


- 1Departments of Psychiatry and Neuroscience, Center of Neuroproteomics and Biomarker Research, McKnight Brain Institute, University of Florida, Gainesville, FL, USA
- 2Laboratory of Neurodegenerative Diseases and Central Nervous System Biomarkers, Departments of Neurology and Physiology/Pharmacology, State University of NewYork Downstate Medical Center, Brooklyn, NY, USA
- 3Banyan Laboratories, Banyan Biomarkers Inc., Alachua, FL, USA
The susceptibility of the normal cellular prion protein isoform, cellular prion protein (PrPC), to proteolytic digestion has been well documented. In addition, a link between PrPC and the cytosolic protease, calpain, has been reported although the specifics of the interaction remain unclear. We performed in vitro and in cell-based studies to examine this relationship. We observed that human recombinant PrP (HrPrP) was readily cleaved by calpain-1 and -2, and we have identified and defined the targeted cleavage sites. In contrast, HrPrP was resistant to caspase-3 digestion. Unexpectedly, when brain lysates from PrPC-expressing mice were treated with calpain, no appreciable loss of the intact PrPC, nor the appearance of PrPC breakdown products (BDPs) were observed, even though alpha II-spectrin was converted to its signature calpain-induced BDPs. In addition, when rat cerebrocortical neuronal cultures (RtCNC) were subjected to the two neurotoxins at subacute levels, maitotoxin (MTX) and N-methyl-D-aspartate (NMDA), PrPC-BDPs were also not detectable. However, a novel finding from these cell-based studies is that apparently full-length, mature PrPC is released into culture media from RtCNC challenged with subacute doses of MTX and NMDA. Calpain inhibitor SNJ-1945 and caspase inhibitor IDN-6556 did not attenuate the release of PrPC. Similarly, the lysosomal protease inhibitor, NH4Cl, and the proteasome inhibitor, lactacystin, did not significantly alter the integrity of PrPC or its release from the RtCNC. In conclusion, rat neuronal PrPC is not a significant target for proteolytic modifications during MTX and NMDA neurotoxic challenges. However, the robust neurotoxin-mediated release of full-length PrPC into the cell culture media suggests an unidentified neuroprotective mechanism for PrPC.
Introduction
The cellular prion protein (PrPC) is a host-coded, glycosylphosphatidylinositol (GPI)-anchored (linked through Serine-231) transmembrane glycoprotein found in all mammalian cells with relatively high levels in the central nervous system (CNS; Kretzschmar et al., 1986; Moudjou et al., 2001) particularly at neuronal synapses (Sales et al., 1998). Following prion agent infection, the protease-sensitive PrPC is converted into the conformationally altered, disease-specific PrPSc isoform which is partially protease-resistant (Riesner, 2003). In contrast to PrPSc, PrPC is soluble in detergents and sensitive to proteolytic digestion by PK.
Although PrPC has been associated with numerous cellular roles associated with cell signaling and/or neuroprotective functions, the specific physiologic function of PrPC remains unclear. PrPC neuroprotective functions that have been demonstrated both in vivo and in vitro (Westergard et al., 2007; Linden et al., 2008) include: cytoprotective activity against internal or environmental stresses that initiate apoptosis and oxidative stress, ionic channel modulation, transmembrane signaling, as well as formation and maintenance of synapses. PrPC not only protects neurons in vitro and in vivo from N-methyl-D-aspartate (NMDA)-evoked excitotoxicity (Khosravani et al., 2008) but also against oxidative stress and plays a role in Cu/Zn superoxide dismutase activity (Brown and Besinger, 1998; Wong et al., 2000; Brown et al., 2002).
PrPC and PrPSc are subject to diverse intracellular proteolytic processing events (Pan et al., 1992; Harris et al., 1993; Taraboulos et al., 1995). PrPC undergoes proteolytic cleavage at amino acids 110/111 within a segment of conserved hydrophobic amino acids to produce a ∼17 kDa C-terminal fragment referred to as C1. Studies suggest that ADAM/TACE (a disintegrin and metalloprotease/tumor necrosis factor α-converting enzyme) matrix metalloproteases may be responsible for the generation of the C1 fragment (Vincent et al., 2001). Previous studies using post-mortem human brain extracts demonstrated that the disease-associated PrP in Creutzfeldt–Jakob disease (CJD) brains is cleaved by a cellular protease to generate a C-terminal fragment, referred to as C2, which has the same molecular weight as PrP27–30, the protease-resistant core of PrPSc (Chen et al., 1995). Dron et al. (2010) showed that the full-length PrPSc to C2 ratio varied such that uncleaved PrPSc accumulated in primary neurons and brain whereas PrPSc processing occurred in infected Rov and MovS cells. They also found that cathepsin, but not calpain, inhibitors markedly reduced C2 formation. In contrast, studies by Yadavalli et al. (2004) using persistently infected scrapie mouse brain cells or persistently infected scrapie N2a cells indicate that endoproteolytic cleavage of PrPSc is facilitated by calpains.
Wang et al. (2005) showed that in several cell lines and in primary mouse cortical neurons PrPC was normally degraded by the proteasome but could alternatively be targeted by calpain. Furthermore, Hachiya et al. (2011) reported that the calpain inhibitor, calpastatin, dramatically inhibited normal endoproteolysis of PrPC in N2a cells. Also, they found that the molecular weight of PrPC fragments generated by spontaneous proteolysis was identical to those produced when PrPC translated in vitro was exposed to exogenously added calpain. Due to these conflicting data regarding the potential vulnerability of PrPC to calpain and other cytosolic proteases, we performed in vitro and in cell-based studies using rat primary cerebrocortical neuronal cultures (RtCNC). We describe the possibility of a novel neuroprotective function for calpain-resistant, full-length PrPC that is shed from neuronal cells in response to neurotoxic challenge.
