

 Christopher C. J. Miller1,2*
Christopher C. J. Miller1,2*
- 1Department of Neuroscience, King’s College London, Institute of Psychiatry, London, UK
- 2Department of Neuroscience and Clinical Neurosciences, King’s College London, Institute of Psychiatry, London, UK
- 3Department of Old Age Psychiatry, Institute of Psychiatry, King’s College London, London, UK
Fibrillar deposits of highly phosphorylated tau are a key pathological feature of several neurodegenerative tauopathies including Alzheimer’s disease (AD) and some frontotemporal dementias. Increasing evidence suggests that the presence of these end-stage neurofibrillary lesions do not cause neuronal loss, but rather that alterations to soluble tau proteins induce neurodegeneration. In particular, aberrant tau phosphorylation is acknowledged to be a key disease process, influencing tau structure, distribution, and function in neurons. Although typically described as a cytosolic protein that associates with microtubules and regulates axonal transport, several additional functions of tau have recently been demonstrated, including roles in DNA stabilization, and synaptic function. Most recently, studies examining the trans-synaptic spread of tau pathology in disease models have suggested a potential role for extracellular tau in cell signaling pathways intrinsic to neurodegeneration. Here we review the evidence showing that tau phosphorylation plays a key role in neurodegenerative tauopathies. We also comment on the tractability of altering phosphorylation-dependent tau functions for therapeutic intervention in AD and related disorders.
Introduction
Characteristic accumulations of highly phosphorylated tau protein aggregates are found in several neurodegenerative tauopathies including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and some forms of frontotemporal lobar dementia (FTLD-tau). It was assumed that these pathological tau aggregates are the toxic form of tau. However, recent studies indicate that soluble and highly phosphorylated tau species are more closely associated with synaptic dysfunction and cell loss (1–4).
Tau is normally a highly soluble protein found predominantly in neurons. A total of six different isoforms of tau are expressed in the adult human CNS via alternative splicing of the MAPT gene, which comprises 16 exons and is found on chromosome 17q21.3. Regulated inclusion of exons 2 and 3 gives rise to tau isoforms with 0, 1, or 2 N-terminal inserts, whereas exclusion or inclusion of exon 10 leads to expression of tau isoforms with three (3R) or four (4R) microtubule-binding repeats (Figure 1A). In normal human brain the ratio of 4R–3R tau is approximately one, whereas in many tauopathies, this ratio is altered; PSP, corticobasal degeneration (CBD), and argyrophilic grain disease all exhibit over-expression of 4R tau isoforms, whereas Pick’s disease is mainly characterized by tau inclusions rich in 3R tau isoforms (5–9).
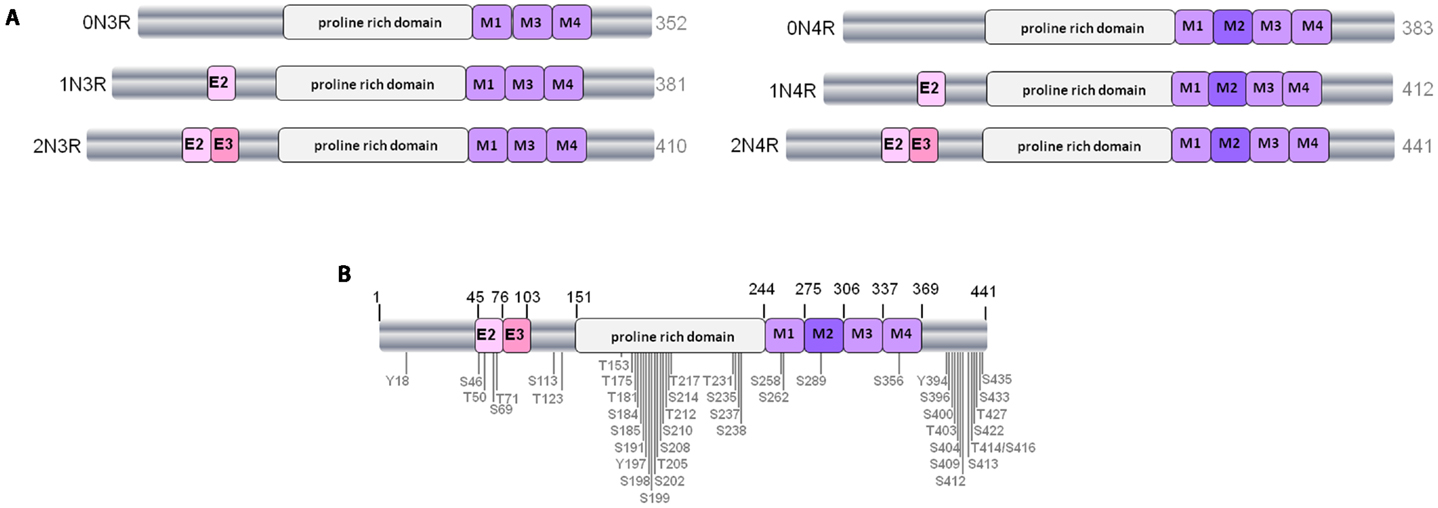
Figure 1. The human tau gene and six protein isoforms. (A) The six isoforms of human CNS tau. Exons 2 and 3 (E2 and E3) encode two different inserts of 28 amino acids near the N-terminus of tau. Absence of E2 and E3 gives rise to 0N tau isoforms, whereas inclusion of E2 produces 1N and inclusion of both E2 and E3 results in 2N tau isoforms. M1–M4 represent the four imperfect-repeat microtubule-binding domains, M2 being encoded by exon 10. Lack of M2 results in the formation of 3R tau and M2 inclusion results in 4R tau isoforms. The proline-rich domain in the center of the tau polypeptide is indicated. Alternative-splicing produces tau isoforms of 352–441 amino acids, as indicated. (B) Positioning of phosphorylation sites on tau from Alzheimer brain. Approximately 45 phosphorylation sites have been identified, these are found predominantly in the proline-rich domain and the regions flanking the microtubule-binding domain.
Tau is a phosphorylated protein, containing 85 potential serine (S), threonine (T), and tyrosine (Y) phosphorylation sites. Many of the phosphorylated residues on tau are found in the proline-rich domain of tau, flanking the microtubule-binding domain (Figure 1B). Both the phosphorylation status and isoform expression of tau are developmentally regulated and both are important factors for cytoskeletal plasticity during embryogenesis and early development. In early developmental stages a single tau isoform, 0N3R, is expressed and tau phosphorylation is elevated relative to adult brain. In contrast, all six tau isoforms are present in normal mature human brain, and at this stage tau phosphorylation is relatively reduced (8, 10).
Despite the significant heterogeneity that exists between and within the various tauopathies, the deposited tau in pathological lesions is invariably highly phosphorylated. Mass spectrometric analysis, combined with Edman sequencing and specific antibody reactivity, shows that approximately ten phosphorylation sites can be detected on soluble tau purified from normal brain (10). In contrast, when insoluble aggregated tau is extracted from tauopathy brain, at least 16 phosphorylated residues have been found in PSP (11–13), and approximately 45 different serine, threonine, and tyrosine phosphorylation sites, representing more than 50% of all phosphorylatable residues, have been found in AD brain (10, 14–17).
