 He Li1*
He Li1* Xin Li2
Xin Li2 Stanley E. Smerin1
Stanley E. Smerin1 Lei Zhang1
Lei Zhang1 Min Jia1
Min Jia1 Guoqiang Xing1
Guoqiang Xing1 Yan A. Su3
Yan A. Su3 Jillian Wen1
Jillian Wen1 David Benedek1
David Benedek1 Robert Ursano1
Robert Ursano1
- 1Department of Psychiatry, Center for the Study of Traumatic Stress, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 2Department of Biostatistics, Bioinformatics, and Biomathematics, Georgetown University Medical Center, Washington, DC, USA
- 3Department of Gene and Protein Biomarkers, GenProMarkers, Rockville, MD, USA
The metabolic mechanisms underlying the development of exaggerated fear in post-traumatic stress disorder (PTSD) are not well defined. In the present study, alteration in the expression of genes associated with mitochondrial function in the amygdala of an animal model of PTSD was determined. Amygdala tissue samples were excised from 10 non-stressed control rats and 10 stressed rats, 14 days post-stress treatment. Total RNA was isolated, cDNA was synthesized, and gene expression levels were determined using a cDNA microarray. During the development of the exaggerated fear associated with PTSD, 48 genes were found to be significantly upregulated and 37 were significantly downregulated in the amygdala complex based on stringent criteria (p < 0.01). Ingenuity pathway analysis revealed up- or downregulation in the amygdala complex of four signaling networks – one associated with inflammatory and apoptotic pathways, one with immune mediators and metabolism, one with transcriptional factors, and one with chromatin remodeling. Thus, informatics of a neuronal gene array allowed us to determine the expression profile of mitochondrial genes in the amygdala complex of an animal model of PTSD. The result is a further understanding of the metabolic and neuronal signaling mechanisms associated with delayed and exaggerated fear.
Introduction
The neurobiology of exaggerated fear is important in understanding traumatic stress and post-traumatic stress disorder (PTSD). Decades of animal studies have shown that the amygdala is key in the neuronal system that orchestrates the fear memory and exaggerated fear evoked by stress. In PTSD patients exposed to an aversive visual stimulus or a fear inducing pulse of sound, fMRI shows an increased blood flow and metabolic rate in the amygdala, indicating a prolonged dysregulation of metabolism and an altered neuronal transmitter system in the amygdala circuitry (1–3). Dysregulation appears to persist throughout life, at least in the frontal cortex, as we found by analyzing a mitochondria gene array from post-mortem frontal cortex samples of PTSD patients. Genes and signaling pathways, as well as pharmacological targets, were significantly dysregulated (4).
Mitochondria are targets for stress hormones such as corticosterone (CORT) and are increasingly recognized as key components in stress-related mental disorders (5, 6). Prolonged and repetitive psychological stress can induce long-lasting neurobiological consequences. However, the exact subcellular mechanisms involved in such long-term neuronal and hormonal impairment remain elusive. The current study used a rat model of PTSD to further examine amygdala function and traumatic stress. In this study, we examined the relationship of altered amygdala mitochondrial-function genes and the molecular signaling pathways associated with a key symptom of PTSD: delayed and exaggerated fear. Amygdala tissues were collected from rats 14 days after stress, a time point at which exaggerated fear manifests. We then screened for 1500 mitochondrial-function-associated genes, including 37 mitochondrial DNA (mtDNA)-encoded genes, 1,098 nuclear DNA (nDNA)-encoded and mitochondria-focused genes, and 365 neuron-related genes (7, 8).
Up until now, the altered subcellular and metabolic molecular markers associated with delayed and exaggerated fear in the amygdala were unknown. Thus, this research was carried out in an attempt to determine whether traumatic stress alters the expression profiles of mitochondrial genes in association with the pathogenesis of delayed and exaggerated fear. A mitochondrial microarray technique was used to examine all mitochondrial-function-associated transcriptomes and to map the susceptible gene clusters in the amygdala complex associated with delayed and exaggerated fear. Our results indicate that signaling pathways, including pre-inflammatory, apoptotic, metabolic, neuronal transmitter system, and genes of G-protein-coupled receptors are up- or downregulated 14 days after the stress paradigm. The impacts of alteration in regulation of these molecular mechanisms are discussed below.
Materials and Methods
Animal Model of PTSD
The animal model of PTSD employs restraint and tail shocks on three consecutive days. This inescapable tail-shock model (ITS) in rats mimics in many ways the pathophysiology of PTSD (9–13). In our model, stress exposure consists of a daily 2-h session of immobilization and tail-shocks for three consecutive days. The animals are restrained in Plexiglas tubes, and 40 electric shocks (2 mA, 3 s duration) are applied at varying intervals (140–180 s). Animals thereby undergo an aversive experience under conditions in which they cannot adaptively respond. We and others have verified that the ITS model induces behavioral and neurobiological alterations similar to those found in PTSD subjects (9–12). Specifically, these stressed rats exhibit (1) a delayed and exaggerated startle response appearing several days after stressor cessation which, given the compressed time scale of the rat’s life compared to a humans, corresponds to the 1–3 months delay of symptoms in PTSD patients (14, 15), (2) enhanced plasma CORT for several days, indicating a compromised hypothalamopituitary axis (HPA), and (3) retarded body weight gain after stressor cessation, indicating dysfunction of gene expression. The gene expression microarray used in this experiment, dubbed the rat mitochondrion-neuron focused microarray (rMNChip), focuses on mitochondrial and mitochondria-related nuclear genes in the rat so as to specifically address the neuronal bioenergetics hypothesized to be involved in arousal, CORT, and growth as addressed in our model of PTSD (13).
Male albino Sprague Dawley rats were used (Taconic Farms, Derwood, MD, USA), weighing 150–200 g at the time of administration of the stress protocol. Two groups of animals (n = 20) were studied. Twenty animals were assigned to each group based on their body weight and baseline startle response. Group 1 received the stress protocol, and Group 2 was the control. Stress exposure consisted of a daily 2-h session of restraint by immobilization in ventilated Plexiglas tubes (“inescapable”) and tail shocks for three consecutive days. Stressing was performed in the morning (between 08:00 and 12:00 hours).
Forty electric shocks (2 mA, 3 s duration; Animal Test Cage Grid Floor Shocker, Coulbourn Instruments, USA) were delivered to the tail of Group 1 rats at semi-random intervals of 150–210 s (Graphic State Notation Software, Habitest Universal Link, Coulbourn Instruments, USA). Electrode gel was applied using Q-tips to form a thin layer of conducting gel between the electrode and the skin of the rat’s tail. The electrode clips were adjusted and connected to the tail to ensure a good connection without affecting the blood circulation of the tail. Amygdala dissection was performed as previously described (13).
Rat Mitochondria Neuronal Chip
The rMNChip was designed and fabricated by GenProMarkers, Inc. (Rockville, MD, USA) using published methods (8). Briefly, the rMNChip contains 1,500 genes, including 37 mtDNA-encoded genes, 1,098 nDNA-encoded and mitochondria-focused genes, and 365 genes associated with neuronal functions, including fear response, circadian rhythms, intraneuronal signal transduction, and neurotransmitters. The oligonucleotides used were designed with the software MacVector v10.6.0 (MacVector) using full-length mRNA sequences as the templates according to published criteria. An amino-C6 modifier was added to the 5′-end of each oligonucleotide probe to enhance the binding of the DNA to glass slides and the accessibility of hybridization with target DNA. The 1,500 test genes (including 80 “housekeeping” genes as positive controls) and 36 negative controls (non-rat DNA) were printed, each in triplicates, onto the N-hydroxysuccinimide ester reactive group-coated glass slides (CodeLink Activated Slide, SurModics, Eden Prairie, MI, USA). DNA probes in the print buffer (50 mM sodium phosphate), at a final concentration of 20 μM of 5′-amino-C6 modified 50-mers, were printed in the Class 100 super-clean environment as described previously (7, 16, 17), using 100 μM pins and the GeneMachine OmniGrid 100 Microarrayer (Genomic Solutions, Ann Arbor, MI, USA).
RNA Purification
Total cellular RNA was purified from rat brain tissues by using a PAXgene RNA Kit (QIAGEN Inc., Valencia, CA, USA) following the manufacturer’s instructions. The amygdalas of rats from both the stress and control groups were dissected immediately after euthanasia (13).
Microarray Hybridization
One microgram of RNA per sample was used for Cy5dUTP labeling of cDNA by use of the express array detection kit (3DNA Array 900, Genisphere, Hatfield, PA, USA) following the manufacturer’s instructions. Slides were scanned using 5 μM resolution and the LOWESS method with a ScanArray Microarray Scanner (PerkinElmer). Triplicate microarray experiments were performed for each RNA sample purified from the amygdala. The background-subtracted mean values of the measured gene expression signal intensities were used for microarray data analysis. All microarray experiments were performed in the same laboratory as GenProMarkers, Inc. (8).
Microarray Database, Bioinformatics, and Systems Biology
A gene expression database was constructed using FileMaker software (FileMaker Pro, Inc., Santa Clara, CA, USA). Database construction, data filtering, selection, exclusion and inclusion procedures, and criteria were performed as described previously (7, 8). The quantile normalization method (18) in software R version 2.15.1 (The R Foundation for Statistical Computing) was used to normalize background-subtracted mean intensities across all intra- and inter-slides of informative microarray data. Correlation and Single linkage methods were used to cluster and to make the heatmap using Cluster version 3.0 and MapleTree softwares1. The normalized data were also used for the calculation of means, SD, fold changes, moderated p-values, and false discovery rates (FDR). Gene IDs, official symbols, and official full names were updated using the NCBI database2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Online Mendelian Inheritance in Man (OMIM) were from DAVID Bioinformatics Resources3.
Statistics
The quantile normalization method (18) in software R/Biocon- ductor version 2.15.1 (The R Foundation for Statistical Computing) was used to normalize data. Means, SDs, and fold changes were calculated from triplicate spots (Amygdala complex) and triplicate experiments using XLSTAT 2006 (XLSTAT, New York, NY, USA). Differentially expressed genes were identified arbitrarily as having a greater than twofold change in the average expression of the background-subtracted mean intensity ratios of a gene between comparisons (19). The moderated p-values and FDR for multiple statistical testing with Benjamini and Hochberg methods (20) were calculated with the software R/Bioconductor version 2.15.1 (The R Foundation for Statistical Computing). The level of statistical significance was set at a p-value <0.01 with a specific FDR indicated. The error bar and plot were generated using Origin Lab version 8.5.
