 Sioui Maldonado Bouchard
Sioui Maldonado Bouchard Michelle A. Hook
Michelle A. Hook- Department of Neuroscience and Experimental Therapeutics, Texas A&M Health Science Center, Texas A&M Institute for Neuroscience, College of Medicine, College Station, TX, USA
There is strong evidence indicating that the social environment triggers changes to the psychological stress response and glucocorticoid receptor function. Considerable literature links the subsequent changes in stress resiliency to physical health. Here, converging evidence for the modulatory role of chronic psychological stress in the recovery process following spinal cord injury (SCI) is presented. Despite the considerable advances in SCI research, we are still unable to identify the causes of variability in patients’ recovery following injury. We propose that individuals’ past and present life experiences (in the form of stress exposure) may significantly modulate patients’ outcome post-SCI. We propose a theoretical model to explain the negative impact of chronic psychological stress on physical and psychological recovery. The stress experienced in life prior to SCI and also as a result of the traumatic injury, could compromise glucocorticoid receptor sensitivity and function, and contribute to high levels of inflammation and apoptosis post-SCI, decreasing the tissue remaining at the injury site and undermining recovery of function. Both stress-induced glucocorticoid resistance and stress-induced epigenetic changes to the glucocorticoid receptor can modulate the nuclear factor-kappa B regulated inflammatory pathways and the Bcl-2 regulated apoptosis pathways. This model not only contributes to the theoretical understanding of the recovery process following injury, but also provides concrete testable hypotheses for future studies.
In 2001, at the 54th World Health Assembly, the 191 Member States of the World Health Organization approved the International Classification of Functioning, Disability, and Health. This classification called for a shift in the way medicine views health and disability. It recognized that an injury occurs in a social context, and thus, that not only physical signs but also environmental elements should be taken into consideration by doctors and medical researchers in the quest to better predict and improve patients’ outcome post-injury (1). Despite this initiative, more than a decade later, outcome prediction following a spinal cord injury (SCI) remains predominantly based on injury-related factors alone, and its accuracy is limited at best (2). We posit that psychological stress inherent to the present and past environments of SCI patients may account, in part, for the unexplained variance in functional recovery.
The link between a stressful environment and disease recovery is well recognized in diverse clinical fields (3). Psychological stress is associated with greater risk of asthma exacerbation (4), increases in cardiovascular disease (5), and faster progression of HIV/AIDS (6). However, despite the fact that decreased psychological well-being post-SCI is strongly documented clinically, understanding of its role in recovery is lacking. As many as 60% of patients suffer from depression (7), anxiety (8), and general decreased quality of life (9). Many patients also experience loneliness; the perception of few people who understand them and in whom they can confide (10). Interestingly, however, not only does the psychological health of SCI patients improve in a positive social environment, but physical health is also benefited. Conducting a systematic review of the literature on patients with SCI, Muller et al. (11) found a positive correlation between social support and physical health, mental health, and pain management post-injury. Patients with a strong social support system showed better health and functioning (11). In 2011, a multilevel modeling, retrospective analysis of cross-sectional survey data also indicated that individual socioeconomic status strongly predicts outcome post-SCI (12). Saunders et al. (13) further found household income to be inversely correlated with pressure ulcers in patients with SCI, even after controlling for demographic and injury variables. Finally, a number of epidemiological studies have found that patients with good coping skills have a more positive experience after SCI, and display a lower incidence of depression (14). These analyses highlight the potential impact of a positive environment, and by extension a stressful environment, on the recovery of patients following SCI.
Both pre and post-SCI environment may influence recovery. Indeed, both a past and present stressful environment, including the traumatic experience of SCI, can alter the function of the glucocorticoid receptor, which is involved in both the stress response and the activity of protein complexes involved in inflammation and apoptosis, processes critical in SCI injury and recovery (Figure 1). Chronic psychological stress can compromise glucocorticoid receptor sensitivity and function (15–17), and this may contribute to high levels of inflammation and apoptosis, decreasing the tissue remaining at the injury site and undermining recovery of function post-SCI. What is more, who is most susceptible to stress as a result of the traumatic injury may depend on the stress experienced in life prior to SCI. While numerous other receptors and proteins involved in SCI and recovery processes (i.e., excitotoxicity, oxidative stress, and glial activation) could be affected by a chronically stressful environment, this review focuses on the impact of glucocorticoid receptors. There is strong evidence for the social environment triggering changes to the psychological stress response, and glucocorticoid receptor function, with considerable literature linking the subsequent changes in stress resiliency to physical health.
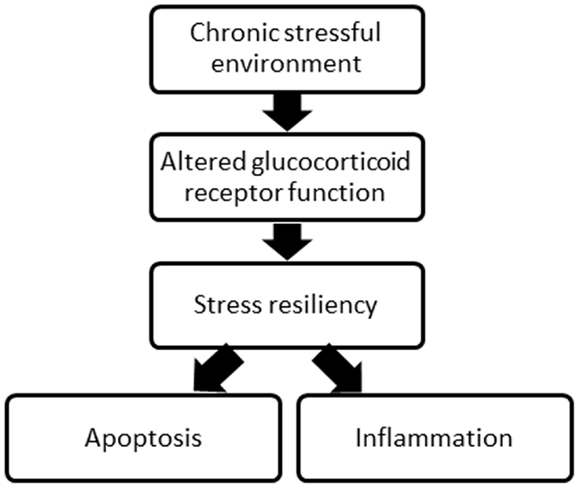
Figure 1. A schematic of the overall concept proposed relating the environment, glucocorticoid receptor function, stress resiliency, and two major processes following SCI.
Stress Resiliency Modulates Inflammation and Apoptosis
Psychological stress is defined as an “emotional experience accompanied by predictable biochemical, physiological, and behavioral changes” (18). Examples include experiences of social isolation, rape, verbal and physical abuse, traumatic injury, or war. A homeostatic challenge triggers a stress response, via both a fast route (neural), involving the autonomic nervous system (often referred to as the fight-or-flight response), and a slower route (endocrine), involving the hypothalamic–pituitary–adrenal axis. When we are exposed to a homeostatic challenge, such as a sign of danger, the brain first reacts via the neural route. It activates the sympathetic nervous system, which prompts the adrenal glands to release catecholamines, namely epinephrine and norepinephrine. Also activated more slowly is the hypothalamic–pituitary–adrenal axis, the endocrine route. An outside challenge will lead the hypothalamus to secrete corticotropin-releasing hormone (CRH) into the pituitary portal circulation. This, in turn, stimulates the pituitary glands to release the adrenocorticotropic hormone (ACTH), which triggers glucocorticoid release by the adrenal glands (19). Glucocorticoids function in a negative neuroendocrine feedback loop; high plasma levels signal the hypothalamus to stop producing CRH, and signal the anterior pituitary to stop producing ACTH. The effects of stress on glucocorticoid receptor signaling and the downstream processes of inflammation and apoptosis, however, depend on whether such stress is acute or chronic.
