

- 1Department of Psychology, Queens College, City University of New York, New York, NY, USA
- 2Department of Neurology, University of Southern California, Los Angeles, CA, USA
- 3Division of Biokinesiology and Physical Therapy, University of Southern California, Los Angeles, CA, USA
Motor dysfunction in Parkinson’s disease is believed to arise primarily from pathophysiology in the dorsal striatum and its related corticostriatal and thalamostriatal circuits during progressive dopamine denervation. One function of these circuits is to provide a filter that selectively facilitates or inhibits cortical activity to optimize cortical processing, making motor responses rapid and efficient. Corticostriatal synaptic plasticity mediates the learning that underlies this performance-optimizing filter. Under dopamine denervation, corticostriatal plasticity is altered, resulting in aberrant learning that induces inappropriate basal ganglia filtering that impedes rather than optimizes cortical processing. Human imaging suggests that increased cortical activity may compensate for striatal dysfunction in PD patients. In this Perspective article, we consider how aberrant learning at corticostriatal synapses may impair cortical processing and learning and undermine potential cortical compensatory mechanisms. Blocking or remediating aberrant corticostriatal plasticity may protect cortical function and support cortical compensatory mechanisms mitigating the functional decline associated with progressive dopamine denervation.
As a primary site of dopamine denervation in Parkinson’s disease (PD) (1–3), pathophysiology in the dorsolateral striatum (DLS, equivalent to the posterior putamen in primates) is widely believed to play a central role in motor symptoms associated with disease progression. In the healthy brain, motor performance relies on the interaction between automatic (habit) and goal-directed (volitional) control of movement (4). Impairment in DLS function arising from denervation may induce degradation of automatic, implicit control of motor movements, and a compensatory shift to goal-directed, cortical control (4, 5). Accumulating imaging studies demonstrate that subjects with PD exhibit altered patterns of cortical activation [reviewed in Ref. (6, 7)]. Although pathophysiological changes related to cortical pathology may also contribute, it is believed that altered cortical activity reflects compensatory cortical circuit changes related to striatal dysfunction observed in early stages of disease progression (7–14). While the focus here is on cortical compensation, alterations in other circuit activity, including cerebellar circuits (15, 16), may also play an important compensatory role in striatal dysfunction.
In this brief perspective, we consider the relationship between the DLS and cortical function and outline a hypothesis suggesting that aberrant corticostriatal plasticity under conditions of dopamine denervation actively degrades cortical processing, gradually undermining cortical function and compensatory mechanisms. The theme of this special topic focuses on plasticity in sensorimotor circuitry involving the primary motor cortex, M1. The DLS and M1 are reciprocally modulated through re-entrant cortical basal ganglia-thalamo-cortical circuits (17, 18). The concepts described will refer to cortex and basal ganglia generally as our focus is the broader architectural and functional relationship between these two neural structures composing a circuit.
The Dorsolateral Striatum: Optimizing Cortical Activity
As the primary input nucleus of the sensorimotor cortico-basal ganglia loop (19), the DLS of the basal ganglia contributes to motor learning and execution (20–24). Associated with stimulus-response learning, the DLS is believed to be a primary substrate for the development of automaticity associated with implicit, procedural learning, particularly sequencing, critical for the fluid execution of complex motor actions [reviewed in Ref. (25)]. M1 exhibits intrinsic activity-dependent synaptic plasticity, important for motor skill and sequence learning (26). This activity is shaped by both intracortical afferents and basal ganglia efferents (27).
The basal ganglia are comprised of GABAergic medium spiny neurons (MSNs) of the striatonigral (direct) and striatopallidal (indirect) projections. These striatal projections modulate inhibition of cortical activity through the output nuclei of the basal ganglia that provide tonic inhibition of excitatory thalamocortical projections (17, 18). In turn, cortical glutamatergic afferents on striatal MSNs can activate the striatonigral or striatopallidal pathways, disinhibiting or inhibiting cortical activity, respectively [(28, 29); Figure 1A]. The responsiveness of striatonigral and striatopallidal MSNs to specific cortical synaptic inputs can be modulated through synaptic strength (Figure 1B). Increased synaptic strength is commonly defined by the emergence of long-term potentiation (LTP) and decreased synaptic strength by long-term depression (LTD). For example, a specific cortical synaptic input may be potentiated (LTP) at striatonigral disinhibitory MSNs while a cortical synaptic input on striatopallidal inhibitory MSNs is depressed (LTD), leading to the combined facilitation of cortical activity (30–34). Increasing evidence supports bidirectional plasticity at corticostriatal synapses (35–41), suggesting the opposite pattern can arise as well. For example, a specific cortical synaptic input might be potentiated, increasing striatopallidal MSN activity in response to that input and increasing cortical inhibition. The basal ganglia, through selective synaptic potentiation and depression, facilitate particular cortical activities while inhibiting others, thus filtering cortical activity through cortico-basal ganglia-thalamo-cortical loops. While the precise function of this filter remains subject to debate, it is hypothesized that the purpose of this basal ganglia filter is to select one action and suppress others (29, 42). An alternative view is that the basal ganglia, by selectively facilitating productive, task-relevant cortical activity and suppressing non-productive and/or irrelevant activity, optimizes cortical processing to increase speed and efficiency.
