 Chin-Hsien Lin
Chin-Hsien Lin Ta-Fu Chen
Ta-Fu Chen Ming-Jang Chiu
Ming-Jang Chiu Han-I Lin
Han-I Lin Ruey-Meei Wu
Ruey-Meei Wu- Department of Neurology, College of Medicine, National Taiwan University Hospital, National Taiwan University, Taipei, Taiwan
Background: The hexanucleotide repeat expansion in intron 1 of the C9orf72 gene is recognized as the most common genetic cause of frontotemporal dementia (FTD). There are overlapping clinical and pathological characteristics between FTD and Parkinsonism syndrome, and some FTD patients may present with Parkinsonism. The aim of this study was to analyze the hexanucleotide repeat numbers of C9orf72 gene in a mixed Taiwanese cohort with FTD, Parkinsonism syndrome, Parkinson’s disease (PD), and Alzheimer’s dementia (AD).
Method: The number of hexanucleotide repeats was estimated in a total of 482 patients with mixed neurodegenerative disorders and 485 control subjects, using a two-step repeat-primed polymerase chain reaction-based genotyping strategy. The individual groups of patients included patients with Parkinsonism syndrome (n = 95), familial PD (n = 109), young-onset PD (n = 201), FTD (n = 9), sporadic AD (n = 61), and early-onset AD (n = 7).
Results: We did not identify any pathogenic repeats (>30 repeats) of C9orf72 in either the patients or control subjects. However, we found one young-onset PD patient and one control subject that each had an intermediate number of repeats (25 and 21 repeats, respectively). The clinical phenotype of the young-onset PD in this patient was similar to typical idiopathic PD without additional features, and the patient responded well to levodopa treatment.
Conclusion: The repeat expansion in C9orf72 is not a common cause of PD, Parkinsonism syndrome, or dementia in our population. Further studies are needed to investigate the clinical and biological significance of intermediate repeats in C9orf72.
Introduction
A massive expansion of the GGGGCC hexanucleotide repeat in the intron between non-coding exons 1a and 1b of the chromosome 9 open reading frame 72 (C9orf72) gene was recently found to be a major genetic cause of familial frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (1, 2). Patients who carry the repeat expansion have an earlier onset, shorter survival, and familial aggregation of dementia or other neurodegenerative disorders than patients with normal repeat number of C9orf72 (3). In addition to FTD and ALS, other features in patients carrying the C9orf72 mutation can include isolated Parkinsonian movement disorder, memory impairment, and signs of cerebellar dysfunction (4). These symptoms tend to accumulate and phenotypes may converge with disease progression. Most patients eventually develop some behavioral abnormalities, as well as language and motor disabilities (5, 6).
Recently, a number of studies have found that Parkinsonism may precede, coincide, or follow the behavioral or language-predominant variant of FTD. FTD with Parkinsonism is part of a growing spectrum of the dementia–Parkinsonism continuum (7–9). The clinical phenotypes include the Parkinsonism syndrome of progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), diffuse Lewy body dementia (DLBD), and typical Parkinson’s disease (PD) (10). The pathological markers of these phenotypes include TAR DNA-binding protein 43 (TDP-43), phosphorylated tau, and tau-negative but ubiquitin-positive neuronal inclusions (6, 11). Therefore, given the clinical and pathological overlaps between FTD, Parkinsonism syndrome, PD, and Alzheimer’s dementia (AD), the aim of this study was to determine whether the abnormal C9orf72 hexanucleotide repeat expansions found in FTD patients is also associated with these other neurodegenerative disorders in a Taiwanese population.
Materials and Methods
Subjects
A total of 482 patients with mixed neurodegenerative disorders and 485 ethnicity matched control subjects were enrolled in the study. The individual groups included patients with Parkinsonism syndrome (n = 95), familial PD (n = 109), young-onset PD (age at onset <50 years, n = 201), FTD (n = 9), sporadic late-onset AD (n = 61), and early-onset AD (age at onset <50 years, n = 7). Mutations in the a-synuclein, Parkin, PINK1, DJ-1, LRRK2, SCA2, SCA3, ATP13A2, and HTRA2 genes were excluded in all young-onset PD patients (12–16).
Patients with PD were diagnosed using the UK brain bank diagnosis criteria (17). Patients with FTD were diagnosed using standard criteria by the Work Group on FTD and Pick’s Disease (18). AD patients were diagnosed by the NINCDS-ADRDA criteria (19). Unrelated adult volunteers without neurological disease were recruited as controls from the community and from our hospital. Informed consent was given by all study participants, and the study was approved by the institutional ethics board committees of National Taiwan University Hospital.