A large number of different kinases and phosphatases have been shown to regulate tau phosphorylation, and an imbalance in tau kinase and phosphatase activity is believed to result in tau hyperphosphorylation in disease. Tau kinases include:
• The proline-directed kinases glycogen synthase kinase-3 (GSK-3) (18–22), cyclin-dependent kinase 5 (cdk5) (23–25), and 5′ adenosine monophosphate-activated protein kinase (AMPK) (26, 27).
• Non-proline-directed kinases, such as casein kinase 1 (CK1) (10), microtubule affinity-regulating kinases (MARKs) (28–30), cyclic AMP-dependent protein kinase A (PKA) (31, 32), and dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK-1A) (33, 34).
• Tyrosine kinases including Fyn (35, 36), Abl (37, 38), and Syk (39).
In addition, several phosphatases dephosphorylate tau, including protein phosphatase-1, -2A, and -5 (PP1, PP2A, and PP5) (reviewed by (40).
Importantly, many of these enzymes have been implicated in pathways affected by amyloid-beta (Aβ) in models of AD (27, 41–43). It remains to be established if the overall phosphorylation state of tau or phosphorylation at specific residues is important in disease pathogenesis, as suggested by studies in flies (44). However, there is evidence that phosphorylation of individual residues on tau can significantly impact its function, and this is discussed below.
The Relationship between Phosphorylation and Tau Structure
In addition to abnormal phosphorylation, tau protein in neurodegenerative disease brain can be modified in a number of ways, including N- and C-terminal proteolytic cleavage, altered conformation, nitration, glycosylation, acetylation, glycation, ubiquitylation, O-GlcNAcylation, aggregation, and filament formation (45, 46). Much research has focused on elucidating the relationship between phosphorylation and the changes in tau structure that are common in neurodegenerative disease brain. Evidence from this research suggests that phosphorylation occurs either prior to, or at the same time as, these other post-translational modifications and before aggregation occurs. It remains to be seen whether this temporal precendence indicates a causative relationship.
Proteolytic Tau Cleavage
Tau is subject to proteolytic cleavage by caspase-3 at aspartate (D) residue 421 (47), and N-terminal cleavage by calpain-1 (48) and caspase-6 (49). The tau fragments that are generated have been detected in affected regions of human tauopathy brain (47, 50). Caspase-cleaved tau fragments show an increased propensity to aggregate, and these may form a seeding nidus that promotes the aggregation and fibrillization of full-length tau species (51). In contrast, cleavage of tau by calpain may partially inhibit tau aggregation (50). The temporal relationship between tau cleavage and phosphorylation is unclear, with data showing that phosphorylation of different tau residues precedes (52), follows (47), and inhibits (53) the proteolytic cleavage of tau by caspase-3. However, substantial evidence shows that caspase-3-cleaved tau species are particularly prone to phosphorylation in both primary neuronal cells (54) and human tauopathy brain (47), and that phosphorylated and caspase-3-cleaved tau species readily form aggregates in cells (55). These results therefore suggest that phosphorylation and caspase-mediated cleavage of tau are important events during the development of the characteristic tau aggregates that accumulate in AD and other tauopathies.
Altered Tau Conformation
Tau is a natively unfolded protein that adopts abnormal conformations in tauopathy brain. For example, tau cleavage by caspase-3 at D421 occurs early in disease development, following an alteration in tau conformation detected by the Alz50 antibody, and prior to the formation of the conformational Tau-66 epitope (tau residues 155–244 and 305–331) which is detected in late-stage AD (56). Altered tau conformation is suggested to be a major determinant in inducing tauopathy development in vivo (57), and abnormal tau conformers are detected in mouse models of tauopathy where elevated tau phosphorylation is apparent, but prior to the appearance of substantial tau aggregation (22, 58). Thus, caspase-3-induced tau cleavage appears to occur relatively early during the development of tauopathies, contemporaneous with increased phosphorylation and altered conformation of tau.
The Development of Tau Oligomers
A number of soluble and insoluble tau oligomers have been detected in AD and FTLD brain (2). Tau oligomers display altered conformation (59), are formed during the early stages of tau aggregation (59), and are closely associated with neurodegenerative phenotypes (2, 60). For example, transgenic mice that conditionally express a proline to leucine mutation at residue 301 (P301L) in human tau (1) exhibit high molecular weight tau oligomers, prior to the presence of neurofibrillary tangles (NFTs), that correlate with the development of cognitive deficits (2). Similarly, in a Drosophila model of tauopathy, the suppression of tau-induced neurodegeneration is associated with clearance of ubiquitinated and phosphorylated low molecular weight (<250 kDa) tau oligomers, concomitant with increases in ubiquitinated tau monomers and high molecular weight (>250 kDa) tau oligomers (61). It should be noted that protection from tau-associated toxicity in this latter study was also accompanied by reduced phosphorylation of soluble monomeric tau. Phosphorylation of tau by GSK-3 promotes the formation of insoluble oligomeric tau species that can constitute both full-length and truncated tau species (62, 63). The majority of insoluble tau in AD brain is intact (13). However, cleaved tau species are prominent in insoluble tau preparations from PSP, CBD, and FTLD-tau brain (13). The increased propensity of caspase-cleaved tau to aggregation (47), and the close association of tau fragments with cell death (64), suggests that although present as a relatively small pool of total tau, cleaved tau may also play an important role in disease. The presence of phosphorylated oligomeric tau species in cortical synapses extracted from AD brain (65) supports a role for highly phosphorylated tau multimers in tau-associated neuronal dysfunction.
The Formation of Insoluble Tau Aggregates
In cell-free systems, soluble tau is a hydrophilic, unstructured, and dynamic protein (66). However, highly ordered aggregated tau filaments constitute the characteristic neurofibrillary lesions observed in tauopathy brain, including NFTs in AD and FTLD-tau, astrocytic plaques in CBD and tufted astrocytes in PSP (67).
There is substantial evidence that tau phosphorylation precedes its aggregation. Highly phosphorylated mouse and human tau undergoes self-assembly in vitro (68, 69), and dephosphorylation of soluble tau from AD brain inhibits its polymerization and restores the ability of tau to stabilize microtubules (70). Transgenic mice in which tau kinase activity is increased display increased tau phosphorylation prior to the presence of tau aggregates (24, 25, 58, 71). Furthermore, treating tau transgenic mice with kinase inhibitors results in reduced tau phosphorylation and also a reduced tau aggregate load (22, 72, 73). It should be noted, however, that reduction of tau aggregate load in tau transgenic mice following lithium treatment could result from enhanced autophagy in addition to reduced GSK-3-mediated tau phosphorylation (74). The relationship between tau phosphorylation and aggregation is clearly complex since phosphorylation of tau at specific sites, that are known to result in tau detachment from microtubules, can prevent tau aggregation (75). In addition, disruption to tau phosphatase activity in transgenic mice leads to the development of early disease-like tau abnormalities (76, 77). In particular, tau phosphorylation at the AT100 epitope is apparent in mice with reduced PP2A activity (77), which show cdk5-mediated enhanced activation of GSK3. Phosphorylation at the AT100 site has previously been shown to precede NFT formation (78), thus these findings may also suggest that changes in tau phosphorylation precede its aggregation. However, NFT formation was lacking in mice with reduced PP2A activity, an event attributed to increased clearance of abnormal tau conformers (77).