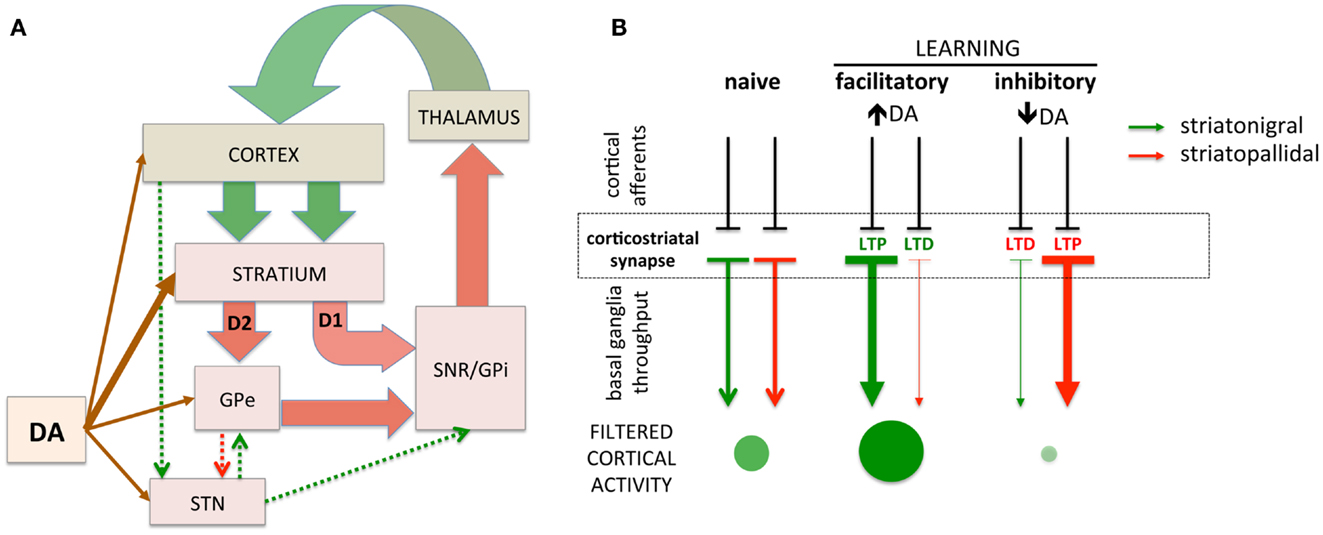
Figure 1. Simplified schematic of cortico-basal ganglia circuitry and corticostriatal filtering of cortical activity. (A) Dual corticostriatal architecture showing the direct and indirect pathways that express D1 and D2 dopamine receptors, respectively. Arrow colors reflect excitatory and inhibitory neurotransmitters. (B) Schematic diagramming selective facilitatory and inhibitory corticostriatal learning. LTP and LTD in the striatonigral and striatopallidal pathways, respectively (middle panel), facilitate cortical activity while the converse (LTD in striatonigral and LTP in striatopallidal) inhibit cortical activity. Green/red arrows represent striatonigral direct and striatopallidal indirect pathways, respectively. Direction of plasticity (LTP vs. LTD) colored red/green to indicate functional facilitation/inhibition of cortical throughput. The size/intensity of green circles represent the increase/decrease in activity of a synapse-specific cortical afferent induced by basal ganglia modulation.
Dopamine Regulates Cortical Filtering through the Basal Ganglia
Bidirectional corticostriatal plasticity is regulated by dopamine (31, 33, 34, 37, 43), widely believed to encode positive and negative reward prediction errors (RPEs) by firing bursts of action potentials or pausing tonic activity, respectively (44, 45). This RPE provides a teaching signal indicating positive and negative (better or worse than expected) outcomes. By regulating bidirectional corticostriatal synaptic plasticity, a dopamine-mediated teaching signal selectively enhances or diminishes cortical inputs associated with positive and negative outcomes, respectively. Through dopamine regulation, corticostriatal plasticity can selectively filter and highlight cortical activity determined to be relevant and beneficial (Figure 2, top). The net result is that cortical activity that is productive and yields a positive outcome will be selectively processed and amplified to complete a motor task.
