 Yase Chen
Yase Chen Shan Bian2†
Shan Bian2† Haijun Zhang
Haijun Zhang Tao Sun
Tao Sun- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY, USA
- 3School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
Expansion of the neural progenitor pool in the developing cerebral cortex is crucial for controlling brain size, since proliferation defects have been associated with the pathogenesis of microcephaly in humans. Cell cycle regulators play important roles in proliferation of neural progenitors. Here, we show that the cyclin-dependent kinase inhibitor p21 (also called Cdkn1a and Cip1) negatively regulates proliferation of radial glial cells (RGCs) and intermediate progenitors (IPs) in the embryonic mouse cortex. MicroRNA-17 (miR-17) displays reciprocal expressions with p21 in the developing cortex. Opposite to p21, miR-17 promotes expansion of RGCs and IPs, as demonstrated by overexpressing miR-17 precursors and miR-17 sponges that can knock down the endogenous miR-17. Moreover, p21 is a putative target normally silenced by miR-17. Co-expression of miR-17 with p21 is sufficient to rescue the negative regulation of p21 on progenitor proliferation. Our results indicate a mechanism of controlling the neural progenitor pool, which is to suppress p21 by miR-17 in the developing cortex.
Introduction
Proper proliferation and differentiation of neural stem cells (NSCs) and neural progenitors (NPs) are crucial for the development and function of the mammalian cerebral cortex. Radial glial cells (RGCs) mainly reside in the cortical ventricular zone (VZ), represent early NPs and normally divide symmetrically to generate two RGCs, and asymmetrically to generate one RGC and one intermediate progenitor (IP) or one differentiated neuron (1–7). IPs mostly reside in the subventricular zone (SVZ) and divide symmetrically to generate two differentiated neurons (2, 8–10). The rate and timing of NP proliferation and differentiation are tightly regulated by various cell cycle regulators (11, 12). p21 (also known as Cdkn1a and Cip1) is a cyclin-dependent kinase (CDK) inhibitor that has been shown to mediate the p53-induced G1 checkpoint in cell cycle regulation (13–15). Lack of p21 promotes self-renewal of embryonic and adult NSCs via suppressing the cell cycle exit (16–19). Because proper regulation of NP proliferation is important for controlling brain size, p21 may play a role in determining cortical growth (20). However, how p21 expression levels are precisely controlled in cortical NSCs and NPs remains unclear.
The endogenous small non-coding RNAs, microRNAs (miRNAs), have been shown to play crucial roles in many aspects of cell cycle regulation in the developing nervous system (21–24). miRNAs normally bind to target genes and negatively regulate their expression through imperfect complementary sequence in the 3′-untranslated region (3′UTR) of messenger RNAs (mRNAs) (25, 26). Studies have shown that miRNAs have either reciprocal or overlapping expression patterns with target genes in specific cells and tissues and silence their expression at a posttranscriptional level (27, 28). An important miRNA family is the miR-17–92 cluster, which produces miR-17, 18, 19, and 92, each with conserved seed sequences, and regulates proliferation and survival of various cells (29–33). Our own work and others have shown that miR-17–92 promotes proliferation of cortical NSCs and NPs (34, 35). Since there are six miRNAs produced from the miR-17–92 cluster, and each miRNA usually has multiple targets, the molecular mechanisms of miR-17–92 function in cortical development remain an exciting research topic.
In this study, we show that while p21 negatively regulates NP proliferation by reducing the numbers of both RGCs and IPs in the developing mouse cortex, miRNA miR-17 has an opposite effect on NP development. miR-17 promotes proliferation of RGCs and IPs through suppressing p21 expression. Our studies have identified a mechanism that controls p21 expression levels in NPs in vivo by miR-17 during cortical development.
Materials and Methods
Animals
CD-1 mice were used for in utero electroporation. For staging of embryos, midday of the day of vaginal-plug formation was considered as E0.5; the first 24 h after birth were defined as P0. Animals were maintained at the facility of Weill Cornell Medical College. Animal use was overseen by the Animal Facility and approved by the IACUC at the Weill Cornell Medical College.
Tissue Preparation and Immunohistochemistry
Mouse brains were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 1 h at room temperature (RT), incubated in 30% sucrose in PBS overnight at 4°C, embedded in OCT, and stored at −80°C until use. Brains were sectioned (14 μm) using a cryostat. For antigen recovery, sections were incubated in heated (95–100°C) antigen recovery solution (1 mM EDTA, 5 mM Tris, pH 8.0) for 20 min, and cooled down for 20–30 min. Before applying antibodies, sections were blocked in 10% normal goat serum (NGS) in PBS with 0.1% Tween-20 (PBT) for 1 h. Sections were incubated with primary antibodies at 4°C overnight and visualized using goat anti-rabbit IgG-Alexa-Fluor-488, goat anti-chicken IgG-Alexa-Fluor-488, and/or goat anti-mouse or anti-rabbit IgG-Alexa-Fluor-546 (1:300, Molecular Probes) for 1.5 h at RT. Images were captured using a Leica digital camera under a fluorescent microscope (Leica DMI6000B) or a Zeiss LSM510 confocal microscope.
Primary antibodies against the following antigens were used: green fluorescent protein (GFP) (1:1000, Abcam), bromodeoxyuridine (BrdU) (1:50, DSHB), Pax6 (1:500, Covance), and Tbr2 (1:500, Abcam). Cell counting in the mouse cortical tissue was performed on a representative column with the width of 200 pixels in the cortical wall. All sections analyzed were selected from a similar medial point on the anterior–posterior cortical axis.
Quantitative Real-Time Reverse Transcription PCR
Total RNA was isolated from the dorsal cortex of E15.5 mice using RNeasy® Mini kit (Qiagen) according to manufacturer’s instructions, and all samples were treated with DNase to remove genomic DNA. Reverse transcription was performed using Random Hexamer primer (Roche). The quantitative real-time reverse transcription PCR (qRT-PCR) was performed using Power SYBR® Green PCR Master Mix (Life Science) on an Mx4000™ Multiplex Quantitative PCR System (Stratagene) according to manufacturer’s instructions. The RT primers to detect primary transcripts for miR-17 are: F-5′-gaacctcaccttgggactga-3′; R-5′-tgctacaagtgccctcactg-3′. The RT primers to detect p21 are: F-5′-cggtggaactttgacttcgt-3′; R-5′-caatctgcgcttggagtgat-3′.
