 Gregory A. Elder
Gregory A. Elder James R. Stone
James R. Stone Stephen T. Ahlers
Stephen T. Ahlers- 1Neurology Service, James J. Peters Department of Veterans Affairs Medical Center, Bronx, NY, USA
- 2Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 3Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 4Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 5Department of Radiology, University of Virginia, Charlottesville, VA, USA
- 6Department of Neurosurgery, University of Virginia, Charlottesville, VA, USA
- 7Department of Neurotrauma, Operational and Undersea Medicine Directorate, Naval Medical Research Center, Silver Spring, MD, USA
High-pressure blast waves can cause extensive CNS injury in human beings. However, in combat settings, such as Iraq and Afghanistan, lower level exposures associated with mild traumatic brain injury (mTBI) or subclinical exposure have been much more common. Yet controversy exists concerning what traits can be attributed to low-level blast, in large part due to the difficulty of distinguishing blast-related mTBI from post-traumatic stress disorder (PTSD). We describe how TBI is defined in human beings and the problems posed in using current definitions to recognize blast-related mTBI. We next consider the problem of applying definitions of human mTBI to animal models, in particular that TBI severity in human beings is defined in relation to alteration of consciousness at the time of injury, which typically cannot be assessed in animals. However, based on outcome assessments, a condition of “low-level” blast exposure can be defined in animals that likely approximates human mTBI or subclinical exposure. We review blast injury modeling in animals noting that inconsistencies in experimental approach have contributed to uncertainty over the effects of low-level blast. Yet, animal studies show that low-level blast pressure waves are transmitted to the brain. In brain, low-level blast exposures cause behavioral, biochemical, pathological, and physiological effects on the nervous system including the induction of PTSD-related behavioral traits in the absence of a psychological stressor. We review the relationship of blast exposure to chronic neurodegenerative diseases noting the paradoxical lowering of Abeta by blast, which along with other observations suggest that blast-related TBI is pathophysiologically distinct from non-blast TBI. Human neuroimaging studies show that blast-related mTBI is associated with a variety of chronic effects that are unlikely to be explained by co-morbid PTSD. We conclude that abundant evidence supports low-level blast as having long-term effects on the nervous system.
Introduction
While an uncommon cause of traumatic brain injury (TBI) in civilian life (1), blast-related TBI has been of long-standing interest in military head trauma. During World War I, it was first recognized that blast exposure could be associated with psychological and neurological symptoms that were reminiscent of both the post-concussion syndrome and what would now be called post-traumatic stress disorder (PTSD) (2–9). Recently, there has been renewed interest in blast-related TBI because of its frequency in the conflicts in Iraq and Afghanistan (8, 10–32). Indeed, estimates are that 10–20% of veterans returning from these conflicts have suffered a TBI with blast exposure most commonly related to improvised explosive devices (IEDs) being the most frequently attributed cause (10, 33).
Most attention focused initially on the moderate to severe end of the TBI spectrum (34), the type of TBI that would be recognized in theater and the war in Iraq has resulted in the highest number of service-related severe TBIs since the Vietnam era (35). However, what soon became apparent was that many returning veterans began appearing at Veterans Affairs (VA) Hospitals and other facilities with symptoms suggestive of the residual effects of mild TBIs (mTBIs) that were never recognized prior to discharge. In fact, mTBIs vastly outnumber moderate to severe TBIs in this population (10, 33).
This renewed interest has lead to a rapid expansion of clinical as well as animal studies related to blast. Despite these efforts, questions remain concerning how blast exerts its effects on the nervous system and even what effects can reasonably be attributed to blast. Indeed, a recent Institute of Medicine report (36) concluded that in terms of long-term adverse health outcomes in human beings, there was sufficient evidence of a causal relationship to blast only for penetrating eye injuries and some long-term effects on the genitourinary system. For post-concussion symptoms and persistent headaches following blast-related mTBI, the report concluded that there was only sufficient evidence for an association (36).
Questions have also emerged as to whether pathophysiologically blast-related TBI is different from the type of non-blast TBI (nbTBI) typical of civilian trauma where injury is caused by inertial and rotational forces along with the effects of blunt impact (37, 38). The most direct physical effects of these forces are bleeding, direct tissue damage, and mechanical shear stress along white matter tracts, which in turn leads to activation of a variety of pathophysiological cascades that are associated with further tissue damage (37, 39). Blast injuries by contrast result from a pressure wave generated at a distance and transmitted though air, which may induce stresses in the brain without significant global motions being imparted. Damage to the nervous system is thought to occur through biophysical mechanisms related to the traveling shock wave’s interaction with the brain (40–42), although it has also been suggested that a blast wave striking the body can induce oscillating pressure waves, which can be transmitted through the systemic circulation to the brain (12, 43).
Blast injuries have typically been divided into four or five categories (13, 19, 44). Injuries directly related to the blast wave are referred to as primary blast injuries. The blast explosion can propel objects including shrapnel contained within the IED causing what is referred to as secondary injury. The blast wave may also cause victims to be knocked down or thrown into solid objects resulting in what is called tertiary injury. Quaternary injuries comprise a group of miscellaneous injuries not classifiable into other categories including burns or the effects of inhaling noxious gases. Finally, the term quinary injury is sometimes used to refer to illnesses that result from chemical, biologic, or radiologic substances released during an explosion (19). Development of this blast taxonomy began during World War II (45) and remains useful today. However, it does not encompass all injury mechanisms encountered today such as platform shock acceleration where energy is transferred through the ground or a vehicular structure (46). This phenomenon, which is particularly relevant to the common situation where injury occurs to personnel in a vehicle hit by an IED occurs due to complex mechanisms unrelated to the primary blast wave itself (44).
Close exposure to a high-pressure blast wave can cause extensive CNS injuries in human beings with high-level blast exposures appearing to be particularly prone to inducing hemorrhagic lesions (47). High-level blast exposures in animals can also produce a variety of gross as well as histological pathology (11, 26). However, the relative importance of the blast injury mechanisms discussed above varies depending on the severity of injury. Blast injuries severe enough to cause moderate to severe TBI are without doubt a mix of secondary and tertiary injuries combined with the contribution of the primary blast wave. In both human beings and animals, secondary and tertiary injuries likely activate many of the same pathophysiological cascades seen in nbTBI. What is less clear is how the primary blast wave damages the brain. Blast-related mTBI differs in that secondary and tertiary injuries are less prominent with most cases presumed to reflect mostly the direct effects of the primary blast wave.
Defining Blast-Related mTBI in Human Beings
Blast-related TBI is currently defined using the same criteria used for defining nbTBI. In human beings, TBI severity is defined in relation to the degree of neurological dysfunction at the time of injury. TBI severity is thus considered separate from TBI outcome, i.e., the chronic signs and symptoms as well as functional impairment that may persist after a TBI. TBI also requires an event. In the case of nbTBI, the head is struck or strikes an object or in blast-related TBI is enveloped by the blast overpressure wave. Historically, TBI has been graded as mild, moderate, or severe based primarily on the immediate effects of the TBI on consciousness.