Experimental Paradigm (Figure 1)
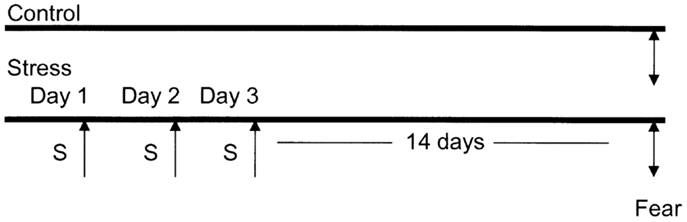
Figure 1. Diagrammatic representation of the animal stress protocol. Amygdala complex (A) from control group (n = 10) and stressed group (n = 10) were dissected 14 days after cessation of 3 days stress (S).
Results
Heatmap Presentation Following Microarray Data Collection and Analysis
The amygdala tissues were dissected from the rat brain 14 days after the cessation of the 3 days of stress. Total RNA samples were extracted from the amygdala complex of stressed rats (n = 10), and controls (n = 10) (13). Microarray experiments were triplicated using our recently developed fourth generation rat mitochondria-focused cDNA chip rMitChip3. With three spots in one cDNA microarray chip and three chips per each amygdala sample, the resulting analysis of nine data values from each single specimen greatly reduces misclassification rates (8). The measurement of expression of individual genes on the rMitChip3 (rMNChip) in one group was analyzed using 90 values (3 identical probes per microarray, 3 microarray experiments per specimen, and 10 specimens per gene), which generated reliable expression data for further pathway analysis. In this present study, the mitochondria-focused gene expression profilings in the amygdala complex 14 days after traumatic stress were analyzed using the cDNA microarray chip containing 1500 genes associated with mitochondria functions. Based on the unsupervised cluster results, we calculated the average expression level of each gene in the stressed group (n = 10) and in the non-stressed control group (n = 10). The microarray data of 270,000 spots across all 60 microarray chips used for 20 RNA samples were filtered by uniform statistic and bioinformatic criteria (7), which generated 85 genes with informative expression profiles.
We identified a cluster and heatmap of 85 genes with significantly expressed RNA derived from the amygdala complex of the 10 control rats and the 10 stressed rats (Figure 2). The resultant dendrograms for all of these 85 genes and 20 amygdala complex specimens are classified as control group from CON-01 to CON-10 and stressed group from STR-1 to STR-10 in Figure 2; these analyses separated the stressed amygdala from those of the controls by their gene expression profiles. Figure 2A reveals 37 downregulated gene profiles and Figure 2B reveals 48 upregulated gene profiles.
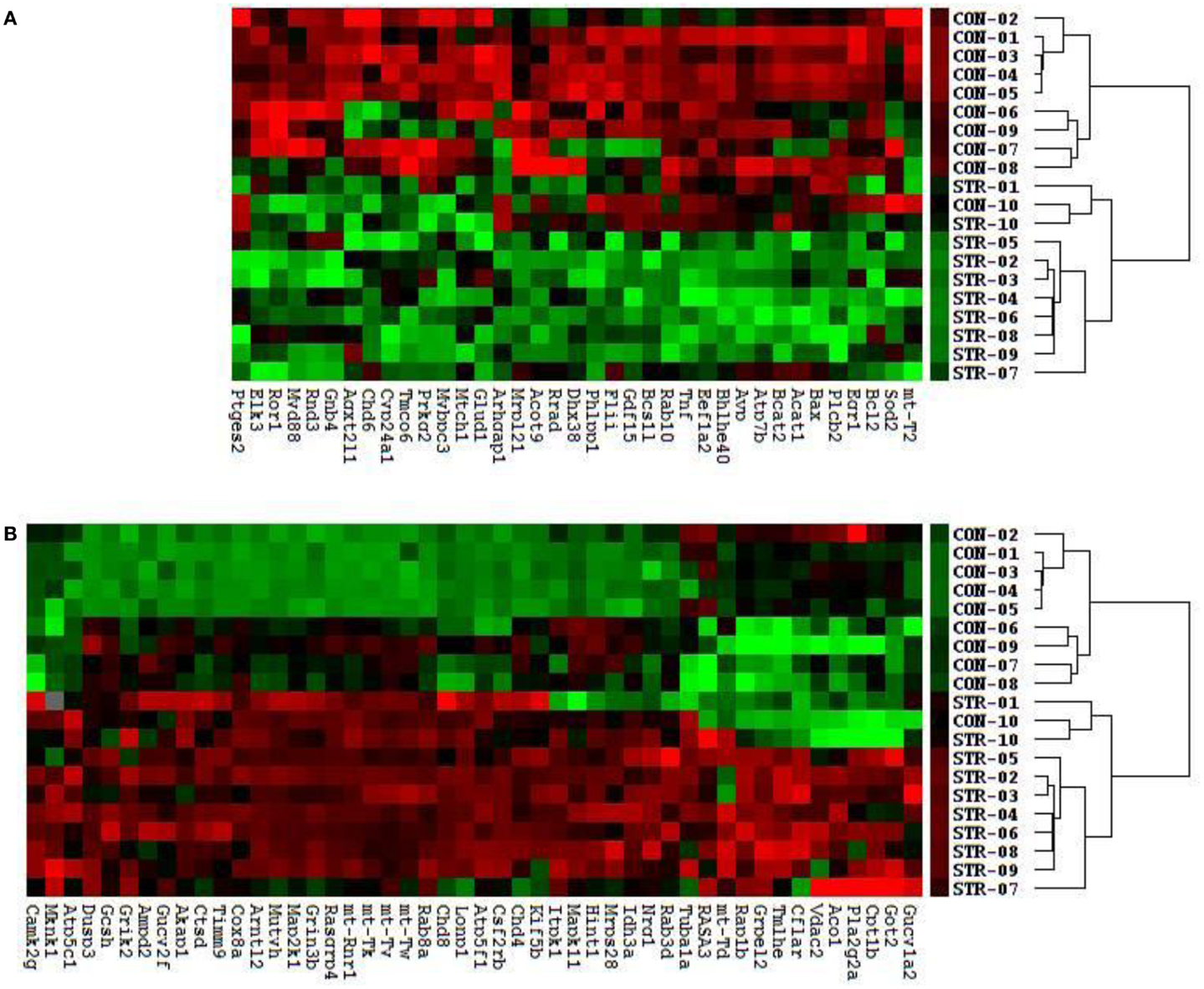
Figure 2. Clustering analysis of 48 upregulated and 37 downregulated genes across all 20 amygdala samples generates double dendrograms and heat maps that distinguish the stressed amygdala specimens from the controls (A,B). The identification of these up- and downregulated genes provides candidates for network analysis. The ratios and p-values (t-test) were calculated between the stressed (n = 10) and control specimens (n = 10), resulting in the identification of 85 genes (p < 0.01) and among these 55 altered genes (p < 0.01) were further selected using a high stringency algorithm that includes fold change and fault discovery rate in the stressed amygdala as compared to the controls (Tables 1 and 2).
Pathway Analysis of Up- and Downregulated Genes
Pathways containing protein products of the up- and downregulated genes were determined using the Ingenuity Pathway Analysis (IPA) program. Table 1 illustrates the 34 upregulated genes and 21 downregulated genes in the amygdala after stress. A high stringency algorithm was used, including fold changes and FDR comparing stressed to controls (Tables 1 and 2). The tables include gene symbols, full names of genes listed in Genecards, fold change [ratio of stress and control, log(2) of the ratio], p-value, and fault discover rate (FDR).
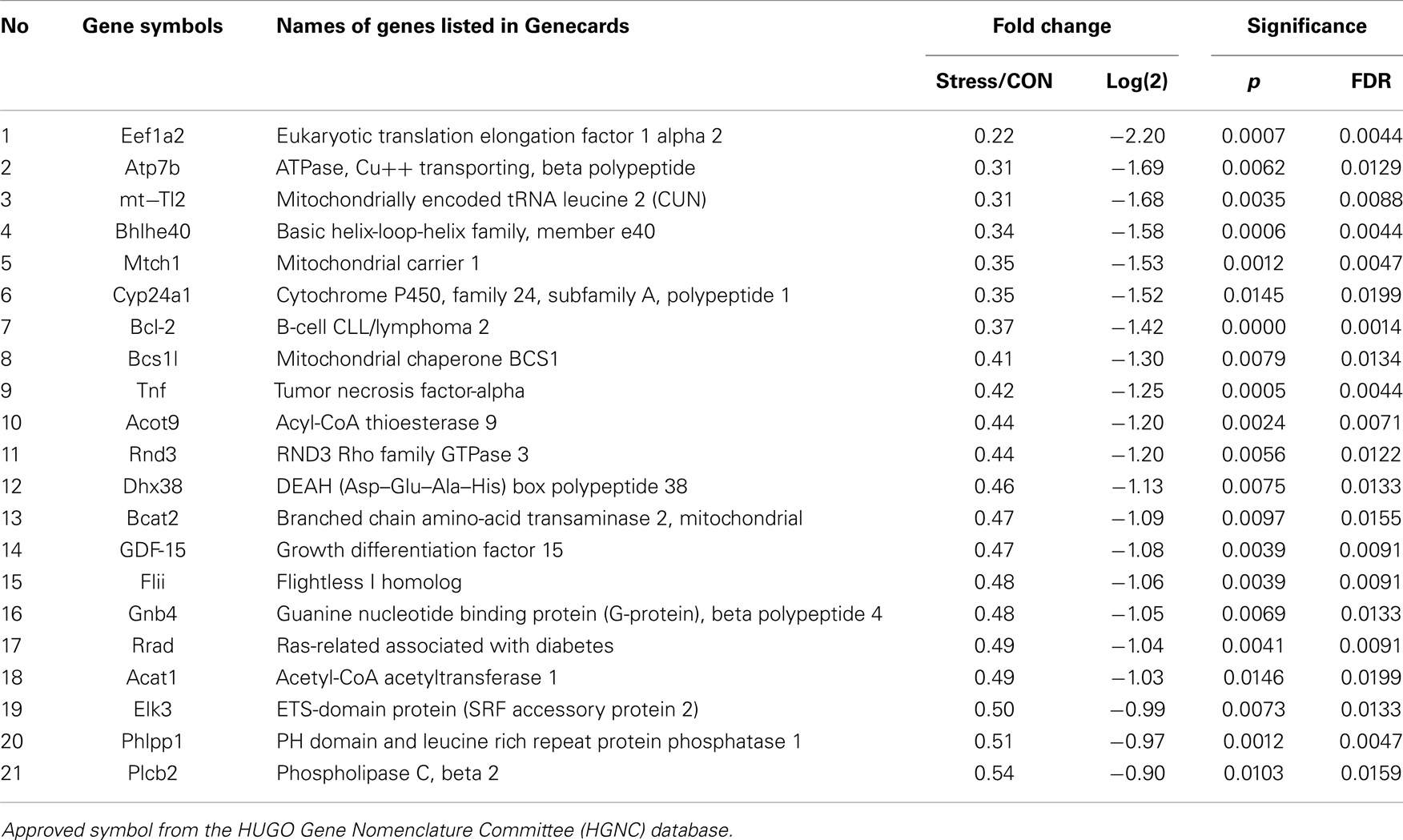
Table 1. Downregulated genes in amygdala (14 days after stress).
