 Cláudia V. Maurer-Morelli1
Cláudia V. Maurer-Morelli1 Rodrigo Secolin1
Rodrigo Secolin1 Márcia E. Morita2
Márcia E. Morita2 Romênia R. Domingues1 Rafael B. Marchesini1 Neide F. Santos1 Eliane Kobayashi2†
Romênia R. Domingues1 Rafael B. Marchesini1 Neide F. Santos1 Eliane Kobayashi2† Fernando Cendes2
Fernando Cendes2 Iscia Lopes-Cendes1*
Iscia Lopes-Cendes1*- 1 Department of Medical Genetics, Faculty of Medical Sciences, University of Campinas, Campinas, São Paulo, Brazil
- 2 Department of Neurology, Faculty of Medical Sciences, University of Campinas, Campinas, São Paulo, Brazil
We aimed to identify the region harboring a putative candidate gene associated with hippocampal abnormalities (HAb) in a family with mesial temporal lobe epilepsy (MTLE). Genome-wide scan was performed in one large kindred with MTLE using a total of 332 microsatellite markers at ∼12 cM intervals. An additional 13 markers were genotyped in the candidate region. Phenotypic classes were defined according to the presence of hippocampal atrophy and/or hyperintense hippocampal T2 signal detected on magnetic resonance imaging. We identified a significant positive LOD score on chromosome 18p11.31 with a Zmax of 3.12 at D18S452. Multipoint LOD scores and haplotype analyses localized the candidate locus within a 6-cM interval flanked by D18S976 and D18S967. We present here evidence that HAb, which were previously related mainly to environmental risk factors, may be influenced by genetic predisposition. This finding may have major impact in the study of the mechanisms underlying abnormalities in mesial temporal lobe structures and their relationship with MTLE.
Introduction
The association between mesial temporal lobe epilepsy (MTLE) and hippocampal sclerosis (HS) has been well established; as well as the use of hippocampal atrophy (HA) and abnormal hippocampal signal, observed on magnetic resonance imaging (MRI), as an in vivo surrogate marker of HS (Berkovic et al., 1991; Cendes et al., 1993; Van Paesschen et al., 1997). Although the precise pathogenesis of mesial temporal sclerosis (MTS) and its relationship with MTLE is not completely clarified, it has been frequently associated with environmental predisposing factors, mainly an increased incidence of prolonged childhood febrile seizures (Abou-Khalil et al., 1993; VanLandingham et al., 1998).
We have described previously clinical and imaging features of a type of MTLE associated with HA, showing familial recurrence and low frequency of febrile seizures (Kobayashi et al., 2001, 2002, 2003). Most affected individuals in familial MTLE have a mild phenotype with a benign form of MTLE (Kobayashi et al., 2001). HA with or without hyperintense T2 signal on MRI was observed in most patients, including those with a single focal seizure, seizure remission (Kobayashi et al., 2003) as well as in 34% of asymptomatic first-degree relatives of patients with familial MTLE (Kobayashi et al., 2002). These reports suggest that hippocampal abnormalities (HAb) identified by MRI in familial MTLE have a genetic predisposition and it is not necessarily associated to seizures in all patients (Kobayashi et al., 2001; Coan et al., 2004).
In the present study we report the results of a genome-wide linkage study, which identifies the candidate region harboring a putative gene associated with MRI signs of HAb in a family with MTLE.
Materials and Methods
Family
Familial MTLE was identified when two or more individuals presented the diagnosis of MTLE, which was based on clinical and electroencephalographic (EEG) findings as defined by the International League against Epilepsy (ILAE) criteria (ILAE, 1989). For this study we selected one single informative kindred for linkage analysis, named F-10, in order to avoid the risk of genetic heterogeneity.
Magnetic Resonance Imaging
Magnetic resonance imagings were performed in a 2-T scanner including thin (1–3 mm) coronal T1-weighted images and T2-weighted coronal images, perpendicular to long axis of the hippocampal formation as described in Cendes et al. (1998) and Kobayashi et al. (2001, 2002).
Manual volumetry was performed using software developed by the National Institutes of Health (NIH-Image, National Institutes of Health, Bethesda, MD, USA) and anatomic guidelines were based on a standard protocol. Hippocampal volumes and asymmetry index (AI) for each patient (defined as the ratio of the smaller by the larger hippocampus) were compared to 30 healthy controls. Values that were 2 SDs below the mean values of control group were considered abnormal. Visual analysis was also performed looking for increased T2 signal and abnormal shape and axis of the hippocampus. Presence of HAb was identified if any abnormality was present.