The first widely used staging system for TBI was the Glasgow Coma Scale (48), which divides TBIs into mild, moderate, and severe based on three clinical criteria: eye opening, best motor, and best verbal response (48–50). Subsequently, several classifications have been proposed (51–54). Four of the more commonly used criteria are those developed by the American Congress of Rehabilitation Medicine (51), the Centers for Disease Control and Prevention (CDC) (52), the World Health Organization (WHO) (53), and the Department of Defense (DoD)/Department of Veterans Affairs (54). The features of these are summarized in Table 1.
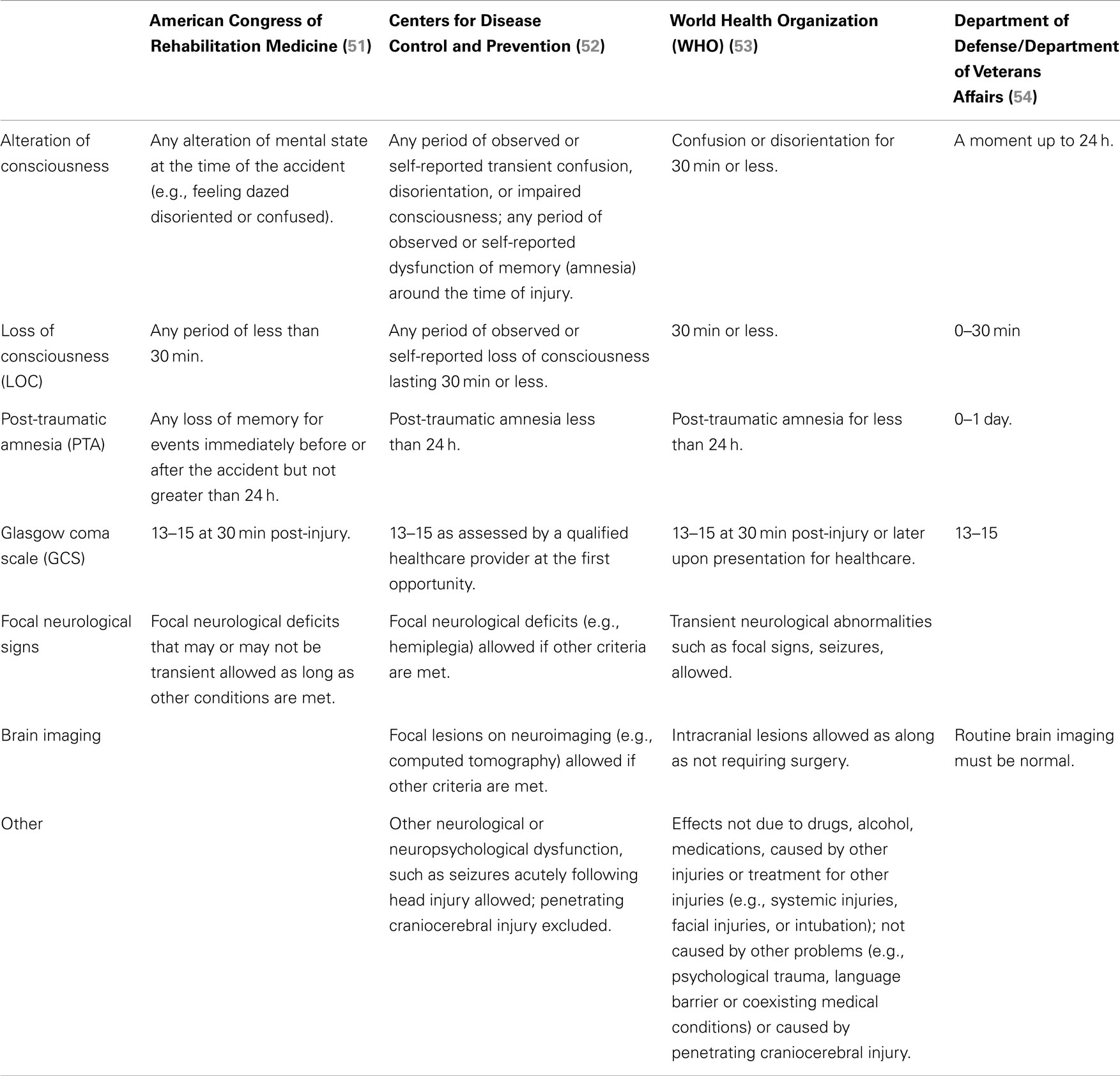
Table 1. Consensus criteria for the classification of human mTBI.
All use alteration or loss of consciousness as the primary indicator that a TBI occurred. This alteration may range from any transient alteration of consciousness including being as little as momentarily dazed or confused up to loss of consciousness for 30 min. All incorporate the Glasgow Coma Scale and exclude cases with scores less than 13 as well as allow for a period of post-traumatic amnesia lasting up to 24 h. The CDC criteria include pre-injury as well as post-injury amnesia, while the DoD/VA and WHO criteria only consider post-traumatic amnesia. The American Congress of Rehabilitation Medicine, CDC, and WHO guidelines allow focal neurological deficits to be present as long as other criteria are met while focal symptoms or signs are not mentioned in the DoD/VA guidelines. Brain imaging results are not considered as a factor in the American Congress of Rehabilitation Medicine guidelines. The CDC and WHO guidelines allow focal lesions on brain imaging if other criteria are met or in the case of the WHO guidelines if surgery is not required. By contrast, the DoD/VA guidelines state that routine neuroimaging must be normal. In the CDC guidelines, penetrating craniocerebral injury is excluded and the WHO guidelines add the stipulation that any neurological dysfunction must not be due to drugs, alcohol, medications, or be caused by other injuries, treatments, psychological trauma or other factors that could confound diagnosis.
Thus, while there is no universally accepted definition of mTBI in human beings, all four sets of criteria use some level of alteration or loss of consciousness as the primary indicator that a TBI occurred. However, the definitions encompass a wide range of severity ranging from as little as being momentarily dazed or confused up to loss of consciousness for 30 min. Three of the four allow focal findings on exam or focal lesions on imaging as long as other criteria for mTBI are met. Inclusion of such cases broadens the mTBI spectrum. Indeed, other criteria separate off into a category labeled “complicated” mTBI that subset of patients who could clinically be classified as mTBI but in whom neuroimaging reveals focal intracranial lesions (55). While the concept of complicated mTBI has not made its way into the major consensus criteria, a number of studies suggest that complicated mTBI is associated with more prolonged recovery and residual neurocognitive impairments (56–66). While a variety of ancillary studies including advanced neuroimaging techniques and serum markers such as S-100 protein have been investigated as markers of brain injury severity (67, 68), none has gained general acceptance as diagnostic markers of mTBI.
Difficulties in Applying Current Definitions of mTBI to Blast Injury
Within the TBI model, blast exposures are dichotomized into TBI or not TBI i.e., either an event has occurred that can be labeled a “TBI” or it has not. If there is no TBI event, then it is concluded that no clinically significant blast exposure occurred. The first difficulty posed by this approach is the defining of a TBI event. The term “concussion” is today treated as being largely synonymous with mTBI. Yet concussion once required a transient loss of consciousness as part of its definition. However, largely due to research in the sports medicine literature (69), it has become clear that loss of consciousness is not necessary for a clinically significant TBI event to have occurred. Thus, current definitions of mTBI, as discussed above, include even the most transient alteration of consciousness including being as little as momentarily dazed, confused, or “seeing stars.” These newer criteria greatly expand the definition of mTBI and most blast-related mTBIs in veterans returning from Iraq and Afghanistan do not involve loss of consciousness (10, 70–73).