Genetic Analysis
DNA was extracted from venous blood using standard protocols (12). The size of the hexanucleotide repeats in the C9orf72 alleles was detected using a two-step repeat-primed polymerase chain reaction-based genotyping strategy, as previously described (1).
Statistical Analysis
Descriptive statistics were expressed as the mean ± the standard deviation. Differences in the distributions of repeat number between the individual patient groups and controls were tested using a two-tailed Mann–Whitney U-test and significance was set at p = 0.05. Statistical analysis was performed using the STATA, version 8.0.
Results
The demographic data of all tested subjects are summarized in Table 1. We used a previously suggested cutoff (>30 repeats) to distinguish the pathogenic expansion from the normal allele (2). We did not detect any pathological repeat expansions of C9orf72 in either patients or control subjects.
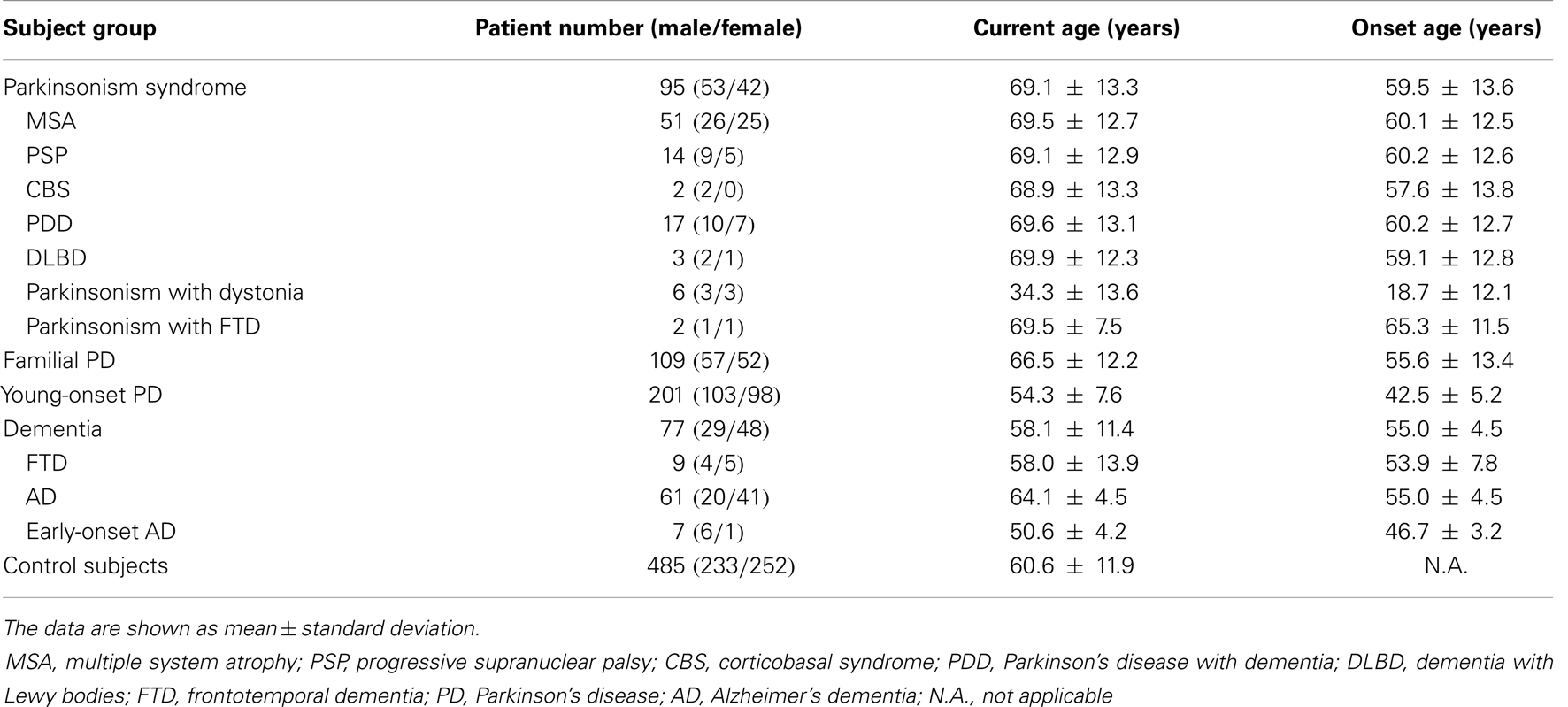
Table 1. Demographic data of the enrolled subjects in each disease group.