Prior to genetic analysis, family members were classified into three phenotypic classes according to MRI findings: (1) affected: individuals with HAb detected by MRI, defined as: (i) HA, (ii) hyperintense T2 signal, (iii) abnormal shape or axis of the hippocampus, or (iv) any combination of these three findings (Kobayashi et al., 2002); (2) unaffected: individuals with no HAb detected by MRI; (3) unknown: individuals with no MRI information. Previous to enrollment, all family members provided informed consent which was approved by the Research Ethics Committee of our Institution.
Genotyping
DNA was isolated from peripheral lymphocytes of fresh blood by standard methods (Sambrook et al., 1989) and genotyped for an in-house customized panel of 332 microsatellite markers regularly spaced at ∼12 cM intervals throughout the 22 autosomal chromosomes. Microsatellite markers were chosen from the sex-average map of the Marshfield Human Genetic Map1 with a polymorphism information content (PIC) >75%. The correct orientation of markers was obtained using the NCBI Map Viewer2. Genotyping was carried out using capillary electrophoresis on MegaBACE™ 1000 96-capillary sequencers (GE Healthcare, Buckinghamshire, UK). The fluorescent dye-labeled primers (FAM™, VIC™, NED™) were customized or chosen from the MD10 panel set (Applied Biosystems, Foster City, CA, USA). Samples amplified by PCR and labeled with different dyes were pooled in a final volume of 40 μl H2O (1:20 for FAM™, 1:20 for VIC™, and 1:10 for NED™) in a 96-wells plate. Two microliters were transferred to a MegaBACE 96-wells plate and mixed with 8 μl of a loading solution (for each reaction: 7.75 μl of 0.1% Tween 20 in H2O + 0.25 μl of internal size standard labeled with ROX™ dye, ET-550R; GE Healthcare, Buckinghamshire, UK). Automatic calling of genotypes was based on the Fragment Profiler Software v1.2 (GE Healthcare, Buckinghamshire, UK). In addition we used a software developed in-house for processing output data from Fragment Profiler and to estimate allelic frequency (Secolin et al., 2008). Mendelian inconsistencies were evaluated by PEDCHECK program (O’Connell and Weeks, 1998).
Fine Mapping
Thirteen additional markers were genotyped on chromosome 18p in order to refine the candidate region: telomere, D18S476, D18S1098, D18S481, D18S1154, D18S52, D18S1132, D18S976, D18S1376, D18S967, D18S1163, D18S464, D18S1150, and D18S1158, centromere.
Linkage Analysis
Linkage analysis was performed under the assumption of a dominant mode of inheritance (Secolin et al., 2010) with 85% penetrance which was calculated based on data obtained directly from F-10 using a method proposed by Wang et al. (2006). Gene frequency of the mutated allele was set at 0.00025. Two-point LOD scores were calculated using the MLINK program, version 5.2 (CEPH, University of Utah and Columbia University, 1990), from the LINKAGE computer package (Lathrop and Lalouel, 1984; Terwilliger and Ott, 1994). In order to further narrow down the candidate region on chromosome 18p we carried out a multipoint linkage analysis using the LINKMAP support program from the LINKAGE package (Lathrop et al., 1985).
Results
A total of 28 individuals, including 14 patients, were genotyped. Clinical and MRI data of F-10 are shown in Table 2. The results of the genome-wide linkage study are summarized in Figure 1. Overall, we found only one single significant positive LOD score on chromosome 18p (Figure 1). Fine mapping of the candidate region identified a Zmax of 3.12 at þeta = 0.0 at the D18S452 locus (Table 1).
