 Asha Kishore
Asha Kishore Sabine Meunier
Sabine Meunier Traian Popa
Traian Popa- 1Department of Neurology, Comprehensive Care Centre for Movement Disorders, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Kerala, India
- 2Institut du Cerveau et de la Moelle epiniere (ICM), INSERM U1127, CNRS UMR 7225, Université Pierre et Marie Curie-Paris 6 UMR S975, Paris, France
- 3Centre de Neuroimagerie de Recherche (CENIR), l’Institut du Cerveau et de la Moelle epiniere (ICM), Paris, France
Normal motor behavior involves the creation of appropriate activity patterns across motor networks, enabling firing synchrony, synaptic integration, and normal functioning of these networks. Strong topography-specific connections among the basal ganglia, cerebellum, and their projections to overlapping areas in the motor cortices suggest that these networks could influence each other’s plastic responses and functions. The defective striatal signaling in Parkinson’s disease (PD) could therefore lead to abnormal oscillatory activity and aberrant plasticity at multiple levels within the interlinked motor networks. Normal striatal dopaminergic signaling and cerebellar sensory processing functions influence the scaling and topographic specificity of M1 plasticity. Both these functions are abnormal in PD and appear to contribute to the abnormal M1 plasticity. Defective motor map plasticity and topographic specificity within M1 could lead to incorrect muscle synergies, which could manifest as abnormal or undesired movements, and as abnormal motor learning in PD. We propose that the loss of M1 plasticity in PD reflects a loss of co-ordination among the basal ganglia, cerebellar, and cortical inputs which translates to an abnormal plasticity of motor maps within M1 and eventually to some of the motor signs of PD. The initial benefits of dopamine replacement therapy on M1 plasticity and motor signs are lost during the progressive course of disease. Levodopa-induced dyskinesias in patients with advanced PD is linked to a loss of M1 sensorimotor plasticity and the attenuation of dyskinesias by cerebellar inhibitory stimulation is associated with restoration of M1 plasticity. Complimentary interventions should target reestablishing physiological communication between the striatal and cerebellar circuits, and within striato-cerebellar loop. This may facilitate correct motor synergies and reduce abnormal movements in PD.
Introduction
Motor cortex, basal ganglia, and cerebellum have unique architectures and synaptic mechanisms that allow specialized forms of information processing (1). Traditionally, motor cortex is considered to be specialized for unsupervised learning through Hebbian plasticity mechanisms (2). Basal ganglia are considered necessary for selection of wanted movements and inhibition of unwanted movements (3). They are specialized for reinforcement learning, based on reward signals encoded in dopaminergic fibers (4). The cerebellum is thought to fine tune movements in real time and to be specialized for supervised learning, based on error signals encoded in the climbing fibers (5). Such compartmentalized roles, though not clear-cut, were perhaps assigned to these structures because of the then-prevailing model that basal ganglia and cerebellum were distinct, parallel systems, each with reciprocal connections through the thalamus to multiple cortical areas (6). In recent years, direct bisynaptic connections between the motor areas of the dentate nucleus and striatum via thalamus were demonstrated in monkeys (7). Later, topographically organized connections that link the subthalamic nucleus and cerebellum through the pontine nuclei (8) were also identified in monkeys. The subthalamic nucleus projections to the motor area VIIB of cerebellum arise from the sensorimotor territory of subthalamic nucleus, which in turn, is under the influence of M1 and pre-motor areas. These well-organized, topography-specific connections among the motor and pre-motor cortices, basal ganglia, and cerebellum indicate that these structures may interact synergistically in humans. This could mean that the information processed by each of them may significantly influence the functioning of all related structures. If so, abnormal dopaminergic release as in Parkinson’s disease (PD), could affect macro- and micro-circuit functions in the striatum, cerebellum, and sensorimotor cortical areas. Consequently, the burden of symptoms in PD, both motor and non-motor, may result from dysfunctions within this large network. We review here the potential relation between striato-thalamo-cortical and cerebello-thalamo-cortical loops and M1 plasticity impairment in PD, and how their progressive imbalance could lead to continuously deteriorating manifestations.
M1 Plasticity and Motor Learning
M1 is interconnected with the parietal lobe, pre-motor cortex, supplementary motor area, basal ganglia, and cerebellum. This strategic location within a distributed system (9) underlies M1’s pivotal role in motor performance and motor learning. The capacity of M1 to offer behavioral flexibility to motor functions is dependent on its ability to modify its local architecture and promote dynamic motor maps. M1 is organized as motor maps consisting of somatotopically arranged representations of muscle synergies. The muscle synergies can be represented by the weight of neural connections. The cortical areas in which the movement representations are embedded have strong interconnections (10) that are highly dynamic and capable of rapid reorganization (11). Task-specific modification in spatial and temporal organization of muscle synergies results in smooth and accurate movement sequences (12). Encoding of a novel movement sequence in motor cortex results in changes in the weight of connectivity, when sets of movements are performed together (13). The representation strength of a sequence is increased as movements are learned and this comes with an expansion of the cortical motor map encoding the specific movement or skill (14). Such organizational capability of the human M1 was demonstrated using transcranial magnetic stimulation techniques (15, 16). The plastic changes in cortical map are thought to occur through reorganization of cortical micro-circuitry and changes in synaptic efficacy (17). Changes in synaptic efficacy occur first through processes that involve long-term potentiation (LTP) and long-term depression (LTD), and then through changes in synaptic architecture. The pyramidal neurons in M1 have extensive networks of branches that establish horizontal connections within M1 (18). The synaptic efficacy of these interconnections can be enhanced or depressed through LTP and LTD, in response to appropriate activity patterns and contexts (11). M1 is also under the influence of other cortical areas (e.g., parietal, pre-motor) and subcortical structures, including the basal ganglia, cerebellum, and brain stem. The timing of cortico-cortical and subcortico-cortical inputs controls the buildup of LTP and LTD within M1. This translates into change in synaptic strength that facilitates only sensorimotor input from subcortical structures that are relevant to a specific context (11). With repetition, these reversible changes can lead to physical reorganization of intracortical connections. The integrity of motor maps and their topography in M1 are also influenced by neurochemical signals that change the cortical circuitry to encode the motor experience (19). Disrupting cortical circuitry by inhibition of protein synthesis or ischemic injury results in a loss of motor maps and degradation of skilled movement (20). These observations suggest that motor maps and their recruitment pattern required for performance of appropriate movements are based on a high level of synergy within M1, and between M1 and other structures.