Acute Stress Response
In response to acute stress, glucocorticoids and their receptors enhance cellular resiliency and plasticity through inhibition of inflammation and apoptosis. Glucocorticoids are found in the bloodstream, where 90% of these molecules are bound to glucocorticoid-binding globulin. Only unbound glucocorticoids, the remaining 10%, can cross the blood–brain barrier and cell membranes (19). The molecules that do cross the membrane of cells, and reach the cytoplasm, bind to two types of receptors: mineralocorticoid receptors and glucocorticoid receptors. Mineralocorticoid receptors have an affinity to glucocorticoids 10 times greater than that of glucocorticoid receptors (20). Thus, of the percentage of glucocorticoid molecules that are able to cross cell membranes, only a fraction actually bind to glucocorticoid receptors (19).
Glucocorticoid receptors are found in virtually all cells of the body, including, importantly, in the microglia of the spinal cord (21, 22). In normal circumstances, glucocorticoids are anti-inflammatory. In the cytoplasm, they can bind to glucocorticoid receptors, and trigger conformational changes, which allow the glucocorticoid receptors to translocate to the cell nucleus via microtubular highways (23). Once in the nucleus, the glucocorticoid receptors can act as ligand-binding transcription factors (24). Nuclear glucocorticoid receptors regulate transcription in conjunction with a number of transcription factors, among them the activator protein 1 (AP-1) and nuclear factor-kappa B (NF-κB), which are major initiators of inflammation (see Figure 2). In the spinal cord, ligand-bound glucocorticoid receptors inhibit the activity of NF-κB, thereby impeding immune cells from expressing certain pro-inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), or IL-1β (25, 26). This occurs in microglia, but also neurons, as they can both express NF-κB (27). The inverse is also true; transcription factors involved in inflammation, such as NF-κB, can regulate the expression of the glucocorticoid receptor gene, by repressing certain promoters of this gene (26). This is referred to as reciprocal repression.
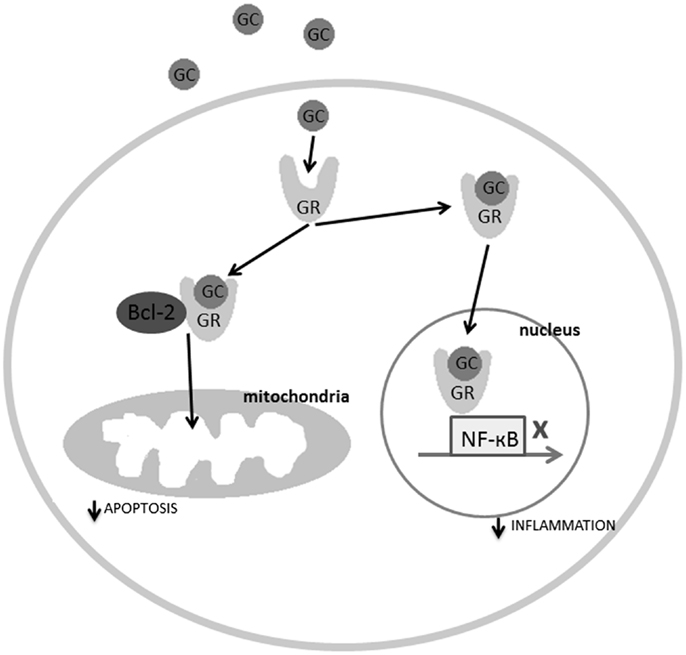
Figure 2. The molecular mechanisms explaining the regulatory role of the ligand-bound glucocorticoid receptors in inflammation and apoptosis. GC, glucocorticoids; GR, glucocorticoid receptor.
Glucocorticoid receptors, in addition to being involved in reciprocal repression with inflammation-related transcription factors, regulate the activity and expression of kinases and phosphatases such as mitogen-activated protein kinases (MAPKs), cyclin-dependent kinases (Cdks), dual-specificity phosphatases (DUSPs), and protein Y phosphatases (26). Kinases and phosphatases are essential in inflammation. They transfer and remove phosphate groups from specific substrates involved in inflammation signaling pathways, and play an important role in initiating the expression of pro-inflammatory genes. For example, in order for the IKK-NF-κB (inhibitors of kappa B kinase-NF-κB) pathway to be activated, a pathway which triggers tissue inflammation, the inhibitor of NF-κB must be dissociated (28). This is done by NF-κB-specific kinases which phosphorylate this inhibitor, thus freeing NF-κB from inhibition (26). Glucocorticoid receptors inhibit inflammatory signals by inhibiting the activity of transcription factors that initiate the expression of pro-inflammatory cytokine genes, and by inhibiting the expression of kinases and phosphatases that initiate inflammation signaling pathways.
In addition to anti-inflammatory effects, glucocorticoid receptors also possess anti-apoptotic properties. It is well-known and regular medical practice, to administer one high dose of prednisone (a corticosteroid) to patients who are extremely ill and at-risk of dying, as in the case of severe SCI (29, 30). This treatment has a dramatic effect on the patient, stabilizing his condition within minutes. This is partly because glucocorticoid receptors not only travel to the cell nucleus to inhibit pro-inflammatory cytokine gene expression, but also travel to the mitochondria to inhibit apoptosis. Specifically, ligand-bound glucocorticoid receptors in the cytoplasm can bind to B-cell CLL/lymphoma 2 (Bcl-2), which is an anti-apoptotic regulator inhibited by the gene Bad post-SCI. The ligand-bound glucocorticoid receptor-Bcl-2 complex travels to the mitochondria and halts the apoptosis cascade consisting of cytochrome c release and caspase activation (31) (see Figure 2). Studies have also shown that low doses of glucocorticoids increase mitochondrial oxidation (necessary for ATP synthesis), mitochondrial membrane potential, and mitochondrial calcium holding capacity (32). In short, glucocorticoid receptors inhibit inflammation and apoptosis in response to acute stress.
This modulatory role of glucocorticoids, for inflammation and apoptosis, has been utilized in the clinical treatment of SCI. Methylprednisolone remains the only option for therapeutic intervention in the emergency management of SCI (33), despite conflicting results both in laboratory experiments and clinical trials (34–36). Indeed, some researchers have questioned clinical trial results (37–39), while others have criticized such evaluations for focusing solely on statistical details and theoretical safety concerns (33, 40). A major concern is that the side effects of methylprednisolone outweigh its modest functional benefits. Dose curve and timing studies may be necessary to evaluate other possibly beneficial treatment regimens, such as a low methylprednisolone dose administration over an extended time period. At present, however, the clinical use of methylprednisolone remains controversial, and while a full discussion of this controversy is beyond the scope of this review [for both sides of the issue, the reader is referred to Ref. (33, 40)] empirical data does support the idea that glucocorticoids reduce apoptosis (41).
Indeed, early animal studies on the use of glucocorticoids following SCI posited that methylprednisolone improved recovery by reducing secondary processes of injury, such as secondary ischemia (42), lipid peroxidation, (42, 43) and apoptosis (44). Because the methylprednisolone dose demonstrating some success in the multicenter, double-blind randomized clinical trial NASCIS II was much higher (30 mg/kg bolus, 5.4 mg/kg maintenance per hour) than that required for glucocorticoid receptor activation, it has also been suggested that the beneficial properties of methylprednisolone treatment following SCI may not involve the glucocorticoid receptors, or at least not only involve glucocorticoid receptor related mechanisms (30). Bracken et al. (30) suggested that instead, methylprednisolone at such a high dose may work through its facilitation of blood flow through the injured cord and inhibition of lipid peroxidation. In any case, based on the modulation of inflammation and apoptosis discussed above, there should be potential for the use of glucocorticoids from a theoretical perspective.