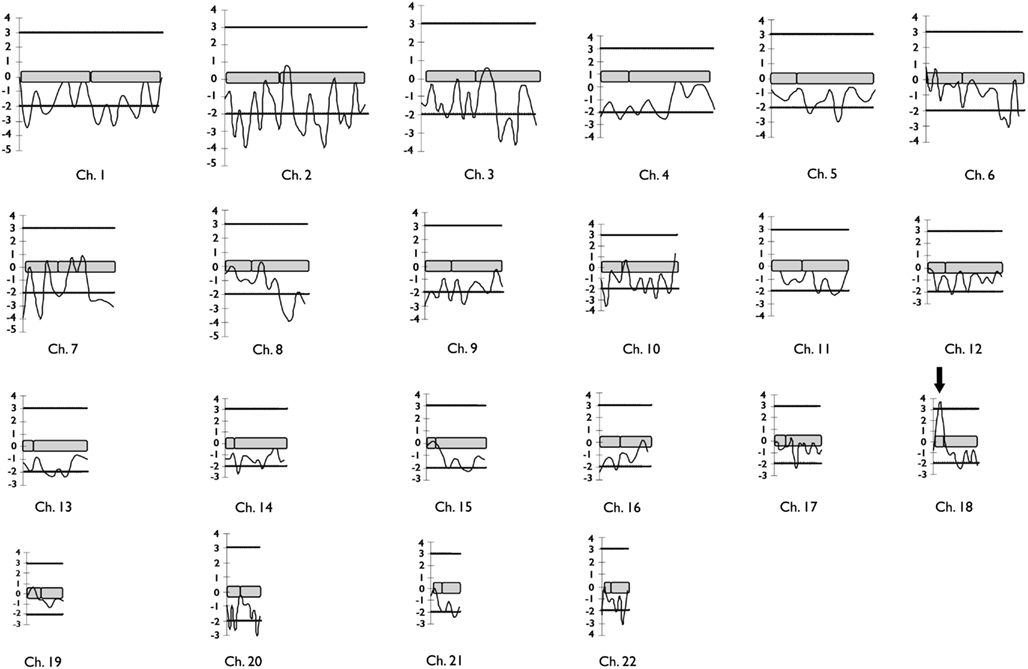
Figure 1. Diagram representing the results of two-point LOD scores for the 332 microsatellites markers genotyped in the genome-wide linkage study of F-10 segregating familial mesial temporal lobe epilepsy. Horizontal lines mark the upper and lower limits for LOD score significance. The arrow indicates the single significant positive LOD score found in the genome scan.
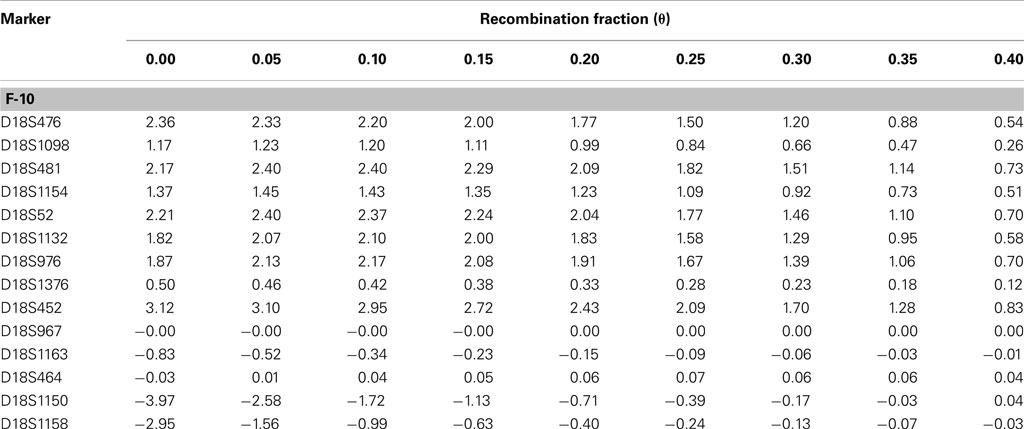
Table 1. Two-point LOD scores for the markers genotyped in the candidate region for familial temporal lobe epilepsy in the family F-10.
Haplotype analysis showed 12 affected individuals segregating the haplotype predicted to carry the candidate gene in F-10, indicating a critical region of 6 cM between markers at D18S976 and D18S967 loci (see individual III-3 in Figure 2).