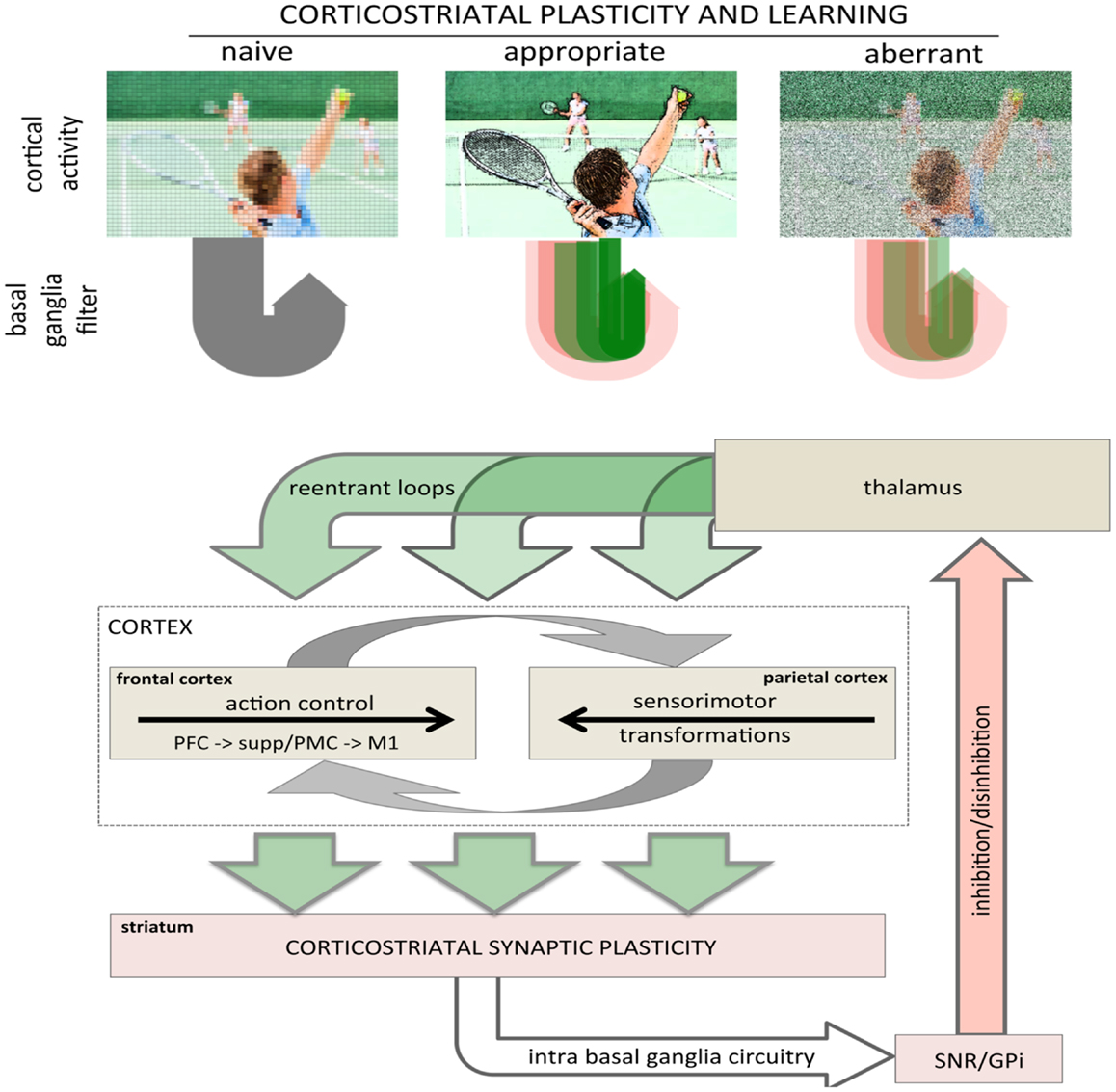
Figure 2. Role of corticostriatal plasticity and learning in basal ganglia filtering of cortical activity. (top) Conceptual illustration of basal ganglia filtering of cortical activity through corticostriatal plasticity and learning. The naïve, pre-learning state is represented in the left panel and processing through the basal ganglia is undifferentiated (gray arrow loop). After appropriate corticostriatal learning (middle panel), task-related elements are sharpened and highlighted (racquet, ball, arm, other players), represented in the corticostriatal loop as a combination of facilitation and inhibition with strong, task-relevant facilitation (sharp, dark green arrow loop). Under aberrant learning (right panel), inappropriate LTP in the inhibitory striatopallidal pathway induces inappropriate inhibition (red shaded arrow loops) and diminished facilitation (green shaded arrow loops) in the corticostriatal filter causing task-related elements become increasingly noisy and less distinct against background compared even to naïve processing (left panel). (bottom) Schematic showing rudimentary architecture for basal ganglia filter of cortical activity highlighting the general loop architecture. Large green arrows represent cortical inputs to the striatum and re-entrant projections returning to the cortex via the thalamus. The intrabasal ganglia circuitry has been collapsed to highlight the basic loop architecture. The cortical schematic has been expanded to represent the two primary intracortical information flows mediating action selection and motor control. The left cortical box represents traditional frontal motor control where information flows rostral to caudal from the prefrontal cortex to M1. The right cortical box represents parietal processing where information flows caudal to rostral mediating sensorimotor transformations specifying movements. These two are intricately interconnected, represented by reciprocal gray arrows. Image used in top panel licensed from Polka Dot Images/Thinkstock.
