 Julia E. Gerson1,2,3
Julia E. Gerson1,2,3 Rakez Kayed1,2,3*
Rakez Kayed1,2,3*
- 1George P. and Cynthia Woods Mitchell Center for Neurodegenerative Diseases, University of Texas Medical Branch, Galveston, TX, USA
- 2Department of Neurology, University of Texas Medical Branch, Galveston, TX, USA
- 3Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, TX, USA
Tau misfolding and aggregation leads to the formation of neurofibrillary tangles (NFTs), which have long been considered one of the main pathological hallmarks for numerous neurodegenerative diseases known as tauopathies, including Alzheimer’s Disease (AD) and Parkinson’s Disease (PD). However, recent studies completed both in vitro and in vivo suggest that intermediate forms of tau, known as tau oligomers, between the monomeric form and NFTs are the true toxic species in disease and the best targets for anti-tau therapies. However, the exact mechanism by which the spread of pathology occurs is unknown. Evidence suggests that tau oligomers may act as templates for the misfolding of native tau, thereby seeding the spread of the toxic forms of the protein. Recently, researchers have reported the ability of tau oligomers to enter and exit cells, propagating from disease-affected regions to unaffected areas. While the mechanism by which the spreading of misfolded tau occurs has yet to be elucidated, there are a few different models which have been proposed, including cell membrane stress and pore-formation, endocytosis and exocytosis, and non-traditional secretion of protein not enclosed by a membrane. Coming to an understanding of how toxic tau species seed and spread through the brain will be crucial to finding effective treatments for neurodegenerative tauopathies.
Tau Oligomers are the Toxic Tau Species in Neurodegenerative Tauopathies
The formation of tau aggregates and neurofibrillary tangles (NFTs) is one of the main pathological hallmarks of numerous diseases termed tauopathies, including the two most common neurodegenerative diseases, Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) (1, 2). However, it is evident that cell death occurs initially prior to NFT formation in AD (3–6) suggesting that NFTs are not the pathogenic species responsible for the spread of the disease. Recent evidence points to the presence of multimeric tau species which are intermediates between tau monomers and NFT – known as tau oligomers – as the toxic species inducing synaptic dysfunction and cell death in neurodegenerative tauopathies (7–12).
Numerous researchers have investigated tau pathology using animal models, yielding a better understanding of the toxicity of different tau structures. A study in aged mice expressing native human tau (htau mice) found that while NFT formation occurred as animals aged, there was no correlation between the presence of tau filaments and cell death (13). Additionally, a study examining the P301S mouse model, which expresses mutant human tau, found that hippocampal synaptic dysfunction occurred prior to NFT formation (14). Studies using the rTg4510 mouse model, which conditionally expresses P301L mutant tau, found that cell death occurred prior to NFT formation and that cell loss and behavioral impairments could be suppressed by inhibiting tau expression without removing NFTs or preventing their continued accumulation (7, 15). In accordance with this finding, it has been shown that NFTs are protective in the same mouse model (16), and only pro-aggregate human tau mice (TauRD) show behavioral deficits (17). Another study in the same mouse model characterized tau oligomers biochemically that appeared early and correlated with cognitive deficits (8, 12). Similar results have also been seen in Drosophila AD models, where expression of mutant tau causes neurodegeneration, synaptic dysfunction, and axonal transport deficiencies in the absence of NFTs (18, 19). Usage of the protein nicotinamide mononucleotide (NAD) adenylyl transferase (NMNAT) was shown to decrease behavioral and morphological deficiencies in a frontotemporal dementia Drosophila model by decreasing levels of tau oligomers (20).
Biochemical analysis of human AD brain tissue has also yielded results suggesting that tau oligomers may initiate toxicity, rather than NFTs. When compared to control brains, levels of tau oligomers were found to be significantly increased in AD brains early in the disease, prior to when NFTs appear and clinical symptoms are evident (9, 21–23). In addition to correlative evidence for the importance of tau oligomers to toxicity, treatment with tau oligomers has also been shown to cause adverse effects in animals. Isolated tau oligomers, but not monomers or NFTs, induced memory impairments, synaptic dysfunction, and mitochondrial dysfunction when given intracerebrally to wild-type mice (24). Therefore, it is possible that NFTs are actually neuroprotective, sequestering toxic forms of tau into large aggregates with less flexibility and surface area to interact with cells. All of these studies form the framework for the model of the progression of neurodegenerative tauopathies beginning with the seeding and propagation of toxic tau oligomers (Figure 1).