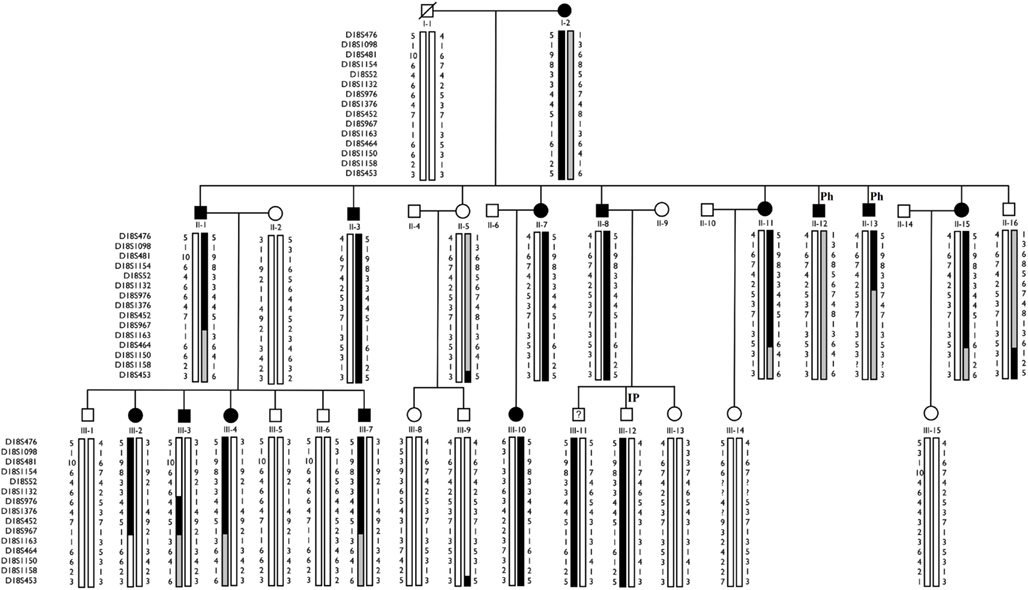
Figure 2. Haplotype analysis of F-10 for 14 markers localized on chromosome 18p. The haplotype inherited by affected individuals is represented by blackened vertical bars. Family members who did not participate in the linkage study are not shown. Individual III-3 shows a critical recombination event. IP, Incomplete Penetrance; Ph, Phenocopy.
However, the “affected” haplotype was also present in two individuals that are obligatory carries of the candidate gene mutation (individuals III-11 and III-12), but who were phenotypically classified as unknown and unaffected, respectively. In addition, haplotype analysis also showed two individuals who were considered affected, but who did not carry the “affected” haplotype (individuals II-12 and II-13). Multipoint analysis (Figure 3) confirms the mapping of the candidate region to a ∼6-cM interval between loci D18S976 and D18S967.
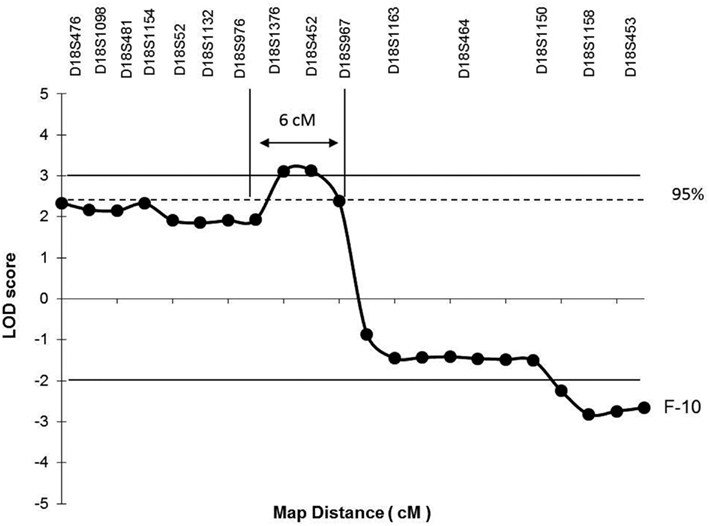
Figure 3. Multipoint linkage analysis on chromosome 18p depicting the candidate region for the putative gene causing the phenotype in F-10. Horizontal lines mark the upper and lower limits for LOD score significance. The single horizontal dotted line indicates the upper limit of the 95% confidence interval calculated for the maximum LOD score achieved. Arrows between vertical lines identify the critical region (6 cM) expected to contain the candidate gene flanked by markers at D18S967 and D18S976 loci.
Discussion
We identified a candidate locus on chromosome 18p11.31 associated with HAb in a family with MTLE.
Over the past 20 years, the progress in molecular genetics has lead to the identification of candidate loci and mutated genes in many epilepsy syndromes (Ottman, 2001). In temporal lobe epilepsy (TLE), a digenic inheritance with loci on 1q25–31 and 18qter and another locus on 12q22–q23.3 were described for familial TLE with febrile seizures (Baulac et al., 2001; Claes et al., 2004). Subsequently, a locus for familial MTLE was identified on chromosome 4q13.2–q21.3 by Hedera et al. (2007), but in contrast with F-10, none of the patients reported had MRI signs of HAb. Therefore, it becomes clear that locus heterogeneity is present even within the so-called familial forms of MTLE confirming the trend already seen in several other types of familial epilepsy syndromes (Baulac et al., 2001; Claes et al., 2004; Hedera et al., 2007).