The timing of a therapeutic intervention, in regulating the glucocorticoid response, would be critical for efficacy. Interestingly, the efficacy of methylprednisolone is also contingent on the timing of the therapeutic intervention (30, 34). Various pro-inflammatory genes are up-regulated immediately following SCI. For example, the NF-κB gene, which acts as a transcription factor to initiate the transcription of various gene types, including genes encoding for pro-inflammatory cytokines such as IL-6 and TNF-α (45, 46), is up-regulated as early as 30 min post-injury in a rodent model of contusion injury (47). This up-regulation continues for at least 72 h, and occurs in neurons, macrophages (microglia), and endothelial cells of the injured spinal cord (47). Methylprednisolone, which is generally considered to have the greatest therapeutic impact when administered in the 8-h following SCI (30), has been found to inhibit the activation of NF-κB and downstream production of pro-inflammatory cytokines such as TNF-α (48). Indeed, in one early rodent study, methylprednisolone administered at 30 mg/kg intravenously inhibited IL-6 expression and NF-κB activity by 55% (48).
Recently, it has been suggested that inhibiting the inhibitor of κB kinase (IKK) might also be useful for post-SCI therapy. IKK, by phosphorylating the IkB, leads to its ubiquitination and degradation by proteases, thus activating the NF-κB complex (49). In a rodent study of SCI, inhibiting IKK was found to prevent neutrophil infiltration as well as inhibiting the activity of caspase-3 (by modulating Bcl-2 expression), thus reducing apoptosis in the injured spinal cord (50). These studies support the potential of methylprednisolone treatment, modulating NF-κB and Bcl-2 activity in the acute phase of SCI. However, methylprednisolone is not always an efficient line of treatment (36). The past and present environment experienced by the patient may critically modulate the molecular cascades activated with glucocorticoid administration. Indeed, exposure to a chronic stressor is linked to glucocorticoid resistance and epigenetic changes that may result in increased unbound glucocorticoid expression and exacerbation of inflammation and apoptosis.
Chronic Stress: Glucocorticoid Resistance and Epigenetic Modifications
Glucocorticoid resistance
While the acute stress response has been linked to reduced inflammation, chronic psychological stress can result in systemic inflammation (15, 51) and the development of glucocorticoid resistance. Chronic stress-induced inflammation has been suggested to play a central role in autoimmune diseases (52), infectious diseases (51, 53), and cardiovascular conditions (54), among others. Glucocorticoid receptors play an integral role in the transition from the protective effects of acute stress to the negative consequences of chronic stress. Chronic stress causes extended activation of the hypothalamic–pituitary–adrenal axis and the autonomic nervous system. This extended exposure to glucocorticoid (55), epinephrine, and norepinephrine hormones (56) results in a diminished expression and function of the glucocorticoid receptor (15). Indeed, chronic exposure to elevated levels of glucocorticoids due to protracted steroid treatment (57), chronic inflammation (58, 59), early-life trauma (60, 61), or chronic psychological stress (15, 51), can lead to the development of glucocorticoid resistance: a decreased glucocorticoid receptor sensitivity. As a result of glucocorticoid resistance, immune cells lose sensitivity to glucocorticoids, the hormone which usually serves to put a stop to inflammatory responses (62).
Glucocorticoid resistance due to chronic psychological stress not only hinders the inhibition of pro-inflammatory gene expression in the cell nucleus by impeding ligand-bound glucocorticoid receptors from inhibiting NF-κB activity, but can also impair the anti-apoptotic role of glucocorticoid receptors in the mitochondria. Recent in vitro and in vivo studies have indicated that whereas low doses or one single high dose of glucocorticoids enable the anti-apoptotic effects of the glucocorticoid receptors, chronically high glucocorticoid levels, due to chronic stress, lead to decreased levels of glucocorticoid receptors and anti-apoptotic protein chaperone Bcl-2 in the mitochondria (32, 63). These mechanisms are of high relevance to SCI patients, as chronic inflammation and apoptosis contribute to neural loss, motor impairment (64), the development of chronic pain (65, 66), anxiety (67), and depression (68, 69).
Although the exact mechanism through which chronic stress leads to glucocorticoid resistance is not fully understood, studies of glucocorticoid resistant animal species point to the role of FK506-binding protein 51 (FKBP51) overexpression and heat shock protein 90 (70). FKBP51 is an immunophilin protein that is part of the heterocomplex of the mature glucocorticoid receptor, and regulates its sensitivity. The FKBP5 gene, which encodes for FKBP51, is up-regulated in response to chronic psychological stress or psychological trauma (71). Recent studies have uncovered an association between FKBP5 dysregulation and individuals’ attention to threats as a result of racial discrimination (71). It also seems that FKBP5 dysregulation could play a role in the vulnerability to post-traumatic stress disorder in war veterans (72) and stress-related psychiatric illness in individuals having experienced childhood trauma (73). Specifically, the ligand-bound glucocorticoid receptors have been shown to up-regulate the expression of FKBP51 (74, 75). When the FKBP51 chaperone interacts with heat shock protein 90, it lowers the affinity of the glucocorticoid receptor heterocomplex to glucocorticoids (76). In other words, chronic psychological stress increases levels of glucocorticoids; ligand-bound glucocorticoid receptors increase levels of FKBP51, and FKBP51 (bound to heat shock protein 90) in turn decreases the affinity of the glucocorticoid receptor to glucocorticoids, further increasing levels of unbound glucocorticoids. Notably in SCI, the expression of heat shock protein 90 is increased (77).
Epigenetic changes
A stressful environment can affect the sensitivity of the glucocorticoid receptors (51), but, critically, it can also alter its expression (60, 61, 78) via epigenetic mechanisms. Epigenetics is the study of changes in gene expression that can be triggered by environmental factors and which do not result in alterations to the DNA itself (79, 80). Environmental factors, such as diet, infections, and drugs, but also present and past psychosocial environmental factors, such as social support, psychotherapy, loneliness, as well as exposure to physical and mental abuse, may trigger epigenetic changes in SCI patients which either promote, or impede their recovery. In a pioneering study, Meaney and colleagues demonstrated that high levels of early-life licking and grooming behavior from a fostering rat mother leads to a specific tableau of stress resilience in the pups as adults (61). They showed that a positive social environment increased expression of the glucocorticoid receptor in the hippocampus, strengthened glucocorticoid feedback sensitivity, reduced the expression of the hypothalamic corticotrophin-releasing hormone, and culminated in a milder hypothalamic–pituitary–adrenal stress response (61, 81–83). At the molecular level, a rich early-life social environment (high maternal care) increases levels of 5-HT, and through a 5-HT signaling cascade, leads to high levels of nerve growth factor-inducible protein A (NGFI-A) (82). Rodent studies have shown that NGFI-A is a transcription factor that binds to the promoter of the glucocorticoid receptor gene NR3C1 at exon 17. NGFI-A recruits histone acetyltransferases (i.e., CREB-binding protein) and together they enable the demethylation of the glucocorticoid receptor promoter region (81). By acetylating the promoter region, and demethylating it, a rich early-life environment thus triggers an epigenetic change at the promoter region of the glucocorticoid receptor gene, which results in its up-regulation. The resulting increase in glucocorticoid receptor concentration in turn translates into a healthier stress response (less circulating glucocorticoids following stress) (84).
