acute phase response to injury (Du Clos, 2000) and its expression is stimulated by the release of cytokines (Okamura et al., 1990); elevated CRP serum levels may reflect a combination of systemic as well as neuronal inflammation. Increased levels of MCP-1 are associated with neurological dysfunction after traumatic axonal injury in rats (Rancan et al., 2001), and are detected in the cerebrospinal fluid in diseases related to neuroinflammation such as stroke, meningitis, and multiple sclerosis (Mastroianni et al., 1998; Losy and Zaremba, 2001; Sindern et al., 2001; Chen et al., 2003; Sorensen et al., 2004). MCP-1 has also been suggested to regulate vascular permeability during CNS inflammation (Tekstra et al., 1999;Stamatovic et al., 2003, 2006).
While tissue levels of Claudin 5 did not significantly change except in the VHC, serum levels were significantly increased in injured-vehicle animals. Claudin 5 is a part of the tight junction complex in brain endothelial cells that contribute to the formation of the BBB (Morita et al., 1999; Liebner et al., 2000); increased serum levels suggest that there may be vascular damage in mbTBI that results in the release of Claudin 5 into systemic blood. Importantly, minocycline treatment normalized Claudin 5 sera levels indicating that vascular changes may be secondary to the inflammatory process or that minocycline possesses cytoprotective effects that also extend to endothelial cells.
Elevated serum levels of neuron- and glia-specific proteins have been found clinically as well as experimentally in various forms of TBI (Povlishock and Christman, 1995; Povlishock and Pettus, 1996; Buki and Povlishock, 2006). Increased serum levels of large neuron-specific molecules also point toward a vascular pathology; heightened BBB permeability is required for the release of large proteins like NF-H from the brain parenchyma and into systemic circulation. In a large animal model of blast TBI, the temporal pattern of serum NF-H levels correlated with clinical and pathological outcomes (Gyorgy et al., 2011). In our current study, minocycline treatment significantly reduced sera levels of NSE, NF-H, Tau, and S100β after injury, but not GFAP, an astroglia-specific intermediate filament (Missler et al., 1999) indicative of brain damage.
Consistent with our behavioral and serum data, we found that minocycline treatment prevented or mitigated injury-induced increases of the selected inflammatory markers CRP, MCP-1, and TLR9 in all four brain regions. TLR9 is member of the toll-like receptor family (Aderem and Ulevitch, 2000; Akira et al., 2001; Takeda and Akira, 2005; Mishra et al., 2006; O’Neill, 2006; Casanova et al., 2011) involved in the induction and the regulation of the inflammatory response in TBI (Hua et al., 2007, 2009) as well as other disorders involving neuroinflammation (Prat and Antel, 2005) and ischemic brain damage (Hua et al., 2007, 2009; Doyle et al., 2008; Gao et al., 2009; Marsh et al., 2009).
Of the vascular markers only FLK-1 and AQP4 tissue levels increased in response to the injury; minocycline treatment mitigated the effect of injury on FLK-1 levels but showed no effect on the tissue levels of AQP4. Increases in AQP4 were only detected in the AD and in the VHC while FLK-1 was in the VHC and the DHC. Elevations in AQP4 expression can contribute to the formation as well as the resolution of edema (Kimelberg, 1995; Papadopoulos et al., 2002; Amiry-Moghaddam and Ottersen, 2003; Neal et al., 2007). The pathology of severe bTBI includes the development of rapid and malignant brain edema (Ling et al., 2009; Ling and Ecklund, 2011) probably involving AQP4 (Neal et al., 2007). However, we currently have no information about water imbalance in mbTBI; if present, it is likely limited to the early phase following injury.
FLK-1 is a membrane-bound tyrosine kinase that mediates the effects of VEGF in the CNS (Sondell et al., 2000; Ogunshola et al., 2002; Rosenstein et al., 2003). Activation of FLK-1 stimulates various intracellular signal transduction pathways including the PI3K/Akt pathway that mediates the neuroprotective function of VEGF (Gerber et al., 1998; Wu et al., 2000; Kilic et al., 2006). VEGF/FLK-1 up-regulation following TBI seems to perform an important endogenous cytoprotective mechanism (Skold et al., 2006; Lee and Agoston, 2009). Interestingly, we did not detect changes in the abundance of VEGF in any of the analyzed brain regions following injury. A potential explanation for this negative finding is the relatively late testing time point (51 days post-injury). In a previous study using another model of TBI, we observed significant increases in VEGF tissue levels in the hippocampus (Lee and Agoston, 2009, 2010); the increases were limited to a few days after the injury.
The tissue levels of NSE, NF-H, Tau, S100β, GFAP, and MBP similarly increased in response to the injury, however, increases were brain region-specific. We measured significant injury-induced increases in sera levels of these proteins indicative of neuronal and glial cell losses. Thus, the detected increases in the tissue levels of these proteins are likely compensatory in nature and can be a part of the repair mechanism (Fawcett, 2009). Importantly, in all cases where injury resulted in an increase in the tissue levels of these markers, minocycline treatment mitigated the effect and tissue levels of these markers were restored to levels measured in sham animals.