Expanding the mTBI definition represents progress if it captures a larger spectrum of clinically significant blast exposures. However, it may be misleading, if it labels clinically insignificant blast exposures as TBI as suggested by some of the TBI/PTSD overlap literature discussed below. Indeed, the expanded definition can sometimes make deciding whether a mTBI has occurred difficult, which is often done based on histories obtained long after an event that occurred in settings where it may be difficult to separate a transient neurological disturbance from a psychologically based stress reaction.
Current definitions of mTBI pose another problem in that if an exposure just sufficient to produce the most minimal alteration of neurological functioning can be considered clinically significant then could exposures beneath the threshold to produce a TBI event matter clinically as well. This issue seems particularly relevant to blast due to the nature of blast exposure that involves a blast wave propagated for considerable distances through air thereby exposing large areas to a continuous range of air-shock conditions. If subclinical blast exposure affects the brain, then any definition of TBI requiring a transient neurological disturbance would fail to capture the full spectrum of blast effects and indeed would suggest that the TBI model may fail to capture a wider spectrum of blast-related brain injury.
Indeed, under such a scenario, multiple subclinical exposures might matter more than a single exposure sufficient to be identified as a TBI event. Multiple blast exposures have been common in the war zones in Iraq and Afghanistan even in subjects not known to have suffered a TBI. For example, in the study of Hoge et al. (10) over 55% of non-injured controls reported two or more occasions in which they were near an IED explosion suggesting that most controls in these settings have been subjected to blast exposure. Comparisons in multivariate regressions are performed along a dichotomy of TBI vs. no TBI. However, if a continuous gradient of subclinical exposures matter, especially cumulative exposures just below the TBI threshold then the dichotomy may not be real and cumulative exposure may matter more.
Separating Blast-Related mTBI and PTSD
One of the striking features of the mTBI cases being seen in veterans from Iraq and Afghanistan is the frequent presence of PTSD (11). Indeed, PTSD or depression is present in over one-third of Iraq veterans with suspected post-concussion syndromes secondary to mTBI (10, 74). The frequent presence of PTSD has complicated diagnosis since the clinical distinction between a post-concussion syndrome and PTSD is often difficult with the two disorders having many overlapping symptoms (11). In both somatic complaints including fatigue, irritability and poor sleep are frequent. Impaired concentration, attention, and memory are also common, and neuropsychological test profiles can look similar with deficits in attention, working memory, executive functioning, and episodic memory present in both (74).
The problem of distinguishing the two disorders is not new and has roots dating back to the entity first recognized during World War I known as “shell shock” (2–8). During this era, a debate raged concerning whether shell shock was a physical injury or the result of psychic trauma. The debate ended without decisive resolution but with the British government clearly favoring psychological over organic explanations. Indeed, the 1922 Southborough report of the War Office’s Committee of Enquiry into “Shell-Shock” concluded that regular units with high morale were virtually immune from shell shock (75). At the beginning of World War II, the British government suppressed the use of the term “shell shock” (76). However, soldiers continued to be exposed to blasts and to present with a similar range of symptoms. The controversy regarding physical vs. psychological injury continued again without any clear resolution (2). Following the Vietnam War, PTSD became conceptualized as a diagnostic entity and was added to the third edition (DSM-III) of the Diagnostic and Statistical Manual of Mental Disorders of the American Psychiatric Association (APA) (77). The role of blast exposure in this symptom complex remerged in the popular press and scientific literature following the onset of the conflicts in Iraq and Afghanistan (78). While similar in many ways to the war exposures of the past, blast exposures in the most recent conflicts have differed in first their high prevalence. Indeed, it has been estimated that 75% of casualties in Iraq and Afghanistan have been due to explosions (36). Improved personal protective equipment seems to have mitigated in particular the severity of blast-related lung injuries. Such factors along with improved frontline medical care have lead to a lower percentage of blast fatalities and thus increased the number of survivors who may live to experience the effects of blast-related TBI.
TBI and PTSD have an interesting relationship in that the two disorders can be considered different ends of a spectrum with TBI being the classic example of an organic brain disease and PTSD a psychologically based reaction to a stressor that was not associated with physical injury. Indeed, it has been suggested that the post-traumatic amnesia associated with TBI may protect against the development of PTSD, based on the notion that amnesia for the event precludes formation of the core affective responses needed to develop PTSD (79). While PTSD can clearly develop after even moderate to severe TBI, evidence does suggest that PTSD rates are higher in subjects who remember the TBI incident compared to those with no memory for the event (80, 81).
The association of PTSD with mTBI might be explained by dual exposures to PTSD stressors as well as independent TBI events. However, other studies have suggested that the apparent epidemic of mTBI in veterans from Iraq and Afghanistan may be an artifact of current definitions of mTBI, which lower the threshold for diagnosing an mTBI to the most transient alteration of consciousness. The study of Hoge et al. (10) first raised such questions. They surveyed over 2700 U.S. Army infantry soldiers from two brigades, 3–4 months after returning from a 1-year deployment in Iraq. In this study, the most frequent cause of TBI was blast and all but 4 of 384 TBIs were mTBI. Among the mTBI, 5% reported loss of consciousness and 10% occurred without loss of consciousness. In soldiers who reported an mTBI complaints of headache, poor memory and concentration were frequent suggesting that a persistent post-concussion syndrome was present. However, using a multivariate logistic regression analysis, after adjusting for the frequent co-existence of PTSD and depression, an mTBI history was no longer associated with any post-concussion symptoms, except for headache.
Hoge et al. (82) argued later that current-screening criteria for mTBI are flawed. Multiple subsequent studies from this group as well as others have supported this interpretation suggesting that post-concussion symptoms and other adverse physical or neuropsychological outcomes following blast-related mTBI are only non-specifically related to blast, better explained by PTSD or only related to blast when there has been loss of consciousness, which is the minority of cases using current definitions of mTBI (70, 72, 73, 83–92). Indeed, the 2014 Institute of Medicine report (36) on the long-term consequences of blast injury concluded that based on human studies, there is “limited/suggestive evidence that most of the shared symptoms are accounted for by PTSD and not a direct result of TBI alone” and that for post-concussion symptoms and persistent headaches following blast-related mTBI, there was only sufficient evidence to suggest an association. While other studies suggest that a blast-related mTBI might be more than a coincidental exposure (93–100), the clear message from many studies is that much of what is presently being called post-concussion syndrome secondary to blast-related mTBI is really PTSD.
Defining Blast-Related mTBI in Animal Models
Animal models should allow effects of primary blast to be determined free of many confounding variables present in natural human exposures (11, 26). Recently, Cernak (101) has discussed the general requirements for animal models of blast-induced neurotrauma. Developing an animal injury model involves both a choice of species as well as exposure simulation. Choice of species has practical as well as theoretical implications. Rodents, mostly rats, have been most commonly used (11, 26), a choice that without doubt has been driven more by practical than theoretical considerations. Besides availability, rodents offer advantages in the power of the genetic systems that exist particularly in mice. Rodents may be less than ideal; however, in that rodent brains lack the gyri and sulci found in human beings, a factor that likely affects the brain’s mechanical response to acceleration impulse (102). The white/gray matter ratio is also less in rodents, another factor that may affect the brain’s reaction to injury. Pigs and non-human primates have brains more similar to human beings but their cost and in the case of non-human primates, availability limits their wider use.