In utero Electroporation
In utero electroporation was performed as described (34). Briefly, electroporation was conducted at E13.5 and the brain tissues were harvested 24 h later. Plasmid DNA was prepared using the EndoFree Plasmid Maxi Kit (QIAGEN) according to manufacturer’s instructions, and diluted to 2 μg/μl. For co-expression of p21 and miR-17, the concentrations of p21 and miR-17 are 0.5 and 1.5 μg/μl, respectively, maintaining a total plasmid concentration of 2 μg/μl. DNA solution was injected into the lateral ventricle of the cerebral cortex, and electroporated with five 50-ms pulses at 35 V using an ECM830 electro square porator (BTX).
Luciferase Assays
Neuro2a cells were transfected using Lipofectamine 2000 (Invitrogen) using the manufacturer’s protocol. Plasmids were quantified by UV spectrophotometry and used for transfection in a 2:1 ratio (miRNA: target luciferase constructs); 8:2:1 ratio (sponge:miRNA:target luciferase constructs). pGL4.13 Firefly luciferase (Promega) was used for 3′UTRs of targets. pGL4.73 Renilla luciferase (Promega) was used as a transfection control. Luciferase was measured using the Dual-Luciferase Reporter Assay kit (Promega) using the manufacturer’s protocol and read on a Victor3 1420 multilabel counter (Perkin Elmer). All conditions were run in triplicate, and all experiments were repeated at least once with similar results. Raw data for each condition were normalized for transfection efficiency as the ratio of Firefly luciferase to Renilla luciferase, normalized to the corresponding empty pGL4.13 column to correct for DNA quantification errors, and finally for each luciferase tested, the empty vector control experiment was set to 1 for display.
Cloning of Constructs
Cloning of constructs was done by standard PCR based methods. cDNA from E15.5 C57Bl/6J mice was used. miR-17 isoform precursors were cloned into the pGEM-T vector (Promega) using the following primers: F-5′-cactcgaggtgacagaatttagagctttgg-3′; R-5′-actggacgcagccagtgccg-3′. Mutations of miR-17 in the seed sequence were generated using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) using the following primers:
F-5′-gtcagaataatgtcgttgtgcttacagtgcaggtagtgatgtgtgcatctactgcagtgagggcacaagtagcattatgctgac-3′;
R-5′-gtcagcataatgctacttgtgccctcactgcagtagatgcacacatcactacctgcactgtaagcacaacgacattattctgac-3′.
Full length cDNA for p21 was cloned using the following primers: F-5′-gactcgaggcaccatgtccaatcctggtgatg-3′; R-5′-cacttcagggttttctcttgcagaag-3′. To knock down p21, the following oligos were used to make short hairpin RNA (shRNA):
m-P21-top1: 5′-agtgtgccgttgtctcttcttcaagagagaagagacaacggcacacttttttt-3′
m-P21-bottom1: 5′-aattaaaaaaagtgtgccgttgtctcttctctcttgaagaagagacaacggcacactggcc-3′
m-P21-top2: 5′-cggtggaactttgacttcgttcaagagacgaagtcaaagttccaccgtttttt-3′
m-P21-bottom2: 5′-aattaaaaaacggtggaactttgacttcgtctcttgaacgaagtcaaagttccaccgggcc-3′
m-P21-top3: 5′-ctttgacttcgtcacggagttcaagagactccgtgacgaagtcaaagtttttt-3′
m-P21-bottom3: 5′-aattaaaaaactttgacttcgtcacggagtctcttgaactccgtgacgaagtcaaagggcc-3′
The 3′UTR fragment for p21 were subcloned into pGL4.13 vector (Promega) using the following primers: F-5′-ataagaatgcggccgccatcttcggccttagccctc-3′; R-5′-gaggactcgggacaatgcag-3′. For luciferase assays, the miR-17 precursor and its mutation were subcloned into the pCDNA3.1 vector.
For cloning miR-17 sponge, the following primers were used: 5′-gtaggaatcttcgaaagctatacaccgctcgagactagtctacctgcactgccgcactttggttatcctacctgcactgccgcactttggttatcctacctgcactgccgcactttgtctagagcttacgttagaatcgcattcg-3′
For cloning miR-17 sponge mutations, the following primers were used: 5′-gtaggaatcttcgaaagctatacaccgctcgagactagtctacctgcactgccgccgcttggttatcctacctgcactgccgccgcttggttatcctacctgcactgccgccgcttgtctagagcttacgttagaatcgcattcg-3′
They were then subcloned into the pCAGIG vector for electroporation, and pCDNA3.1 vector for luciferase assays.
Western Blot Analysis
Expression levels of p21 were analyzed by the western blot analysis. Protein extracts were harvested by lysing N2a cells transfected with combinations of p21 and shRNA for p21 with RIPA lysis buffer (150 mM NaCl, 1 mM Na4P2O7, 1 mM NaF, 1 mM EDTA, 1 mM PMSF, 2 mM Na3VO4, 1% NP-40, 50 mM Tris, pH 7.5) with complete™ EDTA-free protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN, USA). The protein samples were boiled in SDS sample buffer for 10 min before loading onto 10% Tris-Glycine gels as 10 μg for each lane and transferred onto PVDF membrane (Pall Corporation, Pensacola, FL, USA). For immunoblotting, membranes were blocked with 5% (w/v) non-fat milk powder in 0.05% TBST [50 mM Tris-Cl, pH 7.5, 150 mM NaCl, with 0.05% (v/v) Tween-20] and incubated at 4°C overnight with the following primary antibodies, which were diluted in 0.05% TBST with 5% non-fat milk: p21 and actin. After washing with TBST, membranes were incubated with specific HRP-conjugated secondary antibodies for 1 h at RT followed with extended washes with TBST. Immunoblot reactions were visualized using chemiluminescent substrate (Pierce, Rockford, IL, USA) on Kodak BioMax light films (Kodak, Rochester, NY, USA).
Results
p21 Negatively Regulates Expansion of RGCs and IPs in Embryonic Cortices
Previous studies have shown that p21 usually inhibits self-renewal of NSCs (16–19). To test whether p21 specifically affects proliferation of RGCs and IPs, we examined p21 effects by altering its expression levels in mouse embryonic cortices utilizing in utero electroporation (Figure 1A). To overexpress p21, mouse p21 full length cDNA was cloned into a pCAGIG vector with a GFP reporter gene. The electroporation was conducted at embryonic day 13.5 (E13.5) and brain tissues were harvested 24 h later, with a single pulse of BrdU 1 h before tissue collection (Figure 1A). The electroporated cells in E14.5 cortices were detected by the GFP activity in green (Figure 1B).