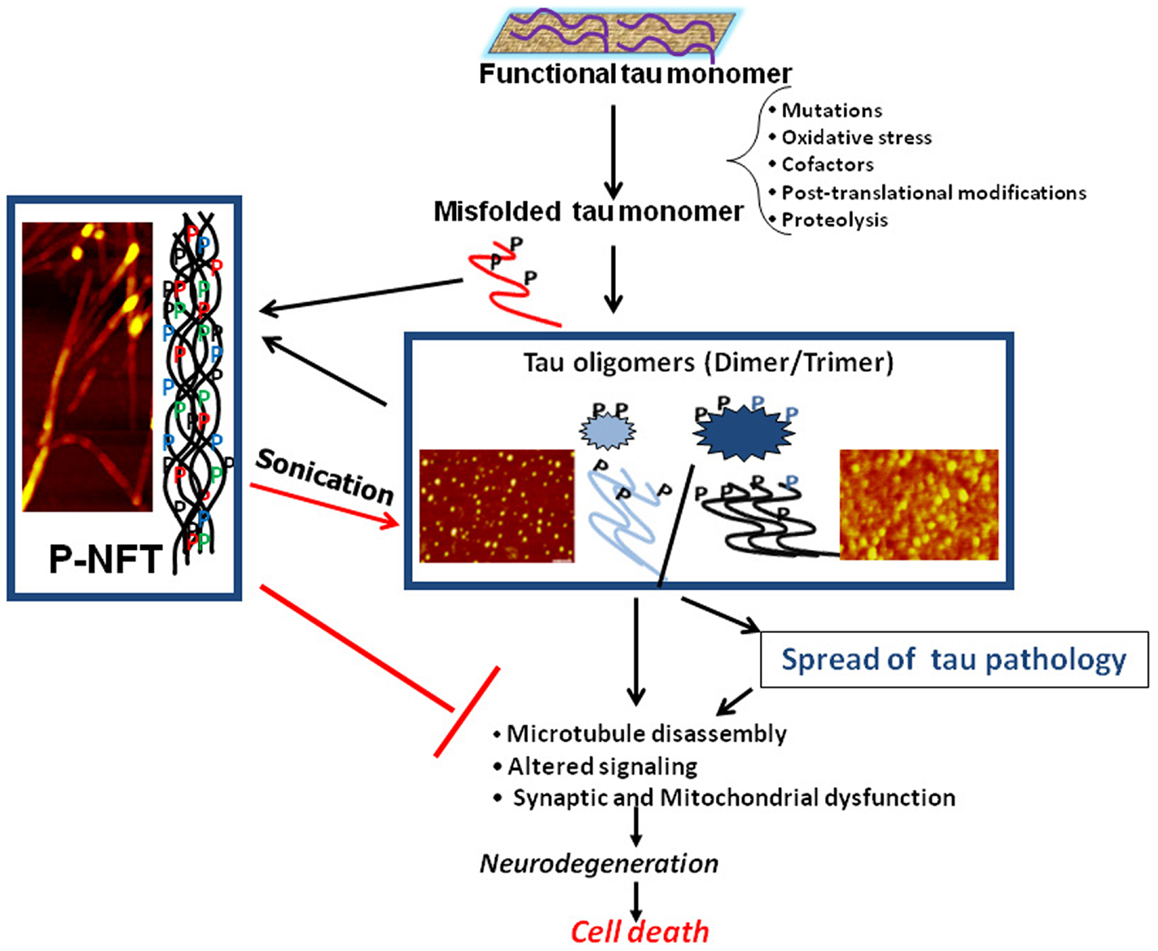
Figure 1. Schematic illustrating the central role of tau oligomers in tauopathies. Tau intermediate soluble aggregates (tau oligomers) are the toxic tau entities and initiators of tau pathology and propagation in tauopathies, rather than monomeric tau or hyperphosphorylated NFTs (p-NFTs). Sonication of fibrillar tau generates toxic tau oligomers. Thus, tau oligomers represent the ideal target for anti-tau therapeutic approaches. AFM images are of brain-derived tau oligomers and NFT (72, 138).
Tau Oligomers are Seeds for the Propagation of Pathological Tau
Recently, researchers have begun to make comparisons between the spread of neurodegenerative disease and prion disease, as studies suggest that misfolded protein templating, known as seeding, may underlie the progression of disease (25). Understanding how tau seeds pathological forms of the protein which propagate to different brain regions is critical to devising a solution to stop the spread of disease. There are two main models for the formation and seeding of pathological tau, oligomer-nucleated conformational induction – based upon the mechanism of action of prion protein, Sup35 (26) and amyloid β (Aβ) (27) – and template-assisted growth. Template-assisted growth proposes that tau fibrils act as template molecules for unfolded monomers. When monomers come in contact with filaments, they are integrated into the filament in organized, parallel stacked β sheets, optimizing hydrogen bonding for stabilization (28). It has been difficult, however, to find spontaneous tau aggregation which occurs experimentally. When fibrils are cleaved, leaving only three microtubule binding repeats, the fragments aggregate spontaneously in vitro (29). However, on its own, tau will not polymerize in vitro without the addition of certain reagents, post-translational modifications, such as phosphorylation, or induction of mutations.
In order for aggregation to occur, tau must be released from microtubules to reach a high concentration of free cytosolic tau, conformational changes must occur to allow for aggregation, possibly by increasing β sheet content, and dimerization must occur (30, 31). The addition of polyanions, such as heparin or RNA can induce fibrillization of tau (32), causing a conformational change from random coil structure to β sheet structure (33). Free fatty acids, such as arachidonic acid can also increase aggregation (34, 35) due to the presence of an alkyl chain, which induces micellization, and a negatively charged head group on the fatty acid to create a negatively charged surface on the micelle. In the presence of tau, the critical concentration for micelle formation is greatly decreased, allowing anionic micelles to attract tau to the negatively charged surface and thereby compensate for positive charges in tau and enable tau aggregation (36, 37). Phosphorylation may also play a role in fibrillization. Paired helical filaments (PHF) and straight filaments (SF), which make up the NFTs found in the brains of patients with AD, are comprised of hyperphosphorylated tau (38, 39). Phosphorylated tau has a higher tendency toward aggregation than unphosphorylated tau and kinases involved in the phosphorylation of these sites in tau have been shown to be altered in AD (40). Hyperphosphorylated tau has been shown to aggregate in vitro, possibly due to the addition of negative charge which would increase aggregation, similarly to the addition of polyanions and free fatty acids. Furthermore, this process can be inhibited by dephosphorylation (41, 42). Phosphorylation may also induce aggregation by reducing the interaction of tau with microtubules and allowing it to interact instead with unphosphorylated tau and form aggregates (43–45). Mutations, such as those that lead to frontotemporal lobar degeneration (FTLD), can increase tau aggregation through different mechanisms. Many mutations lead to a decrease in microtubule assembly kinetics, which could lead to more free cytosolic tau and increase aggregation (46–49). Some mutations lead to a decrease in the dissociation constants (Kd) for dimer and filament formation, while others increase the rate of nucleation without affecting Kd (30). Mutations that cause increased formation of β-sheets lead to heightened aggregation due to an increase in hydrophobic interactions, deviating from the highly hydrophilic native tau protein (50, 51). Fibrillar tau can thereby be recognized by dyes which interact with β-sheets, such as Congo Red and thioflavin S (52). However, at high concentrations, these dyes can induce fibrillization due to an attraction between positive charges formed in the core of PHFs and negative charges of the anionic dyes (53–55).
In amyloid proteins in which seeding has been well-established, such as prion proteins and Aβ, oligomers have been shown to be the most potent seeds (56, 57), working by way of oligomer-nucleated conformational induction (26, 27). Due to the increased interest in the toxicity of tau oligomers, evidence has emerged in support of the oligomer-nucleated conformational induction model as more studies have begun to explore the importance of tau oligomers in the initialization of tauopathies. Oligomer-nucleated conformational induction entails oligomers or conformational changes irreversibly stabilizing the highest energy protein states, known as the nucleus, allowing stable monomers to aggregate into oligomeric structures. Oligomers are driven to further elongation to form lower energy, stable filaments (58). As opposed to template-assisted growth, monomers are not incorporated directly into fibrils, but are instead entirely aggregated into oligomers prior to filament formation (59). Tau dimerization increases the tendency to aggregate and can be induced by oxidation (60), which suggests that tau oligomerization may be an important step in the fibrillization pathway. The appearance of oligomeric species of other amyloid proteins has been observed on the path to fibril formation (61–63).
