


- Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
In normal adult brain the microtubule associated protein (MAP) tau contains 2–3 phosphates per mol of the protein and at this level of phosphorylation it is a soluble cytosolic protein. The normal brain tau interacts with tubulin and promotes its assembly into microtubules and stabilizes these fibrils. In Alzheimer disease (AD) brain tau is three to fourfold hyperphosphorylated. The abnormally hyperphosphorylated tau binds to normal tau instead of the tubulin and this binding leads to the formation of tau oligomers. The tau oligomers can be sedimented at 200,000 × g whereas the normal tau under these conditions remains in the supernatant. The abnormally hyperphosphorylated tau is capable of sequestering not only normal tau but also MAP MAP1 and MAP2 and causing disruption of the microtubule network promoted by these proteins. Unlike Aβ and prion protein (PrP) oligomers, tau oligomerization in AD and related tauopathies is hyperphosphorylation-dependent; in vitro dephosphorylation of AD P-tau with protein phosphatase 2A (PP2A) inhibits and rehyperphosphorylation of the PP2A-AD P-tau with more than one combination of tau protein kinases promotes its oligomerization. In physiological assembly conditions the AD P-tau readily self-assembles into paired helical filaments. Missense tau mutations found in frontotemporal dementia apparently lead to tau oligomerization and neurofibrillary pathology by promoting its abnormal hyperphosphorylation. Dysregulation of the alternative splicing of tau that alters the 1:1 ratio of the 3-repeat: 4-repeat taus such as in Down syndrome, Pick disease, and progressive supranuclear palsy leads to the abnormal hyperphosphorylation of tau.
In Alzheimer disease (AD) the oligomer states of Aβ and tau pathologies are believed to cause the neurodegeneration. Oligomer is an intermediate stage between monomer and a large polymer. It consists of a relatively small and identifiable number of monomers, which is usually 3–10 in the case of most proteins. Unlike a polymer, if one of the monomers is removed from an oligomer, its chemical properties are altered. Protein oligomers may be formed by the polymerization of a number of monomers or the depolymerization of a large protein polymer. Protein polymerization is employed by the cell to perform several useful functions, such as neurofilaments and actin filaments serve as cytoskeleton of a neuron and maintain the cell shape. Microtubules that are polymers of tubulin facilitate axoplasmic flow, a vital function of a neuron. Some protein polymerization reactions are very efficient and almost all the protein in the cell is seen as polymers, as is the case with neurofilaments. In contrast, microtubule assembly and disassembly are extremely dynamic to meet the axoplasmic transport needs of a neuron. The oligomers of neurofilaments and microtubules are apparently very short-lived and are, to date, of no known deleterious consequence.
In AD, Aβ and, in the case of tau also in tauopathies, the protein polymerization is apparently employed as a detoxifying process to get rid of the toxic protein oligomers, which seem to stay in the diseased brain and have been isolated and studied. Tau oligomerization is increasingly being suspected as a prion-like phenomenon. This article, which is an update of our previous article on this subject (1), discusses the tau oligomers seen in AD brain and how they differ from Aβ and PrP oligomers.
In human brain tau is alternatively spliced into six isoforms and the ratio of the 3-repeat: 4-repeat protein is altered in different tauopathies. The alternative splicing of human tau pre-mRNA results in six molecular isoforms of the protein (2). These six tau isoforms differ in containing three (3R) or four (4R) microtubule binding repeats (R) of 31–32 amino acids in the carboxy-terminal half and one (1N) or two (2N) amino-terminal inserts (N) of 29 amino acids each; the extra repeat in 4R tau is the second repeat (R2) of 4R taus. This alternative splicing of tau pre-mRNA results in the expression of three 3R taus (0N3R, 1N3R, 2N3R) and three 4R taus (0N4R, 1N4R, 2N4R). The 2N4R tau is the largest size human brain tau with a total of 441 amino acids (tau441) in length. The smallest size tau isoform, which lacks both the two amino-terminal inserts and the extra microtubule binding repeat (0N3R; tau352) is the only isoform that is expressed in fetal human brain. Tau has little secondary structure; it is mostly random coil with β structure in the second and third microtubule binding repeats.
In a normal mature neuron almost all tau is bound to microtubules; tubulin is present in over 10-fold excess of tau. The concentration of tau in a neuron is ∼2 μM (3, 4) and it binds to microtubules at a kd (dissociation constant) of ∼100 nM (5). Overexpression of tau causes microtubule bundling in cultured cells. However, neither in AD nor in any tauopathy has microtubule bundling been reported.
Neurofibrillary degeneration not only is seen in AD and Down syndrome (DS) but also in a family of related neurodegenerative diseases called tauopathies. These include frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) caused by tau mutations, Pick disease, corticobasal degeneration, dementia pugilistica, and progressive supranuclear palsy. In every one of these tauopathies the neurofibrillary pathology is made up of abnormally hyperphosphorylated tau and these pathological changes in the neocortex are associated with dementia; in a large number of supranuclear palsy cases the tau pathology in the brain stem is associated with motor dysfunction.
Oligomerization of Tau and How it Differs from That of Aβ and PrP
In 1986 we discovered that not only Alzheimer neurofibrillary tangles were made up from abnormally hyperphosphorylated tau protein (6) but also the altered tau was present in AD brain cytosol and was responsible for the inhibition of microtubule assembly (7). In subsequent studies we showed that the cytosolic AD abnormally hyperphosphorylated tau (AD P-tau) sequestered some of the normal tau and sedimented at 200,000 × g, whereas most of the non-hyperphosphorylated tau from the same AD brains remained in the 200,000 × g supernatant (8, 9). The AD P-tau showed up as globular particles by negative stained electron microscopy (Figure 1). The sedimentable AD P-tau is increasingly being referred to as the oligomeric tau or granular tau (10). In situ demonstration of oligomeric tau seen immunohistochemically as amorphous aggregates in the neuronal cytoplasm was described at “stage 0” tangles for the first time by Bancher et al. (11). Biochemical analysis of AD P-tau sedimented from AD brain showed that it co-sedimented some of the non-hyperphosphorylated tau, suggesting that the AD P-tau oligomers are hetero-oligomers of hyperphosphorylated and non-hyperphosphorylated tau (8). Furthermore, normal tau was found to co-aggregate with and promote the aggregation of AD P-tau into filaments (12). As much as 40% of abnormally hyperphosphorylated tau in AD brain is seen as AD P-tau (8).