Cortico-Striatal Plasticity
Striatum is a major input station of glutamatergic projections from the motor cortices and the thalamus. It is also densely innervated by dopaminergic terminals arising from the substantia nigra pars compacta. The integration of informational flow within the striatum determines the final output to other basal ganglia structures. LTP and LTD are the key cellular substrates for motor control and learning subserved by the striatum (21). Induction of plasticity at the cortico-striatal synapses with the medium spiny neurons (MSNs) requires interaction between dopamine and NMDA receptors. NMDA receptor’s complex modification linked to dopamine D1 receptor activation eventually leads to postsynaptic insertion of AMPA receptors that underlies LTP (22). Both D1 and D2 receptor activation and metabotropic NMDA receptors are involved in LTD. Dopamine has a rapid, reversible action that can transiently alter synaptic integration and microcircuit function to enhance the transfer of specific types of information through the striatum. Additionally, dopamine also has a slow action that can induce persistent changes that outlast the dopaminergic signal and translate to long-term motor memory (23). The striatal MSNs are connected to the pallidal output neurons though the direct and indirect pathways. D1 receptor activation has excitatory effects on striatal MSNs in the direct-pathway while D2 receptor activation has inhibitory effects on MSNs in the indirect pathway (24). It has been proposed that activation of direct-pathway circuits facilitates or selects appropriate movements, while activity in the indirect pathway inhibits inappropriate movements (3, 25). Since the striatal release of dopamine can signal a “reward prediction error” (4, 26), basal ganglia could participate in motor learning through the selection of a motor routine by maintaining wanted and eliminating unwanted movements in a precise temporal sequence. Correctly performed actions would lead to micro self-rewarding results, which reinforce the choice of actions that led to the successful outcome. In course of time, the action sequences associate with each other, allowing the rapid selection of motor routines independent of reward values, thus becoming automatic (23). Therefore, basal ganglia can render movements more efficiently, by comparing the input from the motor cortex with the locally stored motor routines, and predict likely future actions. This type of learning is thought to occur through long-term changes in the strength of striatal synapses (27).
Dopamine and M1 Plasticity
In rats, there is a large dopaminergic projection to M1 through the mesocortical system arising in the ventral tegmental area and the medial substantia nigra. Larger and similar motor cortical innervations are also reported in primates (28), which is not different from humans (29). In rats, mesocortical dopaminergic signaling is necessary for the intracortical and cortico-cortical connections of M1 to form LTP. Blocking dopamine D1 and D2 receptors reduce this ability of M1 (30). In humans, mesocortical projections from the ventral tegmental area to the prefrontal cortex and their role in motor function have been studied (30–32). Both dopamine D1 and D2 receptors are present in human M1 (18) and D2 receptor-blocking drugs can prevent LTP (33). The role of dopaminergic transmission in the ventral tegmental area to M1 projection described in humans (34), in M1 plasticity and motor function (both in health and PD) still need to be explored. Plasticity of human M1 is thought to be influenced by striatal dopaminergic system through the glutamatergic striato-thalamo-cortical pathway. This conclusion is supported by the observation that the lost M1 plasticity in PD recovers after exposure to dopaminergic drugs (35). However, cortical dopaminergic denervation by itself could cause the loss of M1 plasticity in PD and dopaminergic drugs could also act directly through the cortical dopamine receptors restore M1 plasticity in early stages of the disease.
Cerebellar Plasticity
Cerebellum controls and co-ordinates complex movements and is important for adapting movements to changes in feedback. It receives sensory and motor information from descending cortical pathways and ascending peripheral pathways. It has also connections to the parietal, pre-motor, and frontal cortices. The two major excitatory afferents to cerebellum are the climbing fibers and mossy fiber–parallel fiber systems, both of which eventually converge on the Purkinje cells, which are the only efferent output from the cerebellar cortex. The exteroceptive and proprioceptive inputs from the spinal cord and the pontine input convey information from brain stem nuclei via mossy fibers to the granule cells. The axons of granule cells form the parallel fibers network. Climbing fibers originate in the inferior olive and directly relay to the Purkinje cells. Plastic changes in the strength of synapses relaying from the climbing and parallel fibers to the Purkinje cells are important in motor learning (36–40). Moreover, plasticity in both granule cell and Purkinje cell networks are required for motor learning and consolidation (41). In addition, there is inhibitory plasticity at the inhibitory interneuron-PC synapses as well intrinsic plasticity mechanisms within the cerebellum (42). There are several theories on the role of cerebellum in motor learning. It was considered that LTD of parallel fibers–Purkinje cell synapses, which in turn required simultaneous co-activation of parallel fibers and climbing fibers inputs to Purkinje cells, provided the cellular correlate of motor learning (36). More recent observations suggest that climbing fibers signaling has a more complex role. Plasticity of climbing fibers input can additionally fine tune complex spike-associated calcium signaling in Purkinje cells and bi-directionally adjust the plasticity of parallel fibers–Purkinje cells synapses (43). This suggests a role of climbing fibers as an error detector preceding motor learning, which signals the need for adjusting the gain of sensory inputs and/or motor output within the cerebellum (44). Based on the circuit architecture around the Purkinje cells, Penhune and Steele proposed that cerebellum participates in sensorimotor integration, error correction, and formation of internal models (45). In this context, internal models were defined as a set of input–output relations between motor commands and their sensory consequences, the input being the motor command and the output being the predicted sensory consequence of that action. Internal models subsequently allow comparison between predicted and actual consequences of a movement. This would enable gauging the movement error signal that guides learning. The authors suggest that internal models in the parallel fibers–Purkinje cells complexes may be modified based on the information about motor plans from motor cortex and on the error signals transmitted from the inferior olive. This can update the relationship between the command to move and the expected sensory consequence. Any disturbance in this cerebellar processing function may result in maladjusted information delivered to M1, leading to abnormal, undesired or ineffective movement sequences.