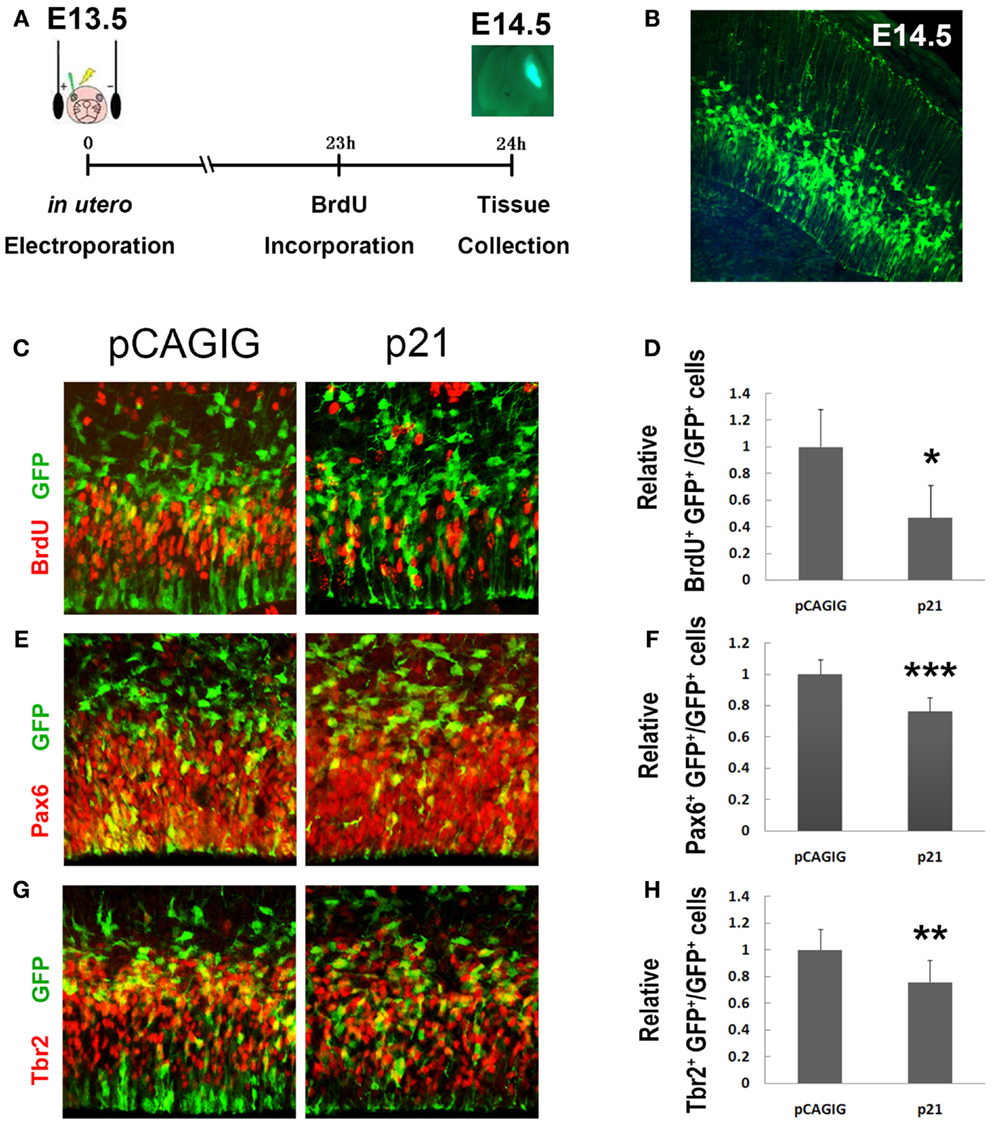
Figure 1. p21 negatively regulates proliferation of cortical neural progenitors. (A) The electroporation was conducted at E13.5 and the brain tissues were harvested 24 h later. A single intraperitoneal injection of BrdU was performed 1 h before tissue collection. (B) The cells located in the ventricular zone (VZ) of mouse cortices were electroporated with exogenous plasmids carrying green fluorescent protein (GFP) at E13.5. The GFP cells can be visualized under microscope at E14.5. (C,D) Overexpressing p21 in E13.5 mouse cortices (n = 4), analyzed at E14.5, decreased the number of proliferating cells co-labeled with GFP and BrdU compared with the empty vector pCAGIG (n = 4). (E,F) Overexpressing p21 (n = 6) decreased the number of radial glial cells (RGCs) co-labeled with GFP and Pax6 compared with the empty vector pCAGIG (n = 4). (G,H) Overexpressing p21 (n = 5) decreased the number of intermediate progenitors (IPs) co-labeled with GFP and Tbr2 compared with the empty vector pCAGIG (n = 4). Data are presented as mean ± SEM; n ≥ 3 in all constructs; p values in relation to the control (*p < 0.05, **p < 0.01, ***p < 0.001).
A significantly decreased percentage of BrdU+/GFP+ cells versus total GFP+ cells in cortices was detected, suggesting a reduced proliferating NP population when p21 is overexpressed (Figures 1C,D). We then examined the impact of overexpressed p21 on producing RGCs and IPs, by labeling with antibodies against Pax6 – a marker for RGCs, and Tbr2 – a marker for IPs, respectively. There was a significant decrease in percentages of Pax6+/GFP+ and Tbr2+/GFP+ cells versus total GFP+ cells, indicating a suppression of maintenance of RGCs and IPs by p21 overexpression (Figures 1E–H).
We next used a short hairpin RNA to knock down endogenous p21 expression (Figure 2A). When the endogenous p21 expression was knocked down using p21 short hairpin RNA-3 (p21–shRNA-3), we found a significant increase in the percentage of BrdU+/GFP+ cells (Figures 2B,C). The percentage of Pax6+/GFP+ cells was also greatly increased, suggesting expanded RGC populations (Figures 2D,E). Interestingly, the percentage of Tbr2+/GFP+ cells was decreased, suggesting that knockdown of p21 affects transition of RGCs to IPs (Figures 2F,G). Our results indicate that p21 negatively regulates proliferation of cortical NPs, and affects expansion and transition of RGCs and IPs.