Critically, the reverse is also true. A stressful early-life environment such as that caused by poverty, abuse, rape, or war can induce powerful epigenetic changes. For example, epigenetic modifications to the glucocorticoid receptor have been found in individuals that have suffered childhood abuse, and these modifications dramatically increased their risk of suicide (60). Stressful environments lead to a decrease in glucocorticoid receptor expression, since the signaling cascade described above cannot be initiated, and the promoter of the glucocorticoid receptor gene is therefore not demethylated (81, 82). This epigenetic alteration remains until adulthood. The resulting down-regulation of the glucocorticoid receptor leads to chronically increased levels of circulating glucocorticoids, reduced glucocorticoid feedback sensitivity, and increased anxiety among other effects (61, 84, 85). The implications of this tableau are higher susceptibility to stress-related health problems, such as autoimmune diseases, cardiovascular diseases, anxiety, and depression – all observed following SCI.
Epigenetics is already used in the development of anti-cancer therapeutic drugs in oncology (86), and neuropsychiatry (87, 88). Proposals have also been presented highlighting the potential of epigenetics in controlling chronic pain (89–91). Even in injury recovery research, scientists have begun to recognize the potential of epigenetics, namely, in traumatic brain injury and stroke (92–94). Yet, little is currently known about the role of environmentally triggered epigenetic changes in SCI and recovery.
Chronic Stress and SCI
Glucocorticoid resistance and epigenetics explain two different modifications of glucocorticoid receptor function, but one can safely argue that both may occur in synergy. When individuals suffer an SCI, for example, placing them at high-risk of suffering from chronic psychological stress (95, 96), past exposure to a stressful environment resulting in epigenetic changes that compromise glucocorticoid receptor sensitivity and function would lead to high levels of inflammation and apoptosis post-SCI, decreasing the tissue remaining at the injury site and undermining recovery of function.
Figure 3 illustrates the proposed glucocorticoid receptor mechanism through which changes triggered by the early-life and adult life environment could affect patients’ psychological stress response, and it in turn could influence processes of inflammation and apoptosis important to SCI injury and recovery. In early-life, a non-stressful environment (indicated by the happy face icon in Figure 3), can promote stress resilience. This in turn leads to decreased levels of circulating glucocorticoids, and a decreased psychological stress response. Conversely, a stressful environment (indicated by the sad face icon in Figure 3) caused, for example, by physical or emotional abuse, poverty, or racism can decrease stress resilience. As a result, there are increased levels of circulating glucocorticoids, and an increased psychological stress response. Likewise, following SCI in adult life, a “non-stressful” environment (i.e., strong social support and good coping skills) may contribute to decreased psychological stress response. Again, in this case too, a stressful environment (for example due to lack of social support, perceived loneliness, or discrimination) may contribute to an increased psychological stress response. Importantly, the glucocorticoid receptors are not only involved in the stress response, but also in the inflammation and apoptosis processes. Therefore, as shown in Figure 3, these mechanisms do not only impact the psychological stress response, but also the processes of inflammation and apoptosis.
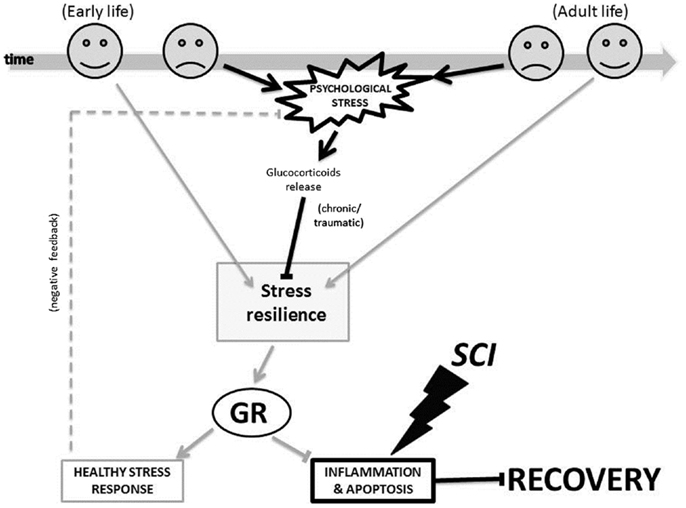
Figure 3. The glucocorticoid receptor mechanism proposed for how environment-triggered epigenetic changes affect SCI recovery. The bold black lines indicate a negative process and the light gray lines indicate a positive process in our model. Arrows (→) indicate that a process is enabled, and bars (–|) indicate that a process is inhibited. The dotted line indicates a feedback loop. GR, glucocorticoid receptor; SCI, spinal cord injury.
In addition to the increase in inflammation due to chronically elevated glucocorticoids levels and the glucocorticoid resistance that ensues, protracted exposure to cytokines, as a result of illness (i.e., SCI) or chronic stress, can also affect glucocorticoid receptor function and lead to increased inflammation. The signaling pathways of pro-inflammatory cytokines, such as NF-κB, MAPKs, and cyclooxygenase can affect the translocation of glucocorticoid receptors from the cytoplasm of the cell to the nucleus, or the ability of the glucocorticoid receptor to function as a transcription factor (97, 98). The result is increased production of pro-inflammatory cytokines.
This is paramount to SCI research, as chronic elevated pro-inflammatory cytokine levels can harm functional recovery, and contribute to the development of chronic pain (64, 99). Indeed, chronic inflammation resulting from the injury leads to a wide range of clinical conditions such as decreased pulmonary function (100), neuropathic pain (101–103), compromised physical recovery (104, 105), and exacerbated depressive symptoms (106). Moreover, SCI-induced chronic inflammation can result in infections and cardiovascular diseases, two leading causes of death after SCI (107).
Clinical Significance
We have illustrated one mechanism through which environmentally triggered changes in stress resilience can affect the health of patients with SCI. We have reviewed how the environment can affect the function of the glucocorticoid receptor, which in turn will affect anti-inflammatory and anti-apoptotic cellular processes in the injured spinal cord. When the glucocorticoid receptors’ function is compromised by environment-triggered changes in stress resiliency, the protective effects of glucocorticoids is diminished, resulting in increased inflammation and apoptosis.
Critically, these mechanisms involving both pre- and post-SCI environments may work in synergy to influence the function of the glucocorticoid receptor. It can be predicted, for example, that a stressful pre-SCI environment combined with a stressful post-SCI environment may both increase the psychological stress response, triggering changes in stress resilience, which will result in decreased glucocorticoid receptor function. Glucocorticoid receptors’ function to inhibit inflammation and apoptosis by inhibiting NF-κB and aiding Bcl-2, respectively, will therefore be compromised. This may increase inflammation and apoptosis, both processes harmful to SCI recovery, as they can contribute to chronic pain, depression, neural loss, and motor impairment. In sum, given the modulatory role of the glucocorticoid receptor in the NF-κB and Bcl-2 pathways, patients’ environment and their psychological stress response are likely to play a role in their recovery after SCI. Indeed, these factors may partially explain individual differences in functional gain.