Conclusion
Our study demonstrates that acute minocycline treatment substantially improve the neurobehavioral outcome in a rodent model of mbTBI likely through mitigating the neuroinflammatory response to injury. The strength of our study lies in combining neurobehavioral tests performed at two different time points after injury with determining changes in serum and brain tissue levels of protein biomarkers. The limitations of the current study are the limited types of neurobehavioral and a single terminal time point of proteomics analyses. Based on these promising results, additional neurobehavioral testing shall be performed in future studies along with obtaining blood at several clinically relevant time points for protein assays. Nevertheless, our findings provide a rationale for exploring the viability of using acute minocycline treatment in mbTBI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Neurotrauma Team (WRAIR) for their technical help during the blast exposures. This work was supported by Veterans Affairs Grant B5044R.
References
Abdel Baki, S. G., Schwab, B., Haber, M., Fenton, A. A., and Bergold, P. J. (2010). Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS ONE 5, e12490. doi:10.1371/journal.pone.0012490
Aderem, A., and Ulevitch, R. J. (2000). Toll-like receptors in the induction of the innate immune response. Nature 406, 782–787.
Agostinho, P., Cunha, R. A., and Oliveira, C. (2010). Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 16, 2766–2778.
Agoston, D. V., Gyorgy, A., Eidelman, O., and Pollard, H. B. (2009). Proteomic biomarkers for blast neurotrauma: targeting cerebral edema, inflammation, and neuronal death cascades. J. Neurotrauma 26, 901–911.
Akira, S., Takeda, K., and Kaisho, T. (2001). Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2, 675–680.
Alano, C. C., Kauppinen, T. M., Valls, A. V., and Swanson, R. A. (2006). Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc. Natl. Acad. Sci. U.S.A. 103, 9685–9690.
Amiry-Moghaddam, M., and Ottersen, O. P. (2003). The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 4, 991–1001.
Antonenko, Y. N., Rokitskaya, T. I., Cooper, A. J., and Krasnikov, B. F. (2010). Minocycline chelates Ca2+, binds to membranes, and depolarizes mitochondria by formation of Ca2+-dependent ion channels. J. Bioenerg. Biomembr. 42, 151–163.
Arvin, B., Neville, L. F., Barone, F. C., and Feuerstein, G. Z. (1996). The role of inflammation and cytokines in brain injury. Neurosci. Biobehav. Rev. 20, 445–452.
Baldwin, D. S., Anderson, I. M., Nutt, D. J., Bandelow, B., Bond, A., Davidson, J. R., Den Boer, J. A., Fineberg, N. A., Knapp, M., Scott, J., and Wittchen, H. U. (2005). Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. (Oxford) 19, 567–596.
Barnes, C. A. (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104.
Belanger, H. G., Vanderploeg, R. D., Curtiss, G., and Warden, D. L. (2007). Recent neuroimaging techniques in mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 19, 5–20.
Berman, J. W., Guida, M. P., Warren, J., Amat, J., and Brosnan, C. F. (1996). Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J. Immunol. 156, 3017–3023.
Bilousova, T. V., Dansie, L., Ngo, M., Aye, J., Charles, J. R., Ethell, D. W., and Ethell, I. M. (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 46, 94–102.
Blum, D., Chtarto, A., Tenenbaum, L., Brotchi, J., and Levivier, M. (2004). Clinical potential of minocycline for neurodegenerative disorders. Neurobiol. Dis. 17, 359–366.
Bolivar, V. J., Caldarone, B. J., Reilly, A. A., and Flaherty, L. (2000). Habituation of activity in an open field: a survey of inbred strains and F1 hybrids. Behav. Genet. 30, 285–293.
Bremner, J. D. (2005). Effects of traumatic stress on brain structure and function: relevance to early responses to trauma. J. Trauma Dissociation 6, 51–68.
Bremner, J. D. (2007). Functional neuroimaging in post-traumatic stress disorder. Expert Rev. Neurother. 7, 393–405.
Brenner, L. A., Vanderploeg, R. D., and Terrio, H. (2009). Assessment and diagnosis of mild traumatic brain injury, posttraumatic stress disorder, and other polytrauma conditions: burden of adversity hypothesis. Rehabil. Psychol. 54, 239–246.
Brundula, V., Rewcastle, N. B., Metz, L. M., Bernard, C. C., and Yong, V. W. (2002). Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain 125, 1297–1308.
Buki, A., and Povlishock, J. T. (2006). All roads lead to disconnection? – Traumatic axonal injury revisited. Acta Neurochir. (Wien) 148, 181–193; discussion 193–184.
Bye, N., Habgood, M. D., Callaway, J. K., Malakooti, N., Potter, A., Kossmann, T., and Morganti-Kossmann, M. C. (2007). Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 204, 220–233.
Cacci, E., Claasen, J. H., and Kokaia, Z. (2005). Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J. Neurosci. Res. 80, 789–797.
Cao, G., Clark, R. S., Pei, W., Yin, W., Zhang, F., Sun, F. Y., Graham, S. H., and Chen, J. (2003). Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab. 23, 1137–1150.
Carobrez, A. P., and Bertoglio, L. J. (2005). Ethological and temporal analyses of anxiety-like behavior: the elevated plus-maze model 20 years on. Neurosci. Biobehav. Rev. 29, 1193–1205.
Casanova, J. L., Abel, L., and Quintana-Murci, L. (2011). Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 29, 447–491.
Cernak, I., Merkle, A. C., Koliatsos, V. E., Bilik, J. M., Luong, Q. T., Mahota, T. M., Xu, L., Slack, N., Windle, D., and Ahmed, F. A. (2011). The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 41, 538–551.