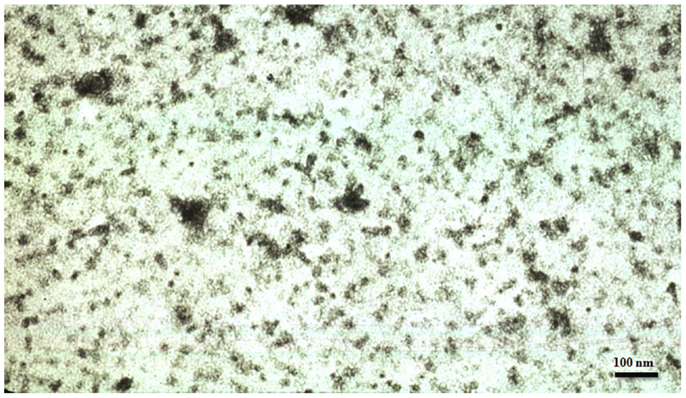
Figure 1. Electron micrograph showing tau oligomers from an Alzheimer disease brain negatively stained with phosphotungstic acid.
Unlike normal tau which binds to tubulin and promotes its assembly into microtubules, the AD P-tau, instead of interacting with tubulin, binds to normal tau as well as MAP1 and MAP2, and causes depolymerization of microtubules (9, 12, 13). In vitro hyperphosphorylation of tau revealed that the oligomeric tau was an intermediate stage between monomeric and filamentous state because at 4–6 mol phosphate/mole protein it became oligomeric and microtubule-assembly inhibitory whereas further hyperphosphorylation made it polymerize into filaments. Neither the in vitro formed hyperphosphorylated tau filaments nor PHF isolated from AD brains had any detectable effect on tau-promoted assembly of microtubules (14– 16). While normal tau promoted GTP binding to tubulin and its assembly into microtubules, the AD P-tau inhibited this activity. AD-PHF had no effect on GTP binding but on in vitro dephosphorylation it promoted GTP binding to tubulin (17). On dephosphorylation with protein phosphatase 2A (PP2A) the AD P-tau oligomers are converted into normal-like non-sedimentable protein that, like normal tau, promotes microtubule assembly (9, 12, 18). PP2A was also found to dissociate Alzheimer neurofibrillary tangles, releasing protein which behaved like normal tau in promoting microtubule assembly (19). Thus, the AD P-tau oligomerization is unique because it is solely induced by abnormal hyperphosphorylation and is reversible on dephosphorylation of the protein (20).
In the AD field the interest in oligomers started with the initial report of Lambert et al. (21) who showed that diffusible, non-fibrillar ligands from Aβ1–42 were potent central nervous system toxins. Though Aβ oligomers are toxic, in contrast to tau oligomerization, they are formed by the strong hydrophobic nature of this peptide and this process is not initiated or promoted by phosphorylation. Similarly, the PrP oligomers are formed at acidic pH and on removal of denaturants such as sodium dodecyl sulfate or salt from the protein solution (22, 23). Unlike AD P-tau and Aβ1–42 oligomers, the PrP filaments are the infective state and their depolymerization into oligomers results in the loss of the infectivity (24). Most recently PrP cellular has been reported to promote the Aβ oligomerization (25).
Role of O-GlcNAcylation in Tau Oligomerization and Neurodegeneration
In addition to phosphorylation, tau is also modified by O-GlcNAcylation, a dynamic protein posttranslational modification, by which O-linked β-N-acetylglucosamine (O-GlcNAc) is transferred enzymatically from a UDP-GlcNAc donor to the hydroxyl group of serine or threonine residues of proteins. In contrast to glycosylation of secreted and membrane proteins, which occurs in the endoplasmic reticulum and Golgi apparatus, O-GlcNAcylation modifies nucleocytoplasmic proteins and is more like protein phosphorylation (26). O-GlcNAcylation and phosphorylation sometimes occur at identical or proximal sites of a protein and thus are reciprocal to each other. The crosstalk between O-GlcNAcylation and phosphorylation has been implicated to be essential for the control of vital cellular processes and for understanding the mechanisms of certain diseases (27, 28). O-GlcNAcylation also serves as a sensor of intracellular glucose metabolism (29), because the UDP-GlcNAc donor for O-GlcNAcylation is formed from glucose metabolism via the hexosamine biosynthetic pathway.
Tau is highly modified by O-GlcNAc, on average, with four O-GlcNAc groups per tau molecule at more than 12 serine/threonine residues (30, 31). Five O-GlcNAcylation sites (Thr123, Ser208, Ser238, Ser400, and one site at Ser409, Ser412, or Ser413) have been mapped to date (32– 34). We previously demonstrated that inhibition of O-GlcNAcylation leads to hyperphosphorylation of tau in cultured cells and in rat brain slices (31). Experimental reduction of brain glucose metabolism leads to decreased O-GlcNAcylation and increased phosphorylation of tau in vivo (27, 35), and inhibition of protein O-GlcNAcylation induces hyperphosphorylation of tau in rat brain (27). Furthermore, we discovered that the global O-GlcNAcylation of proteins, especially of tau, is decreased, which likely results from impaired brain glucose metabolism, and that the decrease in O-GlcNAcylation correlates to hyperphosphorylation of tau in AD brain (27). Furthermore, hyperphosphorylated tau purified from AD brains contains approximately five times less O-GlcNAc than normal tau (27). Therefore, we postulate that tau pathology and neurodegeneration can be caused by impaired brain glucose metabolism via the down-regulation of tau O-GlcNAcylation in AD (27).