Cerebellum and M1 Plasticity
The primary motor cortex is functionally linked specifically with cerebellar lobules V, VI, VIIB, and VIIIA, which are also implicated in motor learning (46, 47). Animal experiments (47–49) and human imaging studies showed that cerebellum is involved in sensory processing besides facilitating motor control and motor learning (50, 51). One view describes the cerebellum function as an adaptive filter (52). It was recently shown that cerebellum plays a very important role in scaling plasticity and influencing topographic specificity of the human M1 through modulation of peripheral sensory afferents (53). In healthy young adults, excitation or inhibition of the posterior cerebellar cortex (using theta-burst transcranial magnetic stimulation) preceding the induction of M1 plasticity, had bidirectional effects on M1 plasticity. Cerebellar cortical excitation led to a loss of response to a subsequent excitatory stimulation protocol pairing somatosensory stimulation paired to TMS applied precisely to target only one muscle representation within M1. In contrast, cerebellar cortical inhibition led to a prolonged plastic response of M1 to the paired-associative stimulation protocol along with a loss of topographic specificity, i.e., changes in both targeted and adjacent, non-targeted muscle representations. This suggested a highly discriminating role of cerebellar excitatory and inhibitory functional outputs to M1. These alterations in the response of M1 following cerebellar modulation were observed for PAS but not for theta-burst transcranial magnetic stimulation (which does not rely on sensory afferent input). This particular behavior highlights the dependence of cerebellar modulation of M1 plasticity on the sensory afferent input. Cerebellar cortical excitation could lead to an enhancement of the normal inhibition of dentate nucleus by the Purkinje cells. This would reduce the normal excitatory control of dentate nucleus on the afferent inflow to M1, probably at the thalamic or olivary nuclear level, thus blocking the sensorimotor-plasticity within M1 (53). The functional relevance of such cerebellar modulation of M1 plasticity could be to prevent the selection of unsuited or new motor programs from sources external to M1 and provide stability to motor maps. In contrast, cerebellar cortical inhibition could lead to disinhibition of dentate nucleus, thus facilitating afferent input to M1 and thereby providing a “controlled instability” of motor maps, which might enable updating the currently selected motor programs by facilitating the insertion of elements of a new motor program.
Cortico-Striatal and M1 Plasticity in Parkinson’s Disease
Parkinson’s disease is characterized by a massive loss of dopaminergic neurons in the midbrain (54) and degeneration of catecholaminergic neurons in other parts of the brainstem (55). Dopamine deficiency at the striatum results in loss of both LTP in the direct pathway and loss of LTD and its replacement by LTP in the indirect pathway (56). Striatal dopamine depletion also leads to enhanced indirect pathway output and decreased direct-pathway output (Figure 1). This results in a decrease in activity in GPe and increase in subthalamic nucleus and GPi both in experimental (57, 58) and human PD (59). As the indirect pathway normally inhibits unwanted movements, the loss of LTD and over-activity in this pathway could result in inhibition of wanted movements and a disruption of learned motor actions. With the loss of LTD, the MSNs in the indirect pathway are liable to increased entrainment to the oscillations in thalamus and cortex through their inputs to striatum (60). As the direct pathway activity normally selects appropriate movements, it’s under activity and loss of LTP could affect initiation and performance of appropriate movements in PD (56, 61). There are intrinsic difficulties in differentiating abnormal motor learning in the presence of abnormal motor performance. Even so, it has been shown that motor learning is abnormal in PD (62, 63). Beeler and colleagues proposed that motor learning may play a significant role in the symptoms of PD and that the long-duration response to chronic levodopa treatment may be a manifestation of rescued motor learning. This was based on observations in the aphakia mouse model that lacked 90% dopamine in the dorsal striatum resulting in impaired new motor learning skills but without motor deficits (64). l-DOPA rescued motor learning and cessation of treatment did not result in an immediate loss of the rescued motor learning skills.