It is possible that the formation of a small pool of cleaved tau may be critically important in mediating the formation of pathological tau aggregates. Caspase-cleaved tau is prone to phosphorylation at specific epitopes (47, 54) and forms aggregation seeds that sequester full-length tau (51). Indeed, in vivo imaging of tau transgenic mice has demonstrated that truncated tau induces the misfolding of soluble tau and leads to the accumulation of hyperphosphorylated tau in tangles (79). Whether or not filamentous tau aggregates are toxic, protective, or inert remains an issue of intense debate (for review, see 80). However, small aggregated tau species have attracted interest recently because of their reported involvement in the propagation/transmission of tau pathology, and this topic is discussed in more detail below.
The Influence of Phosphorylation on Tau Localization and Function
Tau is ubiquitously expressed during early embryonic development, but becomes localized predominantly in axons of mature neurons. The mechanisms underlying the axonal sorting of tau are not fully understood, but might involve selective trafficking of tau mRNA or protein into axons (81, 82), a retrograde transport barrier in the axon initial segment in mice (83), upregulation of tau mRNA translation in axons (84) or selective degradation of tau in dendrites (85). Tau is also found in association with neuronal membranes, in the nucleus, dendrites and synapses, and extracellularly. The localization of tau is altered in disease states. In particular, the redistribution of hyperphosphorylated tau to the somatodendritic compartment is considered a hallmark pathological marker during early tauopathy development (86, 87). The functional consequences of tau phosphorylation-mediated changes in the cellular localization of tau are discussed below.
Cytoplasmic Tau: Cytoskeletal Integrity and Axonal Transport
A large proportion of tau is found in the cytosolic compartment, where it interacts with microtubules through its C-terminal microtubule-binding domain (Figure 1, residues 244–368). The binding of tau with microtubules is regulated by tau phosphorylation status, with in vitro phosphorylation of recombinant tau at S262 and S356, orthologous residues in adjacent microtubule-binding repeats, reducing tau interactions with microtubules and rendering tau less susceptible to aggregation (75). Phosphorylation of tau at residues outside of the microtubule-binding domain of tau, including S214 and T231, have also been shown to reduce its interaction with microtubules (75, 88). These findings suggest that phosphorylation at different tau sites may have opposing effects on the ability of tau to aggregate Furthermore, interaction of the peptidyl-prolyl isomerase Pin1 with phosphorylated T231 mediates the interaction of PP2A with the trans configuration of phosphorylated tau, and results in a conformational change that restores the ability of tau to bind to microtubules (89–91). Regardless of the particular sites involved, increased tau phosphorylation that causes tau to detach from microtubules leads to the disassembly of microtubules and disruption to the structure of the neuronal cytoskeleton. In addition, the accumulation of unbound hyperphosphorylated tau in the cytoplasm could cause further microtubule disassembly by sequestering normal tau and other microtubule-associated proteins (92). When tau is in a filamentous state, its interaction with normal (soluble) tau and its inhibition of microtubule stabilization is disrupted (93). Preventing microtubule instability in tauopathies has become an important target for drug development (94, 95).
Alterations in tau phosphorylation also affect its anterograde axonal transport. In general, reducing tau phosphorylation at S/T residues decreases, whereas mimicking tau phosphorylation increases, the rate of axonal tau transport in fly, rodent, and human neurons (21, 96–98). The influence of tau phosphorylation on its transport appears to be associated with differential binding of S/T phosphorylated tau to the molecular motor protein kinesin-1 (97, 98) and differential degradation rates of phospho-tau species through the lysosomal autophagy system (98).
The interaction of tau with microtubules is critically involved in the regulation of microtubule-dependent axonal transport (99), therefore tau phosphorylation also plays a key role in regulating the transport of other important cargoes. Increasing tau phosphorylation at N-terminal Y residues relieves the inhibition of anterograde axonal transport observed in the presence of highly phosphorylated tau aggregates in squid axons (100). However, tau is not usually highly phosphorylated in squid axons and therefore it is unclear whether this provides a good model to examine mammalian tau functions. In mice over-expressing FTLD-tau mutations, there is impaired anterograde axonal transport of vesicles containing the dopamine-synthesizing enzyme tyrosine hydroxylase, which precede the loss of dopaminergic neurons in the substantia nigra (101). The transport deficits reported in this mouse model were shown to be mediated by interactions between phosphorylated tau and JNK-interacting protein 1 (JIP-1) (102). Since JIP-1 regulates the binding of cargo to kinesin, these results further support the idea that increasing tau phosphorylation disrupts axonal transport. Alternatively, reduced degradation or clearance of aggregated or mutant forms of tau might contribute to a “clogging” of microtubules and consequent disruption in axonal transport (103).
Disruption to axonal transport is predicted to be an early event in several neurodegenerative diseases (104) and recent evidence suggests that dysregulated axonal transport may contribute to tau-induced degeneration. Genetic suppression of Miro, an adapter protein essential for mitochondrial axonal transport, exacerbates the neurodegenerative phenotype in Drosophila expressing human tau, through a mechanism dependent upon phosphorylation of tau at S262 by PAR-1, the Drosophila homolog of MARK kinase (105). Similarly, deletion of kinesin light chain-1 results in accumulation of hyperphosphorylated tau and the appearance of axonal spheroids in mice (106), in line with numerous reports that have characterized the binding of tau to kinesin (21, 96–98).
Finally, alterations in mitochondrial transport and function are intrinsically linked with several neurodegenerative diseases (107). Over-expression of tau in vivo results in alterations to mitochondrial distribution that are associated with soluble, rather than fibrillar, tau species (108). In addition, tau phosphorylation alters the axonal transport and distribution of mitochondria in cultured neuronal cells (109, 110), an effect recently attributed to tau phosphorylation-dependent changes in inter-microtubule spacing (110). Furthermore, highly phosphorylated tau has been shown to interact with the mitochondrial fission protein, Drp1 (111), and DuBoff et al. (112) demonstrated that this relationship is important for neurodegeneration. They show that actin is over-stablised in Drosophila that express human tau, and that this impairs the actin-based translocation of Drp1 and mitochondria, which reduces their interaction and leads to accumulation of Drp1 on F-actin, mitochondrial elongation, and downstream neurotoxicity (112). Thus tau phosphorylation is closely linked to alterations in the localization and/or function of mitochondria. It is therefore likely that phosphorylated tau influences synaptic dysfunction in tauopathies by contributing to the depletion of functional mitochondria from synapses (113).
Membrane-Associated Tau: A Cell Signaling Role for Tau?