While the addition of reagents and mutations used to induce fibrillization has been integral to understanding how tau aggregation occurs, it does not explain how fibril formation may occur spontaneously in sporadic disease. The mechanism by which tau aggregation occurs physiologically has not yet been elucidated, however there have been some advances in the understanding of how certain steps in the process may occur. Release of tau from microtubules may occur following post-translational modifications, such as phosphorylation (43, 44). Localization to anionic surfaces, alternative splicing, and post-translational modifications stabilizing aggregated conformations may all act as enhancers to increase speed of nucleation (64). Under physiological conditions, nucleating cofactors can induce tau aggregation in a similar fashion to agents used in vitro. There is evidence that polyanionic species, such as tubulin, RNA, and α-synuclein can increase the tendency of tau to aggregate (65–67) The formation of disulfide bridges is critical for the initial creation of dimers from monomers, as well as intermolecular crosslinking of the microtubule binding domain independent of cysteine to continue oligomerization of three-repeat tau (68). Prior to monomer aggregation into oligomers, the free energy of solvation decreases, causing a shift in preference for peptide-solvent interactions toward peptide–peptide interactions, as water is evacuated due to poor interaction with the peptide backbone and sidechains. Water release increases entropy of the solvent, thereby balancing the loss in conformational entropy caused by aggregation. The interaction of side chains with the backbone in the form of hydrogen bonding leads to the creation of β-sheet structure and aggregate stabilization. While oligomers form a similar structure to fibrils, they are not as ordered, which likely increases their toxicity (69). Proteolytic processing by endogenous proteases has also been shown to create self-aggregating fragments, which nucleate and co-aggregate with full-length protein effectively enough for a small amount of fragment to seed PHFs (70). Direct interactions between misfolded tau and native protein may be the underlying mechanism of seeding as experiments have shown tau protein–protein interactions occur when tau aggregates enter cells containing native tau (71).
Tau oligomers – identified with the tau oligomer-specific antibody, T22, which does not recognize monomers or fibrils (59) – which have been seeded with oligomers derived from brain tissue have been shown to be highly toxic (23, 72). When tested with Bis-ANS, which recognizes exposed hydrophobic patches, oligomers had higher affinity for Bis-ANS than PHFs, which may underlie toxicity. The toxic effects of tau oligomers formed by seeding recombinant tau with oligomeric seeds, however, can be prevented when pre-treated with T22 (23, 72).
Some tauopathies, such as progressive supranuclear palsy (PSP), only have one pathogenic species involved in disease progression (1). However, most tauopathies contain other amyloid proteins in addition to tau, such as Aβ in AD and α-synuclein in PD. In such diseases, cross-seeding of heterologous protein species is an additional mechanism which is important for tau seeding (59, 67, 73–77).
Tau Oligomers Propagate from Affected Brain Regions to Unaffected Regions
Aβ has been shown to propagate from affected brain areas to unaffected areas in mice over-expressing Aβ precursor protein that have been injected with Aβ isolated from the brains of AD patients and AD transgenic mice (78, 79), suggesting that perhaps tau could spread in a similar fashion. A few years later, a similar mechanism was demonstrated for the propagation of tau. When tau extracted from P301S mice was injected into the brains of mice over-expressing wild-type human tau (ALZ17 mice), which do not form tau aggregates, tau pathology was observed to have spread from the injection site to neighboring brain regions (80). Additionally, in transgenic mice which differentially express pathological tau in the entorhinal cortex, where tau pathology is first observed in AD, human tau has been shown to spread to both neighboring and synaptically connected neurons which do not express human tau mRNA. Translocated human tau was able to seed mouse tau misfolding (81, 82). However, these studies did not specifically investigate which tau species specifically induced seeding and propagation of tau pathology and the usage of transgenic mouse models is not analogous to sporadic forms of AD. When wild-type mice were injected with both tau oligomers and PHFs isolated from AD brains, tau oligomers induced the spread of tau pathology from the injection site to neighboring brain regions and impaired memory, as measured by object recognition. Conversely, mice injected with PHFs only exhibited tau pathology near the injection site and did not exhibit any memory impairments on the behavioral task, suggesting that tau oligomers, but not fibrillar tau, is capable of seeding and propagating pathology (72). Furthermore, similar results have been found using primary neurons. Neurons were exposed to low molecular weight aggregates – recognized by the tau oligomer-specific antibody, T22, and examined via electron microscopy for oligomeric characteristics – as well as to fibrils formed in vitro, filaments formed in vivo, and monomers. Low molecular weight aggregates and short fibrils exhibited uptake into the cell, but monomers and filaments were not internalized (83). Other studies have shown tau aggregate uptake using cell culture, but did not specifically identify the type of aggregates being internalized. Neural stem cells treated with tau monomers and aggregates formed using the tau microtubule binding repeat region induced to fibrillize with arachidonic acid, exhibited significantly more tau aggregate uptake than monomer uptake. Additionally, aggregates, but not monomers, induced seeding of endogenous tau misfolding (84).
On the other hand, Guo and Lee hypothesized that seeding of pathological tau in cultured cells would be able to occur more quickly by seeding with pre-formed tau fibrils, thereby omitting the step where monomer must be converted to oligomer prior to fibril formation. Fibrillization of recombinant tau was induced with the addition of heparin and was verified using thioflavin T. Fibril-treated cells exhibited seeding and propagation of aggregates via endocytosis. However, following fibril confirmation with thioflavin T and prior to cell treatment, fibrils used in this study were sonicated (85). Previous research investigating Aβ seeding found that sonication increased seeding ability by fragmenting fibrils into smaller, soluble species (57) and sonicated prions have also been shown to have more potent seeding potentials than unsonicated fibrils (56). Since it has been shown that both prion and Aβ oligomers, rather than fibrils, are the seeds for pathological protein templating (26, 27), it is likely that sonication partially converts insoluble fibrils into soluble oligomeric forms. Sonication of tau fibrils has also been shown to cause shearing of filaments, particularly those in PHF form (86). Therefore, it is likely that sonicated tau fibrils used to treat cells in the previous study (85) also contained tau in oligomeric form, which may explain why seeding and propagation was successful.