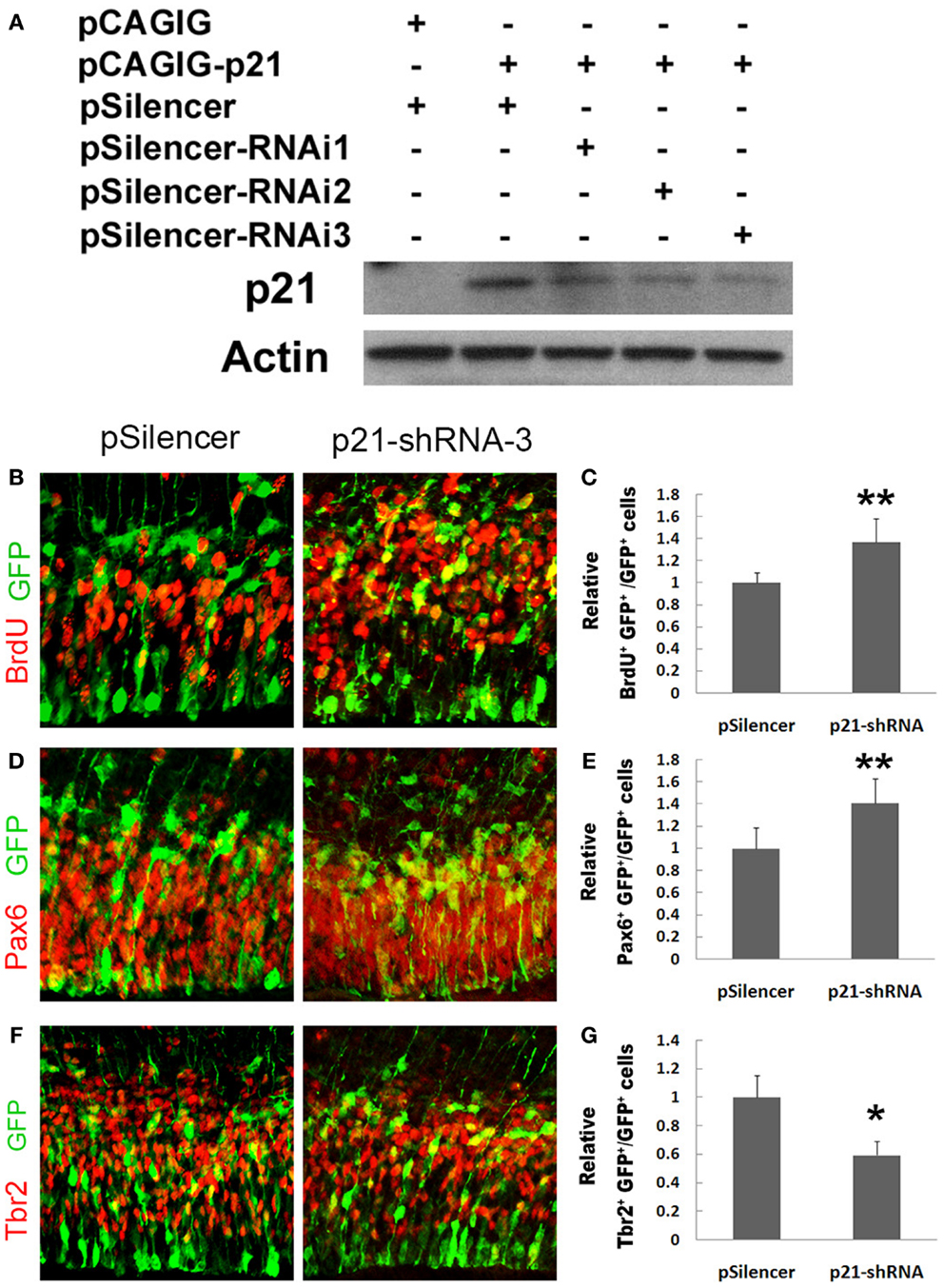
Figure 2. Knockdown of p21 increases proliferation of cortical neural progenitors. (A) Western blotting analyses of p21 short hairpin RNA to knock down endogenous p21 expression levels using RNA interference (RNAi). (B,C) Blocking the p21 expression (n = 4) using a p21 short hairpin RNA-3 (p21–shRNA-3) in E13.5 mouse cortices, analyzed at E14.5, increased the number of proliferating cells co-labeled with GFP and BrdU compared with the empty vector pSilencer (n = 3). (D,E) Blocking the p21 expression (n = 5) using the p21–shRNA increased the number of RGCs co-labeled with GFP and Pax6. (F,G) Blocking the p21 expression (n = 6) using the p21–shRNA decreased the number of IPs co-labeled with GFP and Tbr2. Data are presented as mean ± SEM; n ≥ 3 in all constructs; p values in relation to the control (*p < 0.05, **p < 0.01).
p21 is a Putative Target of miR-17
To maintain a proper population of NPs in early developing cortices, it appears that p21 expression should be precisely modulated, especially repressed. We postulated that miRNA-mediated gene silencing regulation might be one of the mechanisms controlling p21 expression. Based on miRNA prediction algorithm Targetscan (http://www.targetscan.org), we searched the 3′UTR of p21 and found that it contains one binding site for miR-17 (Figure 3A). To prove the targeting effect of miR-17 on p21, we designed a luciferase assay in which the 3′UTR sequence of p21 was cloned into a luciferase vector and co-transfected with miR-17. While relative luciferase activities in constructs containing the 3′UTR of p21 were not affected by a control miRNA miR-9, they were significantly reduced by miR-17 (Figure 3B). However, when a mutated miR-17 containing mutations in the seed sequence, which is responsible for binding to p21 3′UTR, was used, the targeting effect of miR-17 was abolished (Figure 3B). These results indicate that p21 is a specific target of miR-17.
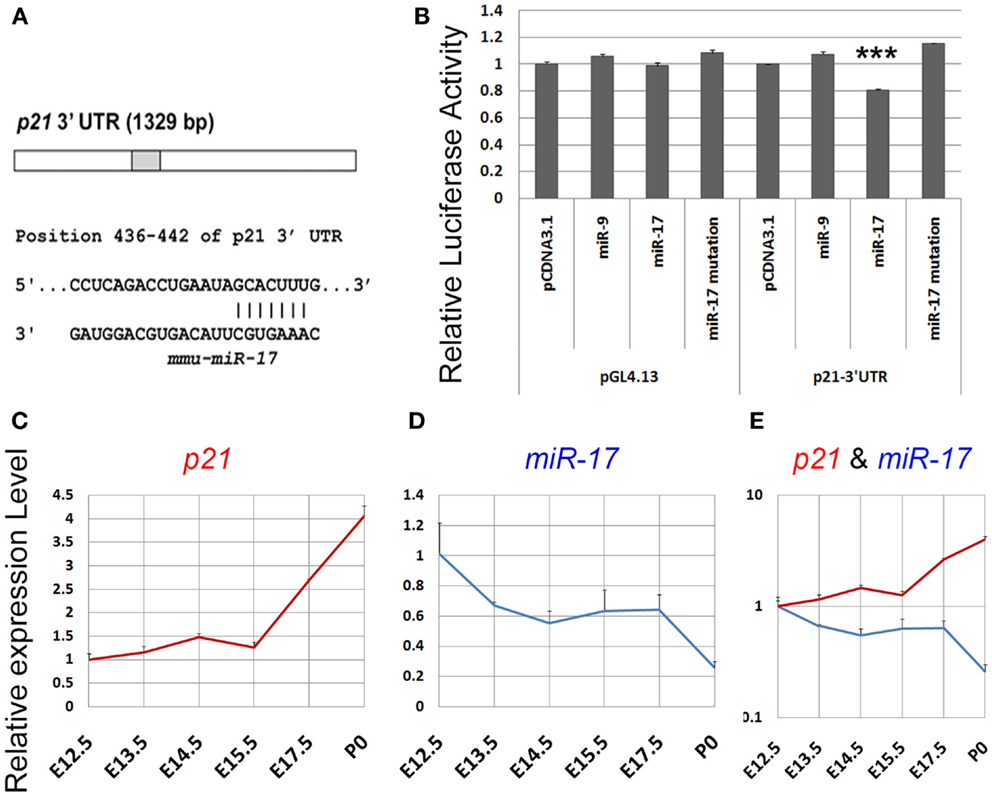
Figure 3. p21 is a putative target of miR-17. (A) A predicted targeting site of miR-17 on the 3′UTR of p21. (B) Luciferase activities of the p21 3′UTR construct were greatly reduced by miR-17, but not by miR-17 mutation or a control microRNA miR-9. miR-17 had no effect on pGL4.13 construct in which the p21 3′UTR had been removed. (C) The expression level of p21 in the mouse cortex continuously increased from E12.5 to P0. (D) The expression level of miR-17 in the mouse cortex continuously decreased from E12.5 to P0. (E) A combined view of the expression levels of p21 and miR-17 at different developing stages. Data are presented as mean ± SEM; n = 3; ***p < 0.001.