Aberrant Plasticity and Learning: Inverting Optimization
Increasing evidence suggest that reduced dopamine may shift corticostriatal plasticity in striatopallidal synapses favoring LTP rather than LTD, inverting plasticity such that conditions that would normally yield LTD produce LTP instead (38–41). The net effect of this would be that everywhere a cortical afferent should be disinhibited it would, instead, be further inhibited. Such a shift or inversion in the directional control of plasticity – the aberrant learning hypothesis – would transform an optimizing substrate into an “anti-optimizing” one that impedes rather than facilitates responding (25, 41, 46–48). Aberrant learning, then, transforms the basal ganglia that normally functions to filter and facilitate cortical activity into a disruptive filter that impedes motor activity.
Aberrant learning arising from reduced striatal dopamine contributes significantly to impaired motor performance over and above the direct motor effects of diminished dopamine (41, 48). The correction of aberrant learning and abnormal corticostriatal plasticity may represent an important component of L-DOPA treatment in PD and underlie the poorly understood long-duration response (LDR), where the benefits of dopamine replacement on motor performance persists beyond the pharmacokinetic half life of L-DOPA (25, 41, 48–50). Given the number of potential modulators of synaptic plasticity within the striatum, that include but are not limited to adenosine, glutamatergic and cholinergic neurotransmission, decreasing inappropriate potentiation at corticostriatal synapses in the striatopallidal pathway may serve as an important therapeutic target for facilitating motor learning and recovery of function in PD. For example, correction of aberrant learning may be an important therapeutic mechanism of adenosine antagonists (36, 41).
The aberrant learning hypothesis can be understood as an extension of the classic model of PD where there is an imbalance between the direct and indirect pathways (28, 29, 51–53). With aberrant learning, this imbalance is structurally encoded as inappropriate synaptic strengths [i.e., inverted corticostriatal plasticity; (25, 41, 46–48, 54)]. Thus, even if dopamine is restored, the inappropriate learning that has already been established will continue to degrade motor performance until appropriate synaptic plasticity and learning has replaced the inappropriate (41). Conversely, if dopamine is reduced (e.g., discontinuation of L-DOPA), performance will initially be partially protected as the appropriate synaptic strengths will facilitate corticostriatal throughput; however, as aberrant plasticity and learning return, synaptic structure will again become inverted and anti-optimal and initially retained function will deteriorate (48).
Reciprocal Relationship between Cortical and Striatal Plasticity and Learning
Learning is the encoding of information through alterations in synaptic strengths that underlie memory formation and skill acquisition. Though the cortex and striatum both exhibit learning and synaptic plasticity, how learning in each substrate affects learning in the other is poorly understood.
Cortical Learning Shapes Basal Ganglia Activity and Learning
As the cortex is a primary afferent to the striatum, alterations in synaptic plasticity that occurs in the cortex will change the afferent input to the basal ganglia, affecting both information flow through the basal ganglia and activity-dependent corticostriatal synaptic plasticity. For example, if the cortex learns to enhance the activity of one ensemble of neurons and diminish the activity of others, this differential will be reflected in the pattern of MSNs activation and the altered synaptic inputs may induce changes in corticostriatal synaptic strength. In this case, alterations in synaptic plasticity of the cortex may directly influence downstream synaptic plasticity within the striatum.
Striatal Learning Shapes Cortical Activity and Learning
Though cortical activity and learning shape activity in the striatum, evidence suggests that the striatum may learn earlier and faster and may shape cortical learning (55), consistent with the idea that the basal ganglia provide a filter for cortical activity. By selectively altering cortico-basal ganglia throughput of specific cortical afferents, corticostriatal plasticity shapes cortical activity; by modulating cortical activity, the basal ganglia can influence activity-dependent cortical learning.
Reciprocal Learning Provides a Gain Mechanism for Learning
Cortical activity that is facilitated through the basal ganglia filter will be more likely to undergo activity-dependent, Hebbian synaptic plasticity, strengthening those particular patterns of cortical activity in the future. Conversely, cortical activity inhibited by the basal ganglia will have lower probability of activity-dependent plasticity (or favor synaptic depression), diminishing that activity in the future. In turn, this altered pattern of cortical afferents to the striatum will differently activate MSNs and further modify corticostriatal synaptic strengths, which further modifies cortical activity and learning.
The Domino Effect: Aberrant Learning and Disrupted Cortical Filtering
Reduction in cortical activity in cortical regions that participate in the dorsal striatal sensorimotor loop (e.g., M1, PMC, posterior parietal) as well as reduced activity in regions associated with the cognitive loop (e.g., DLPFC) have been well documented in PD (6, 56). Reduced cortical activity in these regions is consistent with the classic model of an imbalance between the direct and indirect pathway that results in increased inhibitory tone on cortical activity. However, numerous studies have observed increased activity in these same regions associated with task performance (8–13, 15, 16, 57–62). While task-related increases in cortical activity may be construed as compensatory, it may reflect reduced processing efficiency. With the acquisition of automaticity, healthy normal controls exhibit decreased cortical activity while PD subjects do not show such reductions, consistent with increased cortical load arising from dysfunction in corticostriatal circuits (58). Such increased cortical activity may arise from the loss of appropriate corticostriatal filtering. In a recent study, Ng et al. (57) have shown spatially greater cortical activation (i.e., more spread out) in unmedicated PD subjects performing a simple motor task compared to healthy controls; this spatially greater activation is normalized by L-DOPA, which the authors characterize as a “focusing effect.” In the view adopted here, the increased cortical activity could reflect both compensatory cortical circuits and pathophysiology in the basal ganglia filter. First, denervation in the DLS induces greater inhibitory tone, diminishing cortical activity, which in turn leads to compensatory functional connectivity that increases intracortical and cerebellar drive on cortical activity. Second, this increased activity lacks the filter that “focuses” cortical activity, resulting in overall greater, less efficient cortical activation. The net result is that the cortex has to work harder to maintain behavior and does so with less efficiency.