Here we report a family which is part of a larger cohort showing a distinct type of familial MTLE associated with MRI signs of HAb, which also presents a low frequency of febrile seizures (Kobayashi et al., 2001). In the present study we used MRI signs of HAb to classify individuals as affected, independently of the presence of seizures (individual I-2, F-10; Kobayashi et al., 2002, 2003; Coan et al., 2004).
Although we had a reliable biological maker for phenotypic characterization, there were still difficulties with the MRI characterization of all individuals enrolled in the study. We could not ascertained the MRI-phenotypic status of individual III-11, since he presented claustrophobia and was unable to complete the exam; therefore, individual III-11 was considered unknown despite the fact that he carries the affected haplotype; thus, at this point we cannot completely exclude the possibility that he may have an abnormal MRI. Individual III-12 carries the affected haplotype but he had no MRI abnormalities, even when he was subsequently examined in a 3T MRI scan (data not shown); therefore, he may represent an example of incomplete penetrance. In fact, we estimated the penetrance in our linkage analysis at 85%. Incomplete penetrance is not unusual in familial epilepsies (Johnson et al., 1996; Striano et al., 2008) and it accounts for part of the complex relationship observed between genotype and phenotype in different epilepsy syndromes (Anderson et al., 2002; Ottman, 2005).
In addition, we identified two individuals (II-12 and II-13), who were considered affected, since they had MRI abnormalities (Table 2) but did not present the “affected” haplotype (Figure 2). Individual II-12 had an increased T2 signal and abnormal shape of the hippocampus and individual II-13 had left HA. Individual II-12 has a history of heavy alcohol intake, a well-known cytotoxic agent which can cause hippocampal damage (Sullivan et al., 1995; Agartz et al., 1999); thus, we raise the possibility that one or both individuals could be phenocopies. Therefore, at this point, we cannot completely exclude the possibility that the abnormalities seen on MRI of both individuals may be due to factors other than the mutated gene on chromosome 18p11.31 segregating in this family.