The range of repeat expansions detected in our participants was 2–25 units, and the most frequent repeat number was 2 repeats, followed by 6, 7, and 5 repeats. Notably, we found one young-onset PD patient who had an intermediate number of 25 repeats, and one control subject who harbored 21 repeats (Table 2). The clinical phenotype of the young-onset PD in this patient was similar to typical idiopathic PD without additional features, and the patient responded well to levodopa treatment. The distribution of the estimated repeat numbers for each disease group compared to control subjects is presented in Figure 1.
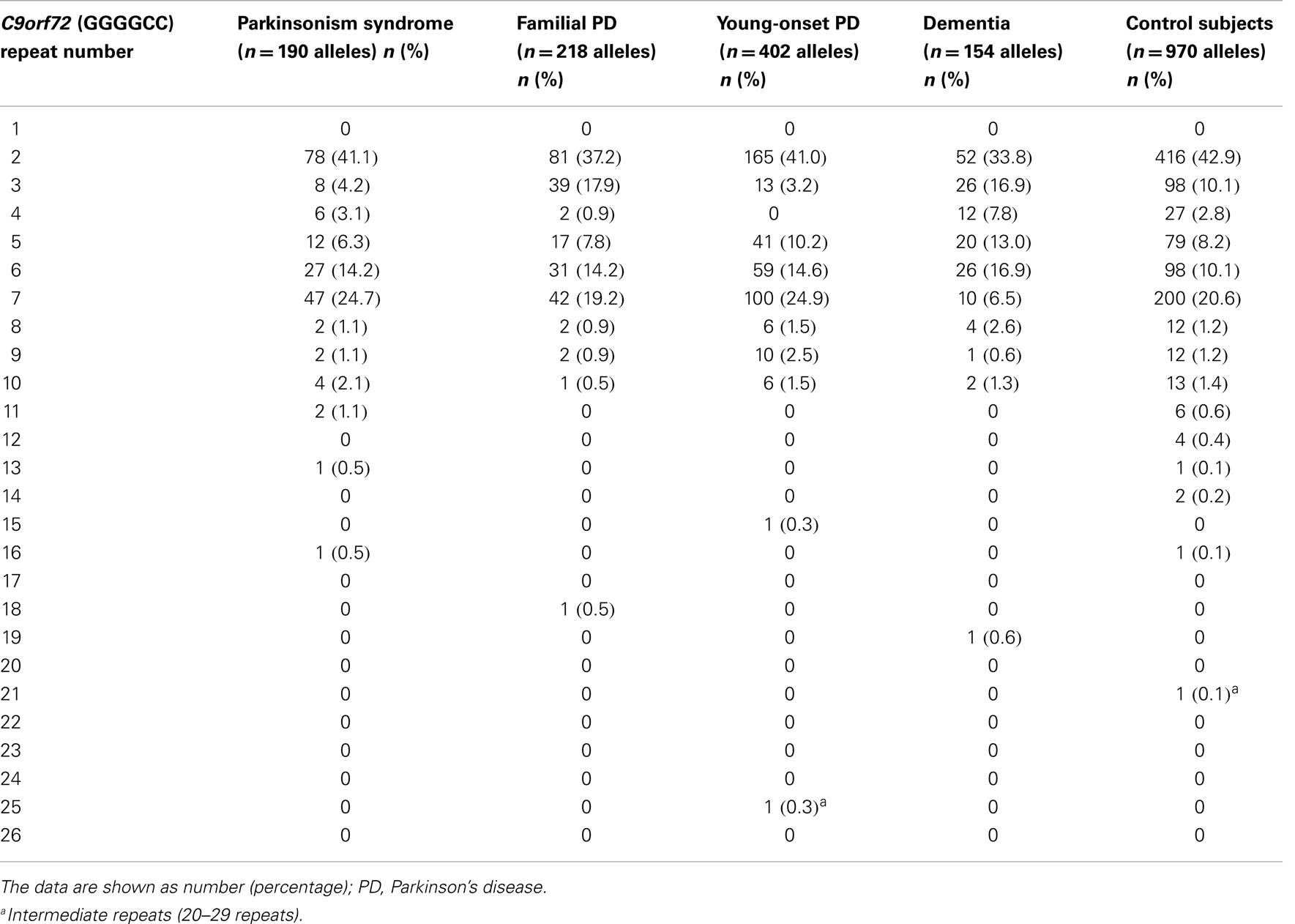
Table 2. Distribution of C9orf72 (GGGGCC) repeat numbers in each disease group and control subjects.