Cernak, I., and Noble-Haeusslein, L. J. (2010). Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J. Cereb. Blood Flow Metab. 30, 255–266.
Chen, M., Ona, V. O., Li, M., Ferrante, R. J., Fink, K. B., Zhu, S., Bian, J., Guo, L., Farrell, L. A., Hersch, S. M., Hobbs, W., Vonsattel, J. P., Cha, J. H., and Friedlander, R. M. (2000). Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 6, 797–801.
Chen, Y., Hallenbeck, J. M., Ruetzler, C., Bol, D., Thomas, K., Berman, N. E., and Vogel, S. N. (2003). Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J. Cereb. Blood Flow Metab. 23, 748–755.
Choi, Y., Kim, H. S., Shin, K. Y., Kim, E. M., Kim, M., Park, C. H., Jeong, Y. H., Yoo, J., Lee, J. P., Chang, K. A., Kim, S., and Suh, Y. H. (2007). Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology 32, 2393–2404.
Cortese, B. M., and Phan, K. L. (2005). The role of glutamate in anxiety and related disorders. CNS Spectr. 10, 820–830.
Czlonkowska, A., and Kurkowska-Jastrzebska, I. (2011). Inflammation and gliosis in neurological diseases – clinical implications. J. Neuroimmunol. 231, 78–85.
Daenen, E. W., Van Der Heyden, J. A., Kruse, C. G., Wolterink, G., and Van Ree, J. M. (2001). Adaptation and habituation to an open field and responses to various stressful events in animals with neonatal lesions in the amygdala or ventral hippocampus. Brain Res. 918, 153–165.
Deacon, R. M., and Rawlins, J. N. (2002). Learning impairments of hippocampal-lesioned mice in a paddling pool. Behav. Neurosci. 116, 472–478.
Doll, H., Truebel, H., Kipfmueller, F., Schaefer, U., Neugebauer, E. A., Wirth, S., and Maegele, M. (2009). Pharyngeal selective brain cooling improves neurofunctional and neurocognitive outcome after fluid percussion brain injury in rats. J. Neurotrauma 26, 235–242.
Doyle, K. P., Simon, R. P., and Stenzel-Poore, M. P. (2008). Mechanisms of ischemic brain damage. Neuropharmacology 55, 310–318.
Du, L., Zhang, X., Han, Y. Y., Burke, N. A., Kochanek, P. M., Watkins, S. C., Graham, S. H., Carcillo, J. A., Szabo, C., and Clark, R. S. (2003). Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 278, 18426–18433.
Du, Y., Ma, Z., Lin, S., Dodel, R. C., Gao, F., Bales, K. R., Triarhou, L. C., Chernet, E., Perry, K. W., Nelson, D. L., Luecke, S., Phebus, L. A., Bymaster, F. P., and Paul, S. M. (2001). Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 14669–14674.
Dunn, A. J., Swiergiel, A. H., and Palamarchouk, V. (2004). Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Ann. N. Y. Acad. Sci. 1018, 25–34.
Fanning, W. L., Gump, D. W., and Sofferman, R. A. (1977). Side effects of minocycline: a double-blind study. Antimicrob. Agents Chemother. 11, 712–717.
Fawcett, J. (2009). Molecular control of brain plasticity and repair. Prog. Brain Res. 175, 501–509.
Festoff, B. W., Ameenuddin, S., Arnold, P. M., Wong, A., Santacruz, K. S., and Citron, B. A. (2006). Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J. Neurochem. 97, 1314–1326.
Floyd, C. L., and Lyeth, B. G. (2007). Astroglia: important mediators of traumatic brain injury. Prog. Brain Res. 161, 61–79.
Fox, G. B., Fan, L., Levasseur, R. A., and Faden, A. I. (1998). Effect of traumatic brain injury on mouse spatial and nonspatial learning in the Barnes circular maze. J. Neurotrauma 15, 1037–1046.
Gage, F. H., Dunnett, S. B., and Bjorklund, A. (1984). Spatial learning and motor deficits in aged rats. Neurobiol. Aging 5, 43–48.
Gao, Y., Fang, X., Tong, Y., Liu, Y., and Zhang, B. (2009). TLR4-mediated MyD88-dependent signaling pathway is activated by cerebral ischemia-reperfusion in cortex in mice. Biomed. Pharmacother. 63, 442–450.
Gerber, H. P., Mcmurtrey, A., Kowalski, J., Yan, M., Keyt, B. A., Dixit, V., and Ferrara, N. (1998). Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 273, 30336–30343.
Glabinski, A. R., Balasingam, V., Tani, M., Kunkel, S. L., Strieter, R. M., Yong, V. W., and Ransohoff, R. M. (1996). Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 156, 4363–4368.
Gonzalez, L. E., and File, S. E. (1997). A five minute experience in the elevated plus-maze alters the state of the benzodiazepine receptor in the dorsal raphe nucleus. J. Neurosci. 17, 1505–1511.
Gump, D. W., Ashikaga, T., Fink, T. J., and Radin, A. M. (1977). Side effects of minocycline: different dosage regimens. Antimicrob. Agents Chemother. 12, 642–646.
Gyorgy, A., Ling, G., Wingo, D., Walker, J., Tong, L., Parks, S., Januszkiewicz, A., Baumann, R., and Agoston, D. V. (2011). Time-dependent changes in serum biomarker levels after blast traumatic brain injury. J. Neurotrauma 28, 1121–1126.