We next examined whether the expression level of p21 is correlated with miR-17 expression in developing cortices by performing qRT-PCR. Total RNAs were extracted from E13.5, E14.5, E15.5, E17.5, and postnatal day 0 (P0) cortices. While the expression level of miR-17 was decreased from E12.5 to P0, p21 expression was continuously increased, suggesting a reciprocal expression between miR-17 and its target p21 during cortical development (Figures 3C–E). These results suggest that miR-17 silencing regulation of p21 likely controls NP population in embryonic cortices.
miR-17 Positively Regulates Proliferation of Cortical Neural Progenitors
We next investigated the effect of miR-17 on NP development in vivo. In E14.5 cortices, electroporated with the miR-17 precursor to overexpress miR-17 at E13.5, there was a significant increase in the percentage of BrdU+/GFP+ cells, which suggests an up-regulation of proliferation of cortical NPs (Figures 4A,B). Moreover, the percentages of Pax6+/GFP+ and Tbr2+/GFP+ cells were also increased, indicating an expansion of RGCs and IPs by miR-17 overexpression (Figures 4C–F). On the other hand, mutations of miR-17 in the seed sequence (miR-17 mut) had no effect on proliferation of RGCs and IPs, indicating a specific effect of miR-17 on NP development (Figures 4C–F). Our results suggest that miR-17 promotes proliferation of cortical NPs.
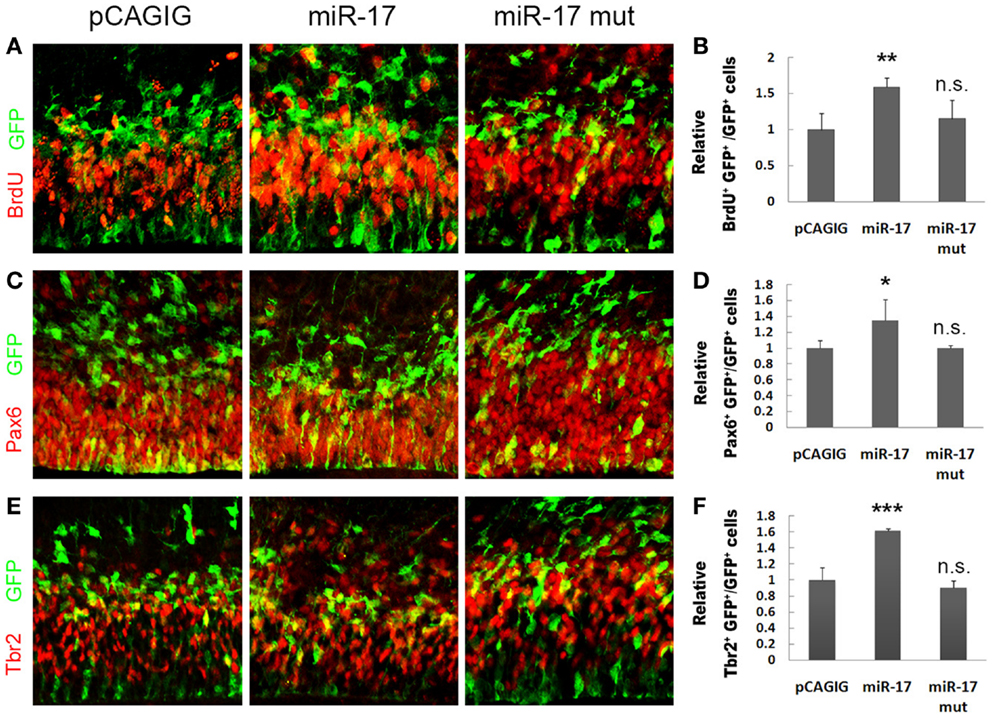
Figure 4. miR-17 positively regulates proliferation of cortical neural progenitors. (A,B) Expression of miR-17 in E13.5 mouse cortices (n = 3), analyzed at E14.5, increased the number of proliferating cells co-labeled with GFP and BrdU. A mutated miR-17 (miR-17 mut) (n = 3) had no effect on cell proliferation. (C,D) Expression of miR-17 in E13.5 mouse cortices (n = 4) increased the number of RGCs co-labeled with GFP and Pax6. A mutated miR-17 (n = 3) had no effect on the number of RGCs. (E,F) Expression of miR-17 in E13.5 mouse cortices (n = 4) increased the number of IPs co-labeled with GFP and Tbr2. A mutated miR-17 (n = 3) had no effect on the number of IPs. Data are presented as mean ± SEM; n ≥ 3 in all constructs; p values in relation to the control (*p < 0.05, **p < 0.01, ***p < 0.001), n.s., not significant.