Animals have been exposed to various forms of blast ranging from direct exposure to live explosives to more commonly controlled blast waves produced by compressed-air generators (11, 26). Although live explosives may best model exposure in the field, this approach affords less experimental control over the physical characteristics of the blast wave as well as depending on the injury paradigm may involve difficulty in separating effects of the primary blast wave from secondary, tertiary, or even quaternary injuries. Pressure generators allow blast overpressure effects to be studied in isolation, offering more experimental control. However, conventional shock tubes can only approximate some aspects of explosive blast conditions. While they replicate the ideal blast wave, they lack the capability of modeling the non-ideal blast wave with its multiple shock and expansion fronts that occur in real-life settings.
Interpreting the shock tube literature is also complicated by there being no standardized conditions for conducting shock tube research (101, 103). Shock tube apparatus and blast insult conditions, as well as specimen mounting, degree of head restraint, and location relative to the tube can vary greatly between labs. Blast waves are characterized by their peak overpressure, duration, and impulse. Different shock tubes may produce pressure waves with differing characteristics leading to different biological effects. For example, large peak pressures delivered over short durations may be better tolerated than lower pressures experienced over long durations (103). Yet, many studies report only peak pressures making it difficult to sometimes compare studies even within species. Sundaramurthy et al. (104) have also shown that the biomechanical loading experienced by rats in a shock tube can vary widely depending on whether the animal is placed inside or outside the tube and at different locations along the length of the tube. Choice of species also affects scaling considerations important in choosing shock wave parameters (105). In addition to size, skull biomechanical properties as well as anatomy of the orbits and sinuses likely affect how external loads become imparted to the brain. Pigs for example have relatively thick skulls compared to human beings while rodents have much thinner skulls, factors that apart from size must be considered in experimental design.
Finally, it is not clear that all injury to the brain occurs through direct effects. For example, it has been suggested that blast waves hitting the thorax or abdomen can be transmitted through the vasculature to the brain causing injury (43, 106). The importance of this mechanism remains unclear. However, it is interesting that in the most recent conflicts, there has been little evidence of “blast lung” implying that the Interceptor Body Armor being provided to soldiers may be effective in mitigating blast injury to the thorax. If so, animal models allowing thoracic exposures may be reproducing effects that are no longer as relevant to current wartime exposures.
However, whatever animal model is chosen, the question arises how to define mTBI given that in human beings TBI severity is defined in relation to the degree of alteration of consciousness at the time of acute injury. While studies of experimental blast injury in animals have been conducted without anesthesia (107, 108), essentially all studies being conducted today are done under general anesthesia, which precludes observing any transient alteration of neurological functioning at the time of exposure. The problem thus becomes how to apply a definition of mTBI in human beings in which severity is judged by alteration of consciousness at the time of injury to animal studies conducted under anesthesia where alteration of consciousness at the time of injury cannot be assessed.
Thus, in animal models, injury severity can only be assessed by the residual effects observed after anesthesia has worn off, i.e., outcomes. Even if animals could be studied without anesthesia, the problem of how to operationalize being “dazed or confused” or “seeing stars” in an animal with no capacity for self-report seems formidable. Given the difficulty of applying definitions of human mTBI to animal studies, the label “mTBI” probably has only limited usefulness in animals. Indeed from here forward, we refer to animal studies thought to be the equivalent of human mTBI based on outcomes as “low-level” blast.
Outcomes that might be considered in animal models include behavioral, pathological, physiological, biochemical, or neuroimaging results. Among these, behavior and pathology are readily available in animals. However, since it is rare that mTBI patients come to postmortem exam, little is known about the pathology of mTBI especially in the acute phase (38). Modern neuroimaging may provide one of the best opportunities for comparative studies since this modality is well developed in human beings (38) and being applied more to animal models (see below).
The issue then becomes how to develop a relevant animal model of low-level blast. One approach taken by Ahlers et al. (107) was to expose animals to sequentially higher blast levels and to determine whether a threshold could be identified where outcomes would mirror those expected of a human mTBI without features inconsistent with mTBI. In these studies, which utilized the pneumatically driven shock tube at the Walter Reed Army Institute of Research (WRAIR) (107), initially unanesthetized rats were exposed to progressively higher blast exposures in different orientations at peak pressures of 36.6 kPa (associated with a duration of 4.1 ms and impulse of 75.2 kPa*ms), 74.5 kPa (duration 4.8, impulse 175.8), and 116.7 kPa (duration 6.8, impulse 335.5). Exposures up to 74.5 kPa (equivalent to 10.8 psi) lead to no persistent neurological impairments, although anterograde memory deficits were observed in rats exposed to 74.5 kPa blasts when facing the blast wave. In addition, 36.6 and 74.5 exposures produced no gross neuropathological effects and histological examination of the lung showed no hemorrhage or other abnormalities. By contrast, 116.7 kPa exposures lead to overt pathology with approximately 30% of rats having subdural hemorrhages and cortical contusions, a finding not consistent with human mTBI. All animals exposed to 116.7 kPa blasts had frequent pulmonary hemorrhages suggesting that these levels of exposure produce a state of polytrauma. Similar studies conducted using anesthetized rats in the WRAIR shock tube exposed to single or multiple blasts have also found that 36.6 and 74.5 level exposures are consistent with a low-level blast exposure (107, 109–111).
Thus, based on studies using the WRAIR shock tube, a dividing line seems to exist between 74.5 and 116.7 kPa that separates low-level blast in rats from moderate to higher level blast exposures that are more equivalent pathologically to human moderate to severe TBI in the context of polytrauma. The findings in the WRAIR studies are consistent with other investigations that have noted similar patterns of injury at pressures of 116.7 kPa and above in rats (112–115). Studies from other labs using exposures of 74.5 kPa or lower also seem largely consistent with the WRAIR findings suggesting that exposures in rats up to 74.5 kPa across a range of durations can reasonably be called low-level blast exposure (116–121). Indeed very similar to the WRAIR studies, Baalman et al. (121) found that while 74 kPa (duration ≈ 4 ms) peak pressures resulted in only subtle pathology, the next highest exposure (98 kPa) resulted in lung trauma and death.
Pressures that mimic low-level blast in other species may of course differ and require validation in relation to outcomes in those species. Of interest are several studies in mice that have examined blast overpressure exposures similar to those that appear consistent with low-level blast in rats. For example, Goldstein et al. (122) studied mice exposed to a single blast exposure of 77 kPa. However, while this exposure is within the range that might be reasonably considered low level in rats, the extent of neuropathology seen in this model seems more equivalent to higher level blast. By contrast, Huber et al. (123) studied mice exposed to 108 kPa pressures and describe what seems like a more mild pathology. In a third set of studies, mice were exposed to 68, 76, or 105 kPa exposures (124, 125). With 68 kPa exposures, scattered axonal pathology was found in multiple CNS tracts (125) with no mortality if the mice were exposed in the prone position although 5% of the mice died if exposed in the supine position. At higher exposures, there was between 11% (76 kPa) and 33% (105 kPa) mortality, if the mice were exposed in the prone position and 37% (76 kPa) to 53% (105 kPa) if exposed supine, levels of mortality that would not be expected of an mTBI exposure. These studies emphasize not only the importance of species specific differences but also that outcomes within the same species may vary with similar pressure exposures. One factor in particular differing between the studies discussed above was that Goldstein et al. (122) used no head restraint allowing the head to be subjected to significant rotational motion as well as likely focal impact injury. Notably, when Goldstein et al. (122) restricted head movement, hippocampal dependent leaning and memory deficits disappeared. Effects on pathology after head restraint were not reported (122). Restricting exposure to the head may also influence effects as Heldt et al. (126) have described a model in which mice exhibited only limited CNS pathology with exposures of 20–60 psi if the blast wave was delivered to a small area on one side of the cranium.