Materials and Methods
Materials
Secondary antibodies were purchased from Novagen (Philadelphia, PA, USA). Maitotoxin (MTX) and clasto-lactacystin-β-lactone from Calbiochem (Billerica, MA, USA). NMDA, NH4Cl, and other chemical reagents were from Sigma-Aldrich (St. Louis, MO, USA). Human recombinant calpain-1, rat calpain-2, and human recombinant caspase-3 were from EMD Millipore Biosciences (Billerica, MA, USA). Calpain inhibitor SNJ-1945 (SNJ) was a gift from Senju Pharmaceutical (Shimazawa et al., 2010) while pan caspase inhibitor IDN-6556 (IDN) was synthesized in-house at Banyan Laboratories according to published methods (Hoglen et al., 2004). Cell culture grade reagents were from Invitrogen (Grand Island, NY, USA). Anti-alpha II (αII)-spectrin monoclonal antibody (Mab) was purchased from Enzo Life Sciences (Farmingdale, NY, USA) while anti-PrP Mabs 7E4, E11, and D8 were generated by Dr. Rubenstein at SUNY Downstate Medical Center. Since rat PrP is not commercially available. We used human recombinant PrP (HrPrP) as a substitute. The human PrP (accession # AAH22532) and rat PrP (accession # BAA08790) show a high level of homology. Purified E. coli HrPrP (residues 23–231 based on the human PrP sequence mimicked signal peptidase removal of the first 22 residues) containing an N-terminal (His)6-tag fusion protein, designated (His)6-HrPrP, was purchased from Abnova (Walnut, CA, USA) and EMD Millipore Bioscience (Billerica, MA, USA).
RtCNC Preparation and Neurotoxin Challenges
The Institutional Animal Care and Use Committee at the University of Florida (Gainesville, FL, USA) approved the use of timed pregnant rats for these studies and all animal use followed the appropriate regulatory standards. RtCNC were prepared from fetal Sprague Dawley rats as previously described (Wang et al., 1996). These neuronal-enriched cultures were maintained in vitro and allowed to mature for 10 days. Cells were then either untreated (control), exposed to dimethylsulfoxide (DMSO) vehicle alone, or challenged with neurotoxins (0.3 nM MTX or 300 μM NMDA) for 24 h in the absence or presence of lysosomal neutralizing agent NH4Cl (10 mM), proteasome inhibitor lactacystin (10 μM), SNJ (30 μM), or IDN (30 μM). Inhibitors were added 1 h prior to addition of neurotoxic drug. All treatments were performed in serum-free Dulbecco’s Minimal Essential Medium in a volume of 300 μl per well of a 12-well cell culture plate. DMSO vehicle alone as well as all drug treatments had no adverse affects on cell morphology and viability for at least 72 h (data not shown). After treatments, conditioned media was collected from each sample into separate tubes on ice and clarified by microcentrifugation at 10,000 × g for 5 min. Supernatants (“conditioned cell culture media”) were immediately frozen at −80°C.
Soluble cell extracts were generated by gently shaking cells for 2 h at 4°C in 1x Triton X-100 lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, 1 mM dithiothreitol (DTT), 1x Roche complete protease inhibitor cocktail, 1x phosphatase inhibitors (Sigma). This treatment resulted in complete cell lysis by microscopic observations. Extracts were transferred to 1.5 ml microcentrifuge tubes, spun at 10,000 × G for 10 min at 4°C to generate insoluble pellets, and both were stored at −80°C. The insoluble pellets were then extracted and resuspended in 1x RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% Igepal CA-630, 0.5% sodium deoxycholate, 0.2% SDS) supplemented with 1x protease inhibitor cocktail and disrupted by cup horn sonication (Sonics Vibracell, Newtown, CT, USA) for three 10 s. on−10 s. off cycles (20% power output). The samples were microcentrifuged at 10,000 × G for 10 min at 4°C and the supernatants, labeled as insoluble cell extracts, were stored at −80°C. Unless otherwise stated, 50 μg each of soluble and insoluble fractions and 20 μl conditioned media were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting.
For Endoglycosidase H (Endo H) treatment, combined 25 μl of samples (soluble or insoluble cell extracts, conditioned cell culture media) with 3 μl of 10x glycoprotein denaturing buffer (5% SDS, 0.4 M DTT) and 2 μl of distilled water. Boiled for 10 min to denature sample and allowed to cool. Next, added a final concentration of 0.05 M sodium citrate (pH 5.5) and 500 units of Endo H (New England BioLabs, Ipswich, MA, USA) in a total final reaction volume of 40 μl. Incubated the reaction for 2 h at 37°C and stopped by boiling for 4 min in 1x SDS-PAGE sample buffer. The samples were electrophoresed on 12% gels and immunoblotted as described below.
Mouse Brain Lysate Preparation
Adult naive mice were anesthetized and immediately sacrificed by decapitation. Brain was exposed and flushed with ice-cold phosphate-buffered saline (PBS). The bilateral cortex was immediately removed, rinsed with ice-cold PBS, and placed into a 1.5 ml microcentrifuge or cryofreeze tube, snap-frozen in liquid nitrogen and stored at −80°C until used. For brain lysates, samples were pulverized to a fine powder with a mortar and pestle set into dry ice. The pulverized brain tissue powder was then treated for 90 min at 4°C with 1X Triton X-100 lysis buffer. The brain lysates were then centrifuged at 8,000 × G for 5 min at 4°C and the supernatants were snap-frozen in liquid nitrogen and stored at −80°C. Protein concentrations of samples were determined using the Bio-Rad DC protein assay.
SDS-PAGE, Immunoblotting, and Statistics
Brain tissue extracts (20 μg), soluble and insoluble RtCNC cell lysate fractions (50 μg), and conditioned media (20 μl) were separated by SDS-PAGE (10–20% gradient gels for HrPrP, 12% gels for PrPC, 4–20% gradient gels for spectrin), and transferred to PVDF membrane by iBlot (Invitrogen) or the semi-dry method. Blots were blocked in 5% non-fat dry milk in Tris-buffered saline containing 0.2% Tween-20, pH 7.4 (TBST), and then probed with primary Mabs (anti-PrP Mabs 7E4, E11, or D8 at 1 μg/ml, and anti-αII-spectrin at 0.05 μg/ml) overnight. The bands were either visualized by chemiluminescence using a goat anti-mouse IgG-horseradish peroxidase conjugate (ECL Super Signal West Dura, Pierce) or by incubation with biotin-conjugated secondary antibody followed by streptavidin-alkaline phosphatase conjugate and developed with NBT-BCIP substrate. Quantification of PrPC, as represented by the non-, mono-, and deglycosylated isoforms of the protein, was performed by densitometric analysis using NIH Image J software v. 1.34. In SDS-PAGE and Western blot experiments, it is noted for in vitro protein digestion, we loaded the same amount of starting protein concentration before digestion and made sure equal sample volume was loaded. For cell lysate studies, we routinely run Ponceau S staining and/or β-actin to ensure protein loading is the same for each lane. Unless stated otherwise, all data shown are representative of three separate experiments.