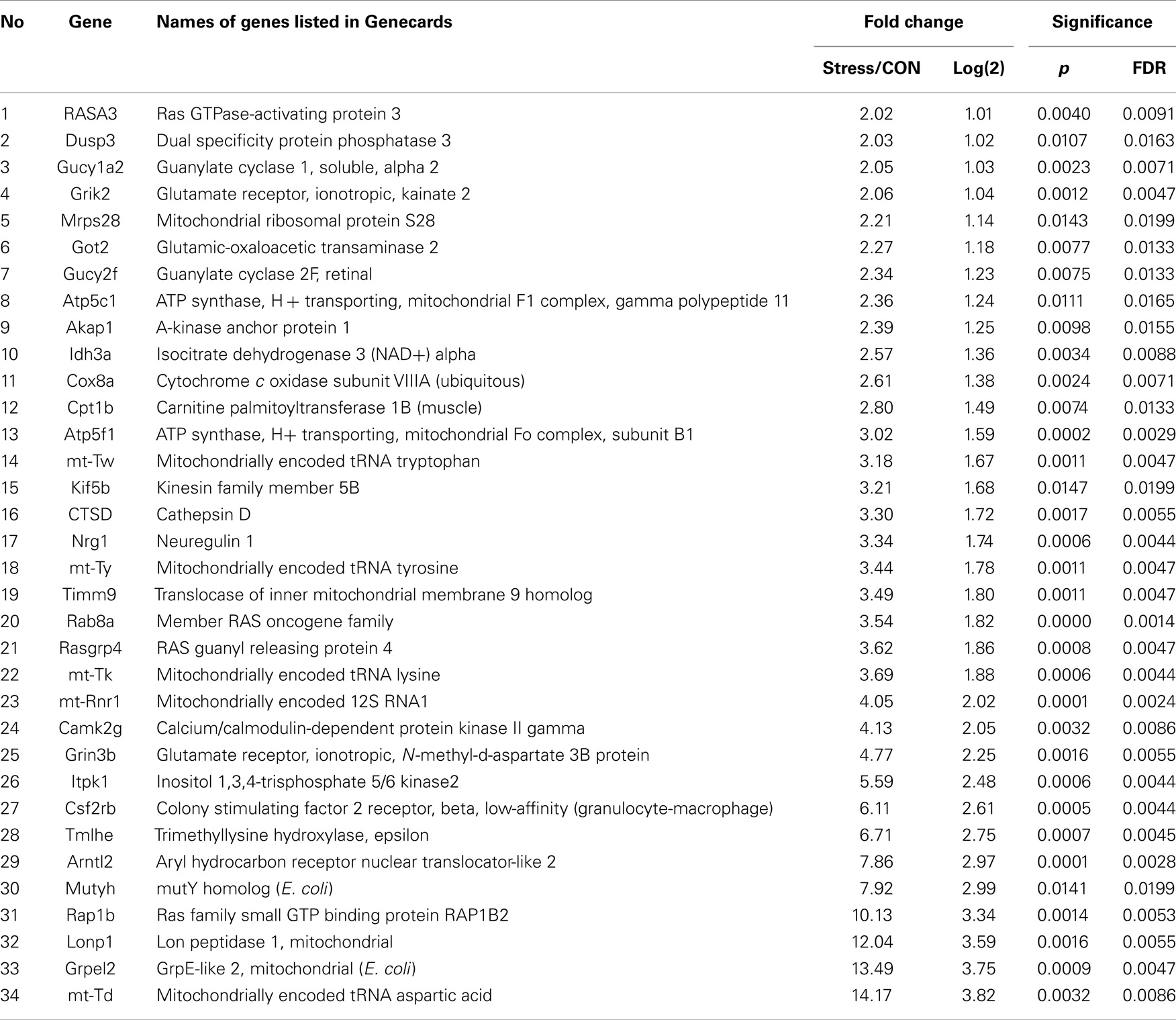
Table 2. Upregulated genes in amygdala (14 days after stress).
Molecular networks associated with mitochondrial functions were constructed based on up- and downregulated genes from the amygdala complex associated with exaggerated fear (13). Network analysis indicated that four reregulated molecular networks were associated with metabolic molecules and neurotransmitter systems, as well as with translational factors in the amygdala complex (Figure 2). Mitochondrial dysfunction involved in endocrine and neuronal signaling was evident in the amygdala. Detailed molecular correlations of these reregulated molecular pathways and the symptoms of PTSD such as exaggerated fear are noted (Figure 2). The results also suggest possible molecular targets for pharmacological intervention following exposure to traumatic stress.
In the network analysis, the networks identified are presented as graphs indicating the molecular relationships between gene products. Gene products are represented as nodes, and a biological relationship between two nodes is indicated by a line. The intensity of the node color indicates the degree of upregulation (red) or downregulation (green). Gene products in uncolored nodes were not identified as differentially expressed in our experiment, but were integrated into the computationally generated networks on the basis of the evidence stored in the IPA knowledge base indicating a relevance to this network.
The node shapes denote enzymes, phosphates, transmembrane receptors, cytokines, channels, transcription factors, G-protein-coupled receptors, growth factors, or nuclear receptor, as shown in Figure 3. Canonical pathway analysis identified the canonical pathways from the IPA library that were most significantly related to the input data set. The significance of the association between the data set and the canonical pathway was determined based on two parameters: (1) a ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway and (2) a p-value calculated using Fischer’s exact test determining the probability that the association between the genes in the data set, and the canonical pathway is due to chance alone.
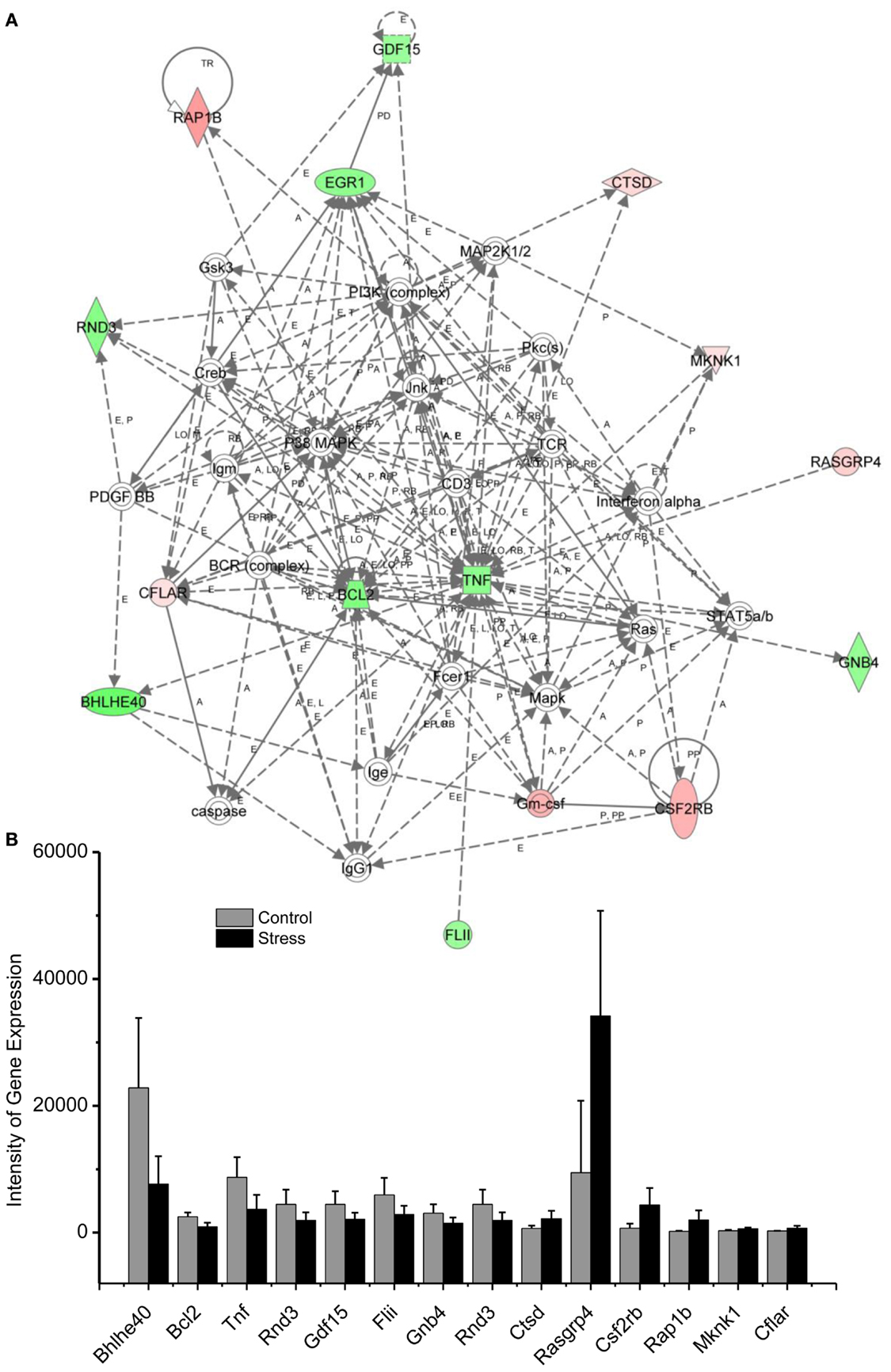
Figure 3. In (A), IPA associates stress reregulated mitochondrial- related genes with the network containing Bcl-2 and TNF are shown. (B) is a bar graph plotted as mean ± SD of the intensity of up- or downregulated genes in the stressed (n = 10) and control amygdala (p < 0.01, n = 10) for the network associated with Bcl-2 and TNF in the amygdala 14 days after stress. The order is plotted according to fold change, from downregulated with lowest negative value to upregulated with highest positive value.
To reveal specific and detailed interactions among up- and downregulated genes in the amygdala, and to further identify gene targets for pharmacological agents, four molecular networks were delineated: (1) Bcl-2 and TNF centered inflammatory and apoptotic signaling networks as shown in Figure 4; (2) energy production and neurotransmitter-mediated signaling pathways as shown in Figure 5; (3) transcriptional factor and chromatin remodeling pathways as shown in Figure 6; (4) immune mediator-related pathways as shown in Figure 7. The genes in the pathways were selected based on recent literature in the IPA database and relevance to neurological dysfunction, psychiatric disorders, and exaggerated fear response associated with amygdala functions as addressed in the Section “Discussion.” Within these networks, 34 (62%) of the genes were significantly upregulated (Table 2; Figure 2B) and 21 (38%) of the genes were significantly downregulated (Table 1; Figure 2A). In the network analysis, these genes display particular relevance to the symptoms of PTSD.