Blocking miR-17 Using Its Sponge Causes Reduced Proliferation of Cortical Neural Progenitors
To further test miR-17 functions in expansion of cortical NPs, we applied a loss-of-function approach by expressing miR-17 sponges (36). A miRNA sponge is a RNA transcript that contains multiple binding sequences complementary to a mature miRNA. It can bind to endogenous miRNAs and block their silencing activity (37, 38). We designed the miR-17 sponge (miR-17 sp), which consists of three narrowly spaced, bulged binding sites for miR-17 (Figure 5A). To verify the blocking effect of miR-17 sponge on the miR-17 silencing activity, we performed the luciferase assay. The miR-17 sponge was cloned in the 3′UTR of a coding gene iCre, and co-transfected with miR-17 and the construct containing the luciferase gene followed by the 3′UTR sequence of p21. The reduction of the relative luciferase activity, which is normally caused by the silencing effect of miR-17, was significantly rescued by the miR-17 sponge (Figure 5B). However, a mutated miR-17 sponge at the seed sequence showed no such rescue effect. These data indicate that the miR-17 sponge is able to block the silencing function of miR-17 to its target gene p21.
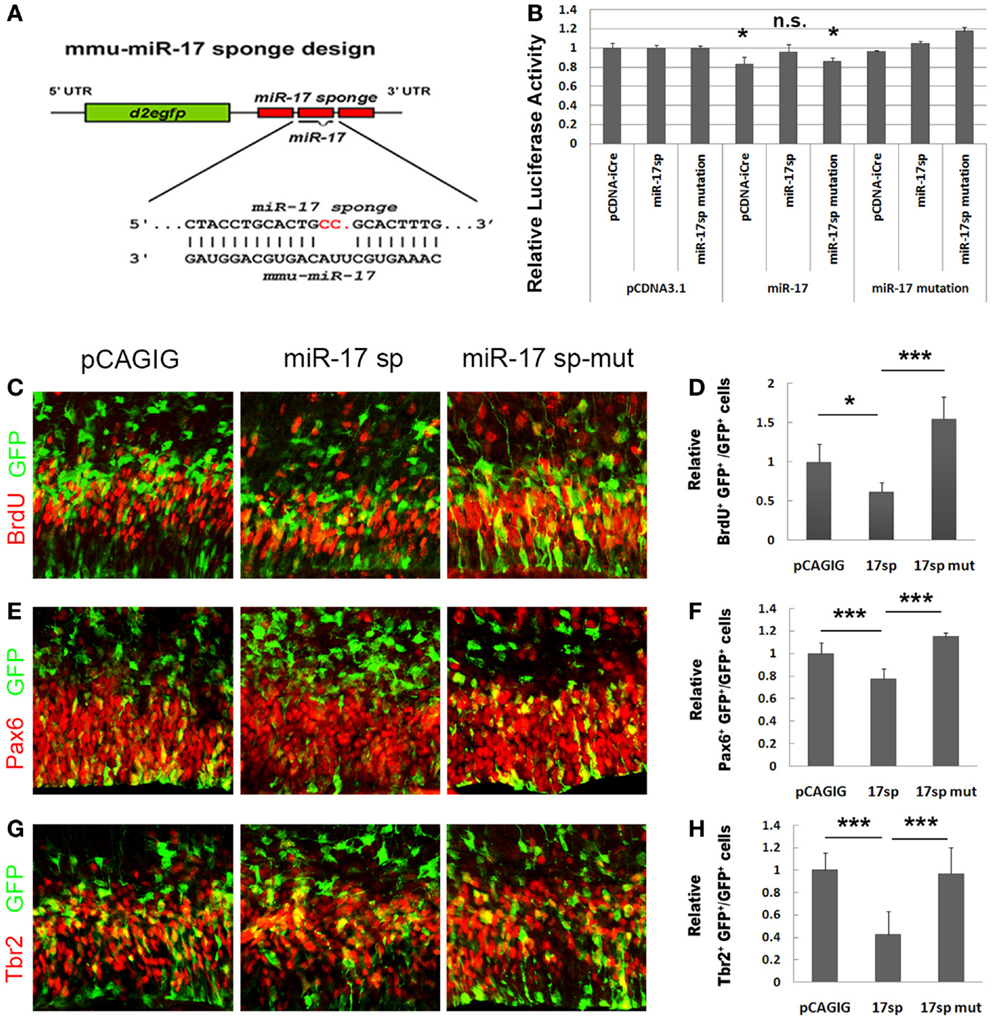
Figure 5. Blocking miR-17 using its sponge causes reduced proliferation of cortical neural progenitors. (A) The design of the miR-17 sponge. (B) The reduction of the relative luciferase activity caused by miR-17 was significantly rescued by the miR-17 sponge, but not by miR-17 sponge mutation. The luciferase activity values were normalized by the average luciferase activity value of the empty vector pcDNA to get a ratio. In all experiments, the pcDNAs luciferase activity value was normalized as 1, and the values of other constructs were the ratios of their luciferase activity values to their pcDNA controls. (C,D) Expression of miR-17 sponge (miR-17 sp) in E13.5 mouse cortices (n = 4), analyzed at E14.5, decreased the number of proliferating cells co-labeled with GFP and BrdU. A mutated miR-17 sponge (miR-17 sp-mut) (n = 3) failed to do so. (E,F) Expression of miR-17 sponge in E13.5 mouse cortices (n = 6) decreased the number of RGCs co-labeled with GFP and Pax6. A mutated miR-17 sponge (n = 3) failed to do so. (G,H) Expression of miR-17 sponge in E13.5 mouse cortices (n = 4) decreased the number of IPs co-labeled with GFP and Tbr2. A mutated miR-17 sponge (n = 4) failed to do so. Data are presented as mean ± SEM; n ≥ 3 in all constructs; p values in relation to the control (*p < 0.05, ***p < 0.001).
To further test the effect of miR-17 sponge on expansion of cortical NPs in vivo, the miR-17 sponge was electroporated into the cortices of E13.5 mice, and analyzed at E14.5. The percentages of BrdU+/GFP+, Pax6+/GFP+, and Tbr2+/GFP+ cells versus total GFP+ cells were all decreased significantly when the miR-17 sponge was expressed (Figures 5C–H). The mutated miR-17 sponge (miR-17 sp-mut), however, did not show effects to the RGCs and IPs, compared to the empty vector and miR-17 sponge (Figures 5C–H). These results indicate that the miR-17 sponge, which can block the endogenous miR-17 silencing activity, suppresses proliferation of cortical NPs.
miR-17 Rescues the Negative Effect of p21 on Proliferation of Neural Progenitors
To directly test the silencing activity of miR-17 on p21 in vivo, we speculated that co-expression of miR-17 with p21 should be able to rescue the negative effect of p21 on proliferation of NPs. The p21 full length cDNA with the 3′UTR was cloned into the pCAGIG vector, co-electroporated with miR-17 into E13.5 cortices, and analyzed at E14.5. Compared to p21 alone, co-expression of miR-17 and p21 significantly rescued NP proliferation, as demonstrated by increased percentages of BrdU+/GFP+, Pax6+/GFP+, and Tbr2+/GFP+ cells, which were compatible to those electroporated with the control vector (Figures 6A–F). However, co-expression of p21 and the mutated miR-17 failed to rescue NP proliferation (Figures 6A–F). These results are consistent with those of the luciferase assay, and prove that miR-17 blocks the negative effect of p21 on proliferation of NPs in vivo, especially RGCs and IPs in developing cortices.