Recently, Wu et al. studied propagation of tau in primary neurons using microfluidic chambers which allow somatodendritic compartments to be isolated from axonal compartments, enabling not only the analysis of tau uptake from the extracellular space into the cell, but also propagation within the neuron. They found that low molecular weight tau aggregates specifically recognized by tau oligomer-specific antibody, T22, propagate between isolated neuronal compartments both anterogradely and retrogradely (83). Importantly, tau is primarily found in the axons of healthy neurons (87), though tau may also be found in the dendrite where it colocalizes with the src kinase, fyn (88, 89). In AD, however, misfolded and hyperphosphorylated tau accumulates in the axon, dendrites, and the cell body (90), suggesting that intracellular transport may also be important for the spread of disease. In lamprey neurons expressing low levels of tau, tau was primarily localized to the axon and proximal dendrites, both regions consistent with tau functioning as a microtubule-associated protein. However, in neurons expressing high levels of tau, tau was found in distal dendrites and near the soma membrane, both areas lacking microtubules. High-expressing tau cells showed more degeneration and secretion of tau. Moreover, as tau can modulate activity of microtubule-associated motor proteins involved in dendritic transport, tau localized to the dendrite may have implications for its propagation (91). Phosphorylated tau localized at the synapse in AD brain samples appears to correlate with ubiquitin-proteasome system (UPS) dysfunction, suggesting that tau oligomer accumulation at the synapse impairs the UPS, which is a crucial player in the breakdown of tau. Accumulation of tau at the synapse may also suggest a mechanism for trans-synaptic tau propagation (92).
Phosphorylation clearly plays a role in the toxicity and localization of tau, however, its exact role in neurodegenerative disease is unknown and appears to be quite complicated. While hyperphosphorylated tau has been shown to have toxic results in the cell, increasing aggregation and abnormal tau localization (40, 90), dephosphorylated tau can also have harmful effects. Phosphorylated tau released into the medium of cultured neuroblastoma cells through muscarinic receptor activation that is dephosphorylated by tissue-non-specific alkaline phosphatase (TNAP) led to excitotoxicity, increasing calcium levels in nearby cells. Additionally, levels of TNAP are heightened in AD brains compared to control brains (93). Another study of primary cortical neurons also found that extracellular tau is largely dephosphorylated (94). Conversely, one study found that phosphorylation of tau increased its secretion from HeLa cells (95). Inflammation and activation of microglia has been shown to increase tau phosphorylation as well as aggregation, but is complicated by the fact that the opposite effect is seen in Aβ (96–99). The localization of tau in the cytosol, cell membrane, and the nucleus also appears to be important for tau toxicity, and is mediated by phosphorylation. Oxidative stress and heat shock induce the dephosphorylation of cytosolic tau and its transport into the nucleus. Once relocated to the nucleus, tau appears to protect neuronal DNA from damage under cell stress (100), which may be important in AD where DNA damage has been shown to occur (101). One possibility is that abnormal phosphorylation of tau in AD may prevent tau from being dephosphorylated and translocated to the nucleus to protect against DNA damage. The localization of tau to the cell membrane may also depend upon its dephosphorylation, as tau with lower levels of phosphorylation in its proline region was shown to be associated with the cell membrane, while phosphorylated tau was found in the cytoplasm (102, 103). However, interaction with membrane bound proteins, such as aforementioned fyn may stabilize association of phosphorylated tau with the membrane.
Mechanism of Tau Propagation
The entry of prion proteins, Aβ, and other amyloid proteins into the cell via different mechanisms has been well-established (104–107). One hypothesis for amyloid oligomer toxicity and entrance into cells is through protein interaction with the cell membrane. One model suggests that oligomers embed themselves into the cell membrane and form pores. However, it appears as though the formation of pore-like annular protofibrils occurs through a separate pathway from fibril formation (108). An alternative model suggests that oligomers interact with lipid rafts in the bilayer, causing membrane thinning and increased membrane permeabilization, which may play a role in oligomer toxicity, allowing non-specific ion entrance, as well as leakage of cellular compartments. Several types of amyloid oligomers have been shown to increase membrane permeability, including Aβ, α-synuclein, and prion protein (109, 110, 107, 111). Tau has been reported to interact with the lipid rafts in the cell membrane and undergo conformational changes leading to membrane stress (112–115). Additionally, permeabilization of the membrane could mediate internalization of oligomers into the cell.
There has also been evidence for endocytosis as a route of amyloid entry into the cell. Propagation of α-synuclein, prion protein, Sup35, and Aβ has been shown to be associated with the endosomal pathway (116–118). One study found that tau aggregates colocalize with dextran in neural stem cells, implying that entry into the cell occurs via macropinocytosis. However, the aggregate type was not specifically tested (84). Aggregates identified specifically as tau oligomers colocalized with fluid-phase endocytosis marker, dextran, as well as with early endosomal marker, Rab5, and late endosomal/lysosomal marker, Lamp1. When endocytosis was inhibited with dynamin inhibitor, Dynasore, tau uptake was blocked, while inhibition of clathrin-mediated endocytosis with Pitstop2B did not impact internalization (83). These studies together suggest a mechanism for tau propagation in which tau is internalized via pinocytosis and enters the endosomal pathway. Tau can move through the endosomal pathway to the lysosome where toxic species may be degraded or recycled back to the cell membrane, where they may be released to be internalized by adjacent neurons. More research is needed to determine how membrane-enclosed tau oligomers are released inside of the cell, though it appears likely that the majority are degraded in the lysosome, while those that avoid degradation may cause the endosomal membrane to burst and be released in the cytoplasm, where they can seed aggregation of healthy tau (83). While clathrin-mediated endocytosis did not appear to be involved in tau propagation, endocytosis inhibitors are often found to be non-specific (119), and therefore, the possibility of other types of endocytosis in tau spread bears more study.