O-GlcNAcylation may also inhibit tau oligomerization directly. The fourth microtubule binding repeat of tau self-aggregates at a slower rate in vitro when it is modified by O-GlcNAc at Ser356 than the unmodified counterpart, as determined by turbidity, precipitation assay, and electron microscopy (36). A recent study showed that O-GlcNAcylation inhibits tau aggregation in rodents (37). O-GlcNAcylation also modulates proteotoxicity in C. elegans models of human neurodegenerative diseases (38, 39). Therefore, decreased O-GlcNAcylation may promote tau-mediated neurodegeneration through promoting tau oligomerization directly and also indirectly by inducing its abnormal hyperphosphorylation.
Abnormal Hyperphosphorylation of Tau Causes Neurodegeneration and Cognitive Impairment
Protein phosphatase 2A accounts for ∼70% of the total tau phosphatase activity in the brain (40). A cause of the abnormal hyperphosphorylation of tau in AD and adults with DS is a decrease in the brain PP2A activity (41– 43). PP2A activity is negatively regulated by two inhibitor proteins, I1PP2A and I2PP2A in a substrate-specific manner (44, 45). Both I1PP2A and I2PP2A inhibit PP2A activity toward AD hyperphosphorylated tau (46) and these inhibitors are predominantly localized in the hippocampus and the cerebellum (47). I1PP2A, which is also known as PHAP-1, is a 239 amino acid long cytoplasmic protein (48). I2PP2A, also known as SETα, PHAP-II, and TAF1β, is primarily a nuclear protein of 277 amino acids in length with an apparent molecular weight of 39 kDa on SDS-PAGE (45, 49, 50). mRNA and protein expression levels of both I1PP2A and I2PP2A are selectively increased in the affected areas of AD brain. I2PP2A, which is a 39 kDa and a primarily nuclear protein, is selectively cleaved at N175 into an amino-terminal (I2NTF) and a carboxy-terminal (I2CTF) fragment and translocated from the neuronal nucleus to the cytoplasm in AD brain (51). Both I2NTF and I2CTF interact with the PP2A catalytic subunit PP2Ac and inhibit its activity toward hyperphosphorylated tau (52). Transduction of the brains of newborn rats with adeno associated virus serotype 1 vector carrying human I2CTF (53) or I2NTF and I2CTF transgenes was found to induce AD-like abnormal hyperphosphorylation and aggregation of tau, a loss of neuronal plasticity, and cognitive impairment in these animals at 5–12 months post-infection (54); however, no neurofibrillary tangles or Aβ plaques were detected in the brains of AAV1-I2NTF-CTF rats up until 13 months. These findings suggest a deleterious role of the abnormally hyperphosphorylated oligomeric tau.
The inhibitory activity of the non-fibrillized abnormally hyperphosphorylated tau has been confirmed in yeast, drosophila, and in mouse models that express human brain tau. The expression of the longest human brain tau (2N4R tau) in yeast produces pathological phosphoepitopes, assumes a pathological conformation, and forms aggregates. These processes are modulated by yeast kinases Mds1 and Pho85, orthologs of GSK-3β and cdk5 (55, 56). In yeast the aggregation of tau increases with increasing hyperphosphorylation and the mobility in SDS-PAGE retards. The hyperphosphorylated tau isolated from the stably transfected yeast is able to assemble into filaments, and nucleate the assembly of the normal non-phosphorylated tau. These yeast studies, like those carried out previously using AD P-tau, suggest that the hyperphosphorylated tau works as a nucleation factor that initiates and promotes the aggregation of tau (12, 15).
In wild-type human tau- and mutated human tau-transgenic Drosophila, the accumulation of the abnormally phosphorylated tau in the absence of its fibrillization into neurofibrillary tangles leads to neurodegeneration (57). In a P301L tau inducible transgenic mouse model, cognitive improvement was observed when expression of human tau, which became abnormally hyperphosphorylated, was suppressed although neurofibrillary tangles continued to form, suggesting that the accumulation of the cytosolic abnormally hyperphosphorylated tau, and not its aggregation, was apparently involved in behavioral impairment in these animals (58). Reduction of soluble Aβ and soluble abnormally hyperphosphorylated tau, but not soluble Aβ alone, was found to ameliorate cognitive decline in 3xTg mice that express both plaque and tangle pathologies (59). Furthermore, in vitro dephosphorylation of neurofibrillary tangles disaggregates filaments and, as a result, the tau released behaves like normal protein in promoting microtubule assembly (19).
Hyperphosphorylation of tau, though not to the same level as in AD, is not only associated with the disease as in tauopathies, but is also employed by the neuron to down regulate its activity transiently and reversibly where required. For instance, during development the level of tubulin in the brain is at its highest, i.e., almost 33% of total cytosolic protein, which is almost 1.5-fold the critical concentration of 4 mg/ml tubulin required for its polymerization into microtubules (60). Probably to avoid microtubule bundling, the fetal tau is transiently hyperphosphorylated during development. However, the level of hyperphosphorylation of tau in fetal brain is far less than that seen in AD brain. Similarly, anesthesia and hypothermia induced by hibernation in animals induces transient hyperphosphorylation of tau (61– 64). The molecular mechanism of the transient hyperphosphorylation of tau observed during development is, at present, not understood. However, during hypothermia the activity of PP2A is transiently and reversibly reduced and is believed to cause the hyperphosphorylation of tau (62, 63). In AD and DS the decrease in brain PP2A activity apparently involves different molecular mechanisms, and occurs in a non-transient and irreversible manner (41– 43). It is the non-reversible nature of the abnormal hyperphosphorylation of tau in AD, DS, and related tauopathies which results in an involuntary slowing down of neuronal activity and a consequent chronic progressive neurodegeneration and its clinical phenotype, the dementia.