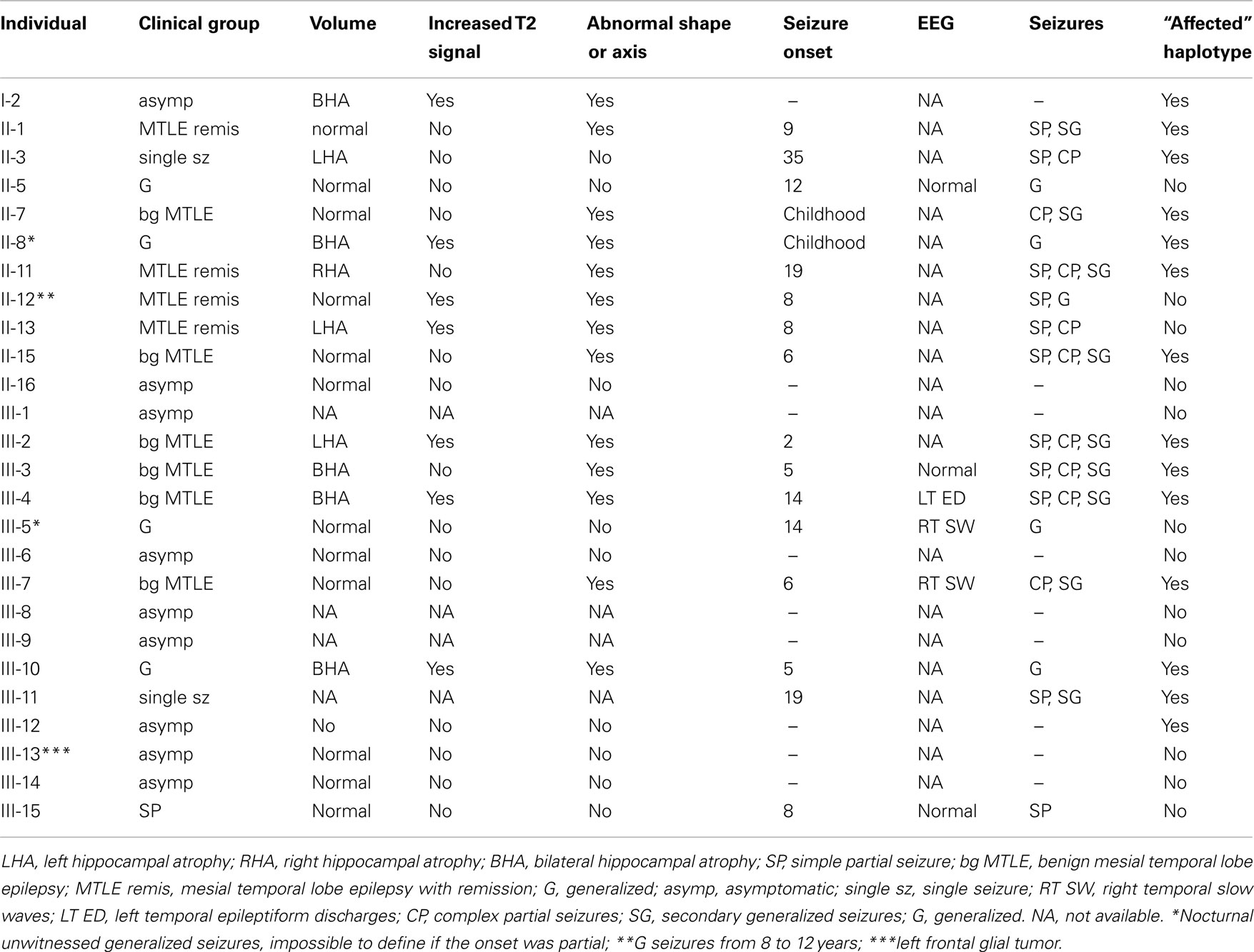
Table 2. Clinical information for family F-10.
The combined results of multipoint LOD scores and haplotype data points to a candidate interval of 6 cM between D18S976 and D18S 967. A search in the NCBI3 and Ensembl database4 showed at least three genes (Figure 4), which could play a role in the pathogenesis of HS such as: ZFP161 (zinc finger protein, 161, MIM 602126) a gene involved in CNS development and recently pointed out as a candidate for holoprosencephaly (Sobek-Klocke et al., 1997; Lee et al., 2004); L3MBTL4 [L(3)mbt-like 4 (Drosophila), gene ID91133], which functions as a cell adhesion molecule and EPB41L3 (erythrocyte membrane protein band 4.1-like3, MIM 605331) that encodes a family of proteins likely to take part in skeletal structures of cells and it is highly expressed in the brain (Peters et al., 1998). In addition, we identified the TTMA gene (two transmembrane domain family member A, gene ID 645369), and five hypothetical proteins localized in this candidate region. Mutation screening in candidate genes located within the 18p11.31 region is under way in order to identify the gene responsible for this disease.
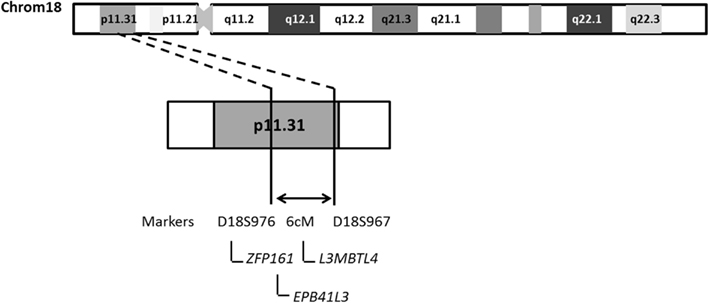
Figure 4. Schematic representation of chromosome 18 indicating the region likely to contain the candidate locus identified in F-10.
In conclusion, we identified a candidate locus for HAb associated with familial MTLE on chromosome 18p11.31 suggesting that a major gene predisposing to HS is present in patients from family F-10. This is strong evidence suggesting that genetic causes leading to hippocampal damage can also be related to MTLE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil), grants 02/10435-0; 03/13424-1; and 05/56578-4. We are grateful to our patients for their helpful cooperation.
Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
References
Abou-Khalil, B., Andermann, E., Andermann, F., Olivier, A., and Quesney, L. F. (1993). Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 34, 878–883.
Agartz, I., Momenan, R., Rawlings, R. R., Kerich, M. J., and Hommer, D. W. (1999). Hippocampal volume in patients with alcohol dependence. Arch. Gen. Psychiatry 56, 356–363.