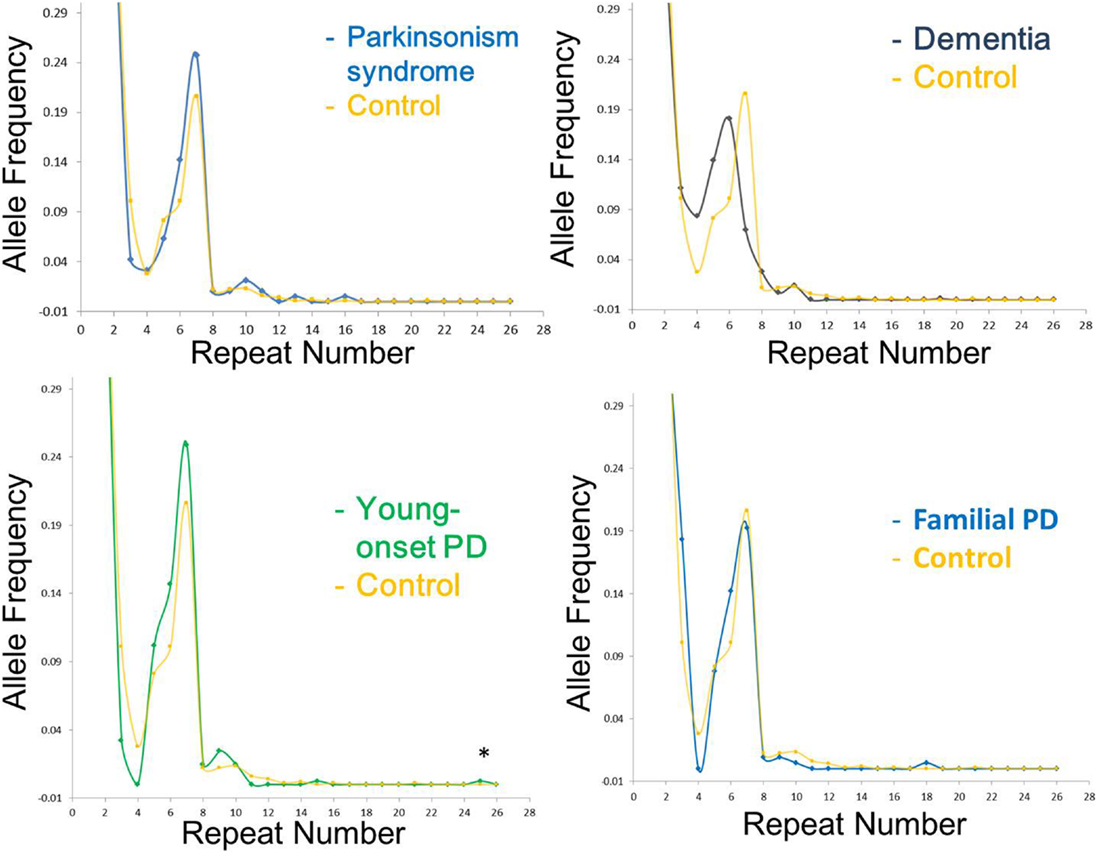
Figure 1. Distribution of C9orf72 (C4G2) repeat number in different disease group as compared to control subjects in the current study. The X-axis represents the estimated repeat number, whereas the Y -axis represents the allele frequency. Asterisk indicates intermediate repeats (20–29 repeats).
Discussion
Numerous studies have supported the concept that various neurodegenerative disorders share overlapping clinical, genetic, and pathologic features. Although the clinical manifestations vary, some patients with FTD can also show features of PD or Parkinsonism syndromes, characterized as PSP, CBS, or DLBD. The identification of a hexanucleotide repeat expansion in the C9orf72 gene as a frequent cause of both FTD and ALS suggests a possible role of this genetic alteration in other dementias or Parkinsonism-related movement disorders. Herein, we demonstrate that the C9orf72 repeat expansion is not a common cause of disease for sporadic AD, FTD, PD, and other related movement disorders in our population.