Effects of Low-Level Blast Exposure in Animal Models
Most studies of blast in animals have utilized relatively high-level exposures that likely more approximate moderate to severe TBI (11, 26). However, particularly in recent years studies have appeared assessing effects of lower level blast. To search for papers reporting low-level blast exposure in animal models, we searched Pubmed using the terms “blast” and “traumatic brain injury.” Articles were selected for review based on the relevance of the title and abstract. References in the studies that were selected for review were examined for additional relevant citations.
In rats, we considered as low-level blast those studies that utilized blast exposures of ≈ 75 kPa or less based on the considerations discussed above and where sufficient pathological information was available to conclude there was no gross pathology particularly hemorrhages that would be incompatible with mTBI. Studies in mice were more difficult to judge due to there being fewer studies and the seeming variability in response to blast in the 68–108 kPa range (122–125) (although see discussion above concerning differences in these studies). However, a series of studies in mice using exposures to live explosives in the 2.5–5.5 psi range seem compatible with a definition of low-level blast (127–129).
Table 2 summarizes studies of exposures in what we term the low-level blast range. These papers first document that low-level blast exposures are transmitted to the brain (130). Chavko et al. (130) showed that pressures as low as 35 kPa are transmitted to the brain in rats although the patterns and durations of pressure traces inside the brain depended on the orientation to the blast with frontal exposures resulting in pressure traces of higher amplitude and longer duration than with other orientations. Peak pressures measured in brain were very similar to those in air (131). Interestingly, the observation of higher pressures with frontal exposures is similar to what has been observed in helmeted manikins (132). Sustained increases in intracranial pressure (ICP) have been documented for 10 h in rats exposed to 30 or 60 kPa blasts (119). While as noted above, it is difficult to directly compare exposures across species, similar observations have been made in pigs subjected to 23–45 kPa exposures from the firing of military weapons (120, 133). As in rats, peak pressures in the pig brain had similar magnitudes to those recorded in air with lower blast wave frequencies being transmitted more readily to brain than higher frequencies (120, 133).
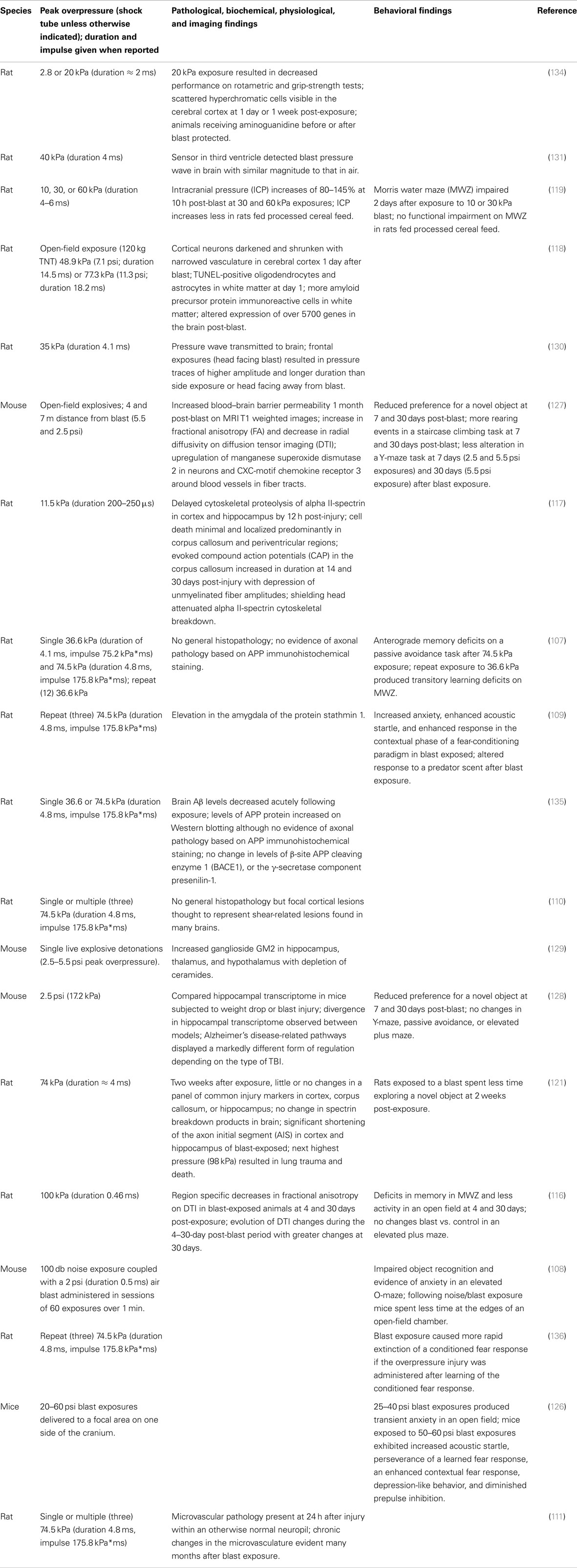
Table 2. Effects of low-level blast exposure in animal models.
Pathologically (Table 2), low-level blast exposure in rats leads to no general histopathology, although most studies have focused on the pathology most commonly seen in moderate to severe nbTBI, which may not be the most relevant to blast. Some studies have described focal cortical lesions that are likely the result of shear-related effects (110), and a selective microvascular pathology has been described at the ultrastructural level (111). Others have described scattered pyknotic neurons (118, 134) with TUNEL-positive oligodendrocytes and astrocytes in white matter and periventricular regions (117, 118). More amyloid precursor protein (APP) immunoreactive cells in the white matter have been observed (118). One study found shortening of the axon initial segment in cortex and hippocampus of blast-exposed rats (121). Biochemically, proteolysis of alpha II-spectrin in cortex and hippocampus has been observed at 12 h post-injury (117) as well as upregulation of manganese superoxide dismutase 2 (127) and increased expression of the CXC-motif chemokine receptor 3 around blood vessels in fiber tracts (127). Increased ganglioside GM2 has been observed in hippocampus, thalamus, and hypothalamus along with depletion of ceramides (129). Physiologically longer duration evoked compound action potentials (CAP) have been observed in the corpus callosum at 14 and 30 days post-injury with a depression in unmyelinated fiber amplitudes (117). Gene expression studies have documented altered expression of over 5700 genes in the post-blast brain (118) as well as differences in the hippocampal transcriptome between blast and non-blast models (128). Neuroimaging studies have found region specific decreases in fractional anisotropy (FA) on diffusion tensor imaging (DTI) (116) and increased blood–brain barrier permeability post-blast (127).