In vitro Proteolysis of (His)6-HrPrP or PrPC in Mouse Brain Lysate
A 100 μg aliquot of (His)6-HrPrP or mouse brain lysate was subjected to calpain-1, calpain-2, or caspase-3 digestion (each at a protease:substrate ratio of 1:50). For (His)6-HrPrP, following SDS-PAGE, the proteins were transferred to PVDF membrane and stained with Coomassie Brilliant Blue (0.6% wt/vol in 1:1 methanol:H2O) for 15 s followed by methanol:H2O (1:1) destaining until protein bands were visible. After soaking in water, PVDF membranes were air-dried. Major breakdown products (BDPs) were identified by N-terminal microsequencing performed at the Interdisciplinary Center for Biotechnology Research (University of Florida, Gainesville, FL, USA). Alternatively, a 5 μg aliquot of (His)6-HrPrP was digested with calpain-2 and 0.5 μg protein were analyzed per lane.
Results
Calpain Processing of HrPrP and Identification of Major Calpain Cleavage Sites
The issue of PrPC vulnerability to calpain proteolysis was first addressed. The E. coli expressed (His)6-HrPrP was subjected to calpain-1, calpain-2, and caspase-3 digestion (Figure 1). Calpain-1 and calpain-2 are the two dominant isoforms of calpain in neurons. Caspase-3 treatment was also examined since it has previously been shown that many calpain substrates also serve as substrates for caspase-3 (Wang, 2000).
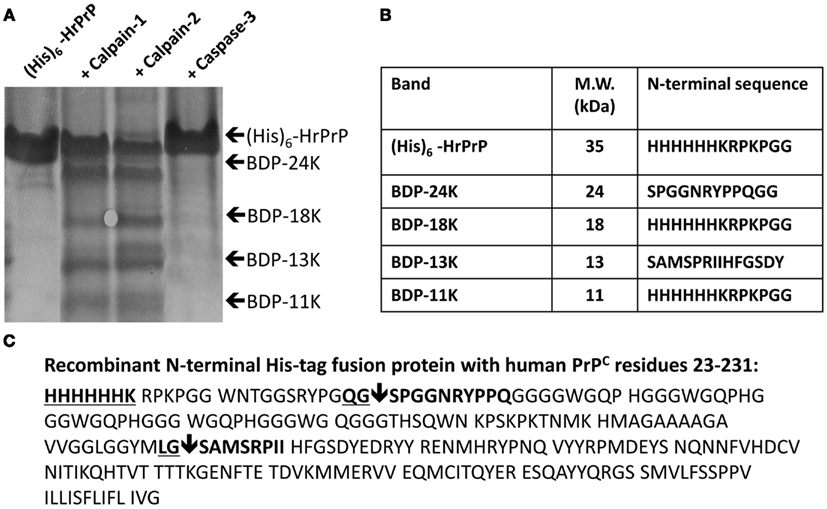
Figure 1. HrPrP vulnerability to calpain-1, -2 digestion with identification of major cleavage sites. E. coli expressed recombinant N-terminal (His)6-tag fusion protein with human PrPC residues 23–231 (Human prion protein; accession # NP 898902) was subjected to calpain-1, calpain-2, and caspase-3 digestion (1:50 protease:substrate ratio). Following SDS-PAGE, proteins were transferred to PVDF membrane and stained with Coomassie blue. (A) Major BDPs were identified by N-terminal microsequencing. (B) Based on the PrPC amino acid sequence the cleavage sites are identified and depicted by downward arrows. The new N-terminal sequences are identified in bold and the calpain recognition P1–P2 sequences are underlined and in bold (C).
Following SDS-PAGE and Coomassie blue staining, intact (His)6-HrPrP migrated as an ∼30 kDa protein, while four major BDPs of molecular weights 24, 18, 13, 11 kDa were observed when (His)6-HrPrP was digested by either calpain-1 or calpain-2 (Figure 1A). In contrast, caspase-3 digestion yielded no BDPs or observable reduction of intact (His)6-HrPrP intensity (Figure 1A). N-terminal microsequencing of these proteins identified PrP-BDP-18 and PrP-BDP-11 kDa as N-terminal PrP products each containing an intact His-tagged N-terminus (Figure 1B). PrP-BDP-24 kDa contained a new N-terminus beginning at residue 40 (S40PGGNRYPPQGG), while PrP-BDP-13 kDa possessed a new N-terminus beginning at residue 131 (S131AMSPRIIHFGSDY; Figures 1B,C). Thus the two major calpain cleavage sites were identified as between PrP residues 40–41 and residues 130–131(Figure 1C).
Immunostaining was performed to further characterize calpain-2 induced proteolytic cleavage of (His)6-HrPrP (Figure 2). (His)6-HrPrP was digested with calpain-2 using three protease:substrate ratios of 1:200, 1:50, and 1:5 with the last condition causing the most extensive PrP hydrolysis. Treated protein samples (500 ng) were analyzed by western blotting using PrP-specific Mabs 7E4 and E11 (Figure 2A) as well as a rabbit polyclonal anti-His tag antibody. As previously reported (Chang et al., 2012) and depicted in Figure 2B, the epitope for Mab 7E4 are PrP residues 29–35. Since one of the targeted calpain cleavage sites is residues 40–41. MAb 7E4 antibody detected epitope residue 29–35 which is N-terminal to the first calpain cleavage site (between residue 40–41), it is thus truncated off as a very small fragment (less than 4 kDa) thus not readily observable on the blot (Figure 2A).