Tau interacts with several neuronal membranes, including the endoplasmic reticulum (114), the Golgi network (114), and the plasma membrane (115, 116). An increasing body of evidence shows that the association of tau with plasma membranes is regulated by phosphorylation (116–118). Plasma membrane-associated tau is dephosphorylated at several sites known to be aberrantly phosphorylated in AD brain (116, 117, 119, 120). Indeed, phosphorylation of tau at N-terminal, but not C-terminal, residues prevents its membrane localization in tau-transfected cells, demonstrating that the phosphorylation state of tau directly impacts its positioning at membranes (116).
Tau has also been detected within cell-surface lipid-rich microdomains of the plasma membrane (35, 41, 121), and the amount of tau associated with these lipid rafts is regulated by tau phosphorylation at N-terminal tyrosine residues (121). Tau interactions with the non-receptor tyrosine kinase Fyn are critical for the interaction of tau with lipid rafts (35, 41, 121) and neuronal plasma membranes (116). Tau can interact with Fyn via its SH2 and SH3 domains (121, 122). Phosphorylation of tau at Y18 is important for tau interactions with Fyn-SH2 (121), whereas phosphorylation of S/T residues on tau negatively influences its interaction with Fyn-SH3 (122). Accumulating evidence therefore suggests that targeting of tau to the plasma membrane may be regulated by the interaction of the tau N-terminal projection domain with the SH3 or SH2 domains of tyrosine kinases such as Fyn (118). Furthermore, these data suggest that by binding to several important signaling molecules in a manner that is regulated by phosphorylation, tau has the potential for a broad role in cell signaling (122).
Dendritic Tau and Synaptic Toxicity
A number of recent cell and animal studies have shown an important role for tau in dendrites leading to the suggestion that tau-mediated synaptic dysfunction may be one of the earliest events in the pathogenesis of tauopathies. Several studies have indicated that the presence of tau aggregates is detrimental to synaptic health (123, 124), however, soluble tau species are associated with synapse loss in mouse models of tauopathy (125) and phosphorylated tau oligomers have also been detected in synapses in postmortem AD brain (126).
A small amount of tau exisits in dendrites under normal conditions, where it acts to target Fyn post-synaptically, regulating N-methyl-D-aspartate (NMDA) receptor subunit 2 phosphorylation and interactions between NMDA receptors and the post-synaptic density protein PSD-95 (3). Disease insults, such as increased concentrations of Aβ in AD, lead to the detachment of highly phosphorylated tau from microtubules and its accumulation in intact dendritic spines (3). This in turn causes local elevations in Ca2+ and disruption of synaptic function through impaired trafficking and/or synaptic anchoring of glutamate receptors (3, 127, 128). In a related study, the redistribution of hyperphosphorylated tau into dendritic spines led to reductions in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subtypes that caused impairments in basal synaptic transmission and long term potentiation (129). Thus, there is increasing evidence that tau-mediated synaptic dysfunction might be one of the earliest events in the pathogenesis of tauopathies (reviewed by 130). Therefore, correction of aberrant tau phosphorylation may be therapeutically beneficial during very early stages of disease progression when synaptic deficits first develop. In this respect, it is worth noting that inhibition of GSK3 has previously been shown to attenuate deficits in LTP (131).
Nuclear Tau – a Role in DNA Protection
It was first suggested that tau might have novel functions mediated by interactions with DNA or RNA following observations that tau is present in the nuclei of human neuroblastoma cells (132). Full-length tau was identified in neuronal nuclei, where it colocalizes with the chromosome scaffold, nuclear and nucleolar organization centers and can exist as SDS-insoluble species (132, 133). Further studies revealed that the microtubule-binding domain of tau can bind RNA (134), and single and double stranded DNA (135, 136). The interaction of tau with RNA enhances tau polymerization; the RNA acting as a nucleation center for tau aggregation (134), whereas interaction of tau with DNA results in conformational changes in DNA (133) and suppression of DNA amplification in vitro (136). Insights into the nuclear function of tau were recently revealed with the observation that tau protects DNA from heat damage and oxidative stress (137). Nuclear tau appears to be largely dephosphorylated (137), suggesting that increased tau phosphorylation in diseased states could interfere with protective functions of non-phosphorylated tau in neuronal nuclei.
Extracellular Tau and the Propagation of Tau Pathology
Tau is present in brain insterstitial fluid in the absence of any neurodegeneration (138). Recent evidence suggests that this extracellular tau is likely to have important functional consequences for neuronal health and for the spread of tau pathology across the brain during disease progression.
To allow investigation of tau pathology spread in vivo, transgenic mice have been created with neuropsin-promoter targeted expression of tau in layer II neurons of the entorhinal cortex. These mice demonstrate an age-dependent spread of phosphorylated and aggregated abnormal tau confomers from the site of transgene expression to neighboring neurons and anatomically connected brain regions (139, 140). There are several mechanisms that could account for this observed spread of tau pathology. Firstly, degenerating neurons with high levels of transgene expression might release pathological forms of tau that subsequently propagate in a “prion-like” fashion through their uptake by neighboring neurons. In support of this process, Frost et al. (141) demonstrated that extracellular tau aggregates, but not tau monomers, are taken up by cultured human embryonic kidney (HEK293) cells and neuronal stem cells, leading to fibrillization of full-length intracellular tau. Similarly, small oligomers of tau, similar to those found in human tauopathy brain, can be taken up by cultured neuronal cells via bulk endocytosis (142). It is possible that this process also underlies the postulated prion-like transmission of tau pathology to distal brain regions observed when pathological forms of human tau are injected into mice expressing wild-type human tau (143, 144). Secondly, tau pathology in the neuropsin-promoter regulated tau transgenic mice appears to spread to anatomically connected pathways in the absence of any notable cell loss (139, 140), suggesting that tau is released from intact neurons and then taken up by connected cells. This process is supported in part by recent findings showing endogenous tau release from cultured neurons in the absence of cell death (145, 146). Interestingly, the release of endogenous full-length tau from rat primary neurons was shown to be a dynamic and physiological process that is calcium-dependent and stimulated by AMPA receptor activation and neuronal activity (146), suggesting that tau release may play a role in signaling between neurons. Indeed, exogenously applied tau can interact with muscarinic receptors on the surface of cultured neuronal cells, promoting increases in intracellular calcium that alter cell signaling pathways (147). It is also possible that tau propagation may be mediated via glial cells, since cytosolic tau accumulations are observed in neurons surrounded by activated microglia (148) and astrocytes promote tau phosphorylation in neighboring neurons (54).
The relationship between tau secretion and tau phosphorylation state is not yet established. However, extracellular tau released from primary neurons, neuroblastoma cells and non-neuronal cells is dephosphorylated at several epitopes known to be highly phosphorylated in AD brain (145, 146, 149) and this has been proposed to result from the action of extracellular tissue non-specific alkaline phosphatase (149). How this relates to the phosphorylation state of intracellular tau is not clear, although the secretion of C-terminally cleaved tau from non-neuronal cells can be enhanced by the increased phosphorylation or cleavage of intracellular tau (150). These studies indicate that changes in tau phosphorylation can modulate its release from neurons, and therefore is also likely to influence the effects of extracellular tau on neuronal health and the spread of tau pathology in diseased brain.