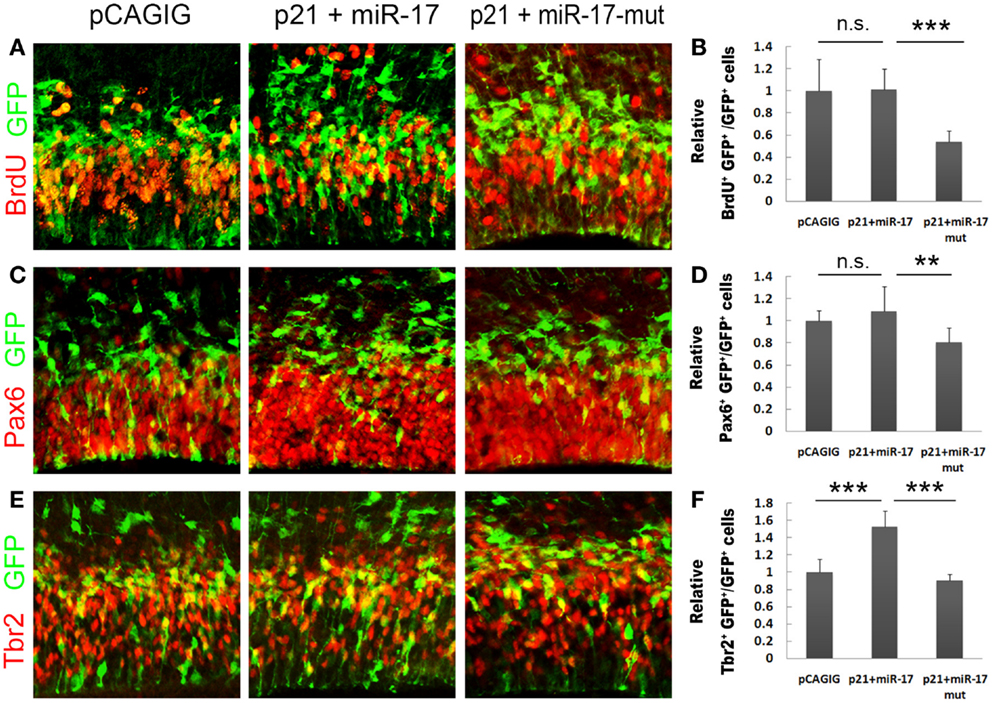
Figure 6. miR-17 rescues the negative effect of p21 on proliferation of neural progenitors. (A,B) Co-expression of miR-17 with p21 in E13.5 mouse cortices (n = 6), analyzed at E14.5, significantly rescued the reduced number of proliferating cells caused by p21 alone, while co-expression of a mutated miR-17 with p21 (n = 3) failed to do so. (C,D) Co-expression of miR-17 with p21 (n = 5) significantly rescued the reduced number of RGCs caused by p21 alone, while co-expression of a mutated miR-17 (miR-17-mut) with p21 (n = 3) failed to do so. (E,F) Co-expression of miR-17 with p21 (n = 5) significantly rescued the reduced number of IPs caused by p21 alone, while co-expression of a mutated miR-17 with p21 (n = 3) failed to do so. Data are presented as mean ± SEM; n ≥ 3 in all constructs; p values in relation to the control (**p < 0.01, ***p < 0.001), n.s., not significant.
Discussions
Accumulating evidence has shown that protein-coding genes and miRNAs play indispensable roles in mammalian neural development. In our studies, we have identified p21 as a negative regulator of RGC and IP expansion in the mouse embryonic cortex. Utilizing in utero electroporation and miRNA sponge, we have found that miR-17 specifically blocks p21 expression, thereby promoting the expansion of the cortical neural progenitor pool. Our studies have revealed a mechanism of suppressing endogenous p21 expression by miR-17 in NPs.
Many studies have shown that p21 functions as a negative cell cycle regulator and induces cell cycle exit (13–15). In the nervous system, p21 has been shown to suppress self-renewal and proliferation of NSCs and NPs (16–19). An important question is how the p21 gene expression level is properly regulated in vivo. In the developing cortex, to produce sufficient numbers of NPs to form a functional brain, p21 expression has to be suppressed. Our study here has shown that miR-17, a member of the miR-17–92 cluster, is an important regulator of p21 expression and expansion of the cortical neural progenitor pool. miR-17 displays decreased expression in mouse cortices at embryonic and postnatal stages, which is opposite to that of p21, suggesting a reciprocal expression between miR-17 and its target p21. These results further suggest that higher expression of miR-17 in embryonic cortices is crucial for suppressing the p21 level and promoting NP proliferation, while low expression of miR-17 in postnatal cortices allows p21 expression to induce differentiation. Furthermore, we have found the direct silencing action of miR-17 on p21 in vivo, since co-expression of p21 and miR-17, but not miR-17 mutations, can block negative effects of p21 on cortical NP proliferation.
It is likely that silencing p21 by miR-17 in proliferative cells is a general rule. The targeting effects of miR-17 and p21 have been observed in oral carcinoma cells, acute myeloid leukemia cells, and Hodgkin’s lymphoma (39–41). Furthermore, miR-17 likely maintains the neural progenitor pool by regulating several targets. In this study, we have demonstrated miR-17 targeting effect on p21 expression. Here, we have not ruled out the likely possibility that the regulation of additional target genes by miR-17 contributes to its ability to promote NP proliferation. For instance, miR-17 has been shown to enhance NSC self-renewal by targeting Trp53inp1, a gene in the p53 pathway, to promote NP proliferation by silencing bone morphogenetic protein type II receptor (42, 43).
Therefore, proper self-renewal of NSCs and expansion of NPs in the developing cortex relies on balanced expression levels of cell cycle regulators such as p21. One of the mechanisms that control precise expression levels of these regulators is through the miRNA silencing regulation such as miR-17.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Sun lab for helpful discussions. This work was supported by the Xiangya Hospital of Central South University from China (Yase Chen and Beisha Tang), the Hirschl/Weill-Caulier Trust (Tao Sun), and an R01-MH083680 grant from the NIH/NIMH (Tao Sun).