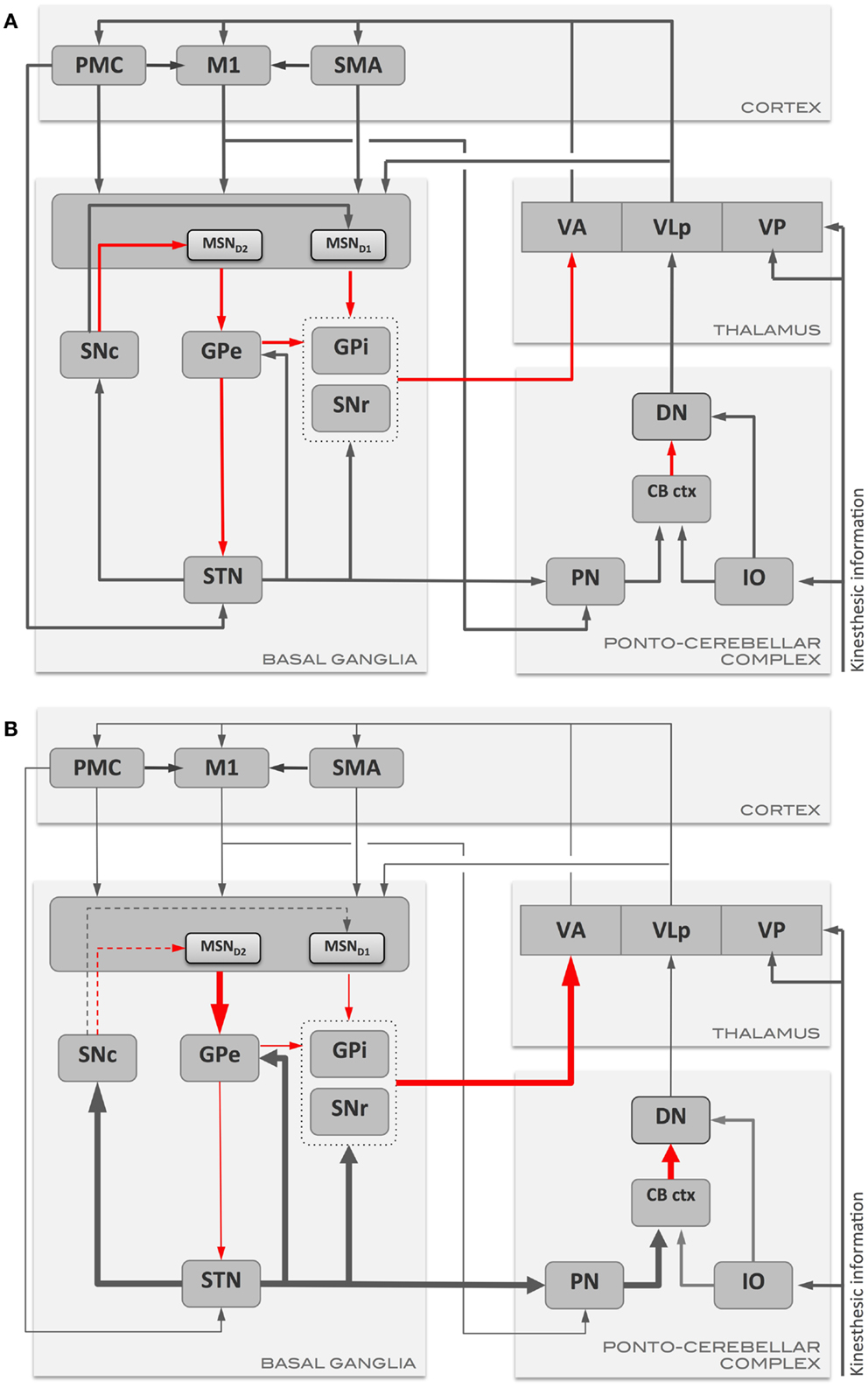
Figure 1. Schematic representation of the main communication pathways between the motor areas, basal ganglia, and ponto-cerebellar complex in (A) health, and in (B) Parkinson’s disease. Gray arrows represent excitatory pathways, and red arrows represent inhibitory pathways. Thickened arrows in (B) represent pathways with augmented output with respect to the normal condition (A), while thinned arrows represent pathways with reduced output. CB ctx, cerebellar cortex; DN, dentate nucleus; GPe, globus pallidus externus; GPi, globus pallidus internus; IO, inferior olive; M1, primary motor cortex; MSN, medium spiny neurons (MSND1/D2: MSN with D1 or D2 dopamine receptors); PMC, pre-motor cortex; PN, pontine nuclei; SMA, supplementary motor area; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus; VA, ventro-anterior thalamic nucleus; VLp, ventro-lateral thalamic nucleus, pars posteriori; VP, ventro-posterior thalamic nucleus.
l-DOPA-responsive, severe impairment of striatal plasticity was first demonstrated in animal models of PD (65). A similar impairment of LTP that responded to l-DOPA was also demonstrated in human substantia nigra pars reticulata (66) in PD patients tested during deep brain stimulation surgery. A decreased response of M1 to LTP- and LTD-inducing protocols was also documented in human M1 in the more affected hemisphere in most studies in PD patients (30, 67, 68). This has been interpreted to reflect an impairment of LTP/LTD mechanisms within M1. The defective plasticity in PD demonstrated at the output neurons of substantia nigra pars reticulata (69) and M1 (67–70) could be secondary to the abnormal signaling in the striatal circuits. However, the loss of direct effects of dopaminergic input from ventral tegmental area on M1 might contribute to the M1 plasticity alterations in PD (67). In de novo PD patients, the intrinsic, plasticity of M1 involving local circuits within M1 (as probed by TMS) was severely and symmetrically impaired in both hemispheres, even when there were only hemi-parkinsonian symptoms (67). The deficient plasticity did not improve with a single dose of l-DOPA even though motor signs of PD improved. Nevertheless, chronic dopamine treatment restored TMS-induced intrinsic plasticity within local M1 circuits in the stable phase of treatment in a manner akin to the long-duration response of motor signs. In the stage of established motor fluctuations and levodopa-induced dyskinesias (LIDs), M1 plasticity reverted to a severe impairment, indicating a loss of long-duration response of M1 plasticity (70). The sensorimotor, M1 plasticity (as probed by paired-associative stimulation protocol) was also impaired in de novo PD patients in the more affected hemisphere, while the less affected hemisphere showed preserved plasticity with loss of topographic specificity of the plastic effect (71). The latter effect was attributed to either the disease process or a compensatory adjustment to reduce the severity of motor signs. In patients with more advanced disease and experiencing motor fluctuations and dyskinesias, both the sensorimotor, plasticity (69) and the plasticity of M1 (70) were lost, and both forms of plastic responses were l-DOPA unresponsive. When the synaptic milieu is unstable and compensatory mechanisms have failed at the cortico-striatal terminals (72), excessive synaptic release of dopamine (73) can swamp the plasticity-inducing intracellular cascade of events by affecting the functioning of key enzymes (74). In animal models of PD with LIDs, besides alterations in synaptic plasticity, there were modifications in the trafficking and subunit composition of NMDA receptors that were attributed to non-physiological dopaminergic stimulation (21). It is likely that the effects of progressive neuro-degeneration and the detrimental effects of non-physiological dopamine replacement therapy affect neural signaling and plasticity mechanisms not only at the striatum but also at the multiple nodes in the interlinked motor circuits that ultimately influence M1 plasticity.