Given the modulatory role of the glucocorticoid receptor in the NF-κB and Bcl-2 pathways of inflammation and apoptosis, respectively, the environment-triggered epigenetic activation of the glucocorticoid receptor gene by acetylation and demethylation may also hold the potential to reduce inflammation and apoptosis post-SCI. The potential of this approach is underscored by findings of neuroprotective properties of valproic acid after SCI. Valproic acid up-regulates the expression of the anti-apoptotic regulator Bcl-2 (108, 109). By inhibiting deacetylation, it increases the expression of the brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF), among others, thus helping regenerate damaged neurons (110). Abdanipour et al. (109) recently found that intraperitoneal administration of valproic acid to spinally contused rats reduced mRNA expression of cytokines that regulate inflammation. Administration of this HDACi thus helped to reduce secondary damage. Subjects who received a dose of 400 mg/kg showed higher locomotor recovery scores (Basso, Beattie, and Bresnahan rating scale) than non-treated contused subjects by day 28 post-injury (109). Research on the epigenetic changes induced by a stressful environment could help develop psychosocial therapies targeted to at-risk patients so that they may cope better with the stress post-SCI and consequently have a more successful recovery.
Indeed, a combination of epigenetic-based pharmacotherapy with psychosocial therapy may be optimal given their possible synergistic effects. In a mouse traumatic brain injury model, an enriched environment post-injury was recently shown to improve memory function. The enriched environment, consisting of housing four mice together in a cage with a running wheel and toys, increased general acetylation and methylation of histone 3 and 4 in the hippocampus and cortex, thus facilitating synaptic plasticity (111). Based on this work, Dash et al. (112) then showed that behavioral training also improves spatial learning and memory post-traumatic brain injury. Furthermore, they found that an HDACi, sodium butyrate, potentiated the level of improvements observed in traumatic brain injured mice, when, and only when, combined with concurrent behavioral training. In other words, histone deacetylase inhibitor treatment alone did not improve memory post-traumatic brain injury, and receiving behavioral training prior to HDACi treatment did not improve outcome either; it is the combination of both behavioral therapy and pharmacotherapy that promoted recovery. Given that the spinal cord is capable of learning (113, 114), it is possible that SCI recovery too may benefit from HDACi treatment administered concurrent with physical training.
Overall, these epigenetic and psychological stress findings underscore the importance of including psychological evaluations and treatments in the post-SCI intervention. Greater collaboration between the various types of health care providers involved in the post-SCI treatment plan, including physicians, psychiatrists, nurses, psychologists, physical therapists, occupational therapists, and speech therapists, will be essential in providing more successful treatment. For example, if a psychologist and psychiatrist, with the help of the patient and his family, can determine whether the patient has experienced traumatic events prior to the injury, or has any history of anxiety or depression, a treatment plan including more immediate and elaborate psychological therapy could be provided to minimize the negative consequences of SCI-associated stress. Likewise, nurses could be informed to encourage a social support team for the patient, composed of family members, friends, and even hospital volunteers, in order to promote the patient’s psychological well-being. These seemingly small changes, in addition to formal psychosocial therapy and drug treatment could be favorable to the patients’ recovery.
Conclusion
We have reviewed one mechanism, via the function of the glucocorticoid receptor and the psychological stress response to the environment, which may account for a part of the unexplained variance found in patients’ recovery following SCI. Based on the current literature, two potential mechanisms involved are the processes of inflammation and apoptosis: the NF-κB and the Bcl-2 pathway, respectively. It must be noted that the action of glucocorticoid receptors is only one pathway that may explain the effect of the environment on spinal cord. Certainly, other relevant pathways involve the reaction of immune cells such as lymphocytes, macrophages, monocytes, natural killer cells, and T cells to the glucocorticoids and catecholamines released by the brain following a stressor. For instance, glucocorticoids and catecholamines inhibit the expression of Substance P (115), a neuropeptide, and its receptor (116). This neuropeptide induces macrophages to release pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, as well as prostaglandin and thromboxane, also involved in inflammation (117). Likewise, in the central nervous system, stress-triggered release of glucocorticoids activates microglia, and sensitizes them to future inflammatory insults (118). The potential synergistic effects of these various glucocorticoid–immune interactions are likely to attenuate physical recovery after SCI.
Regulation of these substrates through epigenetic mechanisms may significantly improve SCI recovery, not only by opening a window of opportunities for new targeted and personalized treatments combining both psychosocial therapies and epigenetic therapies, but also by allowing physicians to identify SCI patients at higher risk for a more difficult recovery – both physical and psychological. Future research will be essential in determining the main epigenetic modifications that positively or negatively affect the recovery from SCI.
Author Contributions
Sioui Maldonado Bouchard wrote the manuscript, conducted the literature review, and proposed the hypothesis put forth. Michelle A. Hook provided extensive feedback and proof-reading, and contributed to the final form of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Jane Welsh, Dr. Sarah Woller, and Miriam Aceves for their comments on a previous version of this manuscript. This study was supported by the National Institute of Drug Abuse (DA31197) to Michelle A. Hook.
References
1. World Health Organization. International Classification of Functioning, Disability, and Health (Resolution WHA 54.21) Geneva: WHO (2001). WHA 5421.