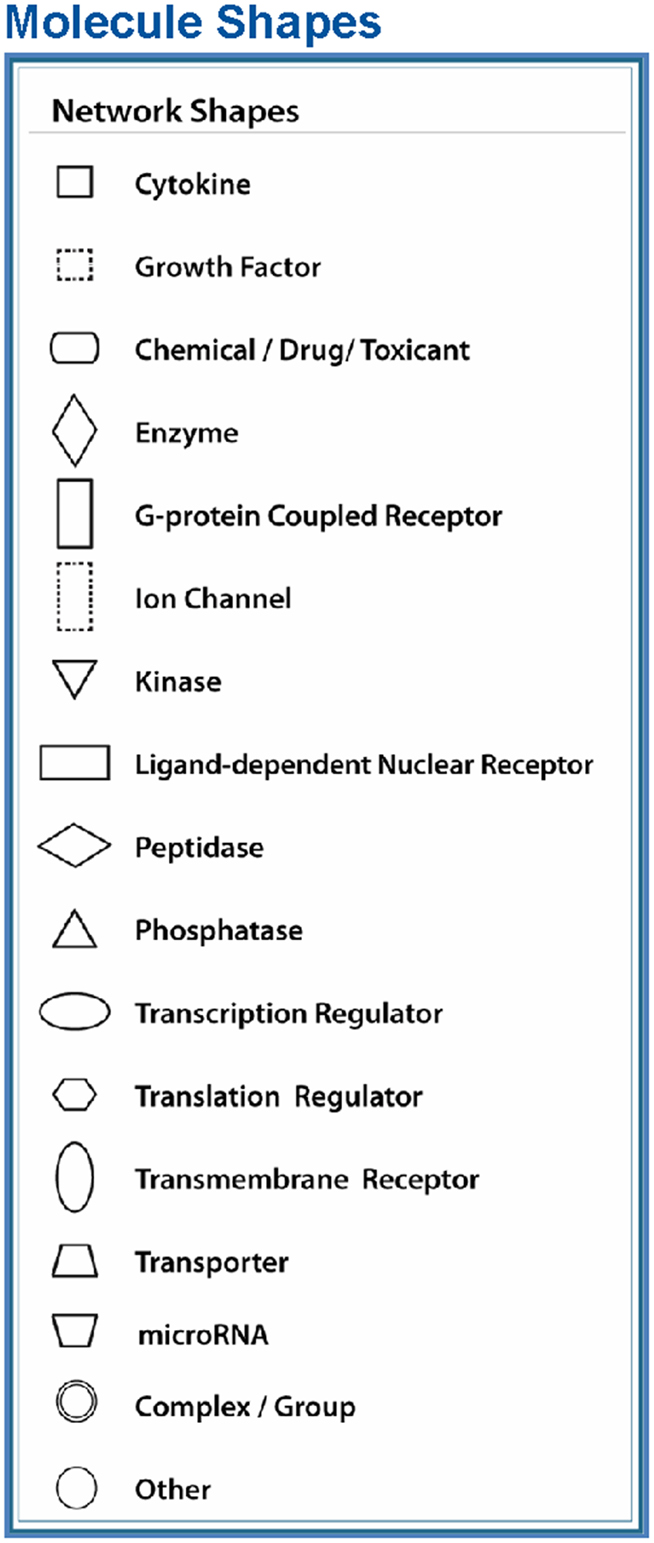
Figure 4. Node (gene) and edge (gene relationship) symbols are described in the figure. The intensity of the node color-(red) indicates the degree of upregulation. Genes in uncolored nodes are not identified as differentially expressed genes in our experiment and are integrated into the computationally generated networks on the basis of the evidence stored in the IPA knowledge memory indicating a relevance to this network. The node shapes denote enzymes, phosphatases, kinases, peptidases, G-protein-coupled receptors, transmembrane receptors, cytokines, growth factors, ion channels, transporters, translation factors, nuclear receptors, and transcription factors.
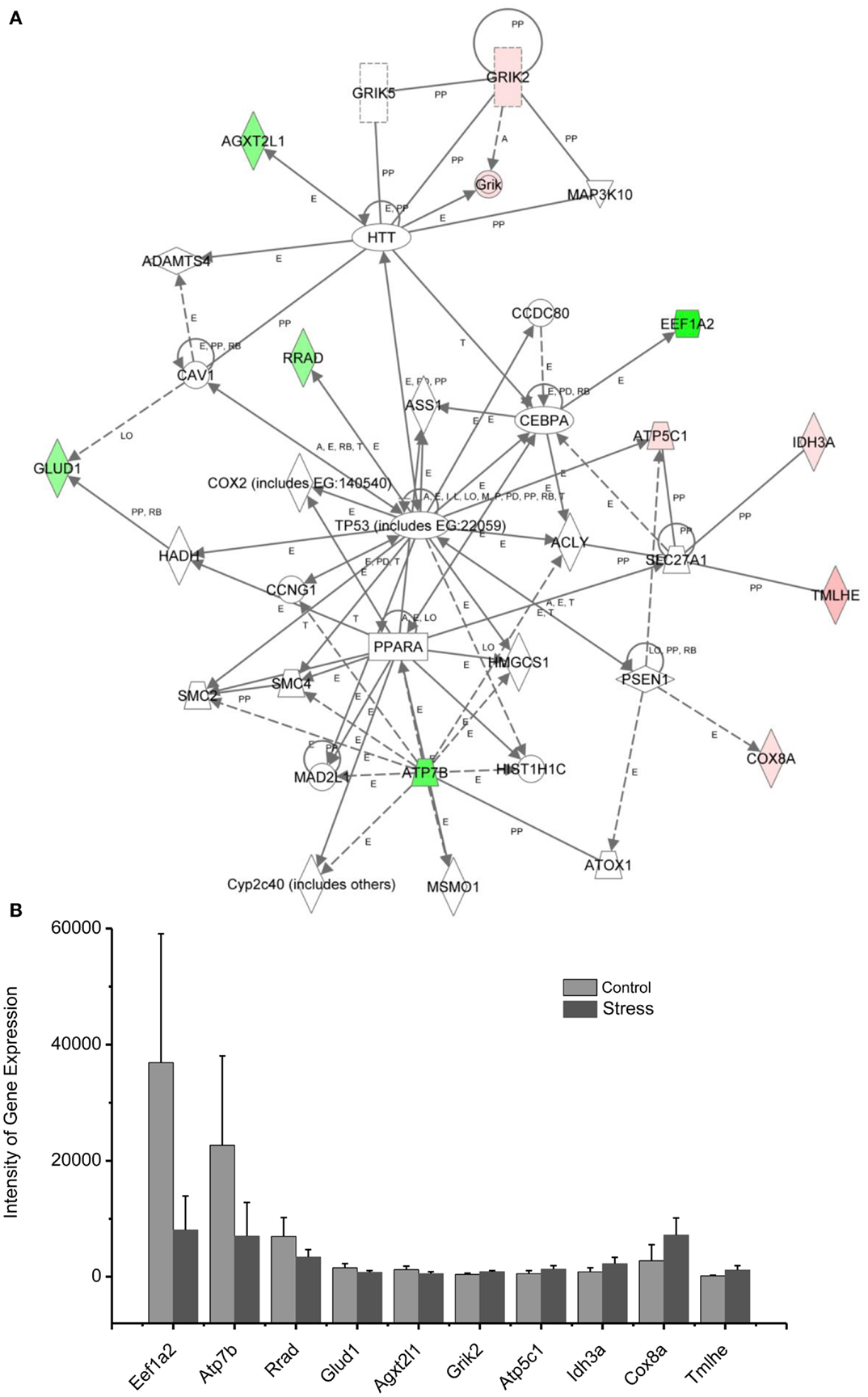
Figure 5. In (A), IPA associates stress up- or downregulated mitochondrial-related genes with the network containing TP53, PPARA, and HTT. (B) is a bar graph plotted with mean ± SD of the intensity of up- or downregulated genes for the stressed (n = 10) and control amygdala (p < 0.01, n = 10) in the network associated with TP53, PPARA, and HTT in the amygdala 14 days after stress. The order is plotted according to fold change, from downregulated with lowest negative value to upregulated with highest positive value.