Cerebellum and Parkinson’s Disease
There is mounting evidence that besides abnormal basal ganglia signaling, cerebellar dysfunction also occurs in PD (75). In animal experiments, the two independent ventral thalamic areas receiving basal ganglia and cerebellar input show decreased neuronal firing following dopamine depletion, indicative of both altered basal ganglia and cerebellar outputs in PD (76). Information about motor plans (delivered by mossy fibers from motor cortices via pontine nuclei) and the error signals (delivered via climbing fibers from inferior olive) are both coded by excitatory inputs that are processed at level of the cerebellar cortex and deep nuclei. It is conceivable that a disturbance in this cerebellar processing function could result in undesired and ineffective movement sequences, by impairing M1 plasticity. A recent study reported that in a chronic model of drug-naïve MPTP-treated parkinsonian monkeys, the level of dopaminergic neuronal loss in substantia nigra pars compacta correlated with a persistent hyper-excitation of the Purkinje cells (77). A similar state of cerebellar over excitation may exist in human PD and could potentially affect the information processing within the cerebellum. In support of the cerebellar hyper-excitation model of PD, a SPECT study found heightened activity of the cerebellum at rest in PD patients compared to controls when off anti-parkinsonian medication but not when the patients were on medication (78). Additionally, resting-state MRI showed that l-DOPA increases the functional connectivity between putamen, cerebellum, and brain stem (79). Functional MRI studies showed hyper-activation of the cerebellum in PD patients during simple motor tasks (79–81). Reciprocally, lesions in the paravermal cerebellum in mice increase D1 receptor levels in the contralateral striatum (82) suggesting that cerebellar cortical and nuclear projections modulate the D1 receptor expression of the striatal direct pathway. Though the neuroimaging evidence of cerebellar over-activity was earlier interpreted as compensatory, the discovery of bidirectional communications between the basal ganglia and cerebellar circuits (7, 8) raises the important question of whether the cerebellar hyper-activation is linked to the abnormal striatal signaling in human PD. It is plausible that the pathologically increased excitatory output from the subthalamic nucleus in PD (83) could propagate to the cerebellum and induce a chronic hyper-excited state. This would prevent any discrete excitatory input from being efficiently processed, thus interfering with the cerebellar tuning of M1 plasticity (84). DBS of subthalamic nucleus that improves the clinical signs of PD may actually also contribute to reduce cerebellar over-activity in PD patients (85).
In late-stage PD, the chronic abnormal excitatory drive from the subthalamic nucleus to the cerebellar cortex might induce physical synaptic reconfigurations that lock the cerebellar cortex in hyper-excited state. Dopamine replacement therapy could normalize the basal ganglia output without being necessarily followed by an immediate and effective reduction of the cerebellar cortex excitation. This would result in a conflict between the normalized output in the basal ganglia–thalamo-cortical circuit and the ongoing abnormal modulation of motor programs by the cerebello-thalamo-cortical circuit, a conflict that could manifest as dyskinetic movements. Such a severe dysfunction of cerebellar sensory processing was demonstrated in advanced PD patients with LIDs (84, 86). Dyskinetic patients with LIDs had severe impairment of both homosynaptic and sensorimotor, heterosynaptic plasticity of M1. However, the sensorimotor M1 plasticity in PD could be temporarily reinstated even by a single session of inhibitory stimulation of cerebellum but not by sham stimulation, when patients were tested while on l-DOPA (84). Repeated sessions of cerebellar inhibitory stimulation had prolonged the antidyskinetic effect (84, 86) and were linked to the resurgence of sensorimotor M1 plasticity. Cerebellar cortical inhibition reinstated only sensorimotor M1 plasticity but not the intrinsic plasticity of M1. This suggested that only the unblocking of cerebellar sensory processing function by inhibition of the cerebellar cortex was involved in the resurgence of sensorimotor M1 plasticity and in the reduction of abnormal movements. Indeed, such inhibition of the cerebellar cortex was shown to reduce blood flow in the cerebellar cortex and dentate nucleus, in PD patients with LIDs (87). Based on these evidences it was proposed that the adjustment of the gain of cerebellar sensory processing is lost in advanced PD. The abnormal cerebello-dentato-thalamic outflow could lead to secondary maladaptive sensorimotor plasticity of M1 (84). It still remains to be elucidated whether the sensory processing dysfunction in PD is due to cerebellar cortical hyper-excitation triggered by the disease itself, as in the animal model of PD (76), or by an abnormal overdrive imposed by chronic non-physiological dopaminergic replacement. Testing the cerebellar plasticity in untreated or de novo PD patients may help resolving this conundrum.
Conclusion and Future Directions
Recent studies establishing strong topography-specific connections among the basal ganglia, cerebellum, and their projections to overlapping areas in the motor cortices suggest that these networks influence each other’s functions. The contribution of the cerebello-thalamo-cortical pathway to tremor (88–90) and to LIDs in PD (84, 86) is now established. Whether there is a similar contribution of cerebellar processing function to other motor or cognitive symptoms in PD remains to be tested in future studies. There is preliminary evidence that DBS of subthalamic nucleus improves sensorimotor plasticity of M1 (91), but whether DBS also restores cerebellar processing function by re-instating normal signaling and plastic mechanisms in the cerebellar networks needs further exploration.