Low-Level Blast Exposure in Animal Models and PTSD
The frequent overlap of TBI and PTSD could represent dual exposures to TBI as well as PSTD-related psychological stressors. Alternatively, TBI might induce PTSD like symptoms if blast damaged brain structures involved in the development of PTSD. Current biological models of PTSD postulate that key frontal and limbic structures, including the prefrontal cortex, amygdala, and hippocampus are involved in PTSD development (137, 138). These models suggest that a key element is an inadequate frontal inhibition of the amygdala, a limbic structure central to controlling fear responses. Exaggerated amygdala responses are thought to heighten responses to psychological threats. A substantial body of functional neuroimaging data is consistent with such models, suggesting that in PTSD there is heightened amygdala activity with decreased hippocampal and orbital frontal activity (137). Damage to the prefrontal cortex by TBI could, therefore, predispose individuals to abnormally sustained responses to psychological stressors.
Animal models should be able to isolate blast effects from psychological stressors and studies have begun to address the behavioral effects of blast in animals across a range of exposures (107, 119, 122, 124, 125, 127, 134, 139–142), although not all studies have been careful to exclude subtle motor or sensory deficits as contributing to apparent behavioral effects. These studies have reported at least short-term effects on anxiety as well as impairments in a variety of learning and memory tasks (107, 119, 122, 124, 125, 127, 134, 139, 141). One study reported impairments in prepulse inhibition immediately after blast exposure, although the changes had mostly recovered 90 days after exposure (142). A few studies have begun to address the issue of dual exposure by examining blast in combination with repeated stress (139) or factors such as transportation stress or anesthesia (140). Studies that have specifically addressed behavioral effects in the context of low-level blast exposure are summarized in Table 2. Abnormalities reflecting impaired memory function have been observed in the Morris water maze (MWZ) (107, 116, 119), novel object recognition (121, 127, 128), Y-maze (127, 128), and passive avoidance (107).
Only a few studies have addressed whether low-level blast exposure alone can induce PTSD-related traits or render animals more sensitive to subsequent PTSD-related stressors. The first of these tested rats that received three 74.5 kPa exposures on a variety of PSTD-related traits several months post-exposure (109). The rats exhibited a variety of PTSD-related traits including increased anxiety in an elevated zero maze and increased acoustic startle. They also showed increased anxiety during a predator scent challenge and an increased cued response in a fear-conditioning paradigm (109).
Subsequently, very similar effects were noted in mice (126). In these studies, mice were exposed to 20–60 psi blast exposures in which the blast wave rather than being allowed to envelope the brain was delivered to a small area on one side of the cranium. While these exposures are in a range that has commonly induced significant CNS pathology in other studies, Heldt et al. (126) reported that using this limited focal exposure, 25–40 psi blasts produced little histological evidence of brain damage and 50–60 psi exposures produced only scattered axonal degeneration. Thus, on pathological grounds, the exposures appeared consistent with low-level blast. Behaviorally, the 25–40 psi blasts produced transient anxiety in an open field. In tests conducted 2–8 weeks after 50–60 psi blast exposures, mice exhibited increased acoustic startle, perseverance of a learned fear response, and an enhanced contextual fear response along with depression-like behavior and diminished prepulse inhibition.
A third study (108) examined non-anesthetized mice exposed to 100 db noise exposures coupled with 2 psi air blasts administered in sessions consisting of 60 exposures over 1 min. The mice exhibited impaired object recognition and anxiety in an elevated O-maze. The mice also spent less time at the edges of an open-field chamber an effect that the authors interpreted as a fear generalization response to the noise/blast exposure having taken place near a chamber wall. No pathology was described to confirm the lack of histological effects and the high frequency of exposure does not model natural human exposures. The levels of exposure are nevertheless within what would be expected to be a low-blast exposure, thus providing additional evidence for cognitive and emotional effects of low-level blast although the lack of anesthesia makes it difficult to determine how much of a psychological stressor may have been involved.
Collectively, the above studies argue that low-level blast exposure alone can induce PTSD-related traits. The increased acoustic startle and altered fear-conditioning responses are of particular interest because of their direct relevance to PTSD (109, 126). The ability of a prior blast exposure to alter subsequent response to a predator scent (109) suggests that blast may render animals more sensitive to later PTSD-related stressors. The fact that the blast overpressure injuries in two of these studies (109, 126) occurred while animals were under general anesthesia, suggests that low-level blast exposures can in the absence of any psychological stressor induce PTSD-related traits. Altered fear responses are suggestive of heightened amygdala function, an effect consistent with current biological models of PTSD (137). Indeed, enhancing fear responses may be a feature of both blast and nbTBI, as recent studies have suggested that increased fear responses can be seen in animal models of nbTBI as well (143, 144).
The studies of blast exposure in rats (109) also found elevation in the amygdala of the protein stathmin 1. Stathmin 1 is highly expressed in the amygdala and mice with null mutations of the stathmin 1 gene are impaired in their ability to lean fear responses (145). Polymorphisms in the stathmin 1 gene have been identified that influence fear and anxiety responses as well as cognitive and affective processing in human beings (146, 147). Heldt et al. (126) also observed in mice reduced numbers of a subpopulation of excitatory projection neurons in the basolateral amygdala that have been linked to fear suppression. Following blast, Xie et al. (108) reported electrophysiological changes in the anterior cingulate cortex, a region implicated in processing emotional memory and inhibitory control. Thus, multiple studies have found blast-associated biochemical, anatomic, or electrophysiological changes in structures relevant to the development of PTSD.
Clearly, questions remain and especially long-term studies are needed to extend these findings. For example, it is not known whether these traits evolve over time. However, the presence of behavioral changes and elevation of stathmin 1, 8 months after blast exposure in one study (109) suggests that blast can induce PTSD-related traits that are chronic and persistent. The relationship to blast may be complex, however, in that another study has suggested that blast exposure causes more rapid extinction of a conditioned fear response if the overpressure injury occurs after learning the conditioned fear response (136).
Low-Level Blast Exposure in Animal Models and Neurodegenerative Diseases
Recently, nbTBI has been linked to the later appearance of progressive neurodegenerative disorders. The two diseases that have attracted the most concern are Alzheimer’s disease (AD) and chronic traumatic encephalopathy (CTE) (148). Single severe nbTBI earlier in life seems to predispose to the later development of AD. By contrast, repetitive non-blast mTBI has been associated with CTE. The disorders differ in that pathologically AD is characterized by amyloid plaques and tau positive neurofibrillary tangles while CTE is primarily a tauopathy that may have diffuse amyloid plaques but not typically the neuritic plaques characteristic of AD (149). What is now called CTE was first described in boxers as dementia pugilistica. However, CTE has now been seen in a variety of other sports including American football, hockey, soccer, and professional wrestling (39, 150–154).
Whether blast exposure causes or predisposes to later development of neurodegenerative diseases is unknown. Indeed, little is known about long-term effects of blast exposure although one study of 167 U.S. military service personnel who were evaluated within 5 years of sustaining an mTBI in Iraq or Afghanistan found that while many improved, a substantial minority (≈20%) reported new symptoms (155). CTE might be of particular concern with respect to blast due to its association with repetitive mTBI, which has been common in Iraq and Afghanistan and cases of CTE have been reported in veterans returning from these conflicts (122, 156, 157).