There is approximately as much tau in the somatodendritic compartment as in the axon (65). In the somatodendritic compartment tau is associated with rough endoplasmic reticulum and Golgi apparatus (7, 8, 66, 67). The abnormal hyperphosphorylation of tau and its accumulation in the somatodendritic compartment in AD might have been responsible for the morphological alterations of the RER and the Golgi apparatus and the abnormal N-glycosylation of tau in AD (68– 71). In AD brain abnormally hyperphosphorylated tau, in addition to forming neurofibrillary tangles, is associated with granulovacuolar changes (6, 72– 74). Overexpression of tau, which results in its hyperphosphorylation, has been found to induce fragmentation of Golgi both in neuronal cultures and in neurons in JNPL3 P301L tau-transgenic mice (66). In P301S tau-transgenic mice, which show abnormal hyperphosphorylation of tau, a selective decrease in mitochondria and RER has been observed (75). The chronic accumulation of the hyperphosphorylated tau as a misfolded protein in the ER could cause neurodegeneration due to protracted ER stress (76). Hyperphosphorylation of tau might also be involved in neurodegeneration through alterations of RER and Golgi and a consequent reduction in RER and mitochondria.
In addition to abnormal hyperphosphorylation, truncation of tau has been found in neurofibrillary tangles in AD and in mutated tau overexpression transgenic mouse models [e.g., (77– 82)]. Of all the proteases that can cleave tau, the role of caspases has been studied the most (83, 84). Caspase 3 and caspase 6 cleave tau at D421 and D13, respectively, and treatment with Aβ can induce the D421 cleavage in cultured neurons (78, 80, 81). Truncation of tau, along with its hyperphosphorylation, promotes its aggregation into fibrils (85, 86). Although only a small fraction of tau is truncated in AD, the truncated protein can apparently recruit the full-length protein to co-aggregate with it in both tau-transgenic rat and mouse models (87, 88). To date, the bulk of the evidence suggests the soluble hyperphosphorylated tau is neurotoxic and upstream of truncation and aggregation of this protein into neurofibrillary tangles [e.g., (89, 90)].
Role of Mutations and Alternative Splicing of Tau in Neurodegeneration
In FTDP-17 several mutations in tau co-segregate with the disease (91– 93). Four of these missense mutations, G272V, P301L, V337M, and R406W, which have been most studied to date, all make tau a preferable substrate for abnormal hyperphosphorylation in vitro (94). Some of the tauopathies are associated with altered alternate splicing of tau. In normal human brain the 3-repeat and 4-repeat taus are expressed in 1:1 ratio.
In some of the FTDP-17 mutations, i.e., tauK257T (95), tauG272V (96), tauΔK280 (97), tauE10+19, and tauE10+29 (98), and in Pick disease most of the tau is 3R isoforms due to the exclusion of axon 10 which codes for the second microtubule binding repeat (R2). In contrast, in other FTDP-17 mutations, cortical basal degeneration and progressive supranuclear palsy, most of the tau is 4R (99, 100).
How the imbalance of 3R tau/4R tau leads to neurofibrillary degeneration and dementia is currently not understood. The 4R taus bind microtubules more readily than 3R taus. Thus, a change in 3R:4R ratio of 1:1 in tauopathies results in free tau that is unbound to microtubules and free tau becomes a favorable substrate for abnormal hyperphosphorylation (101). In DS brain an increase in 3R:4R ratio combined with an extra copy of Dyrk1A, which can hyperphosphorylate tau, results in tau pathology during the fourth decade of life which is almost two decades earlier than the average age of onset of AD (102, 103).
Hyperphosphorylation by brain protein kinases induces the self-assembly of all six human brain tau isoforms into tangles of PHF/SF under physiological conditions of protein concentration, ionic strength, pH, temperature, reducing conditions, and the absence of any cofactor (9). The hyperphosphorylation of tau is catalyzed by one or more combinations of the proline-directed protein kinases (PDPKs) and non-PDPKs. Phosphorylation of tau by non-PDPKs generally primes taus for hyperphosphorylation by PDPKs (20, 104– 106). Tau isoforms in vitro might be phosphorylated differentially. 2N4R tau is a more favorable substrate for phosphorylation by rat brain protein kinases and is phosphorylated faster and to a higher extent than 2N3R tau at Thr181, Ser199, Ser202, Thr205, Thr212, Ser214, Thr217, Thr231, Ser235, Ser262, Ser396, Ser404, and Ser422 (94). The differential phosphorylation of 3R and 4R taus involves a combination of non-PDPKs and PDPKs because, GSK-3β alone phosphorylates tau isoforms similarly (107). Pseudophosphorylation of seven GSK-3β phosphorylation sites S199, S202, T205, T231, S235, S396, and S404, affects the aggregation of tau isoforms differently; the pseudophosphorylation at these seven sites was found to enhance arachidonic acid-induced polymerization of 0N4R tau while greatly inhibiting the aggregation of the 3R isoforms (108). Thus, phosphorylation generated by the same set of kinases could be sufficient to increase the propensity of some isoforms to aggregate while reducing the aggregation of others, resulting in the differential isoform inclusion in pathological tau aggregates (108).
Aggregation of tau isoforms is affected by the type of inducer for aggregation used. Arachidonic acid induces 4R tau to polymerize to a greater extent than 3R tau (107). 0N tau requires higher concentration of arachidonic acid to get maximal polymerization. The concentration of arachidonic acid for reaching a maximal polymerization of 1N tau and 2N tau were reported to be similar, suggesting addition of exon 3 containing isoforms does not further reduce inducer concentrations needed for maximal polymerization (107). Similar results were obtained for the heparin induction of tau isoform polymerization (107). The 2N4R tau required less heparin inducer for maximal polymerization than 1N4R and 0N4R taus. Aggregation of six tau isoforms by thiazine red inducer was also reported in a tau isoform-dependent manner. Tau exons 2 and 10 were found to promote aggregation, whereas exon 3 depressed it with its efficacy dependent on the presence or absence of a fourth microtubule binding repeat (109).