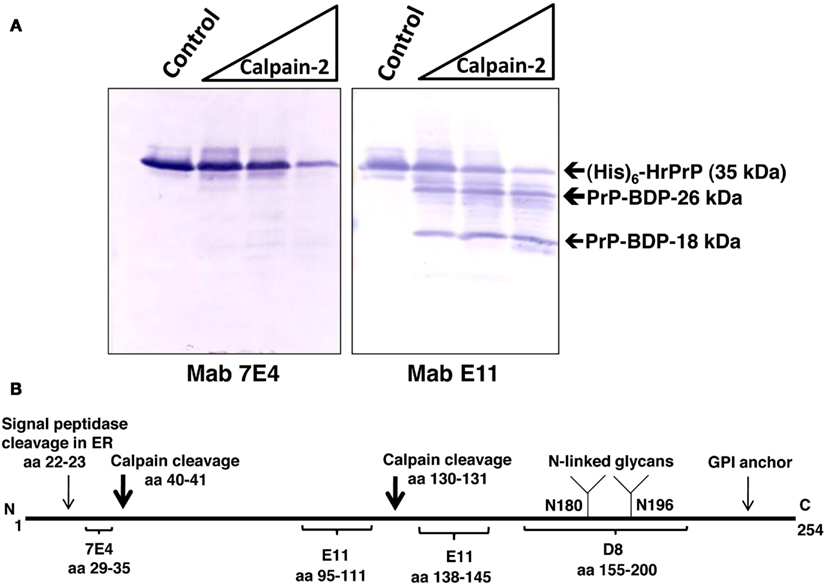
Figure 2. Characterization of HrPrP proteolysis by calpain-2. (A) Recombinant N-terminal (His)6-tag PrP fusion protein was untreated (control) digested with calpain-2 (protease:substrate ratios of 1:200, 1:50, 1:5). Five hundred nanograms of protein was then subjected to SDS-PAGE and western blotting. Three antibodies were used. Anti-PrP Mabs 7E4, E11, and anti-His tag antibody. Rainbow molecular weight markers were used. (B) Schematic of PrP showing the positions of the two calpain cleavage sites and the anti-PrP Mab epitopes.
Instead, we observed a protease dose-dependent reduction of intact (His)6-HrPrP indicative of proteolysis (Figure 2A). Anti-His tag antibody, which detects the N-terminal (His)6-tag, produced essentially the same results as Mab7E4 (data not shown).
In contrast, anti-PrP Mab E11 targets two internal epitopes (aa 95–111 and aa 138–145; Figure 2B) and upon cleavage by calpain-2, two fragments of 26 and 18 kDa are readily observed in addition to the reduction of intact PrP (Figure 2A). These results further confirmed that (His)6-HrPrP can be readily digested by calpain-2.
Vulnerability of Endogenous Brain PrPC to Calpain Proteolysis
Previous studies (Wang et al., 2005; Dron et al., 2010) have showed that PrP in different tissues or cell types might have variable vulnerability to endogenous proteolysis. Thus we next sought to examine the vulnerability of native PrPC to calpain and caspase-3 proteolysis in vitro with mouse brain lysate under digestion conditions similar to that described for (His)6-HrPrP. We observed the characteristic calpain-mediated αII-spectrin BDPs (SBDPs) SBDP150 and SBDP145, while caspase-3 generated the characteristic SBDP150i and SBDP120 (Figure 3A), as has been previously described (Zhang et al., 2009). In contrast, only two very minor BDPs (30 and 28 kDa) of the 33–35 kDa PrPC were observed by western blotting when immunostained with anti-PrP Mab E11 (Figure 3B). No PrPC-BDPs were observed following immunostaining with Mab 7E4 (data not shown). This result strongly suggests that endogenous PrPC, unlike (His)6-HrPrP, is relatively resistant to calpain proteolysis.
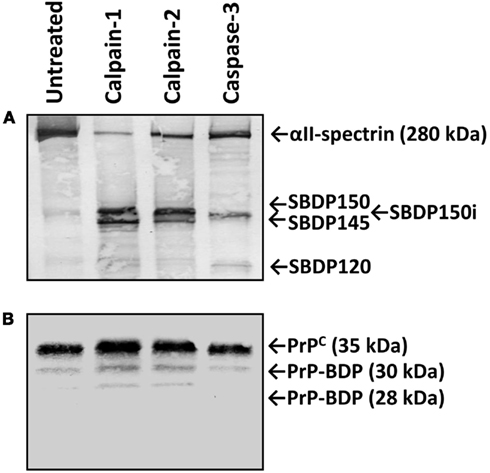
Figure 3. Relative resistance of PrPC from mouse brain lysate to calpain proteolysis. Lysate from naïve mouse brains were prepared and digested with calpain-1, -2, or caspase-3 as described in section “Materials and Methods.” Aliquots of the untreated and treated lysates were then electrophoresed, western blotted, and immunostained with either anti-αII-spectrin Ab (A) or with anti-PrP Mab E11. (B) Characteristic calpain-generated SBDP150 and SBDP145 as well as caspase-3 generated SBDP150i and SBDP120 are indicated. No PrPC-BDPs were observed following the treatments with Mab 7E4 (data not shown).
Integrity and Localization of PrPC in RtCNC Subjected to Neurotoxic Challenges
We next sought to examine the integrity and localization of PrPC in RtCNC after they were subjected to neurotoxic challenges. Neurotoxin calcium channel opener MTX (0.3 nM) or excitotoxin glutamate analog NMDA (300 μM) were selected since it has previously been shown that these challenges induce strong calpain and mixed calpain/caspase-3 activation (Wang et al., 1996; Zhang et al., 2009).
PrPC could reside in membrane-associated fractions or in soluble cytosolic fractions. We therefore performed cell extractions which allowed us to examine soluble and insoluble cellular proteins separately by initially lysing cells with mild neutral detergents followed by extraction of the remaining insoluble proteins with anionic detergents. Thus soluble and insoluble fractions (50 μg each) and conditioned cell culture media (20 μl), were analyzed by western blotting using Mab D8 (Figure 4). In the insoluble fraction of control cortical cultures we observed that the bulk of the PrPC was full-length (33–35 kDa) and a protein that migrated at 20 kDa (most likely the deglycosylated form). Small amount of 33–35 kDa, and to much lesser extent the 20 kDa form, were also detected in the soluble fraction of control cultures. In contrast, there were almost undetectable levels of PrPC in the conditioned cell culture media (Figure 4). MTX has previously been shown to be a potent calpain activator due to the extreme calcium influx it induces (Wang et al., 1996; Zhang et al., 2009). Yet, with MTX treatment of RtCNC, there was no significant alteration of PrPC patterns in either insoluble or soluble fractions as far as the 33–35 and 20 kDa species are concerned (Figure 4A). There was, however, a minor increase of 12 kDa form (possibly a PrPC BDP). Similarly, NMDA challenge did not alter the pattern of PrPC in both soluble or insoluble cellular fractions with the exception that the 20 kDa protein band appeared to be slightly elevated. However, rather unexpectedly, there was a distinct presence of the intact PrPC in conditioned cell culture media only with NMDA or MTX treatment, when compared to control. Given that only 20 μl of a total of 500 μl conditioned cell culture media was examined, the overall increase of PrPC was relatively substantial (Figure 4A). When we added concentrated RIPA buffer to the whole cell culture well to extract and recover the insoluble, soluble fractions, and cell conditioned media with or without MTX or NMDA treatment, we observed that the overall levels of PrPC remained the same, thus ruling out the increased PrP in cell media being due to overexpression and increased production of PrPC by the cells (results not shown). Thus we concluded that both neurotoxin treatments in fact resulted in increased release of 33–35 kDa PrPC into the media (Figure 4). Following MTX exposure, Endo H treatment of the soluble and insoluble cell extracts and the conditioned cell culture media prior to electrophoresis and western blotting did not result in either altered PrPC migration or the immunostaining patterns compared to untreated controls (data not shown) suggesting that these are fully processed and mature forms of the protein. To confirm that calpain was indeed activated following MTX- and NMDA-treatments, soluble fractions were also analyzed with anti-αII-spectrin Mab to assess the formation of SBDPs. In comparison to control conditions, a major reduction of intact αII-spectrin with MTX and NMDA-treatments was observed. Furthermore, calpain-mediated SBDP150/145 bands were prominent under these neurotoxic conditions (Figure 4B).