Both dopaminergic signaling in the basal ganglia and cerebellar sensory processing are necessary for scaling of M1 plasticity and topographic specificity (51). In de novo PD, sensorimotor plasticity is lost in the more affected hemisphere, while topographic specificity is lost in the less affected hemisphere with preservation of plasticity (68). This pattern suggests that loss of topographic specificity occurs prior to the loss of sensorimotor plasticity. These two defects could cause abnormal muscle synergies and thereby abnormal movements in PD. We propose that M1 plasticity, particularly associative sensorimotor plasticity, is an indication of motor map plasticity and therefore its loss or excess may have implications for motor learning and motor performance in disorders affecting the basal ganglia and cerebellum. Therapeutic interventions for such disorders might be more efficient if would attempt to normalize signaling in both striato-thalamo-cortical and cerebello-thalamo-cortical pathways.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Houck JC, Wise SP. Distributed modular architectures linking basal ganglia and the cerebral cortex: their role in planning and controlling action. Cereb Cortex (1995) 2:95–110. doi: 10.1093/cercor/5.2.95
3. Albin R, Young AB, Penny JO. The functional anatomy of basal ganglia disorders. Trends Neurosci (1989) 12:366–75. doi:10.1016/0166-2236(89)90074-X
4. Schultz W. Dopamine neurons and their role in reward mechanisms. Curr Opin Neurobiol (1997) 7:191–7. doi:10.1016/S0959-4388(97)80007-4
6. Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci (1990) 13:266–71. doi:10.1016/0166-2236(90)90107-L
7. Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nat Neurosci (2005) 8:1491–3. doi:10.1038/nn1544
8. Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci U S A (2010) 107:8452–6. doi:10.1073/pnas.1000496107
9. Ito M. Historical significance of the cerebellum and the role of Purkinje cells in motor learning. Ann N Y Acad Sci (2002) 978:273–88. doi:10.1111/j.1749-6632.2002.tb07574.x
11. Hess G, Donoghue JP. Long-term potentiation of horizontal connections provides a mechanism to reorganize cortical motor maps. J Neurophysiol (1994) 71:2543–7.
12. Hammond G. Correlates of human handedness in primary motor cortex: a review and hypothesis. Neurosci Biobehav Rev (2002) 26:285–92. doi:10.1016/S0149-7634(02)00003-9
13. Reitz M, Muller K. Differences between congenital mirror movements and associated movements in normal children: a neurophysiology study. Neurosci Lett (1998) 256:69–72.
14. Schieber MH. Constraints on somatotopic organization in the primary motor cortex. J Neurophysiol (2001) 86:2125–43.
15. Pascaul-Leone A, Nguyet D, Cohen LG, Brasil-Neto JP, Cammarota A, Hallet M. Modulation of muscle responses evoked by transcranial magnetic stimulation during the acquisition of new fine motor skills. J Neurophysiol (1995) 74:1037–45.
16. Siebner HR, Rothwell J. Transcranial magnetic stimulation: new insights into representational cortical plasticity. Exp Brain Res (2003) 148:1–16.
17. Monfils MH, VandenBerg PM, Kleim JA, Teskey GC. Long-term potentiation induces expanded movement representations and dendritic hypertrophy in layer V of rat sensorimotor neocortex. Cereb Cortex (2004) 14:586–93. doi:10.1093/cercor/bhh020
18. Huntley GW. Correlation between patterns of horizontal connectivity and the extend of short-term representational plasticity in rat motor cortex. Cereb Cortex (1997) 7:143–56.
19. Monfils MH, Plautz EJ, Kleim JA. In search of the motor engram; motor map plasticity as a mechanism for encoding motor experience. Neuroscientist (2005) 11:471–83.
20. Kleim JA, Bruneau R, Calder K, Pocock D, VandenBerg PM, MacDonald E, et al. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron (2003) 40:167–76. doi:10.1016/S0896-6273(03)00592-0
21. Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci (2003) 6:501–6. doi:10.1038/nn1040
22. Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson’s disease. Adv Exp Med Biol (2012) 970:553–72. doi:10.1007/978-3-7091-0932-8_24
23. Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron (2008) 60:543–54. doi:10.1016/j.neuron.2008.11.005
24. Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci (2011) 34:441–66. doi:10.1146/annurev-neuro-061010-113641
25. Delong M. Primate models of movement disorders of basal ganglia origin. Trends Neurosci (1990) 13:281–5.
26. Montague PR, McClure SM, Baldwin PR, Philips PE, Budygin EA, Stuber GD, et al. Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci (2004) 24:1754–9. doi:10.1523/JNEUROSCI.4279-03.2004
27. Yin HH, Ostlund SB, Knowlton BJ, Balleine BW. The role of the dorso-medial striatum in instrumental conditioning. Eur J Neurosci (2005) 22:513–23. doi:10.1111/j.1460-9568.2005.04218.x
28. Berger B, Gasoar O, Verney C. Dopaminergic innervations of the cerebral cortex: unexpected differences between rodents and primates. Trends Neurosci (1991) 14:21–7.
29. Raghanti MA, Simpson CD, Marcinkiewicz JL, Erwin JM, Hof PR, Sherwood CC. Cortical dopaminergic innervations among human, chimpanzees, and macaque monkeys: a comparative study. Neuroscience (2008) 155:203–20. doi:10.1016/j.neuroscience.2008.05.008
30. Molina-Luna K, Pekanovic A, Röhrich S, Hertler B, Schubring-Giese M, Rioul-Pedotti MS, et al. Dopamine in motor cortex is necessary for skill learning and synaptic plasticity. PLoS One (2009) 4:e7082. doi:10.1371/journal.pone.0007082
31. Del Arco A, Mora F. Prefrontal cortex-nucleus accumbens interaction: in vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacol Biochem Behav (2008) 90:226–35. doi:10.1016/j.pbb.2008.04.011
32. Hejitz RD, Kolb B, Forssberg H. Motor inhibitory role of dopamine D1 receptors: implications for ADHD. Physiol Behav (2007) 92:155–60. doi:10.1016/j.physbeh.2007.05.024
33. Korchounov A, Ziemann U. Neuromodulatory neurotransmitters influence LTP-like plasticity in human cortex: a pharmaco-TMS study. Psychopharmacology (2011) 36:1894–902. doi:10.1038/npp.2011.75
34. Luft AR, Shwarz S. Dopaminergic signals in primary motor cortex. Int J Neurosci (2009) 27:415–21. doi:10.1016/j.ijdevneu.2009.05.004
35. Ueki Y, Mima T, Kotb MA, Sawada H, Saiki H, Ikeda A, et al. Altered plasticity of human motor cortex in Parkinson’s disease. Ann Neurol (2006) 59:60–71. doi:10.1002/ana.20692