Propagation of Aberrant Learning
As functional connectivity shifts to drive cortical activity and maintain behavior, cortical mechanisms of activity-dependent synaptic plasticity continue to facilitate learning to refine and calibrate cortical networks and ensembles to adapt to new patterns of afferent inputs. In effect, the cortex has to learn the adaptations. If the filtering or “focusing” effect of the DLS and the sensorimotor basal ganglia loop were merely absent, learning might simply be slower and less efficient. From the perspective of the aberrant learning hypothesis, however, the problem is more insidious. Dopamine denervation induces inverted corticostriatal plasticity in the striatopallidal pathway that actively inhibits precisely those high activity cortical afferents that should be facilitated, providing an anti-optimizing filter. Instead of increasing signal-to-noise ratio – enhancing productive and diminishing non-productive/irrelevant cortical activity – aberrant learning would actively diminish signal. Thus, as activity-dependent cortical learning adjust cortical synaptic strengths to improve performance and adapt to shifted functional connectivity, the resulting changes in cortical activity are subsequently opposed by aberrant learning in the DLS effectively filtering out such learning by inappropriately inhibiting precisely those activities the cortex has just learned to enhance. It is not difficult to imagine a progression where inappropriate filtering in conjunction with continued aberrant learning leads to further compensatory cortical activity that is less efficient and, in turn, also degraded by aberrant learning resulting in a vicious cycle of increasingly greater but less efficient cortical activity to maintain motor function.
Clinical Implications and Further Research
Aberrant corticostriatal plasticity, from the perspective of the aberrant learning hypothesis, represents more than a simple loss of function. Rather, it is an active, insidious process that hijacks corticostriatal plasticity, not only impeding the role of the basal ganglia as an effective filter of cortical activity, but structurally encoding inappropriate learning as synaptic strengths that can actively disrupt cortical function. Such aberrant learning will gradually unravel and invert a lifetime of learning – and the millions of finely calibrated synaptic strengths that support that learning – accelerating functional deterioration associated with neurodegeneration.
As dopamine denervation progresses, cortical compensatory activity is engaged. The reciprocal relationship between cortical and basal ganglia structures, however, suggests that aberrant corticostriatal plasticity may increase the burden placed on the cortex while simultaneously interfering with its compensatory capacity. If true, this means that therapeutic agents that ameliorate aberrant learning would have the additional advantage of supporting cortical compensation. In particular, decreasing an inappropriate, “anti-optimizing” basal ganglia filter may facilitate improved cortical learning and adaptation. Therapeutic strategies specifically targeted at correcting or blocking aberrant learning may slow functional deterioration associated with neurodegeneration as well as support adaptive, compensatory mechanisms, providing a form of disease-modifying neuroprotection that may mitigate the functional decline associated with disease progression. Thus, targeting subcellular signaling pathways and cellular mechanisms specific to striatopallidal synaptic plasticity to either diminish inappropriate LTP or enhance appropriate LTD may offer an avenue for the development of alternative disease-modifying, neuroprotective therapeutics (25, 41, 50).
An important aspect of aberrant learning is that it introduces a delay between the pathophysiology (or its correction) and the resulting functional effects on behavior. That is, aberrant learning that occurs now will affect performance in the future. Conversely, improvement associated with corrected aberrant learning will be observed gradually over time as restored corticostriatal plasticity relearns and recalibrates synapses. It is precisely this delay in observing the effects of corrected aberrant learning that we have proposed underlies the LDR to L-DOPA (25, 48). If aberrant corticostriatal plasticity degrades cortical learning, a delay might be expected in the correction of impaired cortical functions as the cortex also undergoes relearning. Interestingly, it has been observed that several cognitive symptoms of PD do not appear to be improved with L-DOPA administration [e.g., Ref. (63)]. Why this might be is unclear, but one possibility is that cognitive symptoms associated with cortical functions may, at least partially, arise from impaired cortical learning induced by aberrant corticostriatal plasticity. If so, L-DOPA would not immediately correct previously degraded or inappropriate cortical learning; however, similar to the LDR, such symptoms might improve gradually over time as cortical learning is protected from the deleterious effects of aberrant corticostriatal plasticity. Conversely, Kishore et al. (64, 65) have recently observed a deficit in plasticity in M1 in newly diagnosed, untreated patients that is corrected under optimal medication (absent fluctuations or dyskinesia). This corrected plasticity is sustained even during the OFF state, suggesting it may represent a LDR (64). The degree to which these findings relate to dopamine replacement and a potential LDR action directly in M1 versus arising secondary to the correction of aberrant corticostriatal plasticity and its effects on cortical processing and learning remain to be determined.