Given the relatively small sample size of the individual patient groups in our series, we cannot definitively exclude the possibility of a role for the C9orf72 repeat expansion in disease risk. In parallel with our findings, other recent studies have also shown an absence of an association between repeat expansion in the C9orf72 gene and disease susceptibility in AD, PD, or related Parkinsonism syndromes, especially in the Asian population (20–24). We speculate that one of the possible reasons why no expansions have been found in our cases or controls may be due to the fact that carriers of the repeat expansion at C9ORF72 arose from a European single common founder, implying that expansions are rare in non-Caucasian populations (25). Another possible reason is that we did not enroll any patients with ALS and the case number in the FTD group in small in our present study. Therefore, although the abnormal repeats of C9orf72 gene account for 23–47% of familial FTD with or without ALS and 4–20% of sporadic ALS (2, 21, 26), our observation combined with previous findings suggest that variation in the C9orf72 does not play a major role in the susceptibility to the wider spectrum of Parkinsonism and dementia syndromes. Pathological expansion of the C9orf72 hexanucleotide repeats may be specific to TDP-43 pathology-associated FTD and ALS.
Based on the allele frequencies in cases and controls, the first studies suggested that expansions with more than 30 repeats should be considered pathological, while alleles with <20 repeats are wild type (2). However, subsequent reports have found that some control subjects may have repeat numbers of 20–30, with 23 being the most frequently reported maximum repeat number (1, 26). Hence, the contribution of the intermediate-size alleles (20–29 repeats) to disease pathology needs to be further evaluated. Notably, recent studies demonstrated that intermediate expansions of the hexanucleotide repeats in C9orf72 may associate with an increased risk of PD, especially those with repeat numbers over 23 (10, 20, 27–29). In our study, we identified one young-onset PD patient harboring a repeat number of 25. The mechanism by which this intermediately expanded repeat number may cause this disease pathophysiology remains unclear. One hypothesis is that the expanded hexanucleotide repeat may bind to other RNAs, resulting in protein sequestration from normal processing and then decreased protein expression (2, 21). Future functional studies are necessary to elucidate the role of intermediate repeats in C9orf72 in neuronal degeneration.
In conclusion, our results do not suggest that an expanded repeat number in the C9orf72 gene plays a major role in the susceptibility to the wider spectrum of movement disorders. Further large-scale studies are required to investigate the clinical and biological significance of intermediate repeats in the C9orf72 gene in degenerative neurological disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the participants of the study. We also thank the staff of the Second Core Lab, Department of Medical Research, National Taiwan University Hospital for providing expert technical support. This study was supported by a grant from the Ministry of Science and Technology (MOST102-2314-B-002-111-MY3).
References
1. DeJesus-Hernandez M, MacKenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
2. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi:10.1016/j.neuron.2011.09.010
3. van Rheenen W, van Blitterswijk M, Huisman MH, Vlam L, van Doormaal PT, Seelen M, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology (2012) 79:878–82. doi:10.1212/WNL.0b013e3182661d14
4. Rademakers R, Neumann M, MacKenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol (2012) 8:423–34. doi:10.1038/nrneurol.2012.117
5. Uitti RJ, Berry K, Yasuhara O, Eisen A, Feldman H, McGeer PL, et al. Neurodegenerative ‘overlap’ syndrome: clinical and pathological features of Parkinson’s disease, motor neuron disease, and Alzheimer’s disease. Parkinsonism Relat Disord (1995) 1:21–34. doi:10.1016/1353-8020(95)00004-P
6. Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry (2011) 82:476–86. doi:10.1136/jnnp.2010.212225
7. Sundar PD, Yu CE, Sieh W, Steinbart E, Garruto RM, Oyanagi K, et al. Two sites in the MAPT region confer genetic risk for Guam ALS/PDC and dementia. Hum Mol Genet (2007) 16:295–306. doi:10.1093/hmg/ddl463
8. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci (2011) 45:343–9. doi:10.1007/s12031-011-9632-1
9. Seltman RE, Matthews BR. Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs (2012) 26:841–70. doi:10.2165/11640070-000000000-00000
10. Nuytemans K, Bademci G, Kohli MM, Beecham GW, Wang L, Young JI, et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann Hum Genet (2013) 77:351–63. doi:10.1111/ahg.12033
11. Josephs KA, Hodges JR, Snowden JS, MacKenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol (2011) 122:137–53. doi:10.1007/s00401-011-0839-6
12. Wu RM, Bounds R, Lincoln S, Hulihan M, Lin CH, Hwu WL, et al. Parkin mutations and early-onset Parkinsonism in a Taiwanese cohort. Arch Neurol (2005) 62:82–7. doi:10.1001/archneur.62.1.82
13. Lin CH, Hwu WL, Chiang SC, Tai CH, Wu RM. Lack of mutations in spinocerebellar ataxia type 2 and 3 genes in a Taiwanese (ethnic Chinese) cohort of familial and early-onset Parkinsonism. Am J Med Genet B Neuropsychiatr Genet (2007) 144B:434–8. doi:10.1002/ajmg.b.30427
14. Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology (2008) 71:1727–32. doi:10.1212/01.wnl.0000335167.72412.68
15. Lee MJ, Mata IF, Lin CH, Tzen KY, Lincoln SJ, Bounds R, et al. Genotype-phenotype correlates in Taiwanese patients with early-onset recessive Parkinsonism. Mov Disord (2009) 24:104–8. doi:10.1002/mds.22093
16. Lin CH, Chen ML, Chen GS, Tai CH, Wu RM. Novel variant Pro143Ala in HTRA2 contributes to Parkinson’s disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum Genet (2011) 130:817–27. doi:10.1007/s00439-011-1041-6
17. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry (1992) 55:181–4. doi:10.1136/jnnp.55.3.181
18. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol (2001) 58:1803–9. doi:10.1001/archneur.58.11.1803
19. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology (1984) 34:939–44. doi:10.1212/WNL.34.7.939
20. Xi Z, Zinman L, Grinberg Y, Moreno D, Sato C, Bilbao JM, et al. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch Neurol (2012) 69:1583–90. doi:10.1001/archneurol.2012.2016
21. Dejesus-Hernandez M, Rayaprolu S, Soto-Ortolaza AI, Rutherford NJ, Heckman MG, Traynor S, et al. Analysis of the C9orf72 repeat in Parkinson’s disease, essential tremor and restless legs syndrome. Parkinsonism Relat Disord (2013) 19:198–201. doi:10.1016/j.parkreldis.2012.09.013
22. Jiao B, Guo JF, Wang YQ, Yan XX, Zhou L, Liu XY, et al. C9orf72 mutation is rare in Alzheimer’s disease, Parkinson’s disease, and essential tremor in China. Front Cell Neurosci (2013) 7:164. doi:10.3389/fncel.2013.00164
23. Ogaki K, Li Y, Takanashi M, Ishikawa K, Kobayashi T, Nonaka T, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord (2013) 19:15–20. doi:10.1016/j.parkreldis.2012.06.019
24. Yeh TH, Lai SC, Weng YH, Kuo HC, Wu-Chou YH, Huang CL, et al. Screening for C9orf72 repeat expansions in Parkinsonian syndromes. Neurobiol Aging (2013) 34(1311):e1313–4. doi:10.1016/j.neurobiolaging.2012.09.002
25. Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. The C9ORF72 expansion mutation is a common cause of ALS±FTD in Europe and has a single founder. Eur J Hum Genet (2013) 21:102–8. doi:10.1038/ejhg.2012.98
26. Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol (2012) 11:54–65. doi:10.1016/S1474-4422(11)70261-7
27. Majounie E, Abramzon Y, Renton AE, Keller MF, Traynor BJ, Singleton AB. Large C9orf72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol Aging (2012) 33(2527):e2521–2. doi:10.1016/j.neurobiolaging.2012.05.007
28. Daoud H, Noreau A, Rochefort D, Paquin-Lanthier G, Gauthier MT, Provencher P, et al. Investigation of C9orf72 repeat expansions in Parkinson’s disease. Neurobiol Aging (2013) 34:.e1717–9. doi:10.1016/j.neurobiolaging.2012.11.025
Keywords: C9orf72, frontotemporal dementia, Alzheimer’s dementia, Parkinson’s disease, Parkinsonism, risk factor
Citation: Lin C-H, Chen T-F, Chiu M-J, Lin H-I and Wu R-M (2014) Lack of C9orf72 repeat expansion in Taiwanese patients with mixed neurodegenerative disorders. Front. Neurol. 5:59. doi: 10.3389/fneur.2014.00059
Received: 10 March 2014; Accepted: 11 April 2014;
Published online: 28 April 2014.
Edited by:
Tibor Hortobágyi, University of Debrecen, HungaryCopyright: © 2014 Lin, Chen, Chiu, Lin and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruey-Meei Wu, Department of Neurology, College of Medicine, National Taiwan University Hospital, National Taiwan University, No. 7 Chung-Shan South Road, Taipei 100, Taiwan e-mail:cm9iaW53dUBudHUuZWR1LnR3