Anderson, E., Berkovic, S., Dulac, O., Gardiner, M., Jain, S., Laue Friis, M., Lindhout, D., Noebels, J., Ottman, R., Scaramelli, A., Serratosa, J., Steinlein, O., Avanzini, G., Bailey-Wilson, J., Cardon, L., Fischbach, R., Gwinn-Hardy, K., Leppert, M., Ott, J., Lindblad-Toh, K., Weiss, K., ILAE Genetics Commission. (2002). ILAE genetics commission conference report: molecular analysis of complex genetic epilepsies. Epilepsia 43, 1262–1267.
Baulac, S., Picard, F., Herman, A., Feingold, J., Genin, E., Hirsch, E., Prud’Homme, J. F., Baulac, M., Brice, A., and LeGuern, E. (2001). Evidence for digenic inheritance in a family with both febrile convulsions and temporal lobe epilepsy implicating chromosomes 18qter and 1q25-31. Ann. Neurol. 49, 786–792.
Berkovic, S. F., Andermann, F., Olivier, A., Ethier, R., Melanson, D., Robitaille, Y., Kuzniecky, R., Peters, T., and Feindel, W. (1991). Hippocampal sclerosis in temporal lobe epilepsy demonstrated by magnetic resonance imaging. Ann. Neurol. 29, 175–182.
Cendes, F., Andermann, F., Gloor, P., Evans, A., Jones-Gotman, M., Watson, C., Melanson, D., Olivier, A., Peters, T., Lopes-Cendes, I., and Leroux, G. (1993). MRI volumetric measurement of amygdala and hippocampus in temporal lobe epilepsy. Neurology 43, 719–725.
Cendes, F., Lopes-Cendes, I., Andermann, E., and Andermann, F. (1998). Familial temporal lobe epilepsy: a clinically heterogeneous syndrome. Neurology 50, 554–557.
Claes, L., Audenaert, D., Deprez, L., Van Paesschen, W., Depondt, C., Goossens, D., Del-Favero, J., Van Broeckhoven, C., and De Jonghe, P. (2004). Novel locus on chromosome 12q22-q23.3 responsible for familial temporal lobe epilepsy associated with febrile seizures. J. Med. Genet. 41, 710–714.
Coan, A. C., Kobayashi, E., Lopes-Cendes, I., Li, L. M., and Cendes, F. (2004). Abnormalities of hippocampal signal intensity in patients with familial mesial temporal lobe epilepsy. Braz. J. Med. Biol. Res. 37, 827–832.
Hedera, P., Blair, M. A., Andermann, E., Andermann, F., D’Agostino, D., Taylor, K. A., Chahine, L., Pandolfo, M., Bradford, Y., Haines, J. L., and Abou-Khalil, B. (2007). Familial mesial temporal lobe epilepsy maps to chromosome 4q13.2-q21.3. Neurology 68, 2107–2112.
ILAE. (1989). Commission on Classification and Terminology of the International League against epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 30, 389–399.
Johnson, W. G., Kugler, S. L., Stenroos, E. S., Meulener, M. C., Rangwalla, I., Johnson, T. W., and Mandelbaum, D. E. (1996). Pedigree analysis in families with febrile seizures. Am. J. Med. Genet. 61, 345–352.
Kobayashi, E., D’Agostino, M. D., Lopes-Cendes, I., Berkovic, S. F., Li, M. L., Andermann, E., Andermann, F., and Cendes, F. (2003). Hippocampal atrophy and T2 weighted signal changes in familial mesial temporal lobe epilepsy. Neurology 60, 405–409.
Kobayashi, E., Li, L. M., Lopes-Cendes, I., and Cendes, F. (2002). MRI evidence of hippocampal sclerosis in asymptomatic first degree relatives of patients with familial mesial temporal lobe epilepsy. Arch. Neurol. 59, 1891–1894.
Kobayashi, E., Lopes-Cendes, I., Guerreiro, C. A., Sousa, S. C., Guerreiro, M. M., and Cendes, F. (2001). Seizure outcome and hippocampal atrophy in familial mesial temporal lobe epilepsy. Neurology 56, 166–172.
Lathrop, G. M., and Lalouel, J. M. (1984). Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 36, 460–465.
Lathrop, G. M., Lalouel, J. M., Julier, C., and Ott, J. (1985). Multilocus linkage analysis in humans: detection of linkage and estimation of recombination. Am. J. Hum. Genet. 37, 482–498.