Receptor-mediated endocytosis could be another route of entry into the cell as amyloids have been reported to bind to cell surface receptors. Internalization of α-synuclein has been shown to be dependent upon receptor-mediated endocytosis, potentially through caveolin-mediated endocytosis (117). Additionally, Aβ binds to NMDA, α7 nicotinic acetylcholine, and APOE receptors, inducing receptor endocytosis (120–124). Aβ oligomers also bind cellular prion protein, PrPc, which is complexed with the Src tyrosine kinase, Fyn. This interaction has been shown to increase tau dysfunction and prevents native tau from binding to fyn (125). Under normal conditions, tau binds to fyn in oligodendrocytes (126) and in neurons, activating the Ras/MAPK pathway (103). Mutations to the microtubule binding region in tau lead to decreases in oligodendrocyte process number and length and disease-related missense mutations increase tau association with Fyn (127). Results indicate that the interaction between tau and fyn may be important for neurodegeneration, both through a loss in native tau interaction and through a gain in toxic tau function. Interaction with PrPc complexed to fyn could also mediate tau entry into the cell as the PrPc complex is associated with and endocytosed with caveolin (128). Tau may also enter the cell through a direct interaction with fyn. Lee et al. used a lamprey ABC tauopathy model in which tau is expressed in specifically identified ABC neurons to investigate the spread of tau. They report that tau phosphorylated at Y18, the site most commonly phosphorylated by fyn kinase, is associated with vesicular organelles. Additionally, when tau is overexpressed and localizes to the dendrite, dendritic vesicle accumulation is observed. Phosphorylated tau is colocalized with vesicles which bear resemblance to endosomes, as well as to fyn. Fyn has also been shown to colocalize with exosomes, suggesting a possible mechanism for fyn-tau transport in which fyn-associated tau is endocytosed, transported from early endosomes to late endosomal compartments, and then transported out of the cell via exosomes (91).
While one mechanism for tau oligomer release is through oligomer toxicity leading to cell death, causing the cell to lyse and release its contents (129), studies show that this likely does not account for the majority of tau release. In primary neurons treated with tau oligomers, extracellular tau only increases once levels as high as 40% cell death are reached, which does not correspond to physiological levels of cell death during the initial spread of neurodegenerative disease (130). Additionally, treatment with tau oligomers in primary neurons does not lead to significant levels of apoptosis (83). There has however been some evidence for non-apoptotic membrane blebbing as a possible secondary mechanism for tau release (91, 131).
Exocytosis has been implicated as a mechanism of amyloid spread as prion proteins and α-synuclein have been shown to be associated with exosomes in cell culture (132, 133). However, investigations of a similar mechanism for tau release have been unclear. Simón et al. found that when tau was overexpressed in kidney-derived cell lines, tau was secreted contained within membrane vesicles (129, 134). While tau secreted by neuroblastoma cells and tau in human CSF was found to be associated with exosomes in one study (135), another reported that tau was not detected in isolated exosomes from neuroblastoma cells (130). However, these studies used cell models where tau was overexpressed. In an attempt to approach more similar conditions to those seen physiologically, researchers cultured primary cortical neurons containing endogenous tau and found that tau was released by a mechanism unrelated to cell death and was regulated by AMPA receptor activation. Inhibition of synaptic vesicle release decreased extracellular tau, while tau was not found to be associated with exosomes, indicating that release of tau through traditional synaptic exocytosis following AMPA receptor activation may be one mechanism of tau release (94). Another study found that cells constitutively release tau which is not contained within a membrane under conditions inhibiting cell death (136). Therefore, more research is warranted to investigate the conditions under which tau is associated with exosomes and the specific tau conformations found in exosomes. Tunneling nanotubes – long, temporary channels that allow for long-distance transport between cells – have recently been discovered as a transport mechanism for prion protein (137). While they have not yet been studied directly in the context of tau, similarities between the spread of prions and tau suggest that tunneling nanotubes may be another potential mode of tau propagation meriting study.
Conclusion
Determining how neurodegenerative tauopathies initiate and propagate toxic species will be crucial to finding a treatment for these diseases. Recent evidence suggests that tau oligomers, not NFTs, are the toxic tau species mediating the initiation, seeding, and propagation of neurodegenerative tauopathies and are the best target for anti-tau therapeutics. The mechanism by which tau seeding occurs remains to be elucidated, but oligomer-nucleated conformational induction, whereby native tau monomers are entirely converted to oligomers prior to aggregation into fibrils, appears to be a likely model. Tau oligomers can effectively enter cells, be transported intracellularly, and be released from cells to affect others. However, the mechanism by which propagation occurs is unclear. Tau likely enters the cell in one of two main ways, stressing the cell membrane or entering via endocytosis. Entrance through interaction with the membrane may occur through formation of pores or by interacting with lipid rafts causing membrane stress. Both macropinocytosis and receptor-mediated endocytosis have been implicated as possible mechanisms for tau entry. Tau secretion is likely not due simply to cell death, but may occur within exosomes, through synaptic vesicle release, or a non-traditional secretion pathway in which tau is not enclosed in a membrane. The elucidation of the mechanisms addressed will lead to a better understanding of neurodegenerative disease and may reveal new targets for treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Diana L. Castillo-Carranza, Marcos J. Guerrero-Munoz, and George R. Jackson for useful suggestions. This work was supported by: The Cullen Family Trust for Health Care, the Alzheimer’s Drug Discovery Foundation (ADDF), the Michael J. Fox Foundation, and the Mitchell Center for Neurodegenerative Diseases.
References
1. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci (2007) 8:663–72.
2. Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener (2009) 4:13. doi:10.1186/1750-1326-4-13
3. Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol (1997) 41:17–24. doi:10.1002/ana.410410106
4. Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol (1999) 58:188–97. doi:10.1097/00005072-199902000-00008
5. Kril JJ, Patel S, Harding AJ, Halliday GM. Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol (2002) 103:370–6. doi:10.1007/s00401-001-0477-5
6. Haroutunian V, Davies P, Vianna C, Buxbaum JD, Purohit DP. Tau protein abnormalities associated with the progression of Alzheimer disease type dementia. Neurobiol Aging (2007) 28:1–7. doi:10.1016/j.neurobiolaging.2005.11.001
7. Spires TL, Orne JD, Santacruz K, Pitstick R, Carlson GA, Ashe KH, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol (2006) 168:1598–607. doi:10.2353/ajpath.2006.050840
8. Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci (2007) 27:3650–62. doi:10.1523/JNEUROSCI.0587-07.2007
9. Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, et al. Granular tau oligomers as intermediates of tau filaments. Biochemistry (2007) 46:3856–61. doi:10.1021/bi061359o
10. Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol (2011) 179:2071–82. doi:10.1016/j.ajpath.2011.07.004
11. Cowan CM, Quraishe S, Mudher A. What is the pathological significance of tau oligomers? Biochem Soc Trans (2012) 40:693–7. doi:10.1042/BST20120135
12. Sahara N, Deture M, Ren Y, Ebrahim AS, Kang D, Knight J, et al. Characteristics of TBS-extractable hyperphosphorylated tau species: aggregation intermediates in rTg4510 mouse brain. J Alzheimers Dis (2013) 33:249–63.
13. Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem (2003) 86:582–90. doi:10.1046/j.1471-4159.2003.01879.x
14. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron (2007) 53:337–51. doi:10.1016/j.neuron.2007.01.010
15. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science (2005) 309:476–81. doi:10.1126/science.1113694
16. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature (2010) 464:1201–4. doi:10.1038/nature08890
17. Sydow A, Van Der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci (2011) 31:2511–25. doi:10.1523/JNEUROSCI.5245-10.2011
18. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science (2001) 293:711–4. doi:10.1126/science.1062382
19. Cowan CM, Bossing T, Page A, Shepherd D, Mudher A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol (2010) 120:593–604. doi:10.1007/s00401-010-0716-8
20. Ali YO, Ruan K, Zhai RG. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet (2012) 21:237–50. doi:10.1093/hmg/ddr449
21. Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci Res (2006) 54:197–201. doi:10.1016/j.neures.2005.11.009
22. Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, et al. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer’s disease. J Biol Chem (2011) 286:23063–76. doi:10.1074/jbc.M111.237974
23. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J (2012) 26:1946–59. doi:10.1096/fj.11-199851
24. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener (2011) 6:39. doi:10.1186/1750-1326-6-39
25. Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol (2013) 70:304–10. doi:10.1001/jamaneurol.2013.1453
26. Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science (2000) 289:1317–21. doi:10.1126/science.289.5483.1317
27. Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol (2011) 7:602–9. doi:10.1038/nchembio.624
28. Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci U S A (2004) 101:10278–83. doi:10.1073/pnas.0401911101
29. Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol (1992) 118:573–84. doi:10.1083/jcb.118.3.573
30. Chang E, Kim S, Yin H, Nagaraja HN, Kuret J. Pathogenic missense MAPT mutations differentially modulate tau aggregation propensity at nucleation and extension steps. J Neurochem (2008) 107:1113–23.
31. Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem (2008) 283:13806–16. doi:10.1074/jbc.M800247200
32. King ME, Ahuja V, Binder LI, Kuret J. Ligand-dependent tau filament formation: implications for Alzheimer’s disease progression. Biochemistry (1999) 38:14851–9. doi:10.1021/bi9911839
33. von Bergen M, Barghorn S, Biernat J, Mandelkow E-M, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta (2005) 1739:158–66. doi:10.1016/j.bbadis.2004.09.010
34. Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol (1997) 150:2181–95.
35. King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem (2000) 74:1749–57. doi:10.1046/j.1471-4159.2000.0741749.x
36. Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry (2002) 41:14885–96.
37. Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. J Biol Chem (2003) 278:25644–50. doi:10.1074/jbc.M301663200
38. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A (1986) 83:4913–7. doi:10.1073/pnas.83.13.4913
39. Lee VMY, Balin BJ, Otvos L Jr., Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science (1991) 251:675–8. doi:10.1126/science.1899488
40. Pérez M, Cuadros R, Smith MA, Perry G, Avila J. Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal. FEBS Lett (2000) 486:270–4. doi:10.1016/S0014-5793(00)02323-1
41. Alonso ADC, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem (2001) 276:37967–73.
42. Alonso ADC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A (2001) 98:6923–8. doi:10.1073/pnas.121119298
43. Biernat J, Gustke N, Drewes G, Mandelkow E, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron (1993) 11:153–63. doi:10.1016/0896-6273(93)90279-Z
44. Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron (1993) 10:1089–99. doi:10.1016/0896-6273(93)90057-X
45. Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys (1998) 357:299–309. doi:10.1006/abbi.1998.0813
46. Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett (1998) 437:207–10. doi:10.1016/S0014-5793(98)01217-4
47. Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, Von Bergen M, Mandelkow EM, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry (2000) 39:11714–21.
48. DeTure M, Ko L-W, Yen S, Nacharaju P, Easson C, Lewis J, et al. Missense tau mutations identified in FTDP-17 have a small effect on tau-microtubule interactions. Brain Res (2000) 853:5–14. doi:10.1016/S0006-8993(99)02124-1
49. Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L τ mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol (2002) 52:511–6. doi:10.1002/ana.10340
50. Harada M, Isersky C, Cuatrecasas P, Page D, Bladen HA, Eanes ED, et al. Human amyloid protein: chemical variability and homogeneity. J Histochem Cytochem (1971) 19:1–15.
51. von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow E-M, et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J Biol Chem (2001) 276:48165–74.