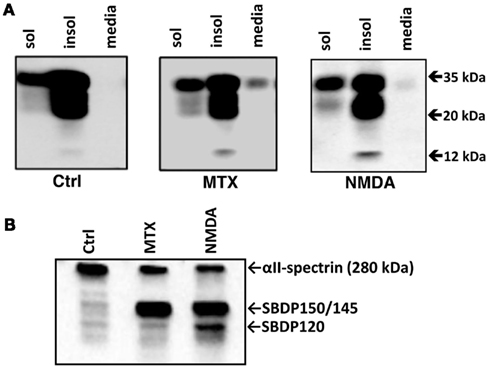
Figure 4. Release of full-length PrPC into culture media from RtCNC challenged with neurotoxins MTX and NMDA. RtCNC were either untreated (control; Ctrl) or challenged with MTX or NMDA. (A) Soluble and insoluble cell fractions and conditioned cell culture media were analyzed by SDS-PAGE followed by western blotting and immunostaining with Mab D8. (B) Soluble cell fractions were also analyzed with anti-αll spectrin Mab to probe the formation of SBDPs (SBDP150 by calpain and SBDP120 by caspase-3). Blots are representative of four separate experiments.
We examined if the unexpected release of full-length PrPC induced by the neurotoxins MTX and NMDA could be attenuated by calpain or caspase-3 inhibition. RtCNC were either untreated (control) or challenged with MTX or NMDA, in the absence or presence of SNJ or IDN (Figure 5). PrPC release was again monitored by immunoblotting with Mab D8. Similar to Figure 4, we confirmed that media from both MTX and NMDA challenged groups contained significantly greater PrPC release compared to control (p < 0.05). We also noted that the MTX-induced PrPC release levels were significantly higher than those by NMDA-induced release (p < 0.05). Neither SNJ nor IDN inhibition significantly altered MTX- or NMDA-induced PrPC release levels, respectively (Figures 5 and 6).
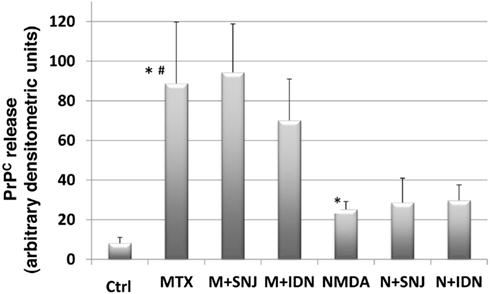
Figure 5. Quantification of release of full-length PrPC into culture media from RtCNC challenged with neurotoxin MTX and NMDA. RtCNC were either untreated (control) or challenged with MTX or NMDA in the absence or presence of SNJ or IDN. Full-length PrPC release was detected by analyzing conditioned cell culture media by SDS-PAGE and western blotting using anti-PrP Mab D8. Quantitation of PrPC immunostaining was performed using Image J as described in the Methods section. Data are representative of up to seven measurements per group from three separate experiments. Values are expressed as mean ± standard error. * indicates NMDA or MTX challenge groups have significantly higher PrPC release compared to control (p < 0.05). # indicates MTX challenge group has significantly higher PrPC release compared to NMDA challenge group (p < 0.05). Neither inhibitor treatment significantly altered MTX- or NMDA-induced PrPC release levels, respectively.
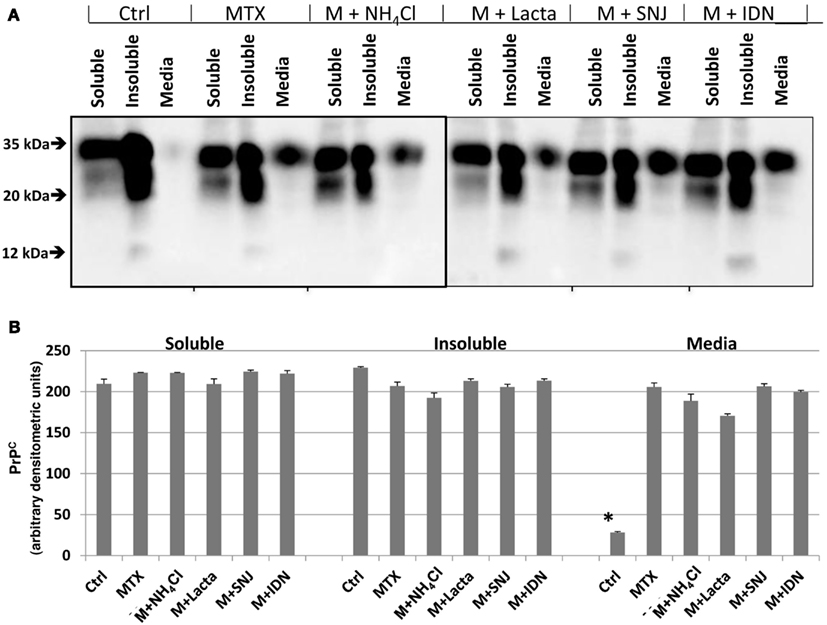
Figure 6. Integrity of PrPC in RtCNC and its release into culture media upon MTX challenge is not significantly altered by protease inhibition. (A) RtCNC were either untreated (control) or challenged with MTX in the absence or presence of NH4Cl, lactacystin (lacta), SNJ, or IDN. Soluble and insoluble cell fractions and conditioned cell culture media were analyzed by SDS-PAGE and western blotting using anti-PrP Mab D8. Blots are representative of three separate experiments. (B) Quantification of PrPC band (33–35 kDa) intensity in the fractions as described in (A). Values are expressed as mean ± standard error. MTX did not alter the levels of PrPC in the soluble or insoluble cell extracts. None of the protease inhibitor treatments altered PrPC levels in either soluble or insoluble cell fractions. The MTX-induced PrPC release into the media was not attenuated by any of the inhibitor treatments.