2. Wilson JR, Cadotte DW, Fehlings MG. Clinical predictors of neurological outcome, functional status, and survival after traumatic spinal cord injury: a systematic review. J Neurosurg (2012) 17(1):11–26. doi:10.3171/2012.4.AOSPINE1245
3. Miller G, Chen E, Cole SW. Health psychology: developing biologically plausible models linking the social world and physical health. Annu Rev Psychol (2009) 60:501–24. doi:10.1146/annurev.psych.60.110707.163551
4. Sandberg S, Paton JY, Ahola S, McCann DC, McGuinness D, Hillary CR, et al. The role of acute and chronic stress in asthma attacks in children. Lancet (2000) 356(9234):982–7. doi:10.1016/S0140-6736(00)02715-X
5. Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA (2007) 298(14):1685–7. doi:10.1001/jama.298.14.1685
6. Leserman J, Petitto JM, Gu H, Gaynes BN, Barroso J, Golden RN, et al. Progression to AIDS, a clinical AIDS condition and mortality: psychosocial and physiological predictors. Psychol Med (2002) 32(6):1059–73. doi:10.1017/S0033291702005949
7. Shin JC, Goo HR, Yu SJ, Kim DH, Yoon SY. Depression and quality of life in patients within the first 6 months after the spinal cord injury. Ann Rehabil Med (2012) 36(1):119–25. doi:10.5535/arm.2012.36.1.119
8. Post MW, van Leeuwen CM. Psychosocial issues in spinal cord injury: a review. Spinal Cord (2012) 50(5):382–9. doi:10.1038/sc.2011.182
9. Boakye M, Leigh BC, Skelly AC. Quality of life in persons with spinal cord injury: comparisons with other populations. J Neurosurg Spine (2012) 17(1):29–37. doi:10.3171/2012.6.AOSPINE1252
10. Lohne V, Severinsson E. Patients’ experiences of hope and suffering during the first year following acute spinal cord injury. J Clin Nurs (2005) 14(3):285–93. doi:10.1111/j.1365-2702.2004.01088.x
11. Muller R, Peter C, Cieza A, Geyh S. The role of social support and social skills in people with spinal cord injury – a systematic review of the literature. Spinal Cord (2012) 50(2):94–106. doi:10.1038/sc.2011.116
12. Botticello AL, Chen Y, Cao Y, Tulsky DS. Do communities matter after rehabilitation? The effect of socioeconomic and urban stratification on well-being after spinal cord injury. Arch Phys Med Rehabil (2011) 92(3):464–71. doi:10.1016/j.apmr.2010.08.028
13. Saunders LL, Krause JS, Acuna J. Association of race, socioeconomic status, and health care access with pressure ulcers after spinal cord injury. Arch Phys Med Rehabil (2012) 93(6):972–7. doi:10.1016/j.apmr.2012.02.004
14. Catalano D, Chan F, Wilson L, Chiu C-Y, Muller VR. The buffering effect of resilience on depression among individuals with spinal cord injury: a structural equation model. Rehabil Psychol (2011) 56(3):200–11. doi:10.1037/a0024571
15. Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol (2002) 21(6):531–41. doi:10.1037/0278-6133.21.6.531
16. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci (2012) 1261:55–63. doi:10.1111/j.1749-6632.2012.06633.x
17. Ladd CO, Thrivikraman KV, Huot RL, Plotsky PM. Differential neuroendocrine responses to chronic variable stress in adult Long Evans rats exposed to handling-maternal separation as neonates. Psychoneuroendocrinology (2005) 30(6):520–33. doi:10.1016/j.psyneuen.2004.12.004
18. Baum A. Stress, intrusive imagery, and chronic distress. Health Psychol (1990) 6:653–75. doi:10.1037/0278-6133.9.6.653
19. Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron (2009) 64(1):33–9. doi:10.1016/j.neuron.2009.09.032
20. Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med (1997) 48:231–40. doi:10.1146/annurev.med.48.1.231
21. González S, Moses DF, De Nicola AF. Glucocorticoid receptors and enzyme induction in the spinal cord of rats: effects of acute transection. J Neurochem (1990) 54(3):834–40. doi:10.1111/j.1471-4159.1990.tb02327.x
22. Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol (2006) 171(1):72–85. doi:10.1016/j.jneuroim.2005.09.012
23. Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Role of molecular chaperones in steroid receptor action. Essays Biochem (2004) 40:41–58.
24. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell (2005) 18(3):331–42. doi:10.1016/j.molcel.2005.03.025
25. Buchheit T, Van de Ven T, Shaw A. Epigenetics and the transition from acute to chronic pain. Pain Med (2012) 13(11):1474–90. doi:10.1111/j.1526-4637.2012.01488.x
26. Beck IME, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev (2009) 30(7):830–82. doi:10.1210/er.2009-0013
27. Carson MJ, Thrash JC, Walter B. The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin Neurosci Res (2006) 6(5):237–45. doi:10.1016/j.cnr.2006.09.004
28. Hayden MS, Ghosh S. Shared principles in NF-kB signaling. Cell (2008) 132(3):344–62. doi:10.1016/j.cell.2008.01.020
29. Yan P, Xu J, Li Q, Chen S, Kim G-M, Hsu CY, et al. Glucocorticoid receptor expression in the spinal cord after traumatic injury in adult rats. J Neurosci (1999) 19(21):9355–63.
30. Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. N Engl J Med (1990) 322(20):1405–11. doi:10.1056/NEJM199005173222001
31. Hunsberger JG, Austin DR, Chen G, Manji HK. Cellular mechanisms underlying affective resiliency: the role of glucocorticoid receptor- and mitochondrially-mediated plasticity. Brain Res (2009) 1293:76–84. doi:10.1016/j.brainres.2009.06.103
32. Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A (2009) 106(9):3543–8. doi:10.1073/pnas.0812671106
33. Bydon M, Lin J, Macki M, Gokaslan ZL, Bydon A. The current role of steroids in acute spinal cord injury. World Neurosurg (2013). doi:10.1016/j.wneu.2013.02.062
34. Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the third national acute spinal cord injury randomized controlled trial. National acute spinal cord injury study. JAMA (1997) 277(20):1597–604.
35. Chikuda H, Yasunaga H, Takeshita K, Horiguchi H, Kawaguchi H, Ohe K, et al. Mortality and morbidity after high-dose methylprednisolone treatment in patients with acute cervical spinal cord injury: a propensity-matched analysis using a nationwide administrative database. Emerg Med J (2013) 31(3):201–6. doi:10.1136/emermed-2012-202058
36. Hugenholtz H. Methylprednisolone for acute spinal cord injury: not a standard of care. CMAJ (2003) 168(9):1145–6.
37. Hurlbert RJ. The role of steroids in acute spinal cord injury: an evidence-based analysis. Spine (Phila Pa 1976) (2001) 26(24 Suppl):S39–46. doi:10.1097/00007632-200112151-00009
38. Nesathurai S. Steroids and spinal cord injury: revisiting the NASCIS 2 and NASCIS 3 trials. J Trauma (1998) 45(6):1088–93. doi:10.1097/00005373-199812000-00021
39. Coleman WP, Benzel E, Cahill DW, Ducker T, Geisler F, Green B, et al. A critical appraisal of the reporting of the national acute spinal cord injury studies (II and III) of methylprednisolone in acute spinal cord injury. J Spinal Disord (2000) 13(3):185–99. doi:10.1097/00002517-200006000-00001
40. Bracken MB. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine (Phila Pa 1976) (2001) 26(24 Suppl):S47–54. doi:10.1097/00007632-200112151-00010
41. Lee J-M, Yan P, Xiao Q, Chen S, Lee K-Y, Hsu CY, et al. Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury. J Neurosci (2008) 28(12):3141–9. doi:10.1523/jneurosci.5547-07.2008
42. Braughler MJ, Hall ED. Acute enhancement of spinal cord synaptosomal (Na+ + K+)-ATPase activity in cats following intravenous methylprednisolone. Brain Res (1981) 219(2):464–9. doi:10.1016/0006-8993(81)90311-5
43. Anderson DK, Saunders RD, Demediuk P, Dugan LL, Braughler JM, Hall ED, et al. Lipid hydrolysis and peroxidation in injured spinal cord: partial protection with methylprednisolone or vitamin E and selenium. Cent Nerv Syst Trauma (1985) 2(4):257–67.
44. Ray SK, Wilford GG, Matzelle DC, Hogan EL, Banik NL. Calpeptin and methylprednisolone inhibit apoptosis in rat spinal cord injury. Ann N Y Acad Sci (1999) 890(1):261–9. doi:10.1111/j.1749-6632.1999.tb08001.x
45. Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A (1994) 91(2):752–6. doi:10.1073/pnas.91.2.752
46. Huang X, Gong R, Li X, Virtue A, Yang F, Yang IH, et al. Identification of novel pre-translational regulatory mechanisms for NF-κB activation. J Biol Chem (2013) 288(22):15628–40. doi:10.1074/jbc.M113.460626
47. Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci (1998) 18(9):3251–60.