36. Ito M, Kao M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett (1982) 33:253–8.
38. Albus J. The theory of cerebellar function. Math Biosci (1971) 10:25–61. doi:10.1016/0025-5564(71)90051-4
39. Gilbert PF. A theory of memory that explains the function and structure of the cerebellum. Brain Res (1974) 70:1–18. doi:10.1016/0006-8993(74)90208-X
40. Ito M. Mechanisms of motor learning in the cerebellum. Brain Res (2000) 886:237–45. doi:10.1016/S0006-8993(00)03142-5
41. Galliano E, Gao Z, Schonewille M, Todorov B, Simons E, Pop AS, et al. Silencing the majority of cerebellar granule cells uncovers their essential role in motor learning and consolidation. Cell Rep (2013) 3:1239–51. doi:10.1016/j.celrep.2013.03.023
42. Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, et al. Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J Neurosci (2010) 30:13630–43. doi:10.1523/JNEUROSCI.3226-10.2010
43. Jontell H, Hasel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron (2006) 52:227–38. doi:10.1016/j.neuron.2006.09.032
44. Ohstuki G, Piochon C, Hansel C. Climbing fiber signaling and cerebellar gain control. Front Cell Neurosci (2009) 3:4. doi:10.3389/neuro.03.004.2009
45. Penhune VB, Steele CJ. Parallel contribution of cerebellar, striatal and M1 mechanisms to motor sequence learning. Behav Brain Res (2012) 226:579–91. doi:10.1016/j.bbr.2011.09.044
46. Gilbert PF, Thach WT. Purkinje cell activity during motor learning. Brain Res (1977) 128:309–28. doi:10.1016/0006-8993(77)90997-0
47. Medina JF, Lisberger SG. Links from complex spikes to local plasticity and motor learning in the cerebellum of awake-behaving monkeys. Nat Neurosci (2008) 11:1185–92. doi:10.1038/nn.2197
48. BenTaib NO, Manto M, Pandolfo M, Brotchi J. Hemicerebellectomy blocks the enhancement of cortical motor output associated with repetitive somatosensory stimulation in the rat. J Physiol (2005) 567:293–300. doi:10.1113/jphysiol.2005.088229
49. Chen XY, Wolpaw JR. Ablation of cerebellar nuclei prevents H-reflex down-conditioning in rats. Learn Mem (2005) 12:28–54. doi:10.1101/lm.91305
50. Nixon PD. The role of the cerebellum in preparing responses to predictable sensory events. Cerebellum (2003) 2:114–22. doi:10.1080/14734220309410
51. Gao JH, Parsons LM, Bower JM, Xiong J, Li J, Fox PT. Cerebellum implicated in sensory acquisition and discrimination than motor control. Science (1996) 272:545–7. doi:10.1126/science.272.5261.545
52. Dean P, Porrill J. The cerebellum as an adaptive filter: a general model? Funct Neurol (2010) 25:173–80.
53. Popa T, Velayudhan B, Hubsch C, Pradeep S, Roze E, Vidailhet M, et al. Cerebellar processing of sensory inputs primes motor cortex plasticity. Cereb Cortex (2013) 23:305–14. doi:10.1093/cercor/bhs016
54. Hornykiewicz O. Basic research on dopamine in Parkinson’s disease and the discovery of the nigrostriatal dopamine pathway: the view of an eyewitness. Neurodegener Dis (2008) 5:114–7. doi:10.1159/000113678
55. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging (2003) 24:197–211. doi:10.1016/S0197-4580(02)00065-9
56. Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature (2007) 445:643–7. doi:10.1038/nature05506
57. Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res (1991) 547:142–51.
58. Mallet N, Ballion B, Le Moine C, Gonon F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J Neurosci (2006) 5:3875–84. doi:10.1523/JNEUROSCI.4439-05.2006
59. Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Macias R, Alvarez L, Guridi J, et al. Pathophysiologic basis of surgery for Parkinson’s disease. Neurology (2000) 55(12 Suppl 6):S7–12.
60. Costa RM, Lin SC, Sotnikova TD, Cyr M, Gainetdinov RR, Caron MG, et al. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron (2006) 52:359–69. doi:10.1016/j.neuron.2006.07.030
61. Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science (2008) 321:845–51. doi:10.1126/science.1160575
62. Moustafa AA, Krishna R, Eissa AM, Hewedi DH. Factors underlying probabilistic and deterministic stimulus-response learning performance in medicated and unmedicated patients with Parkinson’s disease. Neuropsychology (2013) 27:498–510. doi:10.1037/a0032757
63. Muslimovic D, Post B, Speelman D, Schmand B. Motor procedural learning in Parkinson’s disease. Brain (2007) 130:2887–97. doi:10.1093/brain/awm211
64. Beeler JA, Cao ZF, Kheirbek MA, Ding Y, Koranda J, Murakami M, et al. Dopamine-dependent motor learning: insight into levodopa’s long duration response. Ann Neurol (2010) 67:639–47. doi:10.1002/ana.21947
65. Calabresi P, Pisani A, Mercuir NB, Bernardi G. The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci (1996) 9:19–24. doi:10.1016/0166-2236(96)81862-5
66. Prescott IA, Dostrovsky JO, Moro E, Hodaie J, Lozano AM, Hutchison WD. Levodopa enhances synaptic plasticity in the substantia nigra pars reticulate of Parkinson’s disease patients. Brain (2009) 132:309–18. doi:10.1093/brain/awn322
67. Kishore A, Joseph T, Velayudhan B, Popa T, Meunier S. Early, severe and bilateral loss of LTP and LTD-like plasticity in motor cortex (M1) in de novo Parkinson’s disease. Clin Neurophysiol (2012) 123:822–8. doi:10.1016/j.clinph.2011.06.034