Lastly, we investigated if the integrity of PrPC or its distribution in soluble, insoluble, or media fractions could be altered by inhibition of additional cellular proteases. Here we chose the MTX challenge since it produced the most robust PrPC response (Figure 5). RtCNC were either untreated (control) or challenged with MTX in the absence or presence of lysosomal neutralizing agent (10 mM NH4Cl), proteasome inhibitor (10 μM lactacystin), calpain inhibitor (30 μM SNJ), or caspase inhibitor (30 μM IDN). Soluble and insoluble fractions (50 μg each) and conditioned cell culture media (20 μl) were analyzed by western blotting with Mab D8. MTX did not alter PrPC integrity as demonstrated by the unmodified pattern of protein banding in soluble and insoluble cell fractions (Figure 6). However, MTX treatment did cause dramatic release of full-length PrPC into cell culture media, consistent with our previous findings (Figure 6). None of the protease inhibitor treatments significantly altered full-length PrPC levels in either cell fractions or attenuated the MTX-induced PrPC release into the media (Figure 6B). Thus, full-length PrPC release from MTX-challenged cortical neurons into cell culture media appears to be independent of cellular protease activation.
Discussion
There exists a potential vulnerability of PrPC and PrPSc to processing by calpain and other cytosolic proteases (Yadavalli et al., 2004; Wang et al., 2005; Hachiya et al., 2011). However, conflicting data exists especially regarding PrPC. In this study, we performed in vitro and in cell-based studies using primary RtCNC to further examine this issue. (His)6-HrPrP was cleaved by calpain-1 and -2 in vitro and the two targeted cleavage sites were identified between resides Gly40-Ser41 and residues Gly130-Ser131. (His)6-HrPrP, however, was resistant to caspase-3 digestion. In contrast, PrPC in brain cell lysates was fairly resistant to calpain digestion while αII-spectrin was readily converted to its signature calpain-generated BDPs. The amino acid sequences for human and rat PrP are identical at the calpain targeted HrPrP cleavage sites. Therefore, the differences in calpain sensitivity between HrPrP and the rat PrP cell cultures is probably the result of protein folding differences and target site accessibility of the recombinant protein compared to the native rat PrPC. Additionally, the detergent solubilization of HrPrP may contribute to calpain target site availability when compared to the more natural environment of the cell membrane-associated rat PrPC.
Also, when RtCNC were subjected to the MTX and NMDA neurotoxins, proteolysis of PrPC was again minimal. Unexpectedly, these challenges resulted in the release of presumably full-length, mature PrPC into the media. Treatment of the RtCNC with SNJ, IDN, NH4Cl, or lactacystin did not significantly alter the integrity of cellular PrPC or its release into conditioned cell culture media.
Previous studies (Yadavalli et al., 2004) have shown that both PrPSc and PrPC isoforms in CJD brain are cleaved by cellular proteases to generate a C-terminal fragment, referred to as C2 and that one of these proteases is likely to be calpain. Hachiya et al. (2011) found that the molecular weight of PrPC fragments generated by spontaneous proteolysis in mouse N2a cells was identical to the calpain-generated fragments of in vitro-translated PrPC and overexpression of the calpain inhibitor, calpastatin, drastically inhibited normal endoproteolysis of PrPC in N2a cells. In addition, Wang et al. (2005) showed that in several cell lines and primary mouse cortical neurons, a portion of the endogenous PrPC was degraded by the proteasome in the cytosol. Thus we initially hypothesized that PrPC would be vulnerable to calpain cleavage in RtCNC and rat brain lysate but were surprised to find that was not the case. Further, data mining studies by Dron et al. (2010) reported that in primary neurons and brain tissue, PrPSc accumulated predominantly as uncleaved species. Thus, this PrPSc data is consistent with our PrPC data presented here.
Taken together, we observed that PrPC in RtCNC was not a significant target for proteolytic modifications during neurotoxic challenges. Yet, the current study is the first report that describes the release or shedding of full-length PrPC from the cell upon neurotoxic challenges. It is possible that this is a protective response of the cells to neurotoxic threats. Importantly, the release of full-length PrPC as a response to neurotoxins might have a physiological function involved in neuroprotection such as stimulating cell activation and/or release of trophic factors. It has been shown that in transgenic mice, PrPC overexpression is neuroprotective while mice that did not express PrPC were more susceptible to neurotoxicity or neurodegeneration (Westergard et al., 2007).
A protease-mediated physiological cleavage near the C-terminus of PrPC in close proximity to its GPI-anchor has been reported in neurons and lymphoid cells (Borchelt et al., 1993). This resulted in the release of the almost full-length protein from the plasma membrane. This soluble form of shedded PrPC was also found in human CSF (Tagliavini et al., 1992) and blood (Perini et al., 1996; MacGregor et al., 1999; Parizek et al., 2001) indicating physiological relevance. Cell culture experiments identified ADAM10, along with a regulatory role of ADAM9, as the active components responsible for the shedding of nearly full-length PrPC from the cell surface (Cisse et al., 2005; Taylor et al., 2009; Tousseyn et al., 2009; Moss et al., 2011). In vivo studies also confirmed the role of ADAM10 in PrPC shedding (Altmeppen et al., 2011).