48. Xu J, Fan G, Chen S, Wu Y, Xu XM, Hsu CY. Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res (1998) 59(2):135–42. doi:10.1016/S0169-328X(98)00142-9
49. Nelson DE, Ihekwaba AEC, Elliott M, Johnson JR, Gibney CA, Foreman BE, et al. Oscillations in NF-κB signaling control the dynamics of gene expression. Science (2004) 306(5696):704–8. doi:10.1126/science.1099962
50. Han X, Lu M, Wang S, Lv D, Liu H. Targeting IKK/NF-kappaB pathway reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci Lett (2012) 511(1):28–32. doi:10.1016/j.neulet.2012.01.030
51. Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A (2012) 109(16):5995–9. doi:10.1073/pnas.1118355109
52. Meagher MW, Johnson RR, Young EE, Vichaya EG, Lunt S, Hardin EA, et al. Interleukin-6 as a mechanism for the adverse effects of social stress on acute Theiler’s virus infection. Brain Behav Immun (2007) 21(8):1083–95. doi:10.1016/j.bbi.2007.05.001
53. Pedersen A, Zachariae R, Bovbjerg DH. Influence of psychological stress on upper respiratory infection – a meta-analysis of prospective studies. Psychosom Med (2010) 72(8):823–32. doi:10.1097/PSY.0b013e3181f1d003
54. Téo FH, de Oliveira RTD, Mamoni RL, Ferreira MCS, Nadruz W Jr, Coelho OR, et al. Characterization of CD4+CD28null T cells in patients with coronary artery disease and individuals with risk factors for atherosclerosis. Cell Immunol (2013) 281(1):11–9. doi:10.1016/j.cellimm.2013.01.007
55. DeRijk R, Michelson D, Karp B, Petrides J, Galliven E, Deuster P, et al. Exercise and circadian rhythm-induced variations in plasma cortisol differentially regulate interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNFα) production in humans: high sensitivity of TNFα and resistance of IL-6. J Clin Endocrinol Metab (1997) 82(7):2182–91. doi:10.1210/jc.82.7.2182
56. DeRijk RH, Petrides J, Deuster P, Gold PW, Sternberg EM. Changes in corticosteroid sensitivity of peripheral blood lymphocytes after strenuous exercise in humans. J Clin Endocrinol Metab (1996) 81(1):228–35. doi:10.1210/jc.81.1.228
57. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab (2008) 93(5):1563–72. doi:10.1210/jc.2008-0040
58. Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids (2012) 77(11):1041–9. doi:10.1016/j.steroids.2012.05.007
59. Keenan CR, Salem S, Fietz ER, Gualano RC, Stewart AG. Glucocorticoid-resistant asthma and novel anti-inflammatory drugs. Drug Discov Today (2012) 17(17–18):1031–8. doi:10.1016/j.drudis.2012.05.011
60. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12(3):342–8. doi:10.1038/nn.2270
61. Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science (1997) 277(5332):1659–62. doi:10.1126/science.277.5332.1659
62. Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol (2001) 280(6):R1799–805.
63. Feodorova YN, Sarafian VS. Psychological stress – cellular and molecular mechanisms. Folia Med (Plovdiv) (2012) 54(3):5–13.
64. Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res (2009) 175:125–37. doi:10.1016/S0079-6123(09)17508-8
65. DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain (2001) 90(1–2):1–6. doi:10.1016/S0304-3959(00)00490-5
66. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci (2007) 10(11):1361–8. doi:10.1038/nn1992
67. Vogelzangs N, Beekman ATF, de Jonge P, Penninx BW. Anxiety disorders and inflammation in a large adult cohort. Transl Psychiatry (2013) 3:e249. doi:10.1038/tp.2013.27
68. Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci (2009) 34(1):4–20.
69. Anisman H, Merali Z. Cytokines, stress, and depressive illness. Brain Behav Immun (2002) 16(5):513–24. doi:10.1016/s0889-1591(02)00009-0
70. Scammell JG, Denny WB, Valentine DL, Smith DF. Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three new world primates. Gen Comp Endocrinol (2001) 124(2):152–65. doi:10.1006/gcen.2001.7696
71. Fani N, Gutman D, Tone EB. FKBP5 and attention bias for threat: associations with hippocampal function and shape. JAMA Psychiatry (2013) 70(4):392–400. doi:10.1001/2013.jamapsychiatry.210
72. Wald I, Degnan KA, Gorodetsky E, Charney DS, Fox NA, Fruchter E, et al. Attention to threats and combat-related posttraumatic stress symptoms: prospective associations and moderation by the serotonin transporter gene. JAMA Psychiatry (2013) 70(4):401–8. doi:10.1001/2013.jamapsychiatry.188
73. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16(1):33–41. doi:10.1038/nn.3275
74. Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones (2004) 9(3):243–52. doi:10.1379/CSC-32R.1
75. Stechschulte LA, Sanchez ER. FKBP51 – a selective modulator of glucocorticoid and androgen sensitivity. Curr Opin Pharmacol (2011) 11(4):332–7. doi:10.1016/j.coph.2011.04.012
76. Denny WB, Prapapanich V, Smith DF, Scammell JG. Structure-function analysis of squirrel monkey FK506-binding protein 51, a potent inhibitor of glucocorticoid receptor activity. Endocrinology (2005) 146(7):3194–201. doi:10.1210/en.2005-0027
77. Franco MC, Ye Y, Refakis CA, Feldman JL, Stokes AL, Basso M, et al. Nitration of Hsp90 induces cell death. Proc Natl Acad Sci U S A (2013) 110(12):E1102–11. doi:10.1073/pnas.1215177110
78. Szyf M. The early life environment and the epigenome. Biochim Biophys Acta (2009) 1790(9):878–85. doi:10.1016/j.bbagen.2009.01.009
80. Szyf M, McGowan P, Meaney MJ. The social environment and the epigenome. Environ Mol Mutagen (2008) 49(1):46–60. doi:10.1002/em.20357
81. Weaver ICG, D’Alessio AC, Brown SE, Hellstrom IC, Dymov S, Sharma S, et al. The transcription factor nerve growth factor-inducible protein A mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci (2007) 27(7):1756–68. doi:10.1523/jneurosci.4164-06.2007
82. Hellstrom IC, Dhir SK, Diorio JC, Meaney MJ. Maternal licking regulates hippocampal glucocorticoid receptor transcription through a thyroid hormone-serotonin-NGFI-A signalling cascade. Philos Trans R Soc Lond B Biol Sci (2012) 367(1601):2495–510. doi:10.1098/rstb.2012.0223
83. Francis D, Diorio J, Liu D, Meaney M. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science (1999) 286(5442):1155–8. doi:10.1126/science.286.5442.1155
84. Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci (2009) 10(6):446–57. doi:10.1038/nrn2649
85. Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7(8):847–54. doi:10.1038/nn1276
86. Jones PA, Laird PW. Cancer-epigenetics comes of age. Nat Genet (1999) 21:163–7. doi:10.1038/5947
87. Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A (2006) 103(5):1587–92. doi:10.1073/pnas.0510341103