References
1. Chenn A, McConnell SK. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell (1995) 82:631–41. doi: 10.1016/0092-8674(95)90035-7
2. Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci (2005) 25:247–51. doi:10.1523/JNEUROSCI.2899-04.2005
3. Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol (2005) 6:777–88. doi:10.1038/nrm1739
4. Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci U S A (2004) 101:3196–201. doi:10.1073/pnas.0308600100
5. Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature (2001) 409:714–20. doi:10.1038/35055553
6. Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex (2003) 13:541–9. doi:10.1093/cercor/13.6.541
7. Yang YT, Wang CL, Van Aelst L. DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat Neurosci (2012) 15(9):1201–10. doi:10.1038/nn.3171
8. Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, et al. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev (2008) 22:2479–84. doi:10.1101/gad.475408
9. Sessa A, Mao CA, Colasante G, Nini A, Klein WH, Broccoli V. Tbr2-positive intermediate (basal) neuronal progenitors safeguard cerebral cortex expansion by controlling amplification of pallial glutamatergic neurons and attraction of subpallial GABAergic interneurons. Genes Dev (2010) 24:1816–26. doi:10.1101/gad.575410
10. Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron (2008) 60:56–69. doi:10.1016/j.neuron.2008.09.028
11. Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci (2007) 8:438–50. doi:10.1038/nrn2176
12. Kriegstein A, Noctor S, Martinez-Cerdeno V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci (2006) 7:883–90. doi:10.1038/nrn2008
13. Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci U S A (1999) 96:1002–7. doi:10.1073/pnas.96.3.1002
14. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res (2005) 65:3980–5. doi:10.1158/0008-5472.CAN-04-3995
15. Gartel AL, Serfas MS, Tyner AL. p21 – negative regulator of the cell cycle. Proc Soc Exp Biol Med (1996) 213:138–49. doi:10.3181/00379727-213-44046
16. Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell (2007) 1:87–99. doi:10.1016/j.stem.2007.04.001
17. Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev (2005) 19:756–67. doi:10.1101/gad.1272305
18. Marques-Torrejon MA, Porlan E, Banito A, Gomez-Ibarlucea E, Lopez-Contreras AJ, Fernandez-Capetillo O, et al. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell (2013) 12:88–100. doi:10.1016/j.stem.2012.12.001
19. Pechnick RN, Zonis S, Wawrowsky K, Pourmorady J, Chesnokova V. p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc Natl Acad Sci U S A (2008) 105:1358–63. doi:10.1073/pnas.0711030105
20. Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci (2014) 15:217–32. doi:10.1038/nrn3707
21. Bian S, Sun T. Functions of noncoding RNAs in neural development and neurological diseases. Mol Neurobiol (2011) 44(3):359–73. doi:10.1007/s12035-011-8211-3
22. Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron (2009) 64:303–9. doi:10.1016/j.neuron.2009.10.020
24. Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci (2012) 13:528–41. doi:10.1038/nrn3234
25. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi:10.1016/j.cell.2009.01.035
26. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol (2009) 10:126–39. doi:10.1038/nrm2632
28. Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell (2007) 131:136–45. doi:10.1016/j.cell.2007.09.020
29. Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol (2010) 12:372–9. doi:10.1038/ncb2037
30. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell (2008) 133:217–22. doi:10.1016/j.cell.2008.04.001
31. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17 ∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev (2009) 23:2806–11. doi:10.1101/gad.1872909
32. Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev (2009) 23:2839–49. doi:10.1101/gad.1861409
33. Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol (2010) 42:1348–54. doi:10.1016/j.biocel.2010.03.004
34. Bian S, Hong J, Li Q, Schebelle L, Pollock A, Knauss JL, et al. MicroRNA cluster miR-17-92 regulates neural stem cell expansion and transition to intermediate progenitors in the developing mouse neocortex. Cell Rep (2013) 3:1398–406. doi:10.1016/j.celrep.2013.03.037
35. Nowakowski TJ, Fotaki V, Pollock A, Sun T, Pratt T, Price DJ. MicroRNA-92b regulates the development of intermediate cortical progenitors in embryonic mouse brain. Proc Natl Acad Sci U S A (2013) 110:7056–61. doi:10.1073/pnas.1219385110
36. Zhang H, Shykind B, Sun T. Approaches to manipulating microRNAs in neurogenesis. Front Neurosci (2013) 6:196. doi:10.3389/fnins.2012.00196
37. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods (2007) 4:721–6. doi:10.1038/nmeth1079
38. Gentner B, Schira G, Giustacchini A, Amendola M, Brown BD, Ponzoni M, et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods (2008) 6:63–6. doi:10.1038/nmeth.1277
39. Gibcus JH, Kroesen BJ, Koster R, Halsema N, de Jong D, de Jong S, et al. MiR-17/106b seed family regulates p21 in Hodgkin’s lymphoma. J Pathol (2011) 225:609–17. doi:10.1002/path.2958
40. He M, Wang QY, Yin QQ, Tang J, Lu Y, Zhou CX, et al. HIF-1alpha downregulates miR-17/20a directly targeting p21 and STAT3: a role in myeloid leukemic cell differentiation. Cell Death Differ (2013) 20:408–18. doi:10.1038/cdd.2012.130
41. Wu SY, Lin KC, Chiou JF, Jeng SC, Cheng WH, Chang CI, et al. MicroRNA-17-5p post-transcriptionally regulates p21 expression in irradiated betel quid chewing-related oral squamous cell carcinoma cells. Strahlenther Onkol (2013) 189:675–83. doi:10.1007/s00066-013-0347-9
42. Garg N, Po A, Miele E, Campese AF, Begalli F, Silvano M, et al. microRNA-17-92 cluster is a direct Nanog target and controls neural stem cell through Trp53inp1. EMBO J (2013) 32:2819–32. doi:10.1038/emboj.2013.214
Keywords: miRNAs, miR-17, p21, proliferation, cerebral cortex
Citation: Chen Y, Bian S, Zhang J, Zhang H, Tang B and Sun T (2014) The silencing effect of microRNA miR-17 on p21 maintains the neural progenitor pool in the developing cerebral cortex. Front. Neurol. 5:132. doi: 10.3389/fneur.2014.00132
Received: 04 April 2014; Accepted: 03 July 2014;
Published online: 18 July 2014.
Edited by:
Mark Nicholas Ziats, National Institute of Child Health and Human Development, USAReviewed by:
Ruth Luthi-Carter, University of Leicester, UKRenzo Guerrini, University of Florence, Italy
Copyright: © 2014 Chen, Bian, Zhang, Zhang, Tang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beisha Tang, Department of Neurology, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha 410008, China e-mail:YnN0YW5nNzM5OEAxNjMuY29t;
Tao Sun, Department of Cell and Developmental Biology, Weill Medical College of Cornell University, 1300 York Avenue, Box 60, New York, NY 10065, USA e-mail:dGFzMjAwOUBtZWQuY29ybmVsbC5lZHU=
†First co-authorship