52. Friedhoff P, Schneider A, Mandelkow E-M, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry (1998) 37:10223–30. doi:10.1021/bi980537d
53. Lira-De León KI, García-Gutiérrez P, Serratos IN, Palomera-Cárdenas M, Figueroa-Corona MDP, Campos-Peña V, et al. Molecular mechanism of tau aggregation induced by anionic and cationic dyes. J Alzheimers Dis (2013) 35:319–34. doi:10.3233/JAD-121765
54. Kim Y-S, Randolph TW, Manning MC, Stevens FJ, Carpenter JF. Congo red populates partially unfolded states of an amyloidogenic protein to enhance aggregation and amyloid fibril formation. J Biol Chem (2003) 278:10842–50. doi:10.1074/jbc.M212540200
55. Congdon EE, Necula M, Blackstone RD, Kuret J. Potency of a tau fibrillization inhibitor is influenced by its aggregation state. Arch Biochem Biophys (2007) 465:127–35. doi:10.1016/j.abb.2007.05.004
56. Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature (2005) 437:257–61. doi:10.1038/nature03989
57. Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci (2011) 31:14488–95. doi:10.1523/JNEUROSCI.3088-11.2011
58. Ruschak AM, Miranker AD. The role of prefibrillar structures in the assembly of a peptide amyloid. J Mol Biol (2009) 393:214–26. doi:10.1016/j.jmb.2009.06.026
59. Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry (2010) 49:10039–41. doi:10.1021/bi1016233
60. Mandelkow EM, Biernat J, Drewes G, Steiner B, Lichtenberg-Kraag B, Wille H, et al. Microtubule-associated protein tau, paired helical filaments, and phosphorylation. Ann N Y Acad Sci (1993) 695:209–16. doi:10.1111/j.1749-6632.1993.tb23054.x
61. Ahmad A, Uversky VN, Hong D, Fink AL. Early events in the fibrillation of monomeric insulin. J Biol Chem (2005) 280:42669–75. doi:10.1074/jbc.M504298200
62. Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem (2009) 1:326–31. doi:10.1038/nchem.247
63. Frare E, Mossuto MF, De Laureto PP, Tolin S, Menzer L, Dumoulin M, et al. Characterization of oligomeric species on the aggregation pathway of human lysozyme. J Mol Biol (2009) 387:17–27. doi:10.1016/j.jmb.2009.01.049
64. Chirita CN, Congdon EE, Yin H, Kuret J. Triggers of full-length tau aggregation: a role for partially folded intermediates. Biochemistry (2005) 44:5862–72. doi:10.1021/bi0500123
65. Littauer UZ, Giveon D, Thierauf M, Ginzburg I, Ponstingl H. Common and distinct tubulin binding sites for microtubule-associated proteins. Proc Natl Acad Sci U S A (1986) 83:7162–6. doi:10.1073/pnas.83.19.7162
66. Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett (1996) 399:344–9. doi:10.1016/S0014-5793(96)01386-5
67. Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science (2003) 300:636–40. doi:10.1126/science.1082324
68. Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen S-H, et al. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci (2007) 25:3020–9. doi:10.1111/j.1460-9568.2007.05555.x
69. Matthes D, Gapsys V, De Groot BL. Driving forces and structural determinants of steric zipper peptide oligomer formation elucidated by atomistic simulations. J Mol Biol (2012) 421:390–416. doi:10.1016/j.jmb.2012.02.004
70. Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow E-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A (2007) 104:10252–7. doi:10.1073/pnas.0703676104
71. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem (2012) 287:19440–51. doi:10.1074/jbc.M112.346072
72. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep (2012) 2:700. doi:10.1038/srep00700
73. Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron (1995) 14:879–88. doi:10.1016/0896-6273(95)90232-5
74. Gotz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science (2001) 293:1491–5. doi:10.1126/science.1062097
75. Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Gotz J. Beta-amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem (2003) 278:40162–8. doi:10.1074/jbc.M308243200
76. Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci (2011) 31:7604–18. doi:10.1523/JNEUROSCI.0297-11.2011
77. Ono K, Takahashi R, Ikeda T, Yamada M. Cross-seeding effects of amyloid β-protein and α-synuclein. J Neurochem (2012) 122:883–90. doi:10.1111/j.1471-4159
78. Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ. Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides (2002) 23:1241–7. doi:10.1016/S0196-9781(02)00059-1
79. Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral ß -amyloidogenesis is governed by agent and host. Science (2006) 313:1781–4. doi:10.1126/science.1131864
80. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol (2009) 11:909–13. doi:10.1038/ncb1901
81. de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron (2012) 73:685–97. doi:10.1016/j.neuron.2011.11.033
82. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE (2012) 7:e31302. doi:10.1371/journal.pone.0031302
83. Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem (2013) 288:1856–70. doi:10.1074/jbc.M112.394528
84. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem (2009) 284:12845–52. doi:10.1074/jbc.M808759200
85. Guo JL, Lee VM-Y. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem (2011) 286:15317–31. doi:10.1074/jbc.M110.209296
86. Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type tau fibrils specified by templated conformation change. J Biol Chem (2009) 284:3546–51. doi:10.1074/jbc.M805627200
87. Dotti CG, Banker GA, Binder LI. The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience (1987) 23:121–30. doi:10.1016/0306-4522(87)90276-4
88. Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci (1998) 111:3167–77.
89. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, Van Eersel J, et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell (2010) 142:387–97. doi:10.1016/j.cell.2010.06.036
90. Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev (2004) 84:361–84.
91. Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int J Alzheimers Dis (2012) 2012:1–16. doi:10.1155/2012/172837
92. Tai H-C, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol (2012) 181:1426–35. doi:10.1016/j.ajpath.2012.06.033
93. Díaz-Hernández M, Gómez-Ramos A, Rubio A, Gómez-Villafuertes R, Naranjo JR, Miras-Portugal MT, et al. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem (2010) 285:32539–48. doi:10.1074/jbc.M110.145003
94. Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep (2013) 14:389–94. doi:10.1038/embor.2013.15
95. Plouffe V, Mohamed N-V, Rivest-Mcgraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS ONE (2012) 7:e36873. doi:10.1371/journal.pone.0036873