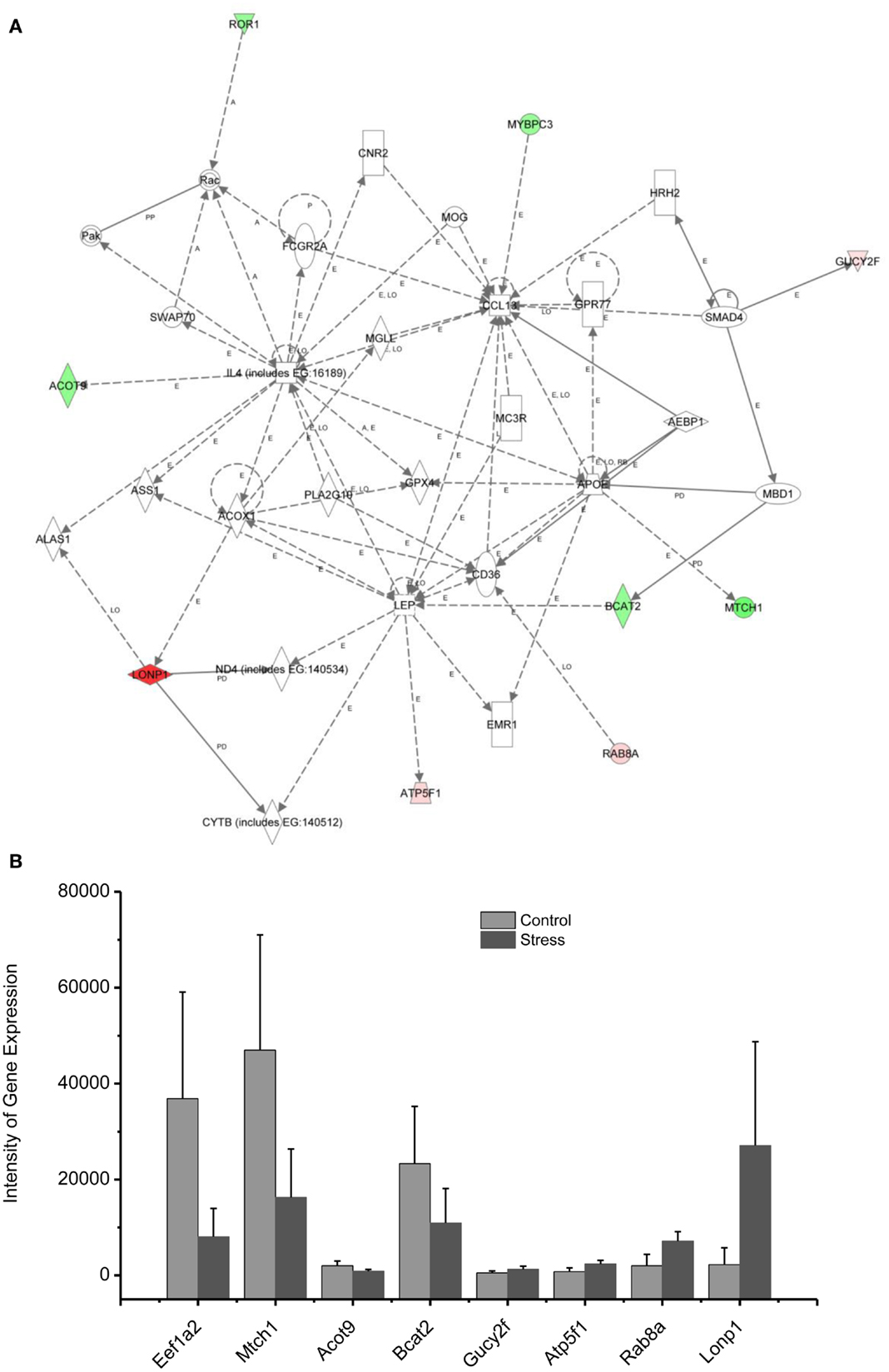
Figure 6. In (A), IPA associates stress up- or downregulated mitochondrial-related genes with the network containing LEP and CL1. (B) is a bar graph plotted with mean ± SD of the intensity of up- or downregulated genes for the stressed (n = 10) and controlled amygdala (p < 0.01, n = 10) in the network associated with LEP and CL1 in the amygdala 14 days after stress. The order is plotted according to fold change, from downregulated with lowest negative value to upregulated with highest positive value.
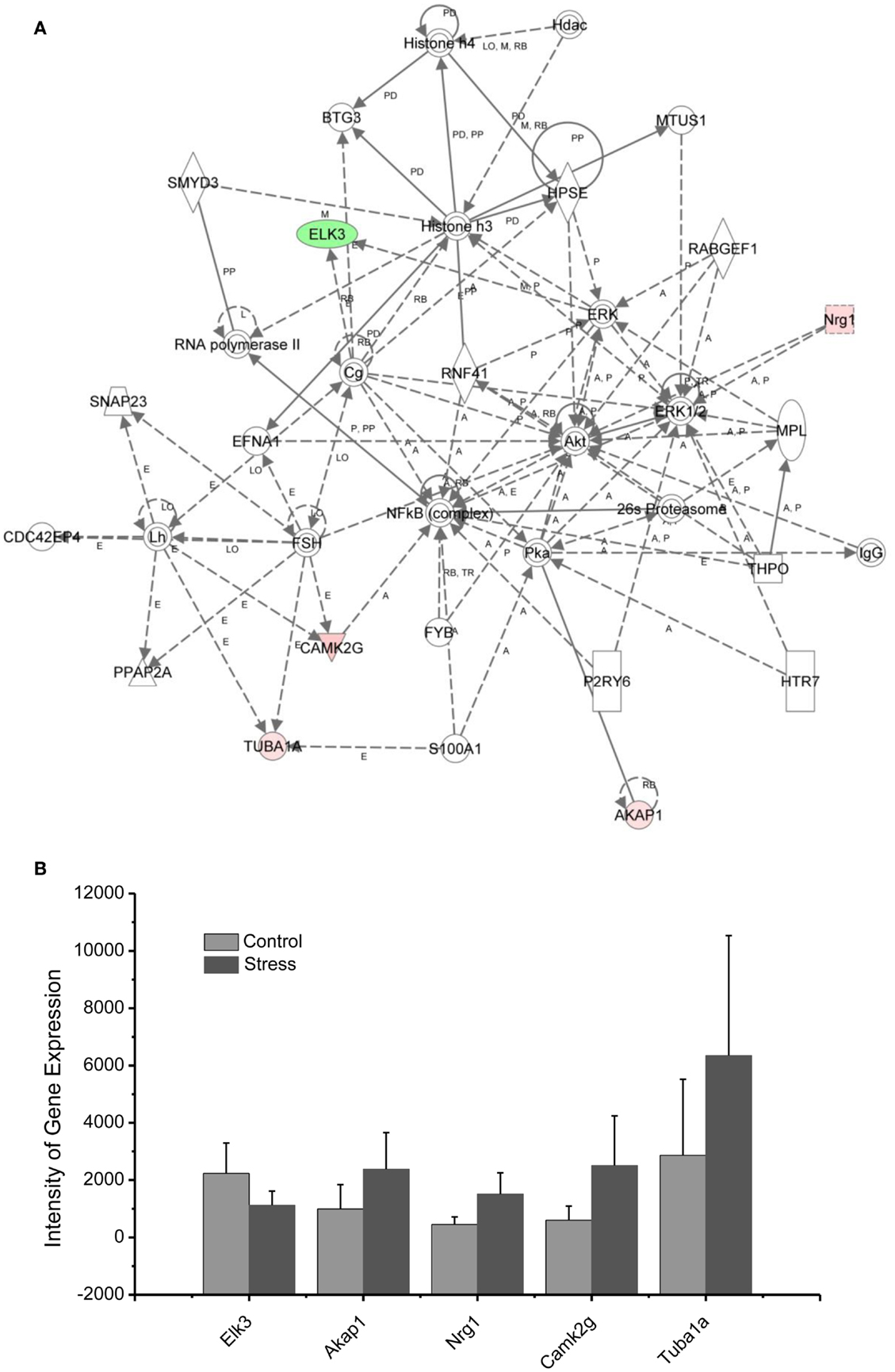
Figure 7. In (A), IPA associates stress reregulated mitochondrial- related genes with the network containing NFkb, Akt, and Histone 3. (B) is a bar graph plotted with mean ± SD of the intensity of up- or downregulated genes for the stressed (n = 10) and controlled amygdala (p < 0.01, n = 10) in the network associated with NFkb, AKt, and Histone 3 in the amygdala 14 days after stress. The order is plotted according to fold change, from downregulated with lowest negative value to upregulated with highest positive value.
Validation of the Rap1b and Arhgap1 Gene Expressions in the Amygdala (Microarray) and Blood (qRT-PCR)
Quantitative real-time PCR (qRT-PCR) was used to validate and to confirm gene expression results obtained from microarray analysis in our previous published results (4). The relative mRNA levels of identified genes measure by qRT-PCR were in agreement with the data detected by microarray from 92 to 67% on five genes examined (4). In an attempt to correlate biomarkers in peripheral tissue (blood) with the profile of dysregulated genes in the amygdala, Rap1b and Arhgap1 were chosen in the current study because of upregulation of (Rap1b) and downregulation of (Arhgap1) expressions observed in stressed amygdala and blood as well as because Rap1b is of known relevance to abnormal fear response (21). The results show that the relative mRNA levels of these genes measured by qRT-PCR (blood, n = 8, p < 0.01) were in agreement with the data detected by microarray (amygdala, n = 10, p < 0.01) experiments (shown in Figures 1A,B in Supplementary Material).
Discussion
Animal models of PTSD offer opportunities to identify mechanisms, potential biomarkers, and treatment targets for this disorder. In this study, we have shown that a substantial number of up- and downregulated genes related to mitochondrial function in the amygdala are associated with an animal model of PTSD based on exaggerated startle and retardation of growth (9, 13). These mitochondrial-function genes have been previously related to neurological dysfunction, psychiatric disorders, and stress response (4, 6). In addition, we have identified novel mitochondrial-function-related genes associated with endocrine and neuronal signaling (4, 5, 22).
Traumatic stress up- or downregulation of anti-inflammatory and anti-apoptotic cellular pathways are evidenced in the amygdala 14 days after stress, as indicated in Figure 4 and Tables 1 and 2. Seven genes, including RAP1B, Cathepsin D (CTSD), MKNK1, RASGRP, CSF2RB, GMCSF, and CFLAR are upregulated and nine genes, including tumor necrosis factor (TNF), Bcl-2, GDF-15, early growth response (Egr1), GNB, FLII, BHLHE40, RND3, and GNB4 are downregulated in the inflammatory and apoptotic signaling networks in the amygdala.
Levels of inflammatory cytokines in blood samples have been used as biomarkers of PTSD vulnerability and resilience in traumatized individuals (23–26). TNF-α and related anti-inflammatory cytokines in the peripheral blood have been correlated with symptoms of PTSD, including re-experiencing, avoidance, and hyperarousal, and with PTSD total symptom score (23, 24, 26). In a single stress rat model, increased neuronal apoptosis has been observed in the medial prefrontal cortex (mPFC) 7 days after stress exposure indicating that apoptotic-related molecules are sensitive to traumatic stress (27). Emerging data indicate that TNF and the anti-apoptotic protein Bcl-2 exert a neuroprotective effect in the brain against traumatic stress (28–30).
Using network analysis, current results reveal for the first time that the 15 genes of the inflammatory pathway in the amygdala complex are up- or downregulated in the exaggerated fear response (Figure 4). These inflammatory signaling molecules may participate in the evolution of PTSD to a chronic disorder because they remain up- or downregulated 14 days after the stress protocol. Bcl-2, an integral outer mitochondrial membrane protein, was initially found to block the apoptotic death of lymphocytes, and over expression of Bcl-2 in transgenic mice reduced the fear response (31, 32). In the current stress model of PTSD, the expression of Bcl-2 is downregulated in the amygdala, indicating that both neuronal inflammatory and cellular apoptosis signaling pathways contribute to the exaggeration of fear following traumatic stress. The network analysis encourages future studies to detail the cellular mechanisms that are evidenced to underlie PTSD (4, 5, 27, 33). For example, the Bcl-2 mediated apoptotic cellular process may contribute not only to the degenerated amygdala structure in PTSD patients but also to the impairment of the controlling function of the amygdala in the fear response of PTSD patients. This process may also be observable in animal models of PTSD (27, 34–36).