As Altmeppen et al. (2012) recently articulated elegantly that when compared to membrane-associated PrPC, shed PrPC may have distinct functions or activities (Harris et al., 1993; Parizek et al., 2001; Altmeppen et al., 2011). Amyloid precursor protein (APP) can be processed either through an amyloidogenic pathway to generate Aβ or through a non-amyloidogenic pathway to block Aβ production and generate soluble APPα (sAPPα). Aβ1-42 accumulation in the brain has been shown to impair neuronal function (Hu et al., 2009), while generation of sAPPα is known to play neuroprotective and neurotrophic roles (Cheng et al., 2002; Stein et al., 2004). De Felice et al. (2007) reported that Aβ oligomers caused the formation of neuronal-damaging reactive oxygen species that was mediated by NMDA receptors (NMDAR). Further, PrPC has been reported to provide a neuroprotective role by its ability to limit NMDAR excitotoxicity (Khosravani et al., 2008) while both the presence of Aβ1-42 and PrPC inactivation caused the NMDAR neurotoxicity (You et al., 2012).
Thus taken these concepts together, and relating them to neurodestructive (Aβ) and neuroprotective (sAPPα) roles of processed APP, the increased release of shed PrPC, could represent a self-defense mechanism to preserve those neurons or neighboring neurons under neurotoxic attacks (Altmeppen et al., 2012). Here, it is tempting to suggest that similar or same neuroprotective mechanisms might be at play in our MTX and NMDA challenge paradigm in RtCNC, thus explaining the observed PrPC release. It is possible that exposure of neuronal cells to neurotoxins activates ADAM10 causing PrPC shedding. This shed PrPC could, in turn, bind to neurotoxins thereby reducing their direct contact with cells. Our laboratories are now in the process of furthering work in this direction.
Roberts et al. (2010) demonstrated that PrPC is significantly increased in the cerebrospinal fluid of HIV-1 infected individuals with cognitive impairment. Their studies indicate that since the increase of PrPC is not a generalized phenomenon of neuronal injury or neuroinflammation, it can therefore serve as a useful biomarker to monitor disease progression in HIV-infected individuals. Although the release of PrPC in response to neurotoxin challenge in vitro is interesting and may offer some insight as to its neuroprotective mechanism, its ability to serve a similar function and/or as a CNS biomarker following brain insult is attractive but speculative and requires further investigation. The release of proteins (e.g., SBDPs, UCH-L1) from neuronal cultures subjected to neurotoxic challenges have not only been reported previously (Dutta et al., 2002; Siman et al., 2004) but have also proven to be useful CNS biomarkers after traumatic or ischemic brain injury (Zhang et al., 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Ming Cheng Liu and Wenrong Zheng (formerly of Banyan Laboratories) for their technical help in cleavage site identification experiments and neuronal culture studies. This work was supported by a grant from the Department of Defense (W81XWH-11-2-0069/DM102153) and the SUNY Downstate Medical Center.
References
Altmeppen, H. C., Prox, J., Puig, B., Kluth, M. A., Bernreuther, C., Thurm, D., et al. (2011). Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in viv.. Mol. Neurodegener. 6, 36.
Altmeppen, H. C., Puig, B., Dohler, F., Thurm, D. K., Falker, C., Krasemann, S., et al. (2012). Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 1, 15–31.
Borchelt, D. R., Rogers, M., Stahl, N., Telling, G., and Prusiner, S. B. (1993). Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid ancho.. Glycobiology 3, 319–329.
Brown, D. R., and Besinger, A. (1998). Prion protein expression and superoxide dismutase activit.. Biochem. J. 334, 423–429.
Brown, D. R., Nicholas, R. S., and Canevari, L. (2002). Lack of prion protein expression results in a neuronal phenotype sensitive to stres.. J. Neurosci. Res. 67, 211–224.
Chang, B., Petersen, R., Wisniewski, T., and Rubenstein, R. (2012). Influence of Mabs on PrPSc formation using in vitro and cell-free system.. PLoS ONE 7, e41626.
Chen, S. G., Teplow, D. B., Parchi, P., Teller, J. K., Gambetti, P., and Autilio-Gambetti, L. (1995). Truncated forms of the human prion protein in normal brain and in prion disease.. J. Biol. Chem. 270, 19173–19180.
Cheng, G., Yu, Z., Zhou, D., and Mattson, M. P. (2002). Phosphatidylinositol-3-kinase-Akt kinase and p42/p44 mitogen-activated protein kinases mediate neurotrophic and excitoprotective actions of a secreted form of amyloid precursor protein. Exp. Neurol. 175, 407–414.
Cisse, M. A., Sunyach, C., Lefranc-Jullien, S., Postina, R., Vincent, B., and Checler, F. (2005). The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activit.. J. Biol. Chem. 280, 40624–40631.
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor dependent mechanism that is blocked by the Alzheimer drug memantin.. J. Biol. Chem. 282, 11590–11601.
Dron, M., Moudjou, M., Chapuis, J., Salamat, M. K., Bernard, J., Cronier, S., et al. (2010). Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependen.. J. Biol. Chem. 285, 10252–10264.
Dutta, S., Chiu, Y. C., Probert, A. W., and Wang, K. K. W. (2002). Selective release of calpain produced αII-spectrin (α-fodrin) breakdown products from acute neuronal cell deat.. Biol. Chem. 383, 785–791.
Hachiya, N., Komata, Y., Harguem, S., Nishijima, K., and Kaneko, K. (2011). Possible involvement of calpain-like activity in normal processing of cellular prion protei.. Neurosci. Lett. 490, 150–155.
Harris, D. A., Huber, M. T., van Dijken, P., Shyng, S. L., Chait, B. T., and Wang, R. (1993). Processing of a cellular prion protein: identification of N- and C-terminal cleavage site.. Biochemistry 32, 1009–1016.
Hoglen, N. C., Chen, L. S., Fisher, C. D., Hirakawa, B. P., Groessl, T., and Contreras, P. C. (2004). Characterization of IDN-6556 (3-[2-(2-tert-butyl- phenylaminooxalyl)-amino]- propionylamino]-4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-pentanoic acid): a liver-targeted caspase inhibito.. J. Pharmacol. Exp. Ther. 309, 634–640.
Hu, N. W., Klyubin, I., Anwy, R., and Rowan, M. J. (2009). GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 20504–20509.
Khosravani, H., Zhang, Y., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptor.. J. Cell Biol. 181, 551–565.
Kretzschmar, H. A., Prusiner, S. B., Stowring, L. E., and DeArmond, S. J. (1986). Scrapie prion proteins are synthesized in neuron.. Am. J. Pathol. 122, 1–5.
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protei.. Physiol. Rev. 88, 673–728.