Following nbTBI, several proteins associated with neurodegenerative diseases accumulate in brain, including tau, APP, and its product the β-amyloid (Aβ) protein (158). The Aβ peptide is most associated with AD where it deposits in senile plaques with many in vitro as well as in vivo studies demonstrating that in particular the longer Aβ 42 species can be neurotoxic and associated with a chain of pathological events (159). Interestingly, changes in Aβ occur rapidly after nbTBI in human beings with diffuse cortical Aβ deposits and increased levels of soluble Aβ appearing as early as 2 h after a severe TBI (148, 160–162).
Elevations of Aβ occur consistently in experimental animal models of nbTBI (163–172) along with increased expression of components of the γ-secretase complex as well as BACE1 (β-site APP cleaving enzyme 1), the principal β-secretase (165, 173–176) both of which process APP toward the Aβ pathway. Studies in nbTBI animal models also consistently find elevated APP protein acutely following TBI (176–179). All these observations are consistent with increased processing of APP toward Aβ production after TBI creating interest in whether APP upregulation following TBI may explain the epidemiological association between a history of prior TBI and the subsequent development of AD (148, 160).
Exploring how blast-related TBI affects expression of proteins related to neurodegenerative diseases is just beginning. However, interestingly the one study that has so far reported on Aβ levels following blast found that in both rat and mouse models of blast injury, rather than being increased, brain Aβ levels were decreased acutely following injury (135). In these studies, rats were exposed to single 36.6, 74.5 kPa or 116.7-kPa-exposures and Aβ 40 and 42 levels were examined 24 h and 1 week after exposure. Blast exposure lead to diminished levels of Aβ 42 in rats with the effect on Aβ 42 most prominent in rats exposed to the lowest level blast exposures (36.6 and 74.5 kPa), while no effects on Aβ 42 were seen at the 116.7 kPa exposure level. There were no consistent effects on Aβ 40 levels in rats, with only the 74.5 kPa exposure showing diminished levels 1 week post-exposure. Levels of APP were increased following blast exposure although there was no evidence of axonal pathology based on APP immunohistochemical staining. Unlike in nbTBI animal models, levels of BACE1 and the γ-secretase component presenilin-1 were unchanged following blast exposure in rats. In mice, only a single blast exposure of 147 kPa was tested, an exposure that is more equivalent to a high-level blast exposure (180). Yet, at this exposure, both Aβ 40 and Aβ 42 were decreased 24 h after the blast.
As noted above, multiple studies in experimental animals have found that APP expression increases acutely following TBI (176–179). Indeed, APP accumulation in axons is widely used as a marker of axonal injury in both human beings and experimental animal models of TBI (181–185). However, while one study has reported APP accumulation in axons following blast exposure (186), multiple others have not (107, 113, 115, 118). Risling et al. (113), for example, noted no APP accumulation in axons of rats exposed to 130 and 260 kPa exposures. Garman et al. (115) studied rats exposed to greater than 240 kPa (35 psi) blast exposures and found evidence of widespread diffuse axonal injury by silver staining. However, despite evident axonal injury by silver staining, APP-stained sections typically showed only minimal axonal staining, except for rats studied at 24 h post-exposure when mild axonal staining was described within the deep cerebellar white matter and adjacent to some foci of acute neuronal degeneration. Thus, while APP accumulation in axons is considered a hallmark of acute axonal injury in both human beings and experimental animal models of nbTBI (181–185), it is at most an inconsistent feature of blast-related TBI.
In contrast to the seemingly paradoxical effects of blast on Aβ, multiple studies have found that aberrant tau species are induced by blast (122, 123, 141, 187). Indeed, Goldstein et al. (122) described CTE-linked neuropathology in wild-type C57BL/6 mice 2 weeks after exposure to a single blast. The blast-exposed mice also expressed multiple species of abnormally phosphorylated tau similar to those seen in human neurodegenerative diseases. Huber et al. (123) studying a mouse model exposed to a single 108.9 kPa (15.8 psi) exposure found multiple aberrantly phosphorylated- and cleaved-tau species present at 24 h post-blast that were still present in hippocampus 30 days after exposure.
Thus, experimental animals provide support for a linkage between blast exposure and aberrant tau processing. Lacking are longitudinal studies to determine whether blast induces a CTE-like neurodegenerative disease with progressive features. Indeed, there are few studies on long-term effects of blast and whether effects are static or can be associated with a progressive neurodegenerative disorder although in one study (116) changes in DTI were followed in a rat model exposed to 100 or 450 kPa exposures. Region specific decreases in FA were found at both exposure levels 4 and 30 days post-exposure. Interestingly, the changes evolved during the 4–30-day post-blast period such that a wider area of decreased FA was present at 30 days, changes suggesting an evolving lesion (116).
Is Blast TBI Different from Non-Blast TBI?
The unexpected lowering of Aβ following blast exposure besides having implications for understanding blast pathophysiology also raises the question of whether blast pathophysiology is different from nbTBI. nbTBI has been so consistently associated with elevated Aβ acutely that decreased levels following blast seem to suggest that blast is pathophysiologically different from nbTBI. The lack of an axonopathy characterized by APP accumulation is also different from nbTBI. These findings have practical implications for treatment of acute blast injury, since blocking Aβ production by a variety of pharmacological or genetic means reduces tissue damage acutely and improves outcome following controlled cortical impact injuries in mice (165, 166, 169). However, such strategies may not be applicable to treatment of acute blast injuries.
Like the issue of longitudinal effects, the question of pathophysiologic distinctness of blast deserves more study although one recent study comparing RNA expression changes between nbTBI and blast TBI models suggests differences as well (128). In these studies, the mouse hippocampal transcriptome was compared following injury using a weight drop model designed to approximate an mTBI or exposure to a 17.2 kPa (2.5 psi) blast. While a common set of upregulated or downregulated RNAs were found, most of the transcriptome changes differed between the two models suggesting that at the molecular level, the TBIs are distinct. Interestingly, in a functional pathway analysis, genes upregulated or downregulated in AD were regulated in similar directions by nbTBI while the opposite was seen following blast with the “Alzheimer’s Disease Up” pathway downregulated by blast and the “Alzheimer’s Disease Down” upregulated by blast (128). Combined with the lowering of Aβ following blast, these studies support the notion that blast-related TBI is pathophysiologically distinct from nbTBI and that in particular multiple pathways related to AD are affected differently by blast and nbTBI.
Evidence for Effects of Blast-Related mTBI in Human Beings
Close exposure to high-pressure blast waves can cause extensive CNS injury in human beings (47). However, this injury is without doubt a combination of primary, secondary, tertiary, and sometimes quaternary effects in which the role of the primary blast wave itself is hard to define. Determining the role of the primary blast wave in mTBI should be more straightforward since secondary, tertiary, and quaternary effects are less prominent. Yet, as discussed throughout this review, defining the residual effects of blast-related mTBI has been complicated by the frequent presence of co-morbid PTSD.