Gyorgy, A. B., Walker, J., Wingo, D., Eidelman, O., Pollard, H. B., Molnar, A., and Agoston, D. V. (2010). Reverse phase protein microarray technology in traumatic brain injury. J. Neurosci. Methods 192, 96–101.
Handley, S. L., and Mithani, S. (1984). Effects of alpha-adrenoceptor agonists and antagonists in a maze-exploration model of “fear”-motivated behaviour. Naunyn Schmiedebergs Arch. Pharmacol. 327, 1–5.
Harrison, F. E., Hosseini, A. H., and Mcdonald, M. P. (2009). Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav. Brain Res. 198, 247–251.
Henke, P. G. (1990). Hippocampal pathway to the amygdala and stress ulcer development. Brain Res. Bull. 25, 691–695.
Heo, K., Cho, Y. J., Cho, K. J., Kim, H. W., Kim, H. J., Shin, H. Y., Lee, B. I., and Kim, G. W. (2006). Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci. Lett. 398, 195–200.
Hewlett, K. A., and Corbett, D. (2006). Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience 141, 27–33.
Hoffer, M. E., Balaban, C., Gottshall, K., Balough, B. J., Maddox, M. R., and Penta, J. R. (2010). Blast exposure: vestibular consequences and associated characteristics. Otol. Neurotol. 31, 232–236.
Hoge, C. W., Mcgurk, D., Thomas, J. L., Cox, A. L., Engel, C. C., and Castro, C. A. (2008). Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 358, 453–463.
Holmes, A., Wrenn, C. C., Harris, A. P., Thayer, K. E., and Crawley, J. N. (2002). Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice. Genes Brain Behav. 1, 55–69.
Hua, F., Ma, J., Ha, T., Kelley, J. L., Kao, R. L., Schweitzer, J. B., Kalbfleisch, J. H., Williams, D. L., and Li, C. (2009). Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 1262, 100–108.
Hua, F., Ma, J., Ha, T., Xia, Y., Kelley, J., Williams, D. L., Kao, R. L., Browder, I. W., Schweitzer, J. B., Kalbfleisch, J. H., and Li, C. (2007). Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J. Neuroimmunol. 190, 101–111.
Hyder, A. A., Wunderlich, C. A., Puvanachandra, P., Gururaj, G., and Kobusingye, O. C. (2007). The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22, 341–353.
Ingram, D. K., London, E. D., and Goodrick, C. L. (1981). Age and neurochemical correlates of radial maze performance in rats. Neurobiol. Aging 2, 41–47.
Kamnaksh, A., Kovesdi, E., Kwon, S. K., Wingo, D., Ahmed, F., Grunberg, N. E., Long, J., and Agoston, D. V. (2011). Factors affecting blast traumatic brain injury. J. Neurotrauma 28, 2145–2153.
Kersel, D. A., Marsh, N. V., Havill, J. H., and Sleigh, J. W. (2001). Psychosocial functioning during the year following severe traumatic brain injury. Brain Inj. 15, 683–696.
Kilic, E., Kilic, U., Wang, Y., Bassetti, C. L., Marti, H. H., and Hermann, D. M. (2006). The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 20, 1185–1187.
Kimelberg, H. K. (1995). Current concepts of brain edema. Review of laboratory investigations. J. Neurosurg. 83, 1051–1059.
Kochanek, P. M., Berger, R. P., Bayir, H., Wagner, A. K., Jenkins, L. W., and Clark, R. S. (2008). Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr. Opin. Crit. Care 14, 135–141.
Korte, S. M., and De Boer, S. F. (2003). A robust animal model of state anxiety: fear-potentiated behaviour in the elevated plus-maze. Eur. J. Pharmacol. 463, 163–175.
Kovesdi, E., Gyorgy, A. B., Kwon, S. K., Wingo, D. L., Kamnaksh, A., Long, J. B., Kasper, C. E., and Agoston, D. V. (2011). The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front. Neurosci. 5:42. doi:10.3389/fnins.2011.00042
Kriz, J., Nguyen, M. D., and Julien, J. P. (2002). Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 268–278.
Kwon, S. K., Kovesdi, E., Gyorgy, A. B., Wingo, D., Kamnaksh, A., Walker, J., Long, J. B., and Agoston, D. V. (2011). Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front. Neurol. 2:12. doi:10.3389/fneur.2011.00012
Lamberty, Y., and Gower, A. J. (1993). Spatial processing and emotionality in aged NMRI mice: a multivariate analysis. Physiol. Behav. 54, 339–343.
Lee, C., and Agoston, D. V. (2009). Inhibition of VEGF receptor 2 increased cell death of dentate hilar neurons after traumatic brain injury. Exp. Neurol. 220, 400–403.
Lee, C., and Agoston, D. V. (2010). Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma 27, 541–553.
Lee, J. H., Tigchelaar, S., Liu, J., Stammers, A. M., Streijger, F., Tetzlaff, W., and Kwon, B. K. (2010). Lack of neuroprotective effects of simvastatin and minocycline in a model of cervical spinal cord injury. Exp. Neurol. 225, 219–230.
Lee, S. M., Yune, T. Y., Kim, S. J., Park Do, W., Lee, Y. K., Kim, Y. C., Oh, Y. J., Markelonis, G. J., and Oh, T. H. (2003). Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J. Neurotrauma 20, 1017–1027.