MacGregor, I., Hope, J., Barnard, G., Kirby, L., Drummond, O., Pepper, D., et al. (1999). Application of a time-resolved fluoroimmunoassay for the analysis of normal prion protein in human blood and its component.. Vox Sang. 77, 88–96.
Moss, M. L., Powell, G., Miller, M. A., Edwards, L., Qi, B., Sang, Q. X., et al. (2011). ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protei.. J. Biol. Chem. 286, 40443–40451.
Moudjou, M., Frobert, Y., Grassi, J., and LaBonnardiere, C. (2001). Cellular prion protein status in sheep: tissue-specific biochemical signature.. J. Gen. Virol. 82, 2017–2024.
Pan, K.-M., Stahl, N., and Prusiner, S. B. (1992). Purification and properties of the cellular prion protein from Syrian hamster brai.. Protein Sci. 1, 1343–1352.
Parizek, P., Roeckl, C., Weber, J., Flechsig, E., Aguzzi, A., and Raeber, A. J. (2001). Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cell.. J. Biol. Chem. 276, 44627–44632.
Perini, F., Vidal, R., Ghetti, B., Tagliavini, F., Frangione, B., and Prelli, F. (1996). PrP27-30 is a normal soluble prion protein fragment released by human platelet.. Biochem. Biophys. Res. Commun. 223, 572–577.
Roberts, T. K., Eugenin, E. A., Morgello, S., Clements, J. E., Zink, M. C., and Berman, J. W. (2010). PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammatio.. Am. J. Pathol. 177, 1848–1860.
Sales, N., Rodolfo, K., Hassig, R., Faucheux, B., Di Giamberardino, L., and Moya, K. L. (1998). Cellular prion protein localization in rodent and primate brai.. Eur. J. Neurosci. 10, 2464–2471.
Shimazawa, M., Suemori, S., Inokuchi, Y., Matsunaga, N., Nakajima, Y., Oka, T., et al. (2010). A novel calpain inhibitor, (1S)-1-(1S)-1-Benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl)-3-methylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester (SNJ-1945), reduces murine retinal cell death in vitro and in viv.. J. Pharmacol. Exp. Ther. 332, 380–387.
Siman, R., McIntosh, T. K., Soltesz, K. M., Chen, Z., Neumar, R. W., and Roberts, V. L. (2004). Proteins released from degenerating neurons are surrogate markers for acute brain damag.. Neurobiol. Dis. 16, 311–320.
Stein, T. D., Anders, N. J., DeCarli, C., Chan, S. L., Mattson, M. P., and Johnson, J. A. (2004). Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J. Neurosci. 24, 7707–7717.
Tagliavini, F., Prelli, F., Porro, M., Salmona, M., Bugiani, O., and Frangione, B. (1992). A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathie.. Biochem. Biophys. Res. Commun. 184, 1398–1404.
Taraboulos, A., Scott, M., Semenov, A., Avrahami, D., Laszlo, L., and Prusiner, S. B. (1995). Cholesterol depletion and modification of COOH-terminal targeting sequence of the priori protein inhibit formation of the scrapie isoform.. J. Cell Biol. 129, 121–132.
Taylor, D. R., Parkin, E. T., Cocklin, S. L., Ault, J. R., Ashcroft, A. E., Turner, A. J., et al. (2009). Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protei.. J. Biol. Chem. 284, 22590–22600.
Tousseyn, T., Thathiah, A., Jorissen, E., Raemaekers, T., Konietzko, U., Reiss, K., et al. (2009). ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretas.. J. Biol. Chem. 284, 11738–11747.
Vincent, B., Paitel, E., Saftig, P., Frobert, Y., Hartmann, D., De Strooper, B., et al. (2001). The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protei.. J. Biol. Chem. 276, 37743–37746.
Wang, K. K. W. (2000). Calpain and caspase: can you tell the difference. Trends Neurosci. 23, 20–26.
Wang, K. K. W., Hajimohammadreza, I., Raser, K. J., and Nath, R. (1996). Maitotoxin induces calpain activation in SH-SY5Y neuroblastoma cells and cerebrocortical culture.. Arch. Biochem. Biophys. 331, 208–214.
Wang, X., Wang, F., Sy, M. S., and Ma, J. (2005). Calpain and other cytosolic proteases can contribute to the degradation of retro-translocated prion protein in the cytoso.. J. Biol. Chem. 280, 317–325.
Westergard, L., Christensen, H. M., and Harris, D. A. (2007). The cellular prion protein (PrPC): its physiological function and role in diseas.. Biochim. Biophys. Acta. 1772, 629–644.
Wong, B. S., Pan, T., and Liu, T. (2000). Differential contribution of superoxide dismutase activity by prion protein in viv.. Biochem. Biophys. Res. Commun. 273, 136–139.
Yadavalli, R., Guttmann, R. P., Seward, T., Centers, A. P., Williamson, R. A., and Telling, G. C. (2004). Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagatio.. J. Biol. Chem. 279, 21948–21956.
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., et al. (2012). Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptor.. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742.
Zhang, Z., Larner, S., Liu, M. C., Zheng, W., Hayes, R. L., and Wang, K. K. W. (2009). Multiple αII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathway.. Apoptosis 14, 1289–1298.
Keywords: cellular prion protein, neurotoxins, maitotoxin, NMDA, Calpain, rat cerebrocortical neurons
Citation: Wang KKW, Zoltewicz JS, Chiu A, Zhang Z and Rubenstein R (2012) Release of full-length PrPC from cultured neurons following neurotoxic challenges. Front. Neur. 3:147. doi: 10.3389/fneur.2012.00147
Received: 07 August 2012; Accepted: 01 October 2012;
Published online: 22 October 2012.
Edited by:
Cameron Bass, Duke University, USAReviewed by:
Bruce P. Capehart, Duke University, USAHana N. Dawson, Duke University Medical Center, USA
Robert Petersen, Case Western Reserve University, USA
Copyright: © 2012 Wang, Zoltewicz, Chiu, Zhang and Rubenstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Richard Rubenstein, Laboratory of Neurodegenerative Diseases and Central Nervous System Biomarkers, Departments of Neurology and Physiology/Pharmacology, State University of New York Downstate Medical Center, 450 Clarkson Avenue, Brooklyn, NY 11203, USA. e-mail:cmljaGFyZC5ydWJlbnN0ZWluQGRvd25zdGF0ZS5lZHU=