A number of clinical studies suggest that the link between mTBI and PSTD may be more than coincidental. Mora et al. (95) reviewed records of over 300 patients admitted consecutively to the United States Army Burn Center for explosion-related injuries and examined the prevalence of PTSD in burn patients with and without primary blast injury or mTBI. They found a greater prevalence of PTSD in burn patients with primary blast injury and mTBI than in burn patients injured by other mechanisms. Walilko et al. (96) in a study of 124 survivors of the Oklahoma City bombing explored the relationship between PTSD and physical injuries. They found an association between PTSD and head/brain injuries while PTSD was not highly correlated with other injuries.
Studies of Vietnam veterans have also suggested that TBI is associated with more severe PTSD (188), and in veterans from Iraq and Afghanistan, PTSD is more prevalent in those reporting an mTBI, as compared to veterans who suffered no injury or injuries not involving the head (10). Indeed, in the study of Hoge et al. (10), mTBI was associated with PTSD even after controlling for intensity of combat experience and a recent study of over 27,000 U.S. Army Special Operations Command personnel found that while a blunt or blast-related mTBI increased the chance of reporting clinically significant levels of PTSD symptoms, residual PTSD symptoms were more prevalent in personnel with blast-related mTBI (91). In addition, a dose–response gradient existed between number of blast-related mTBIs and symptom severity (94). Other clinical studies are also consistent with blast-related mTBI, promoting the development of or worsening of PTSD (97–99).
Thus, collectively a body of literature can be seen as arguing that TBI may predispose to the development of or worsen the symptoms of PTSD. However, their interpretation is limited by their observational nature and complicated by the contrary literature already discussed suggesting that most post-concussive symptoms following blast-related mTBI are only non-specifically related to blast or better explained by PTSD (10, 70, 72, 73, 83–91).
Modern neuroimaging would seem ideal for examining the effects of blast-related mTBI (189). By comparing subjects with blast-related TBI/PTSD to those with PTSD alone, it should be possible to determine whether a blast-related signature exists. A number of neuroimaging studies of mostly blast-related mTBI have appeared utilizing mostly DTI, functional MRI (fMRI), or positron emission tomography (PET) scanning with fluoro-deoxyglucose. These studies are summarized in Table 3. Studies range from case reports or small case series to those having over 60 blast-exposed veterans and include some subjects imaged within weeks of the TBI although most involve subjects studied several years after exposure. Some studies are limited by the lack of a military control group and some involve a mixture of blast as well as non-blast injury.

Table 3. Human imaging studies of blast-related TBI.
Despite these limitations, some common findings emerge across studies with the most consistent being reduced fractional anisotropy (FA) on DTI. Indeed, of 11 DTI studies (Table 3), only one failed to find a reduction in FA (191). The patterns across and even within some studies appear fairly heterogeneous and abnormalities have been found in subjects studied out to 4 years post-blast injury. One study using high angular resolution diffusion imaging (HARDI) found suggestions of lost white matter integrity (197). Fewer studies have been conducted with fMRI and PET. However, both resting-state fMRI studies suggested altered patterns of activation (200, 201) and one task-activated fMRI study suggested that activation patterns differed between blast and nbTBI (198). Two PET studies found reduced cerebral glucose metabolism (192, 202).
Thus, a substantial body of evidence suggests that blast-related TBI including blast-related mTBI is associated with a variety of neuroimaging abnormalities that are chronic and persistent. Yet, many subjects in these studies were known to have or even if not mentioned without doubt had PTSD raising the question of whether abnormalities could be attributed to PTSD. Several studies have examined DTI imaging in PTSD (209) although interpreting these studies is often complicated by their inclusion of subjects whose psychological stressor included TBI. Despite these limitations, a recent meta-analysis of seven DTI studies of adult onset PTSD concluded that PTSD is associated with clusters of both increased and decreased FA in various structures (209) suggesting a pattern different from the consistently reduced FA found in blast-related TBI.
Among the blast-related neuroimaging studies, Petrie et al. (202) examined the effect of PTSD on DTI and fluoro-2-deoxyglucose PET imaging in veterans with blast/impact-mTBI. They found areas of reduced FA in the corpus callosum and decreased cerebral glucose utilization in parietal, somatosensory, and visual cortices compared to controls. Their findings did not differ between veterans with vs. without PTSD suggesting that the abnormalities were not related to co-morbid PTSD. Matthews et al. (196) also found no effect of PTSD on FA in a group of Iraq and Afghanistan veterans all of whom had experienced blast-related mTBI. Likewise, Morey et al. (197) using HARDI while finding a widely distributed pattern of lost white matter integrity with blast-related mTBI found no correlation between their findings and a diagnosis of PTSD or depression. More research is needed but based on available evidence it seems unlikely that co-morbid PTSD can account for the neuroimaging changes found in blast-related mTBI.
Whether any of these findings in blast-related mTBI can be extrapolated to subclinical blast where by definition no TBI event occurred is unclear. Studies of subclinical blast are quite limited at present, although interestingly when Taber et al. (203) compared veterans exposed to primary blast with and without mTBI relative to veterans without blast exposure, they found lower FA and higher radial diffusivity in veterans exposed to blast whether or not they had been diagnosed with an mTBI. Mac Donald et al. (210) in a prospective study involving active duty U.S. military personnel evacuated from Iraq or Afghanistan to Landstuhl, Germany while finding no differences in clinical outcomes based on blast vs. non-blast mechanisms of injury found that blast-exposed controls had worse headaches and more severe PTSD than non-blast-exposed controls. Using data from a DTI study, Bazarian et al. (93) have also argued that subclinical blast may play a role in the genesis of PTSD.
Clearly, future studies directed at in particular neuroimaging of subclinical blast will be important although study of this problem in active duty military personnel where exposures are often a combination of clinically recognized and unrecognized events may be difficult. Among groups that might be studied for effects of subclinical blast, breachers provide perhaps the most accessible population where blast exposure in human beings can be studied in a semi-controlled manner. “Breachers” are a population of military and law enforcement personnel who are routinely exposed to low-level blast during their training and in the course of operations. Repeated exposure in these settings has been associated with symptoms similar to those seen with sports concussion (211). Recently, Tate et al. (211) examined the effects of repeated low-level blast exposure without mTBI during an explosives training course in 21 members of the New Zealand Defense Force. Self-reported symptoms, neurocognitive performance, and three serum biomarkers (ubiquitin C-terminal hydrolase-L1, the alpha II-spectrin breakdown product, and glial fibrillary acidic protein) were measured before, during, and after a 2-week course. Interestingly, those with the highest biomarker loads had longer reaction times, lower cognitive test scores, and reported more symptoms than those with the lowest biomarker loads. Preliminary studies have also observed alterations in white matter and cortical structural MRI measures in a cohort of breachers with 7–21 years of prior blast exposure that were not seen in a group of students attending a 2-week breacher course (212). Indeed, breachers [and similar populations who are exposed to repetitive blast events, e.g., artillery, explosive ordinance disposal, shoulder-fired weapons, etc.] may represent a unique population in which to study how subclinical blast affects human beings.
Conclusion
It has long been appreciated that high-pressure blast waves can cause extensive CNS injury in human beings. Less clear are the effects of lower level blast exposures, the most common exposure in combat settings such as Iraq and Afghanistan. Indeed, the traits that can be attributed to blast-related mTBI have been questioned with suggestions that much of what is presently being called