88. Lee TS, Ng BY, Lee WL. Neuropsychiatry – an emerging field. Ann Acad Med Singapore (2008) 37(7):601–5.
89. Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RW, et al. Epigenetic modulation of mglu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol (2009) 75(5):1014–20. doi:10.1124/mol.108.054346
90. Doehring A, Geisslinger G, Lötsch J. Epigenetics in pain and analgesia: an imminent research field. Eur J Pain (2011) 15:11–6. doi:10.1016/j.ejpain.2010.06.004
91. Crow M, Denk F, McMahon SB. Genes and epigenetic processes as prospective pain targets. Genome Med (2013) 5:12. doi:10.1186/gm416
92. Qureshi IA, Mehler MF. The emerging role of epigenetics in stroke. III. Neural stem cell biology and regenerative medicine. Arch Neurol (2011) 68(3):294–302. doi:10.1001/archneurol.2011.6
93. Qureshi IA, Mehler MF. Emerging role of epigenetics in stroke: part 1. DNA methylation and chromatin modifications. Arch Neurol (2010) 67(11):1316–22. doi:10.1001/archneurol.2010.275
94. Qureshi IA, Mehler MF. The emerging role of epigenetics in stroke. II. RNA regulatory circuitry. Arch Neurol (2010) 67(12):1435–41. doi:10.1001/archneurol.2010.300
95. Rintala DH, Loubser PG, Castro J, Hart KA, Fuhrer MJ. Chronic pain in a community-based sample of men with spinal cord injury: prevalence, severity, and relationship with impairment, disability, handicap, and subjective well-being. Arch Phys Med Rehabil (1998) 79(6):604–14. doi:10.1016/S0003-9993(98)90032-6
96. Peter C, Rauch A, Cieza A, Geyh S. Stress, internal resources and functioning in a person with spinal cord disease. NeuroRehabilitation (2012) 30(2):119–30. doi:10.3233/nre-2012-0735
97. Pace TWW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun (2007) 21(1):9–19. doi:10.1016/j.bbi.2006.08.009
98. Rebeyrol C, Saint-Criq V, Guillot L, Riffault L, Corvol H, Chadelat K, et al. Glucocorticoids reduce inflammation in cystic fibrosis bronchial epithelial cells. Cell Signal (2012) 24(5):1093–9. doi:10.1016/j.cellsig.2012.01.005
99. Stemkowski PL, Smith PA. Sensory neurons, ion channels, inflammation and the onset of neuropathic pain. Can J Neurol Sci (2012) 39(4):416–35.
100. Garshick E, Stolzmann KL, Gagnon DR, Morse LR, Brown R. Systemic inflammation and reduced pulmonary function in chronic spinal cord injury. PM R (2011) 3(5):433–9. doi:10.1016/j.pmrj.2011.02.003
101. Sommer C, Schäfers M. Mechanisms of neuropathic pain: the role of cytokines. Drug Discov Today (2004) 1(4):441–8. doi:10.1016/j.ddmec.2004.11.018
102. Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol (2010) 229(1):26–50. doi:10.1016/j.jneuroim.2010.08.013
103. Tenorio G, Kulkarni A, Kerr BJ. Resident glial cell activation in response to perispinal inflammation leads to acute changes in nociceptive sensitivity: implications for the generation of neuropathic pain. Pain (2013) 154(1):71–81. doi:10.1016/j.pain.2012.09.008
104. Bao F, Liu D. Peroxynitrite generated in the rat spinal cord induces neuron death and neurological deficits. Neuroscience (2002) 115(3):839–49. doi:10.1016/S0306-4522(02)00506-7
105. Moon YJ, Lee JY, Oh MS, Pak YK, Park K-S, Oh TH, et al. Inhibition of inflammation and oxidative stress by Angelica dahuricae radix extract decreases apoptotic cell death and improves functional recovery after spinal cord injury. J Neurosci Res (2012) 90(1):243–56. doi:10.1002/jnr.22734
106. Rawdin BS, Mellon SH, Dhabhar FS, Epel ES, Puterman E, Su Y, et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav Immun (2012) 31:143–52. doi:10.1016/j.bbi.2012.11.011
107. National Spinal Cord Injury Statistical Center. Annual Statistical Report. Birmingham: University of Alabama at Birmingham (2004).
108. Lv L, Han X, Sun Y, Wang X, Dong Q. Valproic acid improves locomotion in vivo after SCI and axonal growth of neurons in vitro. Exp Neurol (2012) 233(2):783–90. doi:10.1016/j.expneurol.2011.11.042
109. Abdanipour A, Schluessener HJ, Tiraihi T. Effects of valproic acid, a histone deacetylase inhibitor, on improvement of locomotor function in rat spinal cord injury based on epigenetic science. Iran Biomed J (2012) 16(2):90–100. doi:10.6091/ibj.1060.2012
110. Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol (2009) 2(1):95–109. doi:10.2174/1874-470210902010095
111. Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai L-H. Recovery of learning and memory is associated with chromatin remodelling. Nature (2007) 447(7141):178–82. doi:10.1038/nature05772
112. Dash PK, Orsi SA, Moore AN. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience (2009) 163(1):1–8. doi:10.1016/j.neuroscience.2009.06.028
113. Gomez-Pinilla F, Huie JR, Ying Z, Ferguson AR, Crown ED, Baumbauer KM, et al. BDNF and learning: evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience (2007) 148(4):893–906. doi:10.1016/j.neuroscience.2007.05.051
114. Grau JW, Crown ED, Ferguson AR, Washburn SN, Hook MA, Miranda RC. Instrumental learning within the spinal cord: underlying mechanisms and implications for recovery after injury. Behav Cogn Neurosci Rev (2006) 5(4):191–239. doi:10.1177/1534582306289738
115. Shang Y-X, Wang Y, Han X-H, Zhao S-Q, Wei K-L. Effect of glucocorticoid on substance P content and mRNA expression in lungs of guinea pigs with asthma. Chinese Journal of Contemporary Pediatrics (2004) 6(1):19–22.
116. Ihara H, Nakanishi S. Selective inhibition of expression of the substance P receptor mRNA in pancreatic acinar AR42J cells by glucocorticoids. J Biol Chem (1990) 265(36):22441–5.
117. Black PH, Berman AS. Stress and inflammation. In: Plotnikoff NP, Faith RE, Murgo AJ, Good RA, editors. Cytokines, Stress and Immunity. Boca Raton: CRC Press LLC (1999). p. 115–32.
Keywords: psychological stress, glucocorticoids, inflammation, glucocorticoid resistance, spinal cord injury, NF-κB, Bcl-2, apoptosis
Citation: Maldonado Bouchard S and Hook MA (2014) Psychological stress as a modulator of functional recovery following spinal cord injury. Front. Neurol. 5:44. doi: 10.3389/fneur.2014.00044
Received: 17 November 2013; Accepted: 23 March 2014;
Published online: 09 April 2014.
Edited by:
Elliot J. Roth, Rehabilitation Institute of Chicago, USAReviewed by:
Alan Anschel, Rehabilitation Institute of Chicago, USAJudith R. Sweet, Rehabilitation Institute of Chicago, USA
Copyright: © 2014 Maldonado Bouchard and Hook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sioui Maldonado Bouchard, Department of Neuroscience and Experimental Therapeutics, Texas A&M Health Science Center, Medical Research and Education Building, 8447 State Highway 47, Bryan, TX 77807-3260, USA e-mail:c2lvdWltYkBnbWFpbC5jb20=