This hypothesis has potentially significant implication for rehabilitative treatments in PD. Rehabilitation fundamentally involves repetition, i.e., practice, and is premised on mechanisms of learning and plasticity. If those underlying mechanisms are not only impaired, but potentially induce counter-productive, aberrant learning and plasticity, the efficacy of rehabilitation could be compromised. Rehabilitative strategies might be most beneficial, then, when conducted under optimal medication to maximally support cortical compensatory learning. The success of physical therapy programs, including programs which patients practice independently (eg., Big and Loud), may be contingent on training and practice being performed during the ON medication state.
In conclusion, agents that target and ameliorate aberrant corticostriatal plasticity in the striatopallidal pathway may represent an important avenue for the development of new therapeutics, potentially yielding a protective LDR-like treatment independent of dopamine replacement with its attendant complications. Remediating an insidious process of inappropriate corticostriatal synaptic plasticity may mitigate circuit deterioration and support cortical compensatory mechanisms, modifying the rate and severity of functional decline associated with disease progression. Further research on aberrant learning and its potential effects on cortical function and learning are needed and may yield new insights and treatment strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was support by NIDA, DA25875 (Jeff A Beeler). The image used in Figure 2 (top) was licensed from Polka Dot Images/Thinkstock.
References
1. Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and huntington. clinical, morphological and neurochemical correlations. J Neurol Sci (1973) 20:415–55. doi:10.1016/0022-510X(73)90175-5
2. Hornykiewicz O. Chemical neuroanatomy of the basal ganglia – normal and in Parkinson’s disease. J Chem Neuroanat (2001) 22:3–12. doi:10.1016/S0891-0618(01)00100-4
3. Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med (1988) 318:876–80. doi:10.1056/NEJM198804073181402
4. Petzinger GM, Fisher BE, McEwen S, Beeler JA, Walsh JP, Jakowec MW. Exercise-enhanced neuroplasticity targeting motor and cognitive circuitry in Parkinson’s disease. Lancet Neurol (2013) 12:716–26. doi:10.1016/S1474-4422(13)70123-6
5. Redgrave P, Rodriguez M, Smith Y, Rodriguez-Oroz MC, Lehericy S, Bergman H, et al. Goal-directed and habitual control in the basal ganglia: implications for Parkinson’s disease. Nat Rev Neurosci (2010) 11:760–72. doi:10.1038/nrn2915
6. Niethammer M, Eidelberg D. Metabolic brain networks in translational neurology: concepts and applications. Ann Neurol (2012) 72:635–47. doi:10.1002/ana.23631
7. Appel-Cresswell S, Fuente-Fernandez de R, Galley S, McKeown MJ. Imaging of compensatory mechanisms in Parkinson’s disease. Curr Opin Neurol (2010) 23:407–12. doi:10.1097/WCO.0b013e32833b6019
8. Baglio F, Blasi V, Falini A, Farina E, Mantovani F, Olivotto F, et al. Functional brain changes in early Parkinson’s disease during motor response and motor inhibition. Neurobiol Aging (2011) 32:115–24. doi:10.1016/j.neurobiolaging.2008.12.009
9. Helmich RC, de Lange FP, Bloem BR, Toni I. Cerebral compensation during motor imagery in Parkinson’s disease. Neuropsychologia (2007) 45:2201–15. doi:10.1016/j.neuropsychologia.2007.02.024
10. Nakamura T, Ghilardi MF, Mentis M, Dhawan V, Fukuda M, Hacking A, et al. Functional networks in motor sequence learning: abnormal topographies in Parkinson’s disease. Hum Brain Mapp (2001) 12:42–60. doi:10.1002/1097-0193(200101)12:1<42::AID-HBM40>3.0.CO;2-D
11. Mentis MJ, Dhawan V, Nakamura T, Ghilardi MF, Feigin A, Edwards C, et al. Enhancement of brain activation during trial-and-error sequence learning in early PD. Neurology (2003) 60:612–9. doi:10.1212/01.WNL.0000044154.92143.DC
12. Wu T, Hallett M. A functional MRI study of automatic movements in patients with Parkinson’s disease. Brain (2005) 128:2250–9. doi:10.1093/brain/awh569
13. Palmer SJ, Ng B, Abugharbieh R, Eigenraam L, McKeown MJ. Motor reserve and novel area recruitment: amplitude and spatial characteristics of compensation in Parkinson’s disease. Eur J Neurosci (2009) 29:2187–96. doi:10.1111/j.1460-9568.2009.06753.x