Li, J., and McCullough, L. D. (2009). Sex differences in minocycline-induced neuroprotection after experimental stroke. J. Cereb. Blood Flow Metab. 29, 670–674.
Liebner, S., Fischmann, A., Rascher, G., Duffner, F., Grote, E. H., Kalbacher, H., and Wolburg, H. (2000). Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 100, 323–331.
Ling, G., Bandak, F., Armonda, R., Grant, G., and Ecklund, J. (2009). Explosive blast neurotrauma. J. Neurotrauma 26, 815–825.
Ling, G. S., and Ecklund, J. M. (2011). Traumatic brain injury in modern war. Curr. Opin. Anaesthesiol. 24, 124–130.
Lobo, S. M., Lobo, F. R., Bota, D. P., Lopes-Ferreira, F., Soliman, H. M., Melot, C., and Vincent, J. L. (2003). C-reactive protein levels correlate with mortality and organ failure in critically ill patients. Chest 123, 2043–2049.
Long, J. B., Bentley, T. L., Wessner, K. A., Cerone, C., Sweeney, S., and Bauman, R. A. (2009). Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840.
Losy, J., and Zaremba, J. (2001). Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke 32, 2695–2696.
Macdonald, H., Kelly, R. G., Allen, E. S., Noble, J. F., and Kanegis, L. A. (1973). Pharmacokinetic studies on minocycline in man. Clin. Pharmacol. Ther. 14, 852–861.
Maegele, M., Lippert-Gruener, M., Ester-Bode, T., Sauerland, S., Schafer, U., Molcany, M., Lefering, R., Bouillon, B., Neiss, W. F., Angelov, D. N., Klug, N., Mcintosh, T. K., and Neugebauer, E. A. (2005). Reversal of neuromotor and cognitive dysfunction in an enriched environment combined with multimodal early onset stimulation after traumatic brain injury in rats. J. Neurotrauma 22, 772–782.
Marchand, F., Tsantoulas, C., Singh, D., Grist, J., Clark, A. K., Bradbury, E. J., and Mcmahon, S. B. (2009). Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur. J. Pain 13, 673–681.
Marsh, B. J., Williams-Karnesky, R. L., and Stenzel-Poore, M. P. (2009). Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158, 1007–1020.
Mastroianni, C. M., Lancella, L., Mengoni, F., Lichtner, M., Santopadre, P., D’Agostino, C., Ticca, F., and Vullo, V. (1998). Chemokine profiles in the cerebrospinal fluid (CSF) during the course of pyogenic and tuberculous meningitis. Clin. Exp. Immunol. 114, 210–214.
Mishra, B. B., Mishra, P. K., and Teale, J. M. (2006). Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J. Neuroimmunol. 181, 46–56.
Missler, U., Wiesmann, M., Wittmann, G., Magerkurth, O., and Hagenstrom, H. (1999). Measurement of glial fibrillary acidic protein in human blood: analytical method and preliminary clinical results. Clin. Chem. 45, 138–141.
Miyakawa, T., Yared, E., Pak, J. H., Huang, F. L., Huang, K. P., and Crawley, J. N. (2001). Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus 11, 763–775.
Morganti-Kossmann, M. C., Rancan, M., Stahel, P. F., and Kossmann, T. (2002). Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105.
Morita, K., Sasaki, H., Furuse, M., and Tsukita, S. (1999). Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 147, 185–194.
Moser, M. B., and Moser, E. I. (1998). Functional differentiation in the hippocampus. Hippocampus 8, 608–619.
Neal, C. J., Lee, E. Y., Gyorgy, A., Ecklund, J. M., Agoston, D. V., and Ling, G. S. (2007). Effect of penetrating brain injury on aquaporin-4 expression using a rat model. J. Neurotrauma 24, 1609–1617.
Neigh, G. N., Karelina, K., Glasper, E. R., Bowers, S. L., Zhang, N., Popovich, P. G., and Devries, A. C. (2009). Anxiety after cardiac arrest/cardiopulmonary resuscitation: exacerbated by stress and prevented by minocycline. Stroke 40, 3601–3607.
Nelson, L. A., Yoash-Gantz, R. E., Pickett, T. C., and Campbell, T. A. (2009). Relationship between processing speed and executive functioning performance among OEF/OIF veterans: implications for postdeployment rehabilitation. J. Head Trauma Rehabil. 24, 32–40.
Ng, S. Y., Semple, B. D., Morganti-Kossmann, M. C., and Bye, N. (2012). Attenuation of microglial activation with minocycline is not associated with changes in neurogenesis after focal traumatic brain injury in adult mice. J. Neurotrauma 29, 1410–1425.
Ogunshola, O. O., Antic, A., Donoghue, M. J., Fan, S. Y., Kim, H., Stewart, W. B., Madri, J. A., and Ment, L. R. (2002). Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. J. Biol. Chem. 277, 11410–11415.
Okamura, J. M., Miyagi, J. M., Terada, K., and Hokama, Y. (1990). Potential clinical applications of C-reactive protein. J. Clin. Lab. Anal. 4, 231–235.
O’Neill, L. A. (2006). How Toll-like receptors signal: what we know and what we don’t know. Curr. Opin. Immunol. 18, 3–9.