68. Suppa A, Marsili L, Belvisi D, Conte A, Iezzi E, Modugno N, et al. Lack of long-term potentiation-like plasticity in primary motor cortex in Parkinson’s disease. Exp Neurol (2011) 227:296–301. doi:10.1016/j.expneurol.2010.11.020
69. Morgante F, Espay AJ, Gunraj C, Lang AE, Chen R. Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain (2006) 129:1059–69. doi:10.1093/brain/awl031
70. Kishore A, Popa T, Velayudhan B, Joseph T, Balachandran A, Meunier S. Acute dopamine boost has a negative effect on the plasticity of motor cortex in advanced Parkinson’s disease. Brain (2012) 135:2074–88. doi:10.1093/brain/aws124
71. Kojovic M, Bologna M, Kassavetis M, Murase N, Palomar FJ, Berardelli A, et al. Functional reorganization of sensorimotor cortex in early Parkinson’s disease. Neurology (2012) 78:1441–8. doi:10.1212/WNL.78.1_MeetingAbstracts.IN6-2.002
72. Nandhagopal R, Kuramotor L, Schulzer M, Mak E, Cragg J, McKenzie J, et al. Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson’s disease. Brain (2011) 134:3290–8. doi:10.1093/brain/awr233
73. de la Fuente-Fernández R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain (2004) 127:2747–54. doi:10.1093/brain/awh290
74. Santini E, Valient E, Usiello A, Carta M, Borgkvist A, Girault JA, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci (2007) 27:6995–7005. doi:10.1523/JNEUROSCI.0852-07.2007
75. Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain (2013) 136:696–709. doi:10.1093/brain/awt100
76. Rolland AS, Herrero MT, Carcia-Martinez V, Ruberg M, Hirsch EC, Francois C. Metabolic activity of cerebellar and basal ganglia-thalamic neurons is reduced in parkinsonism. Brain (2007) 130:265–75. doi:10.1093/brain/awl337
77. Heman P, Barcia C, Gómez A, Ros CM, Ros-Bernal F, Yuste JE, et al. Nigral degeneration correlates with persistent activation of cerebellar Purkinje cells in MPTP-treated monkeys. Histol Histopathol (2012) 27:89–94.
78. Rascol O, Sabtini U, Fabre N, Brefel C, Loubinous I, Celsis P, et al. The ipsilateral cerebellar hemisphere is overactive during hand movements in akinetic patients. Brain (2007) 120:103–10. doi:10.1093/brain/120.1.103
79. Wu T, Hallett M. A functional MRI study of automatic movements in patients with Parkinson’s disease. Brain (2005) 128:2250–9. doi:10.1093/brain/awh569
80. Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. Neuroimage (2007) 35:222–33. doi:10.1016/j.neuroimage.2006.11.047
81. Kelly C, de Zubicaray G, Di Martino A, Copland DA, Reiss PT, Klein DF, et al. L-DOPA modulates functional connectivity in striatal cognitive and motor networks: a double-blind placebo-controlled study. J Neurosci (2009) 29:7364–78. doi:10.1523/JNEUROSCI.0810-09.2009
82. Delis F, Mitsacos A, Giompres P. Lesion of the cerebellar paravermis increases dopamine D1 receptor levels in the contralateral striatum. J Chem Neuroanat (2013) 47:35–41. doi:10.1016/j.jchemneu.2012.10.004
83. Wichmann T, Delong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurg Clin N Am (1998) 9:223–36.
84. Kishore A, Popa T, Balachandran A, Chandran S, Pradeep S, Backer F, et al. Cerebellar sensory processing alteration impact motor cortex plasticity in Parkinson’s disease: clues from dyskinetic patients. Cereb Cortex (2013). doi:10.1093/cercor/bht058
85. Payoux P, Remy P, Damier P, Miloudi M, Loubinoux I, Pidoux B, et al. Subthalamic nucleus stimulation reduces abnormal motor cortical overactivity in Parkinson disease. Arch Neurol (2004) 61:1307–13. doi:10.1001/archneur.61.8.1307
86. Koch G, Brusa L, Carrillo F, Lo Gerfo E, Torriero S, Oliveri M. Cerebellar magnetic stimulation decreases levodopa-induced dyskinesias in Parkinson disease. Neurology (2009) 73:113–9. doi:10.1212/WNL.0b013e3181ad5387
87. Brusa L, Ceravola R, Kiferle L, Monteleone F, Iani C, Schillaci O, et al. Metabolic changes induced by theta burst stimulation of the cerebellum in dyskinetic Parkinson’s disease patients. Parkinsonism Relat Disord (2012) 18:59–62. doi:10.1016/j.parkreldis.2011.08.019
88. Ni Z, Pinto AD, Lang AE, Chen R. Involvement of the cerebellothalamocortical pathway in Parkinson’s disease. Ann Neurol (2010) 68:816–24. doi:10.1002/ana.22221
89. Lewis MM, Du G, Sen S, Kawaguchi A, Truong Y, Lee S, et al. Differential involvement of striato- and cerebello-thalamo-cortical pathways in tremor- and akinetic/rigid-predominant Parkinson’s disease. Neuroscience (2011) 177:230–9. doi:10.1016/j.neuroscience.2010.12.060
Keywords: motor cortex plasticity, Parkinson disease, cerebellum, dopamine, basal ganglia
Citation: Kishore A, Meunier S and Popa T (2014) Cerebellar influence on motor cortex plasticity: behavioral implications for Parkinson’s disease. Front. Neurol. 5:68. doi: 10.3389/fneur.2014.00068
Received: 13 September 2013; Paper pending published: 23 January 2014;
Accepted: 21 April 2014; Published online: 06 May 2014.
Edited by:
Marina Tijssen, Academic Medical Centre, NetherlandsReviewed by:
Anne-Fleur Van Rootselaar, Academic Medical Center, NetherlandsAngelo Quartarone, University of Messina, Italy
Copyright: © 2014 Kishore, Meunier and Popa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asha Kishore, Department of Neurology, Comprehensive Care Centre for Movement Disorders, Sree Chitra Tirunal Institute for Medical Sciences and Technology (SCTIMST), Kerala 695011, India e-mail:YXNoYUBzY3RpbXN0LmFjLmlu