Among the downregulated genes, the basic helix–loop–helix transcription genes, BHLHE, have been associated with symptoms of PTSD such as sleep disturbance. For example, BHLHE40 and BHLHE41 are mainly expressed in the suprachiasmatic nucleus of the hypothalamus, which governs circadian rhythm (37). BHLHE40 synchronizes the circadian rhythm at the intracellular level with the external light–dark cycle. The current study demonstrates for the first time that downregulated BHLHE40 expression in the amygdala is associated with exaggerated fear following traumatic stress. Thus BHLHE40 may be a target for treatment of the disruption of sleep associated with PTSD, a symptom that is often resistant to present pharmacotherapy. Changes in the light/dark cycle have also been shown to disrupt memory of conditioned fear (38–40). Thus, the circadian system may relate to anxiety and altered cognition following traumatic stress.
In addition to the Hub molecules such as Bcl-2 and TNF in the inflammatory and apoptotic pathways, other up- or downregulated gene products in the pathways could contribute to the exaggerated fear response. Although little has been published associating these molecules with exaggerated fear or other PTSD symptoms, they have been related to neuronal regulation. For example, GDF-15 is a potent trophic factor in the recovery of 6-OHDA-lesioned midbrain dopaminergic neurons (41). Downregulation of GDF-15 may jeopardize neuronal survival in the amygdala following traumatic stress. Mitochondrial carrier homolog 1 (Mtch1) is an outer mitochondrial membrane protein and overexpression of Mtch1 causes mitochondrial depolarization and apoptosis, likely via the permeability transition pore (42). Egr transcriptional regulators such as Egr1 have been widely recognized as molecules essential for emotional learning and memory (43, 44). Moreover, Egr1-deficient mice have impairments in late phase hippocampal LTP and consolidation of some forms of amygdala-dependent memory through a protein phosphatase calcineurin-mediated mechanism (43, 44). Thus, downregulated Egr1 following traumatic stress may impair cellular processes associated with emotional learning.
Traumatic Stress Up- and Downregulated Genes in the Amygdala Associated with Suicide
Post-traumatic stress disorder significantly increases risk for suicide, even after adjusting for depression, which is often comorbid. Therefore, it appears that traumatic stress exposure and its consequences are risk factors for suicide in some people (45, 46). Correlation of mechanisms underlying suicide anxiety can help in understanding the cellular and molecular connections between these two mental disorders. Using molecular network analysis of genes up- or downregulated by traumatic stress, as shown in Figure 2 and Tables 1 and 2, genes including TUBA1, CTSD, and Grik2 have been associated with suicidal events (47, 48). Dysregulation of each of these genes produces profound damage to the brain.
Mutations of TUBA1A in this gene cause lissencephaly type 3 (LIS3), a neurological condition characterized by microcephaly, mental retardation, early-onset epilepsy, and defective neuronal migration (49). TUBA1 is also an interacting cytoskeleton protein that has a functional connection to glutamate, GABA, and serotonin receptors (48). Mutations in the CTSD gene are involved in the pathogenesis of AD and Niemann–Pick disease (50–53).
A recent proteomic study on post mortem brains of suicide victims shows that both TUBA1A and CTSD are highly expressed in the prefrontal cortex and amygdala of suicide victims, suggesting that these genes may be a risk biomarker for suicide (48). As shown in Figure 4, S100 Calcium Binding Protein A1 (S100A1; one of the proteins in the super family of S100, also called P11) interacts with TUBA1A and may regulate the function of TUBA1 under stressful conditions (54). In fact, in a recent clinical study, the mRNA level of S100A10 was found to be a potential adjunctive biomarker for the assessment of suicide risk in anxiety disorders (55).
Kainate receptors are members of the ionotropic class of glutamate receptors and are highly expressed in the amygdala neuronal circuitry (56–60). These kainate receptors have been identified both pre- and post-synaptically. They modulate inhibitory and excitatory postsynaptic currents in the basolateral amygdala circuitry and play a role in synaptic plasticity associated with emotional learning and memory (56–60). Thus upregulated Grik2 and Grik genes, as found in the current study, may participate in the emotional hyperarousal following traumatic stress. In addition, recent pharmacogenomic studies of antidepressant treatment-emergent suicidal events report associations with polymorphisms of Grik2 receptor genes in depressed patients (47). The network analysis from the present results suggests that the pharmacologic agents that target specific subtypes of kainate receptors may be therapeutic medications for the treatment of PTSD with suicidal ideation.
In summary, network analysis reveals that signaling pathways associated with TUBA1A, CTSD, and Grik2 are potential biomarkers and therapeutic targets for PTSD with suicidal ideation.
Up/Downregulation of Genes in the Amygdala Subserving Energy Production and Neurotransmitter Synthesis
Diminished interest and participation in activities along with detachment and lack of positive emotions may be seen in PTSD as energy loss or mental fatigue. This decreased energy is a common complaint of service members after repetitive traumatic stress, including PTSD and TBI (61). While the mechanisms underlying the mental fatigue are speculated to be due to a shortage of energetic molecules in the brain, the detailed mechanism remains elusive. The relationship between mitochondrial gene expression in the amygdala and exaggerated fear provides an approach to the cellular and molecular basis of mental fatigue after trauma. As shown in Figure 5, two citrate cycle genes, Idh3a and Cpt1b, were upregulated. Idh3a promotes ATP production by catalyzing the oxidative decarboxylation of isocitrate to 2-oxoglutarate. Cpt1b generates ATP from lipids via beta-oxidation of fatty acids (62–64). Expression of ATP5C1 is enhanced. ATP5C1 is the mitochondrial membrane ATP synthase. Suppression of the ATP5C1 gene caused a decrease in cell proliferation and ATP production (65). Enhanced expression of ATP5C1 may suggest an enhanced demand for energy in the amygdala in response to traumatic stress associated with exaggerated fear. Indeed, enhanced expression of ATP5C1 was also found in the prefrontal cortex of PTSD postmortem brains, indicating that the ATP5C1 gene could be a biomarker for the diagnosis of PTSD (4).
Mitochondrial genes supporting biosynthesis are differentially regulated under conditions of stress. Got2, which is involved in arginine, tyrosine, and tryptophan biosynthesis, is upregulated; Acot1 and Bcat2, two enzymes involved in fatty acid metabolism, as well as synthesis of valine, leucine, and isoleucine, and degradation of lysine, are downregulated. Tryptophan is a precursor of serotonin, which is thought to contribute to emotional well-being and is related to depression, which is highly comorbid with PTSD (66, 67). In addition, selective serotonin reuptake inhibitors (SSRIs) are a first line treatment for PTSD.
These results indicate that altered regulation of expression of genes involved with energy production and biosynthesis in the amygdala may be associated with the diminished interest, overall mental fatigue, and comorbid symptoms of depression in PTSD.
Up/Downregulation of Genes in the Amygdala Associated with Immune Mediators and Energy Expenditure
Proinflammatory chemokines have been suggested to be involved in chronic stress, major depressive disorder, panic disorder, and PTSD (68–73). The chemokines IL4 and CCL13 are interconnected with the up- and downregulated genes shown in the network 3, including Lonp1, ATP5F1, Rab8A, and Gucy2F. Levels of chemokines including IL4, IL2, and TNF alpha in the plasma have been found to be significantly altered in some patients suffering from traumatic stress, including those with PTSD and those suffering from child abuse (74). CCL13 is a chemotactic cytokine that regulates leukocyte migration through interactions with G-protein-coupled receptors (75, 76).
The up- and downregulated genes shown in the network (Figure 6) are connected with Leptin (LEP), which is a key mediator in the regulation of food intake, body weight, and possibly memory. Dysregulation of LEP could be involved in the weight loss resulting from chronic and repetitive stress in our rat model of PTSD (11, 13, 77). Weight loss is a common symptom of depression, highly comorbid with PTSD. Indeed, elevated serum LEP levels were found in PTSD patients suffering from depressive symptoms caused by earthquakes and myocardial infarctions (78, 79). In addition, intraperitoneal injection of LEP mitigated the impairment of fear memory in the mouse deprived of rapid eye movement sleep (80). Thus, it appears that the elevation of LEP in patients with PTSD may be associated with elevated fear, similar to an observation made in the current rat model of PTSD.
Up/Downregulation of Genes in the Amygdala Associated Transcription Factors and Chromatin Remodeling
As indicated in the signaling pathway shown in Figure 7, four genes, including neuregulin 1 (Nrg1), calmodulin-dependent kinases 2 gamma (Camk2g), tubuline (Tuba1A), and the A-kinase anchor protein 1 (AKAP1), are upregulated in the amygdala 14 days after stress, and E-twenty six (ETS) domain-containing transcription factor (ELK3) is downregulated. In addition to the functions of the individual up- or downregulated genes in Figure 7, these genes are interconnected with histone 3 and histone 4. Histone H3 and histone H4 are two of the five main histone proteins involved in the structure of chromatin in eukaryotic cells. Thus, the up- or downregulated genes in the network appear to affect transcription and chromatin remodeling.
Regulation of chromatin structure through post-translational modification of histone proteins is a critical step in the formation of long-term memory (81). As revealed in the network analysis, traumatic and repetitive stress leads to altered regulation of chromatin-related genes in the amygdala, including the upregulation of Nrg1, CamK2g, Tuba1A, and AKAP1, as well as the downregulation of ELK3 interacting with NF-kappa B, histone 3, and histone 4. Histone 3 and histone 4 drive epigenetic modifications and conformational changes in chromatin, stimulating the expression of neuroplasticity-related genes involved in traumatic memory and fear learning (82, 83). Histone acetylation mediated by BDNF gene transcription is involved in the consolidation of fear memory associated with an animal model of PTSD (84). This modulation could promote chromatin remodeling in the amygdala induced by glucocorticoids following traumatic stress. Indeed, activation of the glucocorticoid receptor (GR) under stress has also been shown to modify chromatin structure by post-translational modifications of histones and is considered to be a new therapeutic target for PTSD (85, 86).
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a protein complex that controls the transcription of DNA, interacts with up- or downregulated genes in network 4. Inhibition of NF-κB in the basolateral amygdala impairs the memory reconsolidation associated with auditory fear conditioning (87).
Summary
The current study reveals that multiple signaling networks are up- or downregulated in the amygdala circuitry 14 days after traumatic stress. Traumatic stress significantly alters at least 55 genes associated with at least four pathways related to mitochondria functions in the stressed amygdala. These molecules include well-studied and -documented networks as well as novel networks that have not been well documented in previous literature. These findings provide a guide for further studies associated with diagnostic biomarkers, therapeutic molecular targets, and identification of optimal strategies for fostering resilience before and after traumatic stress. In addition, current analyses suggest new targets for pharmacological intervention to treat PTSD and suicide. Suicide-related genes revealed in the stressed amygdala indicate that dysregulated signaling pathways interweave suicide and fear. These results elucidate the molecular pathways underlying PTSD as a road to diagnosis and treatment.