Overstreet, D. H., Commissaris, R. C., De La Garza, R. II, File, S. E., Knapp, D. J., and Seiden, L. S. (2003). Involvement of 5-HT1A receptors in animal tests of anxiety and depression: evidence from genetic models. Stress 6, 101–110.
Papadopoulos, M. C., Krishna, S., and Verkman, A. S. (2002). Aquaporin water channels and brain edema. Mt. Sinai J. Med. 69, 242–248.
Patel, R. N., Attur, M. G., Dave, M. N., Patel, I. V., Stuchin, S. A., Abramson, S. B., and Amin, A. R. (1999). A novel mechanism of action of chemically modified tetracyclines: inhibition of COX-2-mediated prostaglandin E2 production. J. Immunol. 163, 3459–3467.
Paylor, R., Zhao, Y., Libbey, M., Westphal, H., and Crawley, J. N. (2001). Learning impairments and motor dysfunctions in adult Lhx5-deficient mice displaying hippocampal disorganization. Physiol. Behav. 73, 781–792.
Pellow, S., Chopin, P., File, S. E., and Briley, M. (1985). Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 14, 149–167.
Pitkänen, A., Schwartzkroin, P. A., and Moshé, S. L. (2006). Models of Seizures and Epilepsy. San Diego: Academic Press.
Pompl, P. N., Mullan, M. J., Bjugstad, K., and Arendash, G. W. (1999). Adaptation of the circular platform spatial memory task for mice: use in detecting cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. J. Neurosci. Methods 87, 87–95.
Popovic, N., Schubart, A., Goetz, B. D., Zhang, S. C., Linington, C., and Duncan, I. D. (2002). Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann. Neurol. 51, 215–223.
Povlishock, J. T., and Christman, C. W. (1995). The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J. Neurotrauma 12, 555–564.
Povlishock, J. T., and Pettus, E. H. (1996). Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochir. Suppl. 66, 81–86.
Prat, A., and Antel, J. (2005). Pathogenesis of multiple sclerosis. Curr. Opin. Neurol. 18, 225–230.
Quinn, R. (2005). Comparing rat’s to human’s age: how old is my rat in people years? Nutrition 21, 775–777.
Raber, J., Rola, R., Lefevour, A., Morhardt, D., Curley, J., Mizumatsu, S., Vandenberg, S. R., and Fike, J. R. (2004). Radiation-induced cognitive impairments are associated with changes in indicators of hippocampal neurogenesis. Radiat. Res. 162, 39–47.
Rancan, M., Otto, V. I., Hans, V. H., Gerlach, I., Jork, R., Trentz, O., Kossmann, T., and Morganti-Kossmann, M. C. (2001). Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J. Neurosci. Res. 63, 438–446.
Rapoport, M. J., Mccullagh, S., Shammi, P., and Feinstein, A. (2005). Cognitive impairment associated with major depression following mild and moderate traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 17, 61–65.
Rickels, K., Case, W. G., Schweizer, E., Garcia-Espana, F., and Fridman, R. (1991). Long-term benzodiazepine users 3 years after participation in a discontinuation program. Am. J. Psychiatry 148, 757–761.
Robel, S., Berninger, B., and Gotz, M. (2011). The stem cell potential of glia: lessons from reactive gliosis. Nat. Rev. Neurosci. 12, 88–104.
Rodgers, R. J., Lee, C., and Shepherd, J. K. (1992). Effects of diazepam on behavioural and antinociceptive responses to the elevated plus-maze in male mice depend upon treatment regimen and prior maze experience. Psychopharmacology (Berl.) 106, 102–110.
Rosenstein, J. M., Mani, N., Khaibullina, A., and Krum, J. M. (2003). Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J. Neurosci. 23, 11036–11044.
Saivin, S., and Houin, G. (1988). Clinical pharmacokinetics of doxycycline and minocycline. Clin. Pharmacokinet. 15, 355–366.
Salzberg, M., Kumar, G., Supit, L., Jones, N. C., Morris, M. J., Rees, S., and O’Brien, T. J. (2007). Early postnatal stress confers enduring vulnerability to limbic epileptogenesis. Epilepsia 48, 2079–2085.
Sanchez Mejia, R. O., Ona, V. O., Li, M., and Friedlander, R. M. (2001). Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 48, 1393–1399; discussion 1399–1401.
Silva, R. C., and Brandao, M. L. (2000). Acute and chronic effects of gepirone and fluoxetine in rats tested in the elevated plus-maze: an ethological analysis. Pharmacol. Biochem. Behav. 65, 209–216.
Silveira, M. C., Sandner, G., and Graeff, F. G. (1993). Induction of Fos immunoreactivity in the brain by exposure to the elevated plus-maze. Behav. Brain Res. 56, 115–118.
Silver, J. M., Mcallister, T. W., and Arciniegas, D. B. (2009). Depression and cognitive complaints following mild traumatic brain injury. Am. J. Psychiatry 166, 653–661.
Sindern, E., Niederkinkhaus, Y., Henschel, M., Ossege, L. M., Patzold, T., and Malin, J. P. (2001). Differential release of beta-chemokines in serum and CSF of patients with relapsing-remitting multiple sclerosis. Acta Neurol. Scand. 104, 88–91.
Siopi, E., Cho, A. H., Homsi, S., Croci, N., Plotkine, M., Marchand-Leroux, C., and Jafarian-Tehrani, M. (2011). Minocycline restores sAPPalpha levels and reduces the late histopathological consequences of traumatic brain injury in mice. J. Neurotrauma 28, 2135–2143.