Alzheimer disease P-tau sequesters normal tau, MAP1, and MAP2 and disassembles microtubules and that the dephosphorylation of AD P-tau eliminates this toxic property (9, 13). Tau isoforms bind to AD P-tau deferentially. The binding of AD P-tau to 4R tau tends to be greater than to the corresponding 3R tau and its binding to normal human recombinant tau was found to be 2N4R > 1N4R > 0N4R and 1N4R > 1N3R > 0N3R (110). AD P-tau interacts preferentially with the tau isoforms that have the amino-terminal inserts and four microtubule binding domain repeats and that hyperphosphorylation of tau appears to be sufficient to acquire AD P-tau characteristics. Thus, lack of amino-terminal inserts and extra microtubule binding domain repeat in fetal human brain might be protective from Alzheimer’s neurofibrillary degeneration.
Conclusion
In conclusion, in AD and related tauopathies the abnormal hyperphosphorylation of tau promotes its oligomerization (Figure 2). The tau oligomers sequester normal tau as well as MAP1 and MAP2 and can be separated from normal tau by sedimentation at 200,000 × g. The abnormal hyperphosphorylation of tau seen in AD is different from the normal and from the transient hyperphosphorylation of this protein that occurs during development, anesthesia, or hypothermia. The oligomeric cytosolic AD P-tau probably causes neurodegeneration by sequestering normal MAPs and disrupting the microtubule network. Tau mutations found in frontotemporal dementia may cause neurodegeneration through promoting abnormal hyperphosphorylation of tau. AD P-tau self-assembles into PHF/SF, forming neurofibrillary tangles. Tau truncation found in AD brain promotes its self-assembly into PHF/SF. Unlike AD P-tau, the tangles neither show any detectable activity to sequester normal MAPs nor inhibit microtubule assembly. Inhibition of abnormal hyperphosphorylation of tau offers a promising therapeutic target for AD and related tauopathies. Animal models that recapitulate various disease mechanisms seen in AD and related tauopathies are no less valuable for preclinical studies for drug development than transgenic mouse and rat models in which one or more mutated human proteins are overexpressed to produce Aβ plaques and/or tau neurofibrillary tangles.
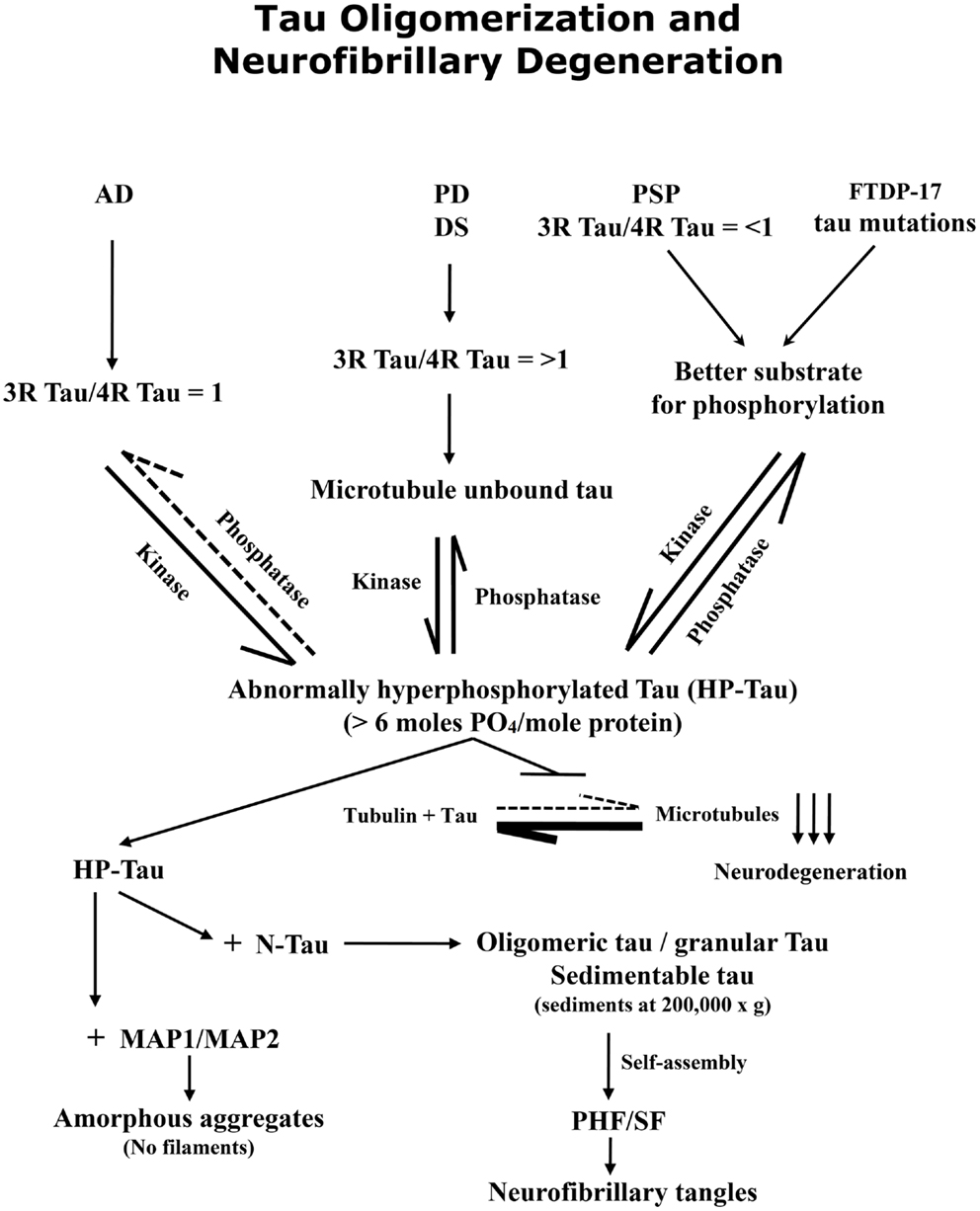
Figure 2. Abnormal hyperphosphorylation of tau promotes its oligomerization and self-assembly into paired helical filaments, forming neurofibrillary tangles. A protein phosphorylation/dephosphorylation imbalance apparently caused by a decrease in protein phosphatase 2A (PP2A) activity leads to abnormal hyperphosphorylation of tau in AD brain. The abnormally hyperphosphorylated tau binds to normal tau (and not to tubulin) and this sequestration leads to the disruption of microtubules and the formation of oligomers which can be sedimented at 200,000 × g; the tau oligomers show up as granular structures by negative stain electron microscopy. The abnormally hyperphosphorylated tau isolated from AD brain cytosol readily self-assembles into paired helical filaments in vitro under polymerizing conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Ms. Janet Murphy for secretarial assistance. Dr. Ezzat El-Akkad helped prepare figures. Studies in our laboratories were supported in part by the New York State Office of People with Developmental Disabilities; NIH grants AG019158, TW008744, and AG27429; Alzheimer Association grants IIRG-00-2002, HRG-05-13095, and NIRG-08-91126 and Zenith Award ZEN-12-231433; ADDF grant 20121203 from Alzheimer’s Drug Discovery Foundation.