Conflict of Interest Statement
The Guest Associate Editor Yumin Zhang declares that, despite being affiliated to the same institution as authors He Li, Stanley E. Smerin, Lei Zhang, Min Jia, Guoqiang Xing, Jillian Wen, David Benedek, and Robert Ursano, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by CSTS and CDMRP grant W81XWH-08-2-006 to He Li.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fneur.2014.00164/abstract
Footnotes
References
1. Rabinak CA, Angstadt M, Welsh RC, Kenndy AE, Lyubkin M, Martis B, et al. Altered amygdala resting-state functional connectivity in post-traumatic stress disorder. Front Psychiatry (2011) 2:62. doi: 10.3389/fpsyt.2011.00062
2. Bryant RA, Kemp AH, Felmingham KL, Liddell B, Olivieri G, Peduto A, et al. Enhanced amygdala and medial prefrontal activation during nonconscious processing of fear in posttraumatic stress disorder: an fMRI study. Hum Brain Mapp (2008) 29:517–23. doi:10.1002/hbm.20415
3. Stein MB, Simmons AN, Feinstein JS, Paulus MP. Increased amygdala and insula activation during emotion processing in anxiety-prone subjects. Am J Psychiatry (2007) 164:318–27. doi:10.1176/appi.ajp.164.2.318
4. Su YA, Wu J, Zhang L, Zhang Q, Su DM, He P, et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci (2008) 4:223–35. doi:10.7150/ijbs.4.223
5. Zhang L, Zhou R, Li X, Ursano RJ, Li H. Stress-induced change of mitochondria membrane potential regulated by genomic and non-genomic GR signaling: a possible mechanism for hippocampus atrophy in PTSD. Med Hypotheses (2006) 66:1205–8. doi:10.1016/j.mehy.2005.11.041
6. Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab (2007) 18:190–8. doi:10.1016/j.tem.2007.04.004
7. Bai X, Wu J, Zhang Q, Alesci S, Manoli I, Blackman MR, et al. Third-generation human mitochondria-focused cDNA microarray and its bioinformatic tools for analysis of gene expression. Biotechniques (2007) 42:365375. doi:10.2144/000112388
8. Su YA, Zhang Q, Su DM, Tang MX. Rat mitochondrion-neuron focused microarray (rMNChip) and bioinformatics tools for rapid identification of differential pathways in brain tissues. Int J Biol Sci (2011) 7:308–22. doi:10.7150/ijbs.7.308
9. Servatius RJ, Ottenweller JE, Natelson BH. Delayed startle sensitization distinguishes rats exposed to one or three stress sessions: further evidence toward an animal model of PTSD. Biol Psychiatry (1995) 38:539–46. doi:10.1016/0006-3223(94)00369-E
10. Braga MF, roniadou-Anderjaska V, Post RM, Li H. Lamotrigine reduces spontaneous and evoked GABAA receptor-mediated synaptic transmission in the basolateral amygdala: implications for its effects in seizure and affective disorders. Neuropharmacology (2002) 42:522–9. doi:10.1016/S0028-3908(01)00198-8
11. Jiang X, Xing G, Yang C, Verma A, Zhang L, Li H. Stress impairs 5-HT2A receptor mediated serotonergic facilitation of GABA release in juvenile rat basolateral amygdala. Neuropsychopharmacology (2009) 34:410–23. doi:10.1038/npp.2008.71
12. Garrick T, Morrow N, Shalev AY, Eth S. Stress-induced enhancement of auditory startle: an animal model of posttraumatic stress disorder. Psychiatry (2001) 64:346–54.
13. Jia M, Meng F, Smerin SE, Xing G, Zhang L, Su DM, et al. Biomarkers in an animal model for revealing neural, hematologic, and behavioral correlates of PTSD. J Vis Exp (2012) 68:e3361. doi:10.3791/3361
14. Dunleavy K, Kubo SA. Emergence of delayed posttraumatic stress disorder symptoms related to sexual trauma: patient-centered and trauma-cognizant management by physical therapists. Phys Ther (2012) 92:339–51. doi:10.2522/ptj.20100344
15. Tomb DA. The phenomenology of post-traumatic stress disorder. Psychiatr Clin North Am (1994) 17:237–50.
16. Su YA, Bittner ML, Chen Y, Tao L, Jiang Y, Zhang Y, et al. Identification of tumor-suppressor genes using human melanoma cell lines UACC903, UACC903(+6), and SRS3 by comparison of expression profiles. Mol Carcinog (2000) 28:119–27. doi:10.1002/1098-2744(200006)28:2<119::AID-MC8>3.0.CO;2-N
17. Su YA, Trent JM. Isolation of tumor suppressor genes in melanoma by cDNA microarray. Methods Mol Med (2001) 61:15–29. doi:10.1385/1-59259-145-0:15
18. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics (2003) 19:185–93. doi:10.1093/bioinformatics/19.2.185
19. Tarca AL, Romero R, Draghici S. Analysis of microarray experiments of gene expression profiling. Am J Obstet Gynecol (2006) 195:373–88. doi:10.1016/j.ajog.2006.07.001
20. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res (2001) 125:279–84. doi:10.1016/S0166-4328(01)00297-2
21. Pan BX, Vautier F, Ito W, Bolshakov VY, Morozov A. Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J Neurosci (2008) 28:2089–98. doi:10.1523/JNEUROSCI.5156-07.2008
22. Choi KH, Le T, McGuire J, Coyner J, Higgs BW, Diglisic S, et al. Expression profiles of mitochondrial genes in the frontal cortex and the caudate nucleus of developing humans and mice selectively bred for high and low fear. PLoS One (2012) 7:e49183. doi:10.1371/journal.pone.0049183
23. Gill J, Vythilingam M, Page GG. Low cortisol, high DHEA, and high levels of stimulated TNF-alpha, and IL-6 in women with PTSD. J Trauma Stress (2008) 21:530–9. doi:10.1002/jts.20372
24. Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry (2013) 13:40. doi:10.1186/1471-244X-13-40
25. Guo M, Liu T, Guo JC, Jiang XL, Chen F, Gao YS. Study on serum cytokine levels in posttraumatic stress disorder patients. Asian Pac J Trop Med (2012) 5:323–5. doi:10.1016/S1995-7645(12)60048-0
26. von KR, Hepp U, Kraemer B, Traber R, Keel M, Mica L, et al. Evidence for lowgrade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res (2007) 41:744–52. doi:10.1016/j.jpsychires.2006.06.009
27. Li XM, Han F, Liu DJ, Shi YX. Single-prolonged stress induced mitochondrial dependent apoptosis in hippocampus in the rat model of post-traumatic stress disorder. J Chem Neuroanat (2010) 40:248–55. doi:10.1016/j.jchemneu.2010.07.001
28. Della FP, Abelaira HM, Reus GZ, Antunes AR, Dos Santos MA, Zappelinni G, et al. Tianeptine exerts neuroprotective effects in the brain tissue of rats exposed to the chronic stress model. Pharmacol Biochem Behav (2012) 103:395–402. doi:10.1016/j.pbb.2012.09.018
29. Rossi-George A, Urbach D, Colas D, Goldfarb Y, Kusnecov AW. Neuronal, endocrine, and anorexic responses to the T-cell superantigen staphylococcal enterotoxin A: dependence on tumor necrosis factor-alpha. J Neurosci (2005) 25:5314–22. doi:10.1523/JNEUROSCI.0687-05.2005
30. Salim S, Asghar M, Taneja M, Hovatta I, Chugh G, Vollert C, et al. Potential contribution of oxidative stress and inflammation to anxiety and hypertension. Brain Res (2011) 1404:63–71. doi:10.1016/j.brainres.2011.06.024
31. Rondi-Reig L, Lemaigre DY, Martinou JC, haye-Bouchaud N, Caston J, Mariani J. Fear decrease in transgenic mice overexpressing bcl-2 in neurons. Neuroreport (1997) 8:24292432. doi:10.1097/00001756-199707280-00004
32. Pinkoski MJ, Green DR. Lymphocyte apoptosis: refining the paths to perdition. Curr Opin Hematol (2002) 9:43–9. doi:10.1097/00062752-200201000-00008
33. Ding J, Han F, Shi Y. Single-prolonged stress induces apoptosis in the amygdala in a rat model of post-traumatic stress disorder. J Psychiatr Res (2010) 44:48–55. doi:10.1016/j.jpsychires.2009.06.001
34. Shucard JL, Cox J, Shucard DW, Fetter H, Chung C, Ramasamy D, et al. Symptoms of posttraumatic stress disorder and exposure to traumatic stressors are related to brain structural volumes and behavioral measures of affective stimulus processing in police officers. Psychiatry Res (2012) 204:25–31. doi:10.1016/j.pscychresns.2012.04.006
35. Majewska MD. Cocaine addiction as a neurological disorder: implications for treatment. NIDA Res Monogr (1996) 163:1–26.
36. Karl A, Malta LS, Maercker A. Meta-analytic review of event-related potential studies in post-traumatic stress disorder. Biol Psychol (2006) 71:123–47. doi:10.1016/j.biopsycho.2005.03.004
37. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature (2002) 419:841844. doi:10.1038/nature01123
38. Amir S, Stewart J. Conditioned fear suppresses light-induced resetting of the circadian clock. Neuroscience (1998) 86:345–51. doi:10.1016/S0306-4522(98)00172-9
39. Loh DH, Navarro J, Hagopian A, Wang LM, Deboer T, Colwell CS. Rapid changes in the light/dark cycle disrupt memory of conditioned fear in mice. PLoS One (2010) 5(9):e12546. doi:10.1371/journal.pone.0012546
40. Chaudhury D, Colwell CS. Circadian modulation of learning and memory in fear conditioned mice. Behav Brain Res (2002) 133:95–108. doi:10.1016/S0166-4328(01)00471-5
41. Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. TGF-beta and the regulation of neuron survival and death. J Physiol Paris (2002) 96:25–30. doi:10.1016/S0928-4257(01)00077-8
42. Lamarca V, Marzo I, Sanz-Clemente A, Carrodeguas JA. Exposure of any of two proapoptotic domains of presenilin 1-associated protein/mitochondrial carrier homolog 1 on the surface of mitochondria is sufficient for induction of apoptosis in a Bax/Bak-independent manner. Eur J Cell Biol (2008) 87:325–34. doi:10.1016/j.ejcb.2008.02.004
43. Baumgartel K, Genoux D, Welzl H, Tweedie-Cullen RY, Koshibu K, Livingstone Zatchej M, et al. Control of the establishment of aversive memory by calcineurin and Zif268. Nat Neurosci (2008) 11:572–8. doi:10.1038/nn.2113
44. Li L, Yun SH, Keblesh J, Trommer BL, Xiong H, Radulovic J, et al. Egr3, a synaptic activity regulated transcription factor that is essential for learning and memory. Mol Cell Neurosci (2007) 35:76–88. doi:10.1016/j.mcn.2007.02.004
45. Capron DW, Cougle JR, Ribeiro JD, Joiner TE, Schmidt NB. An interactive model of anxiety sensitivity relevant to suicide attempt history and future suicidal ideation. J Psychiatr Res (2012) 46:174–80. doi:10.1016/j.jpsychires.2011.10.009
46. Hendin H, Haas AP. Suicide and guilt as manifestations of PTSD in Vietnam combat veterans. Am J Psychiatry (1991) 148:586–91.