96. Li Y, Liu L, Barger SW, Griffin WST. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci (2003) 23:1605–11.
97. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron (2010) 68:19–31. doi:10.1016/j.neuron.2010.08.023
98. Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, Laferla FM, Olschowka JA, et al. Sustained interleukin-1 overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci (2013) 33:5053–64. doi:10.1523/JNEUROSCI.4361-12.2013
99. Nash KR, Lee DC, Hunt JB Jr., Morganti JM, Selenica M-L, Moran P, et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging (2013) 34:1540–8. doi:10.1016/j.neurobiolaging.2012.12.011
100. Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem (2011) 286:4566–75. doi:10.1074/jbc.M110.199976
101. Coppede F, Migliore L. DNA damage and repair in Alzheimer’s disease. Curr Alzheimer Res (2009) 6:36–47. doi:10.2174/156720509787313970
102. Arrasate M, Pérez M, Avila J. Tau dephosphorylation at tau-1 site correlates with its association to cell membrane. Neurochem Res (2000) 25:43–50. doi:10.1023/A:1007583214722
103. Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging (2012) 33:431.e427–38. doi:10.1016/j.neurobiolaging.2011.01.005
104. Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, et al. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol (2002) 12:523–30. doi:10.1016/S0960-9822(02)00722-4
105. Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet (2002) 11:2905–17. doi:10.1093/hmg/11.23.2905
106. Magalhães AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, Prado MAM, et al. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci (2005) 25:5207–16. doi:10.1523/JNEUROSCI.0653-05.2005
107. Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol (2009) 11:219–25. doi:10.1038/ncb1830
108. Lasagna-Reeves CA, Glabe CG, Kayed R. Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem (2011) 286:22122–30. doi:10.1074/jbc.M111.236257
109. Kayed R, Sokolov Y, Edmonds B, Mcintire TM, Milton SC, Hall JE, et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem (2004) 279:46363–6. doi:10.1074/jbc.C400260200
110. Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem (2005) 280:17294–300. doi:10.1074/jbc.M500997200
111. Stockl MT, Zijlstra N, Subramaniam V. Alpha-synuclein oligomers: an amyloid pore? Insights into the mechanisms of alpha-synuclein oligomer-lipid interactions. Mol Neurobiol (2013) 47:613–21. doi:10.1007/s12035-012-8331-4
112. Flach K, Hilbrich I, Schiffmann A, Gartner U, Kruger M, Leonhardt M, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem (2012) 287:43223–33. doi:10.1074/jbc.M112.396176
113. Jones EM, Dubey M, Camp PJ, Vernon BC, Biernat J, Mandelkow E, et al. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemistry (2012) 51:2539–50. doi:10.1021/bi201857v
114. Kunze G, Barre P, Scheidt HA, Thomas L, Eliezer D, Huster D. Binding of the three-repeat domain of tau to phospholipid membranes induces an aggregated-like state of the protein. Biochim Biophys Acta (2012) 1818:2302–13. doi:10.1016/j.bbamem.2012.03.019
115. Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem (2012) 287:32040–53. doi:10.1074/jbc.M112.401240
116. Narayanan S, Bosl B, Walter S, Reif B. Importance of low-oligomeric-weight species for prion propagation in the yeast prion system Sup35/Hsp104. Proc Natl Acad Sci U S A (2003) 100:9286–91. doi:10.1073/pnas.1233535100
117. Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol (2008) 40:1835–49. doi:10.1016/j.biocel.2008.01.017
118. Yu C, Nwabuisi-Heath E, Laxton K, Ladu MJ. Endocytic pathways mediating oligomeric Abeta42 neurotoxicity. Mol Neurodegener (2010) 5:1–11. doi:10.1186/1750-1326-5-19
119. Ivanov A. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? In: Ivanov A editor. Exocytosis and Endocytosis. Totowa: Humana Press (2008). p. 15–33.
120. Wang H-Y, Lee DHS, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid1-42 binds to α7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer’s disease pathology. J Biol Chem (2000) 275:5626–32. doi:10.1074/jbc.275.8.5626
121. Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid1-42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience (2002) 110:199–211. doi:10.1016/S0306-4522(01)00460-2
122. Bu G, Cam J, Zerbinatti C. LRP in amyloid-β production and metabolism. Ann N Y Acad Sci (2006) 1086:35–53.
123. Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, et al. Aβ-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci (2010) 30:5948–57. doi:10.1523/JNEUROSCI.0157-10.2010
124. Kessels HW, Nabavi S, Malinow R. Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc Natl Acad Sci U S A (2013) 110:4033–8. doi:10.1073/pnas.1219605110
125. Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, et al. The complex PrPc-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci (2012) 32:16857–71. doi:10.1523/JNEUROSCI.1858-12.2012
126. Klein C, Krämer E-M, Cardine A-M, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of Fyn kinase with the cytoskeletal protein tau. J Neurosci (2002) 22:698–707.
127. Bhaskar K, Yen S-H, Lee G. Disease-related modifications in tau affect the interaction between fyn and tau. J Biol Chem (2005) 280:35119–25. doi:10.1074/jbc.M505895200
128. Peters PJ, Mironov A, Peretz D, Van Donselaar E, Leclerc E, Erpel S, et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol (2003) 162:703–17. doi:10.1083/jcb.200304140
129. Simón D, García-García E, Gómez-Ramos A, Falcón-Pérez JM, Díaz-Hernández M, Hernández F, et al. Tau overexpression results in its secretion via membrane vesicles. Neurdegener Dis (2012) 10:73–5. doi:10.1159/000334915
130. Karch CM, Jeng AT, Goate AM. Extracellular tau levels are influenced by variability in tau that is associated with tauopathies. J Biol Chem (2012) 287:42751–62. doi:10.1074/jbc.M112.380642
131. Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol (2008) 181:879–84. doi:10.1083/jcb.200802081
132. Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A (2004) 101:9683–8. doi:10.1073/pnas.0308413101
133. Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci (2010) 30:6838–51. doi:10.1523/JNEUROSCI.5699-09.2010
134. Simón D, García-García E, Royo F, Falcón-Pérez JM, Avila J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett (2012) 586:47–54. doi:10.1016/j.febslet.2011.11.022
135. Saman S, Kim W, Raya M, Visnick Y, Miro S, Jackson B, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem (2012) 287:3842–9. doi:10.1074/jbc.M111.277061
136. Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis (2012) 48:356–66. doi:10.1016/j.nbd.2012.05.021
137. Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol (2009) 11:328–36. doi:10.1038/ncb1841
Keywords: tauopathies, tau oligomers, oligomeric seeding, propagation of tau pathology, Alzheimer’s
Citation: Gerson JE and Kayed R (2013) Formation and propagation of tau oligomeric seeds. Front. Neurol. 4:93. doi: 10.3389/fneur.2013.00093
Received: 12 June 2013; Paper pending published: 19 June 2013;
Accepted: 01 July 2013; Published online: 17 July 2013.
Edited by:
Naruhiko Sahara, University of Florida, USAReviewed by:
Naruhiko Sahara, University of Florida, USAIrving E. Vega, University of Puerto Rico-Rio Piedras Campus, Puerto Rico
Copyright: © 2013 Gerson and Kayed. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Rakez Kayed, George P. and Cynthia Woods Mitchell Center for Neurodegenerative Diseases, 301 University Blvd, Medical Research Building, Room 10.138C, Galveston, TX 77555-1045, USA e-mail:cmFrYXllZEB1dG1iLmVkdQ==