References
1. Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res (2010) 7:656–64. doi:10.2174/156720510793611592
2. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron (1989) 3:519–26. doi:10.1016/0896-6273(89)90210-9
3. Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol (1991) 115:717–30. doi:10.1083/jcb.115.3.717
4. Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem (1992) 59:750–3. doi:10.1111/j.1471-4159.1992.tb09432.x
5. Ackmann M, Wiech H, Mandelkow E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J Biol Chem (2000) 275:30335–43. doi:10.1074/jbc.M002590200
6. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A (1986) 83:4913–7. doi:10.1073/pnas.83.13.4913
7. Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet (1986) 2:421–6. doi:10.1016/S0140-6736(86)92134-3
8. Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem (1993) 268:24374–84.
9. Alonso AD, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A (1994) 91:5562–6. doi:10.1073/pnas.91.12.5562
10. Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, et al. Granular tau oligomers as intermediates of tau filaments. Biochemistry (2007) 46:3856–61. doi:10.1021/bi061359o
11. Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res (1989) 477:90–9. doi:10.1016/0006-8993(89)91396-6
12. Alonso AD, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med (1996) 2:783–7. doi:10.1038/nm0796-783
13. Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A (1997) 94:298–303. doi:10.1073/pnas.94.1.298
14. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A (2001) 98:6923–8. doi:10.1073/pnas.121119298
15. Alonso AD, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A (2006) 23:8864–9. doi:10.1073/pnas.0603214103
16. Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol (2007) 113:501–11. doi:10.1007/s00401-007-0207-8
17. Khatoon S, Grundke-Iqbal I, Iqbal K. Guanosine triphosphate binding to beta-subunit of tubulin in Alzheimer’s disease brain: role of microtubule-associated protein tau. J Neurochem (1995) 64:777–87. doi:10.1046/j.1471-4159.1995.64020777.x
18. Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res (1996) 38:200–8. doi:10.1016/0169-328X(95)00316-K
19. Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem (1995) 270:4854–60. doi:10.1074/jbc.270.9.4854
20. Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci (2007) 25:59–68. doi:10.1111/j.1460-9568.2006.05226.x
21. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A (1998) 95:6448–53. doi:10.1073/pnas.95.11.6448
22. Post K, Pitschke M, Schafer O, Wille H, Appel TR, Kirsch D, et al. Rapid acquisition of beta-sheet structure in the prion protein prior to multimer formation. Biol Chem (1998) 379:1307–17. doi:10.1515/bchm.1998.379.11.1307
23. Swietnicki W, Morillas M, Chen SG, Gambetti P, Surewicz WK. Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry (2000) 39:424–31. doi:10.1021/bi991967m
24. Riesner D, Kellings K, Post K, Wille H, Serban H, Groth D, et al. Disruption of prion rods generates 10-nm spherical particles having high alpha-helical content and lacking scrapie infectivity. J Virol (1996) 70:1714–22.
25. Younan ND, Sarell CJ, Davies P, Brown DR, Viles JH. The cellular prion protein traps Alzheimer’s Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J (2013) 27:1847–58. doi:10.1096/fj.12-222588
26. Butkinaree C, Park K, Hart GW. O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta (2010) 1800:96–106. doi:10.1016/j.bbagen.2009.07.018
27. Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain (2009) 132:1820–32. doi:10.1093/brain/awp099
28. Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci (2010) 123:13–22. doi:10.1242/jcs.053678
29. Zachara NE, Hart GW. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophys Acta (2004) 1673:13–28. doi:10.1016/j.bbagen.2004.03.016
30. Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem (1996) 271:28741–4. doi:10.1074/jbc.271.46.28741
31. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A (2004) 101:10804–9. doi:10.1073/pnas.0400348101
32. Wang Z, Udeshi ND, O’Malley M, Shabanowitz J, Hunt DF, Hart GW. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol Cell Proteomics (2010) 9:153–60. doi:10.1074/mcp.M900268-MCP200
33. Smet-Nocca C, Broncel M, Wieruszeski JM, Tokarski C, Hanoulle X, Leroy A, et al. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol Biosyst (2011) 7:1420–9. doi:10.1039/c0mb00337a
34. Yuzwa SA, Yadav AK, Skorobogatko Y, Clark T, Vosseller K, Vocadlo DJ. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids (2011) 40:857–68. doi:10.1007/s00726-010-0705-1
35. Li X, Lu F, Wang JZ, Gong CX. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci (2006) 23:2078–86. doi:10.1111/j.1460-9568.2006.04735.x
36. Yu CH, Si T, Wu WH, Hu J, Du JT, Zhao YF, et al. O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau. Biochem Biophys Res Commun (2008) 375:59–62. doi:10.1016/j.bbrc.2008.07.101
37. Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol (2012) 8:393–9. doi:10.1038/nchembio.797
38. Gong CX, Liu F, Iqbal K. O-GlcNAc cycling modulates neurodegeneration. Proc Natl Acad Sci U S A (2012) 109:17319–20. doi:10.1073/pnas.1215395109
39. Wang P, Lazarus BD, Forsythe ME, Love DC, Krause MW, Hanover JA. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc Natl Acad Sci U S A (2012) 109:17669–74. doi:10.1073/pnas.1205748109
40. Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci (2005) 22:1942–50. doi:10.1111/j.1460-9568.2005.04391.x
41. Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem (1993) 61:921–7. doi:10.1111/j.1471-4159.1993.tb03603.x
42. Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem (1995) 65:732–8. doi:10.1046/j.1471-4159.1995.65020732.x
43. Liang Z, Liu F, Iqbal K, Grundke-Iqbal I, Wegiel J, Gong CX. Decrease of protein phosphatase 2A and its association with accumulation and hyperphosphorylation of tau in Down syndrome. J Alzheimers Dis (2008) 13:295–302.
44. Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry (1995) 34:1988–96. doi:10.1021/bi00006a020
45. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem (1996) 271:11059–62. doi:10.1074/jbc.271.19.11059
46. Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett (2005) 579:363–72. doi:10.1016/j.febslet.2004.11.097
47. Tanimukai H, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A: topography and subcellular localization. Brain Res Mol Brain Res (2004) 126:146–56. doi:10.1016/j.molbrainres.2004.04.003
48. Vaesen M, Barnikol-Watanabe S, Gotz H, Awni LA, Cole T, Zimmermann B, et al. Purification and characterization of two putative HLA class II associated proteins: PHAPI and PHAPII. Biol Chem Hoppe Seyler (1994) 375:113–26. doi:10.1515/bchm3.1994.375.2.113
49. von Lindern M, Van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol Cell Biol (1992) 12:3346–55.
50. Matsuoka K, Taoka M, Satozawa N, Nakayama H, Ichimura T, Takahashi N, et al. A nuclear factor containing the leucine-rich repeats expressed in murine cerebellar neurons. Proc Natl Acad Sci U S A (1994) 91:9670–4. doi:10.1073/pnas.91.21.9670
51. Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol (2005) 166:1761–71. doi:10.1016/S0002-9440(10)62486-8
52. Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I(2)(PP2A)/SET. FEBS Lett (2011) 585:2653–9. doi:10.1016/j.febslet.2011.07.020
53. Wang X, Blanchard J, Kohlbrenner E, Clement N, Linden RM, Radu A, et al. The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J (2010) 24:4420–32. doi:10.1096/fj.10-158477
54. Bolognin S, Blanchard J, Wang X, Basurto-Islas G, Tung YC, Kohlbrenner E, et al. An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol (2012) 123:133–51. doi:10.1007/s00401-011-0908-x
55. Vandebroek T, Vanhelmont T, Terwel D, Borghgraef P, Lemaire K, Snauwaert J, et al. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry (2005) 44:11466–75. doi:10.1021/bi0506775
56. Vandebroek T, Terwel D, Vanhelmont T, Gysemans M, Van Haesendonck C, Engelborghs Y, et al. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J Biol Chem (2006) 281:25388–97. doi:10.1074/jbc.M602792200
57. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science (2001) 293:711–4. doi:10.1126/science.1062382
58. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science (2005) 309:476–81. doi:10.1126/science.1113694
59. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, Laferla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem (2006) 281:39413–23. doi:10.1074/jbc.M608485200
60. Schmitt H, Gozes I, Littauer UZ. Decrease in levels and rates of synthesis of tubulin and actin in developing rat brain. Brain Res (1977) 121:327–42. doi:10.1016/0006-8993(77)90155-X
61. Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van Der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci (2003) 23:6972–81.
62. Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci (2007) 27:3090–7. doi:10.1523/JNEUROSCI.4854-06.2007
63. Su B, Wang X, Drew KL, Perry G, Smith MA, Zhu X. Physiological regulation of tau phosphorylation during hibernation. J Neurochem (2008) 105:2098–108. doi:10.1111/j.1471-4159.2008.05294.x
64. Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX. Anesthesia induces phosphorylation of tau. J Alzheimers Dis (2009) 16:619–26. doi:10.3233/JAD-2009-1003
65. Khatoon S, Grundke-Iqbal I, Iqbal K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett (1994) 351:80–4. doi:10.1016/0014-5793(94)00829-9
66. Liazoghli D, Perreault S, Micheva KD, Desjardins M, Leclerc N. Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am J Pathol (2005) 166:1499–514. doi:10.1016/S0002-9440(10)62366-8
67. Farah CA, Perreault S, Liazoghli D, Desjardins M, Anton A, Lauzon M, et al. Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton (2006) 63:710–24. doi:10.1002/cm.20157
68. Gray EG, Paula-Barbosa M, Roher A. Alzheimer’s disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol (1987) 13:91–110. doi:10.1111/j.1365-2990.1987.tb00174.x
69. Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nat Med (1996) 2:871–5. doi:10.1038/nm0896-871
70. Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol (2003) 32:1091–105. doi:10.1023/B:NEUR.0000021904.61387.95
71. Perreault S, Bousquet O, Lauzon M, Paiement J, Leclerc N. Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J Neuropathol Exp Neurol (2009) 68:503–14. doi:10.1097/NEN.0b013e3181a1fc49
72. Ikegami K, Kimura T, Katsuragi S, Ono T, Yamamoto H, Miyamoto E, et al. Immunohistochemical examination of phosphorylated tau in granulovacuolar degeneration granules. Psychiatry Clin Neurosci (1996) 50:137–40. doi:10.1111/j.1440-1819.1996.tb01678.x
73. Ghoshal N, Smiley JF, Demaggio AJ, Hoekstra MF, Cochran EJ, Binder LI, et al. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am J Pathol (1999) 155:1163–72. doi:10.1016/S0002-9440(10)65219-4
74. Lagalwar S, Berry RW, Binder LI. Relation of hippocampal phospho-SAPK/JNK granules in Alzheimer’s disease and tauopathies to granulovacuolar degeneration bodies. Acta Neuropathol (2007) 113:63–73. doi:10.1007/s00401-006-0159-4
75. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron (2007) 53:337–51. doi:10.1016/j.neuron.2007.01.010
76. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov (2008) 7:1013–30. doi:10.1038/nrd2755
77. Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J (1993) 12:365–70.
78. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A (2003) 100:10032–7. doi:10.1073/pnas.1630428100
79. Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, Leblanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol (2004) 165:523–31. doi:10.1016/S0002-9440(10)63317-2
80. Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci (2004) 24:7895–902. doi:10.1523/JNEUROSCI.1988-04.2004
81. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest (2004) 114:121–30. doi:10.1172/JCI20640
82. Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol (2008) 67:470–83. doi:10.1097/NEN.0b013e31817275c7
83. Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging (2005) 26:1015–22. doi:10.1016/j.neurobiolaging.2004.09.019
84. Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans (2010) 38:955–61. doi:10.1042/BST0380955
85. Cho JH, Johnson GV. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem (2004) 279:54716–23. doi:10.1074/jbc.M403364200
86. Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, et al. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett (2006) 580:3582–8. doi:10.1016/j.febslet.2006.05.029
87. Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci (2008) 28:737–48. doi:10.1523/JNEUROSCI.2824-07.2008
88. Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol (2009) 118:71–86. doi:10.1007/s00401-009-0499-y
89. Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, et al. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol (2008) 172:123–31. doi:10.2353/ajpath.2008.070627
90. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature (2010) 464:1201–4. doi:10.1038/nature08890
91. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature (1998) 393:702–5. doi:10.1038/31508
92. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol (1998) 43:815–25. doi:10.1002/ana.410430617
93. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A (1998) 95:7737–41. doi:10.1073/pnas.95.13.7737
94. Alonso AD, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem (2004) 279:34873–81. doi:10.1074/jbc.M405131200
95. Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, et al. Tau gene mutation K257T causes a tauopathy similar to Pick’s disease. J Neuropathol Exp Neurol (2000) 59:990–1001.
96. Bronner IF, Ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, et al. Hereditary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain (2005) 128:2645–53. doi:10.1093/brain/awh591
97. van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, et al. The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol (2007) 66:17–25. doi:10.1097/nen.0b013e31802c39a4
98. Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, et al. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain (2003) 126:814–26. doi:10.1093/brain/awg090
99. Yoshida M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology (2006) 26:457–70. doi:10.1111/j.1440-1789.2006.00743.x
100. Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener (2008) 3:8. doi:10.1186/1750-1326-3-8
101. Sengupta A, Grundke-Iqbal I, Iqbal K. Regulation of phosphorylation of tau by protein kinases in rat brain. Neurochem Res (2006) 31:1473–80. doi:10.1007/s11064-006-9205-9
102. Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J (2008) 22:3224–33. doi:10.1096/fj.07-104539
103. Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem (2008) 283:28660–9. doi:10.1074/jbc.M802645200
104. Singh TJ, Zaidi T, Grundke-Iqbal I, Iqbal K. Modulation of GSK-3-catalyzed phosphorylation of microtubule-associated protein tau by non-proline-dependent protein kinases. FEBS Lett (1995) 358:4–8. doi:10.1016/0014-5793(94)01383-C
105. Singh TJ, Zaidi T, Grundke-Iqbal I, Iqbal K. Non-proline-dependent protein kinases phosphorylate several sites found in tau from Alzheimer disease brain. Mol Cell Biochem (1996) 154:143–51. doi:10.1007/BF00226782
106. Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys (1998) 357:299–309. doi:10.1006/abbi.1998.0813
107. Voss K, Gamblin TC. GSK-3beta phosphorylation of functionally distinct tau isoforms has differential, but mild effects. Mol Neurodegener (2009) 4:18. doi:10.1186/1750-1326-4-18
108. Combs B, Voss K, Gamblin TC. Pseudohyperphosphorylation has differential effects on polymerization and function of tau isoforms. Biochemistry (2011) 50:9446–56. doi:10.1021/bi2010569
109. Zhong Q, Congdon EE, Nagaraja HN, Kuret J. Tau isoform composition influences rate and extent of filament formation. J Biol Chem (2012) 287:20711–9. doi:10.1074/jbc.M112.364067
Keywords: microtubule associated protein tau, abnormal hyperphosphorylation of tau, O-GlcNAcylation of tau, protein phosphatase 2A, alternate splicing of tau, Alzheimer neurofibrillary degeneration, Alzheimer disease, tauopathies
Citation: Iqbal K, Gong C-X and Liu F (2013) Hyperphosphorylation-induced tau oligomers. Front. Neurol. 4:112. doi: 10.3389/fneur.2013.00112
Received: 21 May 2013; Paper pending published: 24 June 2013;
Accepted: 23 July 2013; Published online: 15 August 2013.
Edited by:
Jesus Avila, Centro de Biología Molecular Severo Ochoa CSIC UAM, SpainReviewed by:
Michal Novak, Slovak Academy of Sciences, SlovakiaNicole Leclerc, Université de Montréal, Canada
Copyright: © 2013 Iqbal, Gong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalid Iqbal, Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, NY 10314, USA e-mail:a2hhbGlkLmlxYmFsLmlickBnbWFpbC5jb20=