47. Brent D, Melhem N, Turecki G. Pharmacogenomics of suicidal events. Pharmacogenomics (2010) 11:793–807. doi:10.2217/pgs.10.64
48. Kekesi KA, Juhasz G, Simor A, Gulyassy P, Szego EM, Hunyadi-Gulyas E, et al. Altered functional protein networks in the prefrontal cortex and amygdala of victims of suicide. PLoS One (2012) 7:e50532. doi:10.1371/journal.pone.0050532
49. Sohal AP, Montgomery T, Mitra D, Ramesh V. TUBA1A mutation-associated lissencephaly: case report and review of the literature. Pediatr Neurol (2012) 46:127–31. doi:10.1016/j.pediatrneurol.2011.11.017
50. Cluzeau CV, Watkins-Chow DE, Fu R, Borate B, Yanjanin N, Dail MK, et al. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum Mol Genet (2012) 21:3632–46. doi:10.1093/hmg/dds193
51. Li XQ, Chen D, Zhang ZX, Qu QM, Zhang JW. Association between cathepsin D polymorphism and Alzheimer’s disease in a Chinese Han population. Dement Geriatr Cogn Disord (2004) 18:115–9. doi:10.1159/000079189
52. Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med (2008) 14:12471255. doi:10.1038/nm.1876
53. May FE, Smith DJ, Westley BR. The human cathepsin D-encoding gene is transcribed from an estrogen-regulated and a constitutive start point. Gene (1993) 134:277–82. doi:10.1016/0378-1119(93)90107-E
54. Hong D, Chen HX, Yu HQ, Wang C, Deng HT, Lian QQ, et al. Quantitative proteomic analysis of dexamethasone-induced effects on osteoblast differentiation, proliferation, and apoptosis in MC3T3-E1 cells using SILAC. Osteoporos Int (2011) 22:2175–86. doi:10.1007/s00198-010-1434-8
55. Zhang L, Su TP, Choi K, Maree W, Li CT, Chung MY, et al. P11 (S100A10) as a potential biomarker of psychiatric patients at risk of suicide. J Psychiatr Res (2011) 45:435–41. doi:10.1016/j.jpsychires.2010.08.012
56. Braga MF, roniadou-Anderjaska V, Li H. The physiological role of kainate receptors in the amygdala. Mol Neurobiol (2004) 30:127–41. doi:10.1385/MN:30:2:127
57. Rogawski MA, Gryder D, Castaneda D, Yonekawa W, Banks MK, Li H. GluR5 kainate receptors, seizures, and the amygdala. Ann N Y Acad Sci (2003) 985:150–62. doi:10.1111/j.1749-6632.2003.tb07079.x
58. Braga MF, Roniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci (2003) 23:442–52. doi:10.1385/MN:30:2:127
59. Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci (2001) 4:612–20. doi:10.1038/88432
60. Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology (1998) 37:1279–86. doi:10.1016/S0028-3908(98)00109-9
61. Weeks SR, McAuliffe CL, Durussel D, Pasquina PF. Physiological and psychological fatigue in extreme conditions: the military example. PM R (2010) 2:438–41. doi:10.1016/j.pmrj.2010.03.023
62. Dunning KR, Cashman K, Russell DL, Thompson JG, Norman RJ, Robker RL. Betaoxidation is essential for mouse oocyte developmental competence and early embryo development. Biol Reprod (2010) 83:909–18. doi:10.1095/biolreprod.110.084145
63. Zhang HN, Wu J, Jin T, Chang M, Sun L. Transient elevation of synaptosomal mitoenergetic proteins and Hsp70 early in a rat model of chronic cerebrovascular hypoperfusion. Neurol Sci (2013) 34:471–7. doi:10.1007/s10072-012-1063-4
64. Huh TL, Kim YO, Oh IU, Song BJ, Inazawa J. Assignment of the human mitochondrial NAD+-specific isocitrate dehydrogenase alpha subunit (IDH3A) gene to 15q25.1- >q25.2by in situ hybridization. Genomics (1996) 32:295–6. doi:10.1006/geno.1996.0120
65. Lwin WW, Park K, Wauson M, Gao Q, Finn PW, Perkins D, et al. Systems biology approach to transplant tolerance: proof of concept experiments using RNA interference (RNAi) to knock down hub genes in Jurkat and HeLa cells in vitro. J Surg Res (2012) 176:e41–6. doi:10.1016/j.jss.2011.12.002
66. Arun P, bu-Taleb R, Oguntayo S, Wang Y, Valiyaveettil M, Long J, et al. Acute mitochondrial dysfunction after blast exposure: potential role of mitochondrial glutamate oxaloacetate transaminase. J Neurotrauma (2013) 30:1645–51. doi:10.1089/neu.2012.2834
67. Young SN. The effect of raising and lowering tryptophan levels on human mood and social behaviour. Philos Trans R Soc Lond B Biol Sci (2013) 368:20110375. doi:10.1098/rstb.2011.0375
68. Hoge EA, Brandstetter K, Moshier S, Pollack MH, Wong KK, Simon NM. Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress Anxiety (2009) 26:447–55. doi:10.1002/da.20564
69. Mommersteeg PM, Vermetten E, Kavelaars A, Geuze E, Heijnen CJ. Hostility is related to clusters of T-cell cytokines and chemokines in healthy men. Psychoneuroendocrinology (2008) 33:1041–50. doi:10.1016/j.psyneuen.2008.05.007
70. Song Y, Zhou D, Guan Z, Wang X. Disturbance of serum interleukin-2 and interleukin-8 levels in posttraumatic and non-posttraumatic stress disorder earthquake survivors in northern China. Neuroimmunomodulation (2007) 14:248–54. doi:10.1159/000112050
71. Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A (1998) 95:14500–5. doi:10.1073/pnas.95.24.14500
72. Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology (2012) 37:137–62. doi:10.1038/npp.2011.205
73. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol (2006) 27:24–31. doi:10.1016/j.it.2005.11.006
74. Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet (2011) 156B:700–8. doi:10.1002/ajmg.b.31212
75. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity (2012) 36:705–16. doi:10.1016/j.immuni.2012.05.008
76. O’Boyle G, Brain JG, Kirby JA, Ali S. Chemokine-mediated inflammation: identification of a possible regulatory role for CCR2. Mol Immunol (2007) 44:1944–53. doi:10.1016/j.molimm.2006.09.033
77. Harris RB, Mitchell TD, Simpson J, Redmann SM Jr., Youngblood BD, Ryan DH. Weight loss in rats exposed to repeated acute restraint stress is independent of energy or leptin status. Am J Physiol Regul Integr Comp Physiol (2002) 282:R77–88.
78. Liao SC, Lee MB, Lee YJ, Huang TS. Hyperleptinemia in subjects with persistent partial posttraumatic stress disorder after a major earthquake. Psychosom Med (2004) 66:23–8. doi:10.1097/01.PSY.0000106880.22867.0E
79. von KR, Begre S, Abbas CC, Saner H, Gander ML, Schmid JP. Inflammatory biomarkers in patients with posttraumatic stress disorder caused by myocardial infarction and the role of depressive symptoms. Neuroimmunomodulation (2010) 17:39–46. doi:10.1159/000243084
80. Chang HF, Su CL, Chang CH, Chen YW, Gean PW. The beneficial effects of leptin on REM sleep deprivation-induced cognitive deficits in mice. Learn Mem (2013) 20:328335. doi:10.1101/lm.030775.113
81. Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, et al. Histone methylation regulates memory formation. J Neurosci (2010) 30:3589–99. doi:10.1523/JNEUROSCI.3732-09.2010
82. Moriya T, Kouzu Y, Shibata S, Kadotani H, Fukunaga K, Miyamoto E, et al. Close linkage between calcium/calmodulin kinase II alpha/beta and NMDA-2A receptors in the lateral amygdala and significance for retrieval of auditory fear conditioning. Eur J Neurosci (2000) 12:3307–14. doi:10.1046/j.1460-9568.2000.00203.x
83. Trollope AF, Gutierrez-Mecinas M, Mifsud KR, Collins A, Saunderson EA, Reul JM. Stress, epigenetic control of gene expression and memory formation. Exp Neurol (2012) 233:3–11. doi:10.1016/j.expneurol.2011.03.022
84. Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S. Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res (2011) 45:460–8. doi:10.1016/j.jpsychires.2010.08.009
85. Zhang L, Li H, Hu X, Li XX, Smerin S, Ursano R. Glucocorticoid-induced p11 overexpression and chromatin remodeling: a novel molecular mechanism of traumatic stress? Med Hypotheses (2011) 76:774–7. doi:10.1016/j.mehy.2011.02.015
86. Deroo BJ, Archer TK. Glucocorticoid receptor-mediated chromatin remodeling in vivo. Oncogene (2001) 20:3039–46. doi:10.1038/sj.onc.1204328
Keywords: PTSD, amygdala, mitochondrial genes, stress, fear, Bcl-2, microarray
Citation: Li H, Li X, Smerin SE, Zhang L, Jia M, Xing G, Su YA, Wen J, Benedek D and Ursano R (2014) Mitochondrial gene expression profiles and metabolic pathways in the amygdala associated with exaggerated fear in an animal model of PTSD. Front. Neurol. 5:164. doi: 10.3389/fneur.2014.00164
Received: 03 December 2013; Accepted: 15 August 2014;
Published online: 23 September 2014.
Edited by:
Yumin Zhang, Uniformed Services University of the Health Sciences, USAReviewed by:
Namas Chandra, New Jersey Institute of Technology, USAFiras H. Kobeissy, University of Florida, USA
Copyright: © 2014 Li, Li, Smerin, Zhang, Jia, Xing, Su, Wen, Benedek and Ursano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: He Li, Department of Psychiatry, Center for the Study of Traumatic Stress, Uniformed Services University of the Health Sciences, Building B, Room B3060, 4301 Jones Bridge Road, Bethesda, MD 20814, USA e-mail:aGUubGlAdXN1aHMuZWR1
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Uniformed Services University, Department of the Navy, Department of Defense, nor the U.S. Government.