Lee, K. H., Kwak, Y. D., Kim, D. H., Chang, M. Y., Lee, Y. S., and Lee, Y. S. (2004). Human zinc finger protein 161, a novel transcriptional activator of the dopamine transporter. Biochem. Biophys. Res. Commun. 313, 969–976.
O’Connell, J. R., and Weeks, D. E. (1998). DEPedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 63, 259–266.
Peters, L. L., Weier, H. U., Walensky, L. D., Snyder, S. H., Parra, M., Mohandas, N., and Conboy, J. G. (1998). Four, paralogous protein 4.1 genes map to distinct chromosomes in mouse and human. Genomics 54, 348–350.
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Secolin, R., Maurer-Morelli, C. V., Cendes, F., and Lopes-Cendes, I. (2010). Segregation analysis in mesial temporal lobe epilepsy with hippocampal atrophy. Epilepsia 51, 47–50.
Secolin, R., Rocha, C. S., Torres, F. R., Santos, M. L., Maurer-Morelli, C. V., Santos, N. F., and Lopes-Cendes, I. (2008). LINKGEN: a new algorithm to process data in genetic linkage studies. Genomics 91, 544–547.
Sobek-Klocke, I., Disqué-Kochem, C., Ronsiek, M., Klocke, R., Jockusch, H., Breuning, A., Ponstingl, H., Rojas, K., Overhauser, J., and Eichenlaub-Ritter, U. (1997). The human gene ZFP161 on 18p11.21-pter encodes a putative c-myc repressor and is homologous to murine Zfp161 (chr 17) and Zfp161-rs1 (X chr). Genomics 43, 156–164.
Striano, P., Gambardella, A., Coppola, A., Di Bonaventura, C., Bovo, G., Diani, E., Boaretto, F., Egeo, G., Ciampa, C., Labate, A., Testoni, S., Passarelli, D., Manna, I., Sferro, C., Aguglia, U., Caranci, F., Giallonardo, A. T., Striano, S., Nobile, C., and Michelucci, R. (2008). Familial mesial temporal lobe epilepsy (FMTLE): a clinical and genetic study of 15 Italian families. J. Neurol. 255, 16–23.
Sullivan, E. V., Marsh, L., Mathalon, D. H., Lim, K. O., and Pfefferbaum, A. (1995). Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol. Clin. Exp. Res. 19, 110–122.
Terwilliger, J. D., and Ott, J. (1994). Handbook of Human Genetic Linkage. Baltimore: Johns Hopkins University Press.
Van Paesschen, W., Revesz, T., Duncan, J. S., King, M. D., and Connelly, A. (1997). Quantitative neuropathology and quantitative magnetic resonance imaging of the hippocampus in temporal lobe epilepsy. Ann. Neurol. 42, 756–766.
VanLandingham, K. E., Heinz, E. R., Cavazos, J. E., and Lewis, D. V. (1998). Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann. Neurol. 43, 413–426.
Keywords: genetics, microsatellites, linkage study, 18p11.31, hippocampal abnormalities
Citation: Maurer-Morelli CV, Secolin R Morita ME, Domingues RR, Marchesini RB, Santos NF, Kobayashi E Cendes F and Lopes-Cendes I (2012) A locus identified on chromosome18p11.31 is associated with hippocampal abnormalities in a family with mesial temporal lobe epilepsy . Front. Neur. 3:124. doi: 10.3389/fneur.2012.00124
Received: 02 June 2012; Paper pending published: 28 June 2012;
Accepted: 18 July 2012; Published online: 10 August 2012.
Edited by:
David M. Labiner, University of Arizona College of Medicine, USAReviewed by:
Larry Baum, The Chinese University of Hong Kong, Hong Kong; Erik K. St. Louis, Mayo Clinic and Foundation, USACopyright: © 2012 Maurer-Morelli, Secolin, Morita, Domingues, Marchesini, Santos, Kobayashi, Cendes and Lopes-Cendes. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Iscia Lopes-Cendes, Department of Medical Genetics, Faculty of Medical Sciences, University of Campinas, Tessália Vieira de Camargo, 126, Campinas, São Paulo 13083-887, Brazil. e-mail:aWNlbmRlc0B1bmljYW1wLmJy
†Current address: Eliane Kobayashi, Montreal Neurological Institute and Hospital, McGill University, Montreal, QC, Canada.