Skold, M. K., Risling, M., and Holmin, S. (2006). Inhibition of vascular endothelial growth factor receptor 2 activity in experimental brain contusions aggravates injury outcome and leads to early increased neuronal and glial degeneration. Eur. J. Neurosci. 23, 21–34.
Sondell, M., Sundler, F., and Kanje, M. (2000). Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur. J. Neurosci. 12, 4243–4254.
Sorensen, T. L., Ransohoff, R. M., Strieter, R. M., and Sellebjerg, F. (2004). Chemokine CCL2 and chemokine receptor CCR2 in early active multiple sclerosis. Eur. J. Neurol. 11, 445–449.
Sprott, R. L., and Eleftheriou, B. E. (1974). Open-field behavior in aging inbred mice. Gerontologia 20, 155–162.
Stamatovic, S. M., Dimitrijevic, O. B., Keep, R. F., and Andjelkovic, A. V. (2006). Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J. Biol. Chem. 281, 8379–8388.
Stamatovic, S. M., Keep, R. F., Kunkel, S. L., and Andjelkovic, A. V. (2003). Potential role of MCP-1 in endothelial cell tight junction “opening”: signaling via Rho and Rho kinase. J. Cell Sci. 116, 4615–4628.
Stirling, D. P., Khodarahmi, K., Liu, J., Mcphail, L. T., Mcbride, C. B., Steeves, J. D., Ramer, M. S., and Tetzlaff, W. (2004). Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J. Neurosci. 24, 2182–2190.
Stirling, D. P., Koochesfahani, K. M., Steeves, J. D., and Tetzlaff, W. (2005). Minocycline as a neuroprotective agent. Neuroscientist 11, 308–322.
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M., and Kroemer, G. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446.
Tanielian, T., and Jaycox, L. H. (2008). Invisible Wounds of War: Psychological and Cognitive Injuries, their Consequences, and Services to Assist Recovery. Santa Monica: RAND Corporation.
Tekstra, J., Beekhuizen, H., Van De Gevel, J. S., Van Benten, I. J., Tuk, C. W., and Beelen, R. H. (1999). Infection of human endothelial cells with Staphylococcus aureus induces the production of monocyte chemotactic protein-1 (MCP-1) and monocyte chemotaxis. Clin. Exp. Immunol. 117, 489–495.
Teng, Y. D., Choi, H., Onario, R. C., Zhu, S., Desilets, F. C., Lan, S., Woodard, E. J., Snyder, E. Y., Eichler, M. E., and Friedlander, R. M. (2004). Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc. Natl. Acad. Sci. U.S.A. 101, 3071–3076.
Tenovuo, O. (2006). Pharmacological enhancement of cognitive and behavioral deficits after traumatic brain injury. Curr. Opin. Neurol. 19, 528–533.
Terrio, H., Brenner, L. A., Ivins, B. J., Cho, J. M., Helmick, K., Schwab, K., Scally, K., Bretthauer, R., and Warden, D. (2009). Traumatic brain injury screening: preliminary findings in a US Army Brigade Combat Team. J. Head Trauma Rehabil. 24, 14–23.
Thurman, D. J., Alverson, C., Dunn, K. A., Guerrero, J., and Sniezek, J. E. (1999). Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 14, 602–615.
Tikka, T. M., and Koistinaho, J. E. (2001). Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 166, 7527–7533.
Walf, A. A., and Frye, C. A. (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2, 322–328.
Wang, X., Zhu, S., Drozda, M., Zhang, W., Stavrovskaya, I. G., Cattaneo, E., Ferrante, R. J., Kristal, B. S., and Friedlander, R. M. (2003). Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 10483–10487.
Warden, D. (2006). Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil. 21, 398–402.
Wells, J. E., Hurlbert, R. J., Fehlings, M. G., and Yong, V. W. (2003). Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 126, 1628–1637.
Wixey, J. A., Reinebrant, H. E., Spencer, S. J., and Buller, K. M. (2011). Efficacy of post-insult minocycline administration to alter long-term hypoxia-ischemia-induced damage to the serotonergic system in the immature rat brain. Neuroscience 182, 184–192.
Wu, D. C., Jackson-Lewis, V., Vila, M., Tieu, K., Teismann, P., Vadseth, C., Choi, D. K., Ischiropoulos, H., and Przedborski, S. (2002). Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 22, 1763–1771.
Wu, L. W., Mayo, L. D., Dunbar, J. D., Kessler, K. M., Baerwald, M. R., Jaffe, E. A., Wang, D., Warren, R. S., and Donner, D. B. (2000). Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J. Biol. Chem. 275, 5096–5103.
Xu, L., Fagan, S. C., Waller, J. L., Edwards, D., Borlongan, C. V., Zheng, J., Hill, W. D., Feuerstein, G., and Hess, D. C. (2004). Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol. 4, 7. 12. doi:10.3389/fneur.2011.00012
Yrjanheikki, J., Keinanen, R., Pellikka, M., Hokfelt, T., and Koistinaho, J. (1998). Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. U.S.A. 95, 15769–15774.
Yrjanheikki, J., Tikka, T., Keinanen, R., Goldsteins, G., Chan, P. H., and Koistinaho, J. (1999). A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc. Natl. Acad. Sci. U.S.A. 96, 13496–13500.