14. Wu AD, Petzinger GM, Lin C-HJ, Kung M, Fisher B. Asymmetric corticomotor excitability correlations in early Parkinson’s disease. Mov Disord (2007) 22:1587–93. doi:10.1002/mds.21565
15. Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. Neuroimage (2007) 35:222–33. doi:10.1016/j.neuroimage.2006.11.047
16. Cerasa A, Hagberg GE, Peppe A, Bianciardi M, Gioia MC, Costa A, et al. Functional changes in the activity of cerebellum and frontostriatal regions during externally and internally timed movement in Parkinson’s disease. Brain Res Bull (2006) 71:259–69. doi:10.1016/j.brainresbull.2006.09.014
17. Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev (1995) 20:91–127. doi:10.1016/0165-0173(94)00008-D
18. Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci (1990) 13:266–71. doi:10.1016/0166-2236(90)90107-L
19. Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat (2000) 196(Pt 4):527–42. doi:10.1046/j.1469-7580.2000.19640527.x
20. Costa RM, Cohen D, Nicolelis MAL. Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr Biol (2004) 14:1124–34. doi:10.1016/j.cub.2004.06.053
21. Jin X, Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature (2010) 466:457–62. doi:10.1038/nature09263
22. Yin HH, Mulcare SP, Hilário MRF, Clouse E, Holloway T, Davis MI, et al. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci (2009) 12:333–41. doi:10.1038/nn.2261
23. Doyon J, Bellec P, Amsel R, Penhune V, Monchi O, Carrier J, et al. Contributions of the basal ganglia and functionally related brain structures to motor learning. Behav Brain Res (2009) 199:61–75. doi:10.1016/j.bbr.2008.11.012
24. Lehéricy S, Benali H, Van de Moortele P-F, Pélégrini-Issac M, Waechter T, Ugurbil K, et al. Distinct basal ganglia territories are engaged in early and advanced motor sequence learning. Proc Natl Acad Sci U S A (2005) 102:12566–71. doi:10.1073/pnas.0502762102
25. Beeler JA. Preservation of function in Parkinson’s disease: what”s learning got to do with it? Brain Res (2011) 1423:96–113. doi:10.1016/j.brainres.2011.09.040
26. Matsuzaka Y, Picard N, Strick PL. Skill representation in the primary motor cortex after long-term practice. J Neurophysiol (2007) 97:1819–32. doi:10.1152/jn.00784.2006
27. Kelly RM, Strick PL. Macro-architecture of basal ganglia loops with the cerebral cortex: use of rabies virus to reveal multisynaptic circuits. Prog Brain Res (2004) 143:449–59.
28. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci (1989) 12:366–75. doi:10.1016/0166-2236(89)90074-X
29. Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol (1996) 50:381–425. doi:10.1016/S0301-0082(96)00042-1
30. Lerner TN, Kreitzer AC. Neuromodulatory control of striatal plasticity and behavior. Curr Opin Neurobiol (2011) 21:322–7. doi:10.1016/j.conb.2011.01.005
31. Wickens JR. Synaptic plasticity in the basal ganglia. Behav Brain Res (2009) 199:119–28. doi:10.1016/j.bbr.2008.10.030
32. Costa RM. Plastic corticostriatal circuits for action learning: what’s dopamine got to do with it? Ann N Y Acad Sci (2007) 1104:172–91. doi:10.1196/annals.1390.015
33. Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci (2007) 30:211–9. doi:10.1016/j.tins.2007.03.001
34. Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol (2009) 19:621–8. doi:10.1016/j.conb.2009.10.003
35. Schiffmann SN, Dassesse D, d’Alcantara P, Ledent C, Swillens S, Zoli M. A2A receptor and striatal cellular functions: regulation of gene expression, currents, and synaptic transmission. Neurology (2003) 61:S24–9. doi:10.1212/01.WNL.0000095207.66853.0D
36. Peterson JD, Goldberg JA, Surmeier DJ. Adenosine A2a receptor antagonists attenuate striatal adaptations following dopamine depletion. Neurobiol Dis (2012) 45:409–16. doi:10.1016/j.nbd.2011.08.030
37. Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology (2010) 58:951–61. doi:10.1016/j.neuropharm.2010.01.008
38. Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science (2008) 321:848–51. doi:10.1126/science.1160575
39. Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, et al. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci (1997) 17:4536–44.
40. Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci (2003) 6:501–6.