Tau Phosphorylation as a Therapeutic Target
As summarized above, tau phosphorylation plays a key role in regulating tau function at different neuronal locations, including the involvement of cytosolic tau in stabilizing the neuronal cytoskeleton and influencing axonal transport; the role of membrane tau and extracellular tau in cell signaling and neurofibrillary pathology spread through diseased brains; the relationship between nuclear tau and protection from DNA damage; and dendritic functions of tau that are involved in synaptotoxicity (Figure 2). These data suggest that inhibition of tau phosphorylation could have widespread disease-modifying effects in tauopathies. Therapeutic strategies aimed at targeting tau phosphorylation have been widely reviewed elsewhere (e.g., 8, 9, 67, 151), therefore we will comment only briefly here.
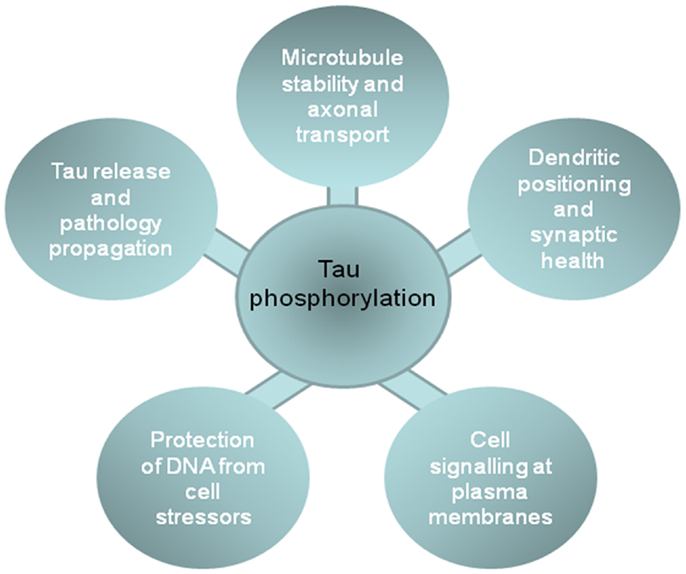
Figure 2. The impact of phosphorylation on tau functions in different cell locations. The figure shows the functions of tau in different cellular compartments that are influenced by tau phosphorylation, and that likely contribute to the development or progression of neurodegenerative tauopathies.
Although several kinases and phosphatases regulate tau phosphorylation, only GSK-3 inhibitors have entered clinical trials for the treatment of AD or rarer tauopathies such as PSP. Based on promising data from animal models (21, 22, 152), the relatively non-specific GSK-3 inhibitor, lithium, was tested in small-scale clinical trials for mild to moderate AD. Whilst lithium did not cause significant adverse effects in an open label study of a year (153), neither did it have any beneficial effects in a short-term trial (154). However, a small trial of lithium in patients with mild cognitive impairment reduced phosphorylated tau in CSF and reported better performance of treated patients in cognitive and attention tasks (155), suggesting that administration of lithium during the early stages of disease could have some therapeutic benefit in defined patient populations.
Tideglusib (NP-12) is a non-ATP competitive inhibitor of GSK3 that has entered clinical trials. Tideglusib has disease-modifying effects when administered to transgenic mice that develop both tau and amyloid pathology (156). Pilot trials for tideglusib in AD and PSP showed good tolerance of tideglusib (157) and phase II studies are underway.
Kinase inhibitors have entered clinical use for conditions unrelated to neurodegeneration (158). However, kinases make for complex therapeutic targets, and probably because of incomplete drug specificity, off-target effects are problematical. An alternative strategy may be to modulate the activity of proteins that directly affect the activity of tau kinases. One interesting target in this respect is lemur tyrosine kinase-2 (LMTK2). LMTK2 phosphorylates PP1C on T320, thereby inhibiting PP1C activity (159–161). PP1 regulates phosphorylation of GSK3β at the inhibitory phosphorylation site S9 (162, 163), and therefore, via its effect on PP1C, LMTK2 regulates GSK-3β phosphorylation at S9, and ultimately GSK-3 activity (160, 161). Therefore, an alternative strategy for inhibiting GSK-3 activity may be to increase LMTK2 expression or activity. Small molecule allosteric agonists for a variety of kinases have now been described, and the development of kinase agonists has been identified as key area for the development of new therapies (164).
Finally, biomarkers are increasingly used to follow the progression of AD, and in some cases to support early diagnosis of the disease (165). However, to accelerate the clinical translation of therapeutics that modify tau phosphorylation, it is essential that sensitive and specific biomarkers are available to allow the measurement of drug–target interactions, and the impact of treatment on downstream pathophysiology. The development of such target validation biomarkers will allow a faster selection of candidate treatments, and appropriate dose ranges. This should accelerate the clinical development of tau phosphorylation inhibitors that are likely to have wide-ranging benefit for the treatment of AD and related tauopathies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work is funded by Alzheimer’s Research UK, Biotechnology, and Biological Sciences Research Council, Motor Neuron Disease Association, Medical Research Council, National Council for the Replacement, Refinement and Reduction of Animals in Research, the NIHR Biomedical Research Centre for Mental Health at the Maudsley, and the Wellcome Trust.
References
1. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science (2005) 309:476–81. doi:10.1126/science.1113694
2. Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci (2007) 27:3650–62. doi:10.1523/JNEUROSCI.0587-07.2007
3. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell (2010) 142:387–97. doi:10.1016/j.cell.2010.06.036
4. Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol (2010) 223:385–93. doi:10.1016/j.expneurol.2009.07.029
5. Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, et al. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol (2002) 61:547–56.
6. Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, et al. Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol (2002) 51(6):730–9. doi:10.1002/ana.10222
7. Connell JW, Rodriguez-Martin T, Gibb GM, Kahn NM, Grierson AJ, Hanger DP, et al. Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Brain Res Mol Brain Res (2005) 137:104–9. doi:10.1016/j.molbrainres.2005.02.014
8. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med (2009) 15:112–9. doi:10.1016/j.molmed.2009.01.003
9. Noble W, Pooler AM, Hanger DP. Advances in tau-based drug discovery. Expert Opin Drug Discov (2011) 6:797–810. doi:10.1517/17460441.2011.586690
10. Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem (2007) 282:23645–54. doi:10.1074/jbc.M703269200
11. Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem (1999) 72:1243–9. doi:10.1046/j.1471-4159.1999.0721243.x
12. Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K, et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol (2004) 55:72–9. doi:10.1002/ana.10793
13. Wray S, Saxton M, Anderton BH, Hanger DP. Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J Neurochem (2008) 105:2343–52. doi:10.1111/j.1471-4159.2008.05321.x
14. Hanger DP, Brion JP, Gallo JM, Cairns NJ, Luthert PJ, Anderton BH. Tau in Alzheimer’s disease and Down’s syndrome is insoluble and abnormally phosphorylated. Biochem J (1991) 275(Pt 1):99–104.
15. Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem (1995) 270:823–9. doi:10.1074/jbc.270.2.823
16. Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem (1998) 71:2465–76. doi:10.1046/j.1471-4159.1998.71062465.x
17. Tavares IA, Touma D, Lynham S, Troakes C, Schober M, Causevic M, et al. Prostate-derived Sterile 20-like Kinases (PSKs/TAOKs) Phosphorylate Tau Protein and Are Activated in Tangle-bearing Neurons in Alzheimer Disease. J Biol Chem (2013) 288(21):15418–29. doi:10.1074/jbc.M112.448183
18. Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett (1992) 147:58–62. doi:10.1016/0304-3940(92)90774-2
19. Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol (1994) 4:1077–86. doi:10.1016/S0960-9822(00)00246-3
20. Lovestone S, Hartley CL, Pearce J, Anderton BH. Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience (1996) 73:1145–57. doi:10.1016/0306-4522(96)00126-1
21. Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry (2004) 9:522–30. doi:10.1038/sj.mp.4001483
22. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A (2005) 102(19):6990–5. doi:10.1073/pnas.0500466102
23. Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett (1993) 336:417–24. doi:10.1016/0014-5793(93)80849-P
24. Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron (2003) 40:471–83. doi:10.1016/S0896-6273(03)00627-5
25. Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron (2003) 38:555–65. doi:10.1016/S0896-6273(03)00259-9
26. Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J (2011) 434:503–12. doi:10.1042/BJ20101485
27. Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through tau phosphorylation. Neuron (2013) 78:94–108. doi:10.1016/j.neuron.2013.02.003
28. Lee S, Wang JW, Yu W, Lu B. Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Abeta toxicity in Drosophila. Nat Commun (2012) 3:1312. doi:10.1038/ncomms2278
29. Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, et al. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem (1995) 270:7679–88.
30. Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell (1995) 6:1887–902.
31. Andorfer CA, Davies P. PKA phosphorylations on tau: developmental studies in the mouse. Dev Neurosci (2000) 22:303–9. doi:10.1159/000017454
32. Wang D, Fu Q, Zhou Y, Xu B, Shi Q, Igwe B, et al. Beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem (2013) 288:10298–307. doi:10.1074/jbc.M112.415141
33. Sheppard O, Plattner F, Rubin A, Slender A, Linehan JM, Brandner S, et al. Altered regulation of tau phosphorylation in a mouse model of down syndrome aging. Neurobiol Aging (2012) 33(828):e831–44. doi:10.1016/j.neurobiolaging.2011.06.025
34. Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, et al. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J (2001) 355:609–15.
35. Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci (2002) 22:10–20.
36. Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, et al. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci (2004) 24:2304–12. doi:10.1523/JNEUROSCI.4162-03.2004
37. Cancino GI, Perez de Arce K, Castro PU, Toledo EM, Von Bernhardi R, Alvarez AR. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging (2011) 32:1249–61. doi:10.1016/j.neurobiolaging.2009.07.007
38. Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, et al. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci (2005) 25:6584–93. doi:10.1523/JNEUROSCI.1487-05.2005
39. Lebouvier T, Scales TM, Hanger DP, Geahlen RL, Lardeux B, Reynolds CH, et al. The microtubule-associated protein tau is phosphorylated by Syk. Biochim Biophys Acta (2008) 1783:188–92. doi:10.1016/j.bbamcr.2007.11.005
40. Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci (2005) 22:1942–50. doi:10.1111/j.1460-9568.2005.04391.x
41. Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane-bound beta-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J (2008) 22:1552–9. doi:10.1096/fj.07-9766com
42. Killick R, Ribe EM, Al-Shawi R, Malik B, Hooper C, Fernandes C, et al. Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol Psychiatry (2012). doi:10.1038/mp.2012.163. [Epub ahead of print].
43. Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, et al. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron (2012) 75:824–37. doi:10.1016/j.neuron.2012.06.024
44. Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell (2007) 18(12):5060–8. doi:10.1091/mbc.E07-04-0327
45. Yuzwa SA, Yadav AK, Skorobogatko Y, Clark T, Vosseller K, Vocadlo DJ. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids (2011) 40:857–68. doi:10.1007/s00726-010-0705-1
46. Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int (2011) 58:458–71. doi:10.1016/j.neuint.2010.12.023
47. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest (2004) 114:121–30. doi:10.1172/JCI20640
48. Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci (2005) 25:5365–75. doi:10.1523/JNEUROSCI.1125-05.2005
49. Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci (2004) 24:7895–902. doi:10.1523/JNEUROSCI.1988-04.2004
50. Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med (2011) 17:676–85. doi:10.2119/molmed.2010.00220
51. Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, and tangles, Alzheimer’s disease. Biochim Biophys Acta (2005) 1739:216–23. doi:10.1016/j.bbadis.2004.08.014
52. Rametti A, Esclaire F, Yardin C, Terro F. Linking alterations in tau phosphorylation and cleavage during neuronal apoptosis. J Biol Chem (2004) 279:54518–28. doi:10.1074/jbc.M408186200
53. Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem (2006) 97:1005–14. doi:10.1111/j.1471-4159.2006.03784.x
54. Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis (2011) 2:e167. doi:10.1038/cddis.2011.50
55. Cho JH, Johnson GV. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem (2004) 279:54716–23. doi:10.1074/jbc.M403364200
56. Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging (2005) 26:1015–22. doi:10.1016/j.neurobiolaging.2004.09.019
57. Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, et al. Changed conformation of mutant tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of tau-4R/2N transgenic mice. J Biol Chem (2005) 280(5):3963–73. doi:10.1074/jbc.M409876200
58. Kelleher I, Garwood C, Hanger DP, Anderton BH, Noble W. Kinase activities increase during the development of tauopathy in htau mice. J Neurochem (2007) 103:2256–67. doi:10.1111/j.1471-4159.2007.04930.x
59. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J (2012) 26:1946–59. doi:10.1096/fj.11-199851
60. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener (2011) 6:39. doi:10.1186/1750-1326-6-39
61. Ali YO, Ruan K, Zhai RG. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet (2012) 21:237–50. doi:10.1093/hmg/ddr449
62. Chun W, Johnson GV. The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci (2007) 12:733–56. doi:10.2741/2097
63. Nubling G, Bader B, Levin J, Hildebrandt J, Kretzschmar H, Giese A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with alpha-synuclein at the single molecule level. Mol Neurodegener (2012) 7:35. doi:10.1186/1750-1326-7-35
64. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A (2003) 100(17):10032–7. doi:10.1073/pnas.1630428100
65. Henkins KM, Sokolow S, Miller CA, Vinters HV, Poon WW, Cornwell LB, et al. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer’s cortical synapses. Brain Pathol (2012) 22:826–33. doi:10.1111/j.1750-3639.2012.00598.x
66. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med (2012) 2:a006247. doi:10.1101/cshperspect.a006247
67. Lee G,Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci (2012) 107:263–93. doi:10.1016/B978-0-12-385883-2.00004-7
68. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A (2001) 98(12):6923–8. doi:10.1073/pnas.121119298
69. Chohan MO, Haque N, Alonso A, El-Akkad E, Grundke-Iqbal I, Grover A, et al. Hyperphosphorylation-induced self assembly of murine tau: a comparison with human tau. J Neural Transm (2005) 112:1035–47. doi:10.1007/s00702-004-0241-9
70. Iqbal K, Alonso AC, Gong CX, Khatoon S, Pei JJ, Wang JZ, et al. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J Neural Transm Suppl (1998) 53:169–80. doi:10.1007/978-3-7091-6467-9_15
71. Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J (2001) 20:27–39. doi:10.1093/emboj/20.1.27
72. Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A (2006) 103:9673–8. doi:10.1073/pnas.0602913103
73. Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci (2013) 33:334–43. doi:10.1523/JNEUROSCI.3593-12.2013
74. Shimada K, Motoi Y, Ishiguro K, Kambe T, Matsumoto SE, Itaya M, et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol Dis (2012) 46:101–8. doi:10.1016/j.nbd.2011.12.050
75. Schneider A, Biernat J, Von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry (1999) 38:3549–58. doi:10.1021/bi981874p
76. Kins S, Kurosinski P, Nitsch RM, Götz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol (2003) 163(3):833–43. doi:10.1016/S0002-9440(10)63444-X
77. Louis JV, Martens E, Borghgraef P, Lambrecht C, Sents W, Longin S, et al. Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A (2011) 108(17):6957–62. doi:10.1073/pnas.1018777108
78. Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science (2001) 293(5534):1491–5. doi:10.1126/science.1062097
79. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature (2010) 464(7292):1201–4. doi:10.1038/nature08890
81. Kanai Y, Hirokawa N. Sorting mechanisms of tau and MAP2 in neurons: suppressed axonal transit of MAP2 and locally regulated microtubule binding. Neuron (1995) 14:421–32. doi:10.1016/0896-6273(95)90298-8
82. Aronov S, Aranda G, Behar L, Ginzburg I. Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J Cell Sci (2002) 115:3817–27. doi:10.1242/jcs.00058
83. Li X, Kumar Y, Zempel H, Mandelkow EM, Biernat J, Mandelkow E. Novel diffusion barrier for axonal retention of tau in neurons and its failure in neurodegeneration. EMBO J (2011) 30:4825–37. doi:10.1038/emboj.2011.376
84. Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and tau via the mTOR-p70S6K pathway. J Biol Chem (2009) 284:27734–45. doi:10.1074/jbc.M109.008177
85. Hirokawa N, Funakoshi T, Sato-Harada R, Kanai Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J Cell Biol (1996) 132:667–79. doi:10.1083/jcb.132.4.667
86. Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem (2003) 86:582–90. doi:10.1046/j.1471-4159.2003.01879.x
87. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol (2011) 70:960–9. doi:10.1097/NEN.0b013e318232a379
88. Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem (2003) 278:187–93. doi:10.1074/jbc.M206236200
89. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature (1999) 399:784–8. doi:10.1038/21650
90. Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, et al. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell (2000) 6(4):873–83. doi:10.1016/S1097-2765(05)00083-3
91. Landrieu I, Smet-Nocca C, Amniai L, Louis JV, Wieruszeski JM, Goris J, et al. Molecular implication of PP2A and Pin1 in the Alzheimer’s disease specific hyperphosphorylation of Tau. PLoS One (2011) 6(6):e21521. doi:10.1371/journal.pone.0021521
92. Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med (1996) 2:783–7. doi:10.1038/nm0796-783
93. Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A (2006) 103(23):8864–9. doi:10.1073/pnas.0603214103
94. Brunden KR, Ballatore C, Lee VM, Smith AB III, Trojanowski JQ. Brain-penetrant microtubule-stabilizing compounds as potential therapeutic agents for tauopathies. Biochem Soc Trans (2012) 40:661–6. doi:10.1042/BST20120010
95. Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci (2012) 32:3601–11. doi:10.1523/JNEUROSCI.4922-11.2012
96. Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J Cell Sci (2005) 118:4645–54. doi:10.1242/jcs.02558
97. Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, Anderton BH, et al. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J (2008) 22:3186–95. doi:10.1096/fj.08-109181
98. Rodríguez-Martín T, Cuchillo-Ibáñez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging (2013) 34(9):2146–57. doi:10.1016/j.neurobiolaging.2013.03.015
99. Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science (2008) 319:1086–9. doi:10.1126/science.1152993
100. Kanaan NM, Morfini G, Pigino G, Lapointe NE, Andreadis A, Song Y, et al. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging (2012) 33(826):e815–30. doi:10.1016/j.neurobiolaging.2011.06.006
101. Ittner LM, Fath T, Ke YD, Bi M, van Eersel J, Li KM, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci U S A (2008) 105:15997–6002. doi:10.1073/pnas.0808084105
102. Ittner LM, Ke YD, Gotz J. Phosphorylated tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem (2009) 284:20909–16. doi:10.1074/jbc.M109.014472
103. Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging (2003) 24:1079–85. doi:10.1016/j.neurobiolaging.2003.04.007
104. De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci (2008) 31:151–73. doi:10.1146/annurev.neuro.31.061307.090711
105. Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B, et al. Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet (2012) 8:e1002918. doi:10.1371/journal.pgen.1002918
106. Falzone TL, Gunawardena S, McCleary D, Reis GF, Goldstein LS. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum Mol Genet (2010) 19:4399–408. doi:10.1093/hmg/ddq363
107. Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci (2012) 107:355–415. doi:10.1016/B978-0-12-385883-2.00005-9
108. Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol (2011) 179:2071–82. doi:10.1016/j.ajpath.2011.07.004
109. Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol (1998) 143:777–94. doi:10.1083/jcb.143.3.777
110. Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci (2012) 32:2430–41. doi:10.1523/JNEUROSCI.5927-11.2012
111. Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet (2012) 21:2538–47. doi:10.1093/hmg/dds072
112. DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron (2012) 75(4):618–32. doi:10.1016/j.neuron.2012.06.026
113. DuBoff B, Feany M, Gotz J. Why size matters – balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci (2013). doi:10.1016/j.tins.2013.03.002
114. Farah CA, Perreault S, Liazoghli D, Desjardins M, Anton A, Lauzon M, et al. Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton (2006) 63:710–24. doi:10.1002/cm.20157
115. Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol (1995) 131:1327–40. doi:10.1083/jcb.131.5.1327
116. Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging (2012) 33(431):e427–38. doi:10.1016/j.neurobiolaging.2011.01.005
117. Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem (2000) 275:15733–40. doi:10.1074/jbc.M000389200
118. Pooler AM, Hanger DP. Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans (2010) 38:1012–5. doi:10.1042/BST0381012
119. Arrasate M, Perez M, Avila J. Tau dephosphorylation at tau-1 site correlates with its association to cell membrane. Neurochem Res (2000) 25:43–50. doi:10.1023/A:1007583214722
120. Ekinci FJ, Shea TB. Phosphorylation of tau alters its association with the plasma membrane. Cell Mol Neurobiol (2000) 20:497–508. doi:10.1023/A:1007075115574
121. Usardi A, Pooler AM, Seereeram A, Reynolds CH, Derkinderen P, Anderton B, et al. Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J (2011) 278:2927–37. doi:10.1111/j.1742-4658
122. Reynolds CH, Garwood CJ, Wray S, Price C, Kellie S, Perera T, et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem (2008) 283:18177–86. doi:10.1074/jbc.M709715200
123. Hall GF, Lee VM, Lee G, Yao J. Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol (2001) 158:235–46. doi:10.1016/S0002-9440(10)63962-4
124. Katsuse O, Lin WL, Lewis J, Hutton ML, Dickson DW. Neurofibrillary tangle-related synaptic alterations of spinal motor neurons of P301L tau transgenic mice. Neurosci Lett (2006) 409:95–9. doi:10.1016/j.neulet.2006.09.021
125. Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, et al. The beta-propensity of tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem (2007) 282:31755–65. doi:10.1074/jbc.M705282200
126. Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol (2012) 181:1426–35. doi:10.1016/j.ajpath.2012.06.033
127. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci (2010) 30:11938–50. doi:10.1523/JNEUROSCI.2357-10.2010
128. Zempel H, Mandelkow EM. Linking amyloid-beta and tau: amyloid-beta induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener Dis (2012) 10:64–72. doi:10.1159/000332816
129. Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron (2010) 68:1067–81. doi:10.1016/j.neuron.2010.11.030
130. Crimins JL, Pooler A, Polydoro M, Luebke JI, Spires-Jones TL. The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res Rev (2013) doi:10.1016/j.arr.2013.03.002. [Epub ahead of print].
131. Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci (2007) 25:81–6. doi:10.1111/j.1460-9568.2006.05245.x
132. Loomis PA, Howard TH, Castleberry RP, Binder LI. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci U S A (1990) 87:8422–6. doi:10.1073/pnas.87.21.8422
133. Qu MH, Li H, Tian R, Nie CL, Liu Y, Han BS, et al. Neuronal tau induces DNA conformational changes observed by atomic force microscopy. Neuroreport (2004) 15:2723–7.
134. Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett (1996) 399:344–9. doi:10.1016/S0014-5793(96)01386-5
135. Krylova SM, Musheev M, Nutiu R, Li Y, Lee G, Krylov SN. Tau protein binds single-stranded DNA sequence specifically – the proof obtained in vitro with non-equilibrium capillary electrophoresis of equilibrium mixtures. FEBS Lett (2005) 579:1371–5. doi:10.1016/j.febslet.2005.01.032
136. Li W, Wang XS, Qu MH, Liu Y, He RQ. Human protein tau represses DNA replication in vitro. Biochim Biophys Acta (2005) 1726:280–6. doi:10.1016/j.bbagen.2005.08.014
137. Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem (2011) 286:4566–75. doi:10.1074/jbc.M110.199976
138. Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci (2011) 31(37):13110–7. doi:10.1523/JNEUROSCI.2569-11.2011
139. de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron (2012) 73:685–97. doi:10.1016/j.neuron.2011.11.033
140. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One (2012) 7:e31302. doi:10.1371/journal.pone.0031302
141. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem (2009) 284:12845–52. doi:10.1074/jbc.M808759200
142. Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem (2013) 288:1856–70. doi:10.1074/jbc.M112.394528
143. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol (2009) 11:909–13. doi:10.1038/ncb1901
144. Clavaguera F, Lavenir I, Falcon B, Frank S, Goedert M, Tolnay M. “Prion-like” templated misfolding in tauopathies. Brain Pathol (2013) 23:342–9. doi:10.1111/bpa.12044
145. Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis (2012) 48:356–66. doi:10.1016/j.nbd.2012.05.021
146. Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep (2013) 14:389–94. doi:10.1038/embor.2013.15
147. Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci (2008) 37:673–81. doi:10.1016/j.mcn.2007.12.010
148. Gorlovoy P, Larionov S, Pham TT, Neumann H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J (2009) 23:2502–13. doi:10.1096/fj.08-123877
149. Diaz-Hernandez M, Gomez-Ramos A, Rubio A, Gomez-Villafuertes R, Naranjo JR, Miras-Portugal MT, et al. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem (2010) 285:32539–48. doi:10.1074/jbc.M110.145003
150. Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One (2012) 7:e36873. doi:10.1371/journal.pone.0036873
151. Yoshiyama Y, Lee VM, Trojanowski JQ. Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry (2013) 84(7):784–95. doi:10.1136/jnnp-2012-303144
152. Caccamo A, Oddo S, Tran LX, Laferla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol (2007) 170:1669–75. doi:10.2353/ajpath.2007.061178
153. Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int J Geriatr Psychiatry (2008) 23:704–11. doi:10.1002/gps.1964
154. Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry (2009) 70:922–31. doi:10.4088/JCP.08m04606
155. Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry (2011) 198:351–6. doi:10.1192/bjp.bp.110.080044
156. Sereno L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sanchez MB, Gich I, et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis (2009) 35:359–67. doi:10.1016/j.nbd.2009.05.025
157. Del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis (2013) 33:205–15. doi:10.3233/JAD-2012-120805
158. D’Cruz OJ, Uckun FM. Protein kinase inhibitors against malignant lymphoma. Expert Opin Pharmacother (2013) 14:707–21. doi:10.1517/14656566.2013.780031
159. Wang H, Brautigan DL. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. J Biol Chem (2002) 277:49605–12. doi:10.1074/jbc.M209335200
160. Manser C, Guillot F, Vagnoni A, Davies J, Lau KF, McLoughlin DM, et al. Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo. Oncogene (2012) 31:2773–82. doi:10.1038/onc.2011.437
161. Manser C, Vagnoni A, Guillot F, Davies J, Miller CC. Cdk5/p35 phosphorylates lemur tyrosine kinase-2 to regulate protein phosphatase-1C phosphorylation and activity. J Neurochem (2012) 121:343–8. doi:10.1111/j.1471-4159.2012.07650.x
162. Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, Deboer S, et al. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J (2004) 23:2235–45. doi:10.1038/sj.emboj.7600237
163. Hernandez F, Langa E, Cuadros R, Avila J, Villanueva N. Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol Cell Biochem (2010) 344:211–5. doi:10.1007/s11010-010-0544-0
164. Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem (2011) 80:769–95. doi:10.1146/annurev-biochem-090308-173656
Keywords: tau, phosphorylation, oligomers, Alzheimer’s disease, function, extracellular
Citation: Noble W, Hanger DP, Miller CCJ and Lovestone S (2013) The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Received: 25 April 2013; Paper pending published: 20 May 2013;
Accepted: 14 June 2013; Published online: 01 July 2013.
Edited by:
Jesus Avila, Centro de Biología Molecular Severo Ochoa CSIC-UAM, SpainReviewed by:
Jurgen Gotz, The University of Sydney, AustraliaAlejandra Alonso, The City University of New York, USA
Copyright: © 2013 Noble, Hanger, Miller and Lovestone. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Wendy Noble, Diane P. Hanger, Christopher C. J. Miller and Simon Lovestone, King’s College London, Institute of Psychiatry, De Crespigny Park, London SE5 8AF, UK e-mail:d2VuZHkubm9ibGVAa2NsLmFjLnVrLA==ZGlhbmUuaGFuZ2VyQGtjbC5hYy51ayw=Y2hyaXMubWlsbGVyQGtjbC5hYy51ayw=c2ltb24ubG92ZXN0b25lQGtjbC5hYy51aw==