Yune, T. Y., Lee, J. Y., Jung, G. Y., Kim, S. J., Jiang, M. H., Kim, Y. C., Oh, Y. J., Markelonis, G. J., and Oh, T. H. (2007). Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J. Neurosci. 27, 7751–7761.
Zemke, D., and Majid, A. (2004). The potential of minocycline for neuroprotection in human neurologic disease. Clin. Neuropharmacol. 27, 293–298.
Zhang, X., Chen, J., Graham, S. H., Du, L., Kochanek, P. M., Draviam, R., Guo, F., Nathaniel, P. D., Szabo, C., Watkins, S. C., and Clark, R. S. (2002). Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J. Neurochem. 82, 181–191.
Zhu, S., Stavrovskaya, I. G., Drozda, M., Kim, B. Y., Ona, V., Li, M., Sarang, S., Liu, A. S., Hartley, D. M., Wu, D. C., Gullans, S., Ferrante, R. J., Przedborski, S., Kristal, B. S., and Friedlander, R. M. (2002). Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417, 74–78.
Appendix

Table A1. List of animal models of various diseases, dose of minocycline treatment and the observed effects of the treatment.
Table A2. List of antibodies and their respective classifications and dilutions used to measure protein biomarker levels in sera and brain tissues.
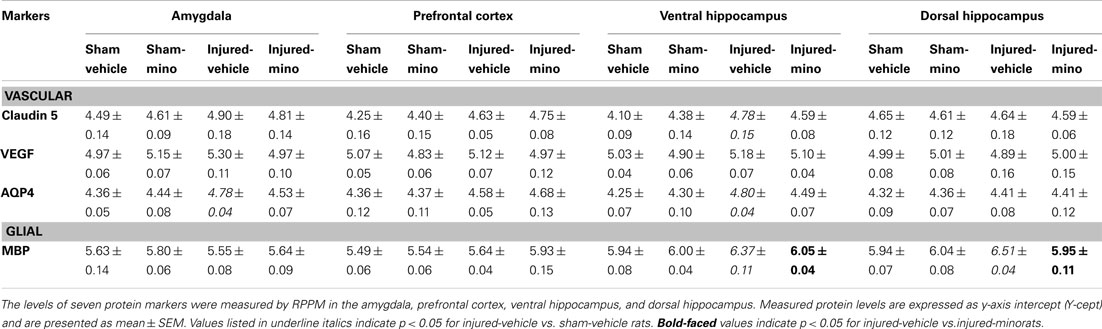
Table A3. The effect of injury and minocycline treatment on tissue levels of the selected protein biomarkers in the different experimental groups.
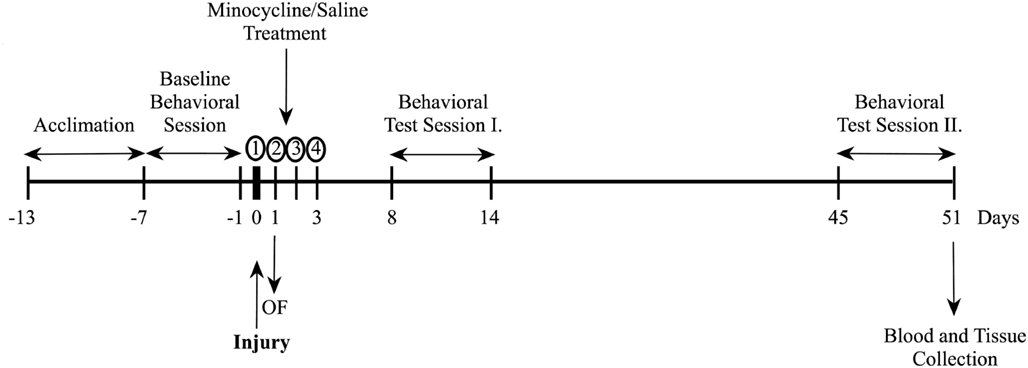
Figure A1. Outline of the experimental schedule. After 1 week of acclimation, baseline behavioral analyses, and injury (or sham), rats were treated intraperitoneally for four consecutive days (marked by circled numbers of 1 through 4) with 50 mg/kg of minocycline or saline starting at 4 hours after injury. Behavioral assessments (Open Field, Elevated Plus Maze, and Barnes Maze) were conducted before injury (Baseline Behavioral Session), and at 1 (open field OF only), 8 (Behavioral Test Session I.), and 45 days (Behavioral Test Session II.) after injury or sham. At the end of the experiment blood and brains were collected, processed, and analyzed using RPPM and ELISA.
Keywords: TBI, anti-inflammatory, treatment, neurobehavior, proteomics
Citation: Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, Kasper CE and Agoston DV (2012) Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front. Neur. 3:111. doi: 10.3389/fneur.2012.00111
Received: 03 January 2012; Accepted: 22 June 2012;
Published online: 16 July 2012.
Edited by:
Mårten Risling, Karolinska Institutet, SwedenReviewed by:
Mattias Sköld, Uppsala University, SwedenCandace L. Floyd, University of Alabama at Birmingham, USA
Copyright: © 2012 Kovesdi, Kamnaksh, Wingo, Ahmed, Grunberg, Long, Kasper and Agoston. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Denes V. Agoston, Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814, USA. e-mail:dmFnb3N0b25AdXN1aHMuZWR1