41. Beeler JA, Frank MJ, McDaid J, Alexander E, Turkson S, Sol Bernandez M, et al. A role for dopamine-mediated learning in the pathophysiology and treatment of Parkinson’s disease. Cell Rep (2012) 2:1747–61. doi:10.1016/j.celrep.2012.11.014
42. Redgrave P, Prescott TJ, Gurney K. The basal ganglia: a vertebrate solution to the selection problem? Neuroscience (1999) 89:1009–23. doi:10.1016/S0306-4522(98)00319-4
43. Reynolds JNJ, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw (2002) 15:507–21. doi:10.1016/S0893-6080(02)00045-X
44. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science (1997) 275:1593–9. doi:10.1126/science.275.5306.1593
45. Schultz W. Behavioral dopamine signals. Trends Neurosci (2007) 30:203–10. doi:10.1016/j.tins.2007.03.007
46. Wiecki TV, Riedinger K, Ameln-Mayerhofer von A, Schmidt WJ, Frank MJ. A neurocomputational account of catalepsy sensitization induced by D2 receptor blockade in rats: context dependency, extinction, and renewal. Psychopharmacology (Berl) (2009) 204:265–77. doi:10.1007/s00213-008-1457-4
47. Wiecki TV, Frank MJ. Neurocomputational models of motor and cognitive deficits in Parkinson’s disease. Prog Brain Res (2010) 183:275–97. doi:10.1016/S0079-6123(10)83014-6
48. Beeler JA, Cao ZFH, Kheirbek MA, Ding Y, Koranda J, Murakami M, et al. Dopamine-dependent motor learning: insight into levodopa’s long-duration response. Ann Neurol (2010) 67:639–47. doi:10.1002/ana.21947
49. Anderson E, Nutt J. The long-duration response to levodopa: phenomenology, potential mechanisms and clinical implications. Parkinsonism Relat Disord (2011) 17:587–92. doi:10.1016/j.parkreldis.2011.03.014
50. Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat Rev Neurol (2013) 9:248–56. doi:10.1038/nrneurol.2013.57
51. Penney JB, Young AB. Striatal inhomogeneities and basal ganglia function. Mov Disord (1986) 1:3–15. doi:10.1002/mds.870010102
52. DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci (1990) 13:281–5. doi:10.1016/0166-2236(90)90110-V
53. Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci (1990) 13:277–80. doi:10.1016/0166-2236(90)90109-N
54. Frank MJ. Dynamic dopamine modulation in the basal ganglia: a neurocomputational account of cognitive deficits in medicated and nonmedicated Parkinsonism. J Cogn Neurosci (2005) 17:51–72. doi:10.1162/0898929052880093
55. Pasupathy A, Miller EK. Different time courses of learning-related activity in the prefrontal cortex and striatum. Nature (2005) 433:873–6. doi:10.1038/nature03287
56. Eckert T, Tang C, Eidelberg D. Assessment of the progression of Parkinson’s disease: a metabolic network approach. Lancet Neurol (2007) 6:926–32. doi:10.1016/S1474-4422(07)70245-4
57. Ng B, Palmer S, Abugharbieh R, McKeown MJ. Focusing effects of L-dopa in Parkinson’s disease. Hum Brain Mapp (2010) 31:88–97.
58. Wu T, Chan P, Hallett M. Effective connectivity of neural networks in automatic movements in Parkinson’s disease. Neuroimage (2010) 49:2581–7. doi:10.1016/j.neuroimage.2009.10.051
59. Samuel M, Ceballos-Baumann AO, Blin J, Uema T, Boecker H, Passingham RE, et al. Evidence for lateral premotor and parietal overactivity in Parkinson’s disease during sequential and bimanual movements. A PET study. Brain (1997) 120(Pt 6):963–76. doi:10.1093/brain/120.6.963
60. Catalan MJ, Ishii K, Honda M, Samii A, Hallett M. A PET study of sequential finger movements of varying length in patients with Parkinson’s disease. Brain (1999) 122(Pt 3):483–95. doi:10.1093/brain/122.3.483
61. Wu T, Long X, Zang Y, Wang L, Hallett M, Li K, et al. Regional homogeneity changes in patients with Parkinson’s disease. Hum Brain Mapp (2009) 30:1502–10. doi:10.1002/hbm.20622
62. van der Vegt JPM, van Nuenen BFL, Bloem BR, Klein C, Siebner HR. Imaging the impact of genes on Parkinson’s disease. Neuroscience (2009) 164:191–204. doi:10.1016/j.neuroscience.2009.01.055
63. Ghilardi MF, Feigin AS, Battaglia F, Silvestri G, Mattis P, Eidelberg D, et al. L-Dopa infusion does not improve explicit sequence learning in Parkinson’s disease. Parkinsonism Relat Disord (2007) 13:146–51. doi:10.1016/j.parkreldis.2006.08.006
64. Kishore A, Popa T, Velayudhan B, Joseph T, Balachandran A, Meunier S. Acute dopamine boost has a negative effect on plasticity of the primary motor cortex in advanced Parkinson’s disease. Brain (2012) 135:2074–88. doi:10.1093/brain/aws124
Keywords: corticostriatal plasticity, striatopallidal pathway, dorsolateral striatum, cortical compensation, basal ganglia
Citation: Beeler JA, Petzinger G and Jakowec MW (2013) The enemy within: propagation of aberrant corticostriatal learning to cortical function in Parkinson’s disease. Front. Neurol. 4:134. doi: 10.3389/fneur.2013.00134
Received: 24 May 2013; Accepted: 28 August 2013;
Published online: 12 September 2013.
Edited by:
Asha Kishore, Sree Chitra Tirunal Institute for Medical Sciences and Technology, IndiaReviewed by:
Benzi Kluger, University of Colorado Denver, USAJaime Kulisevsky, Sant Pau Hospital – Sant Pau Institute of Biomedical Research, Spain
Copyright: © 2013 Beeler, Petzinger and Jakowec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeff A. Beeler, Department of Psychology, Queens College, City University of New York, Science Building E318, 65-30 Kissena Blvd., Queens, New York, NY 11367-1597, USA e-mail:amJlZWxlckBxYy5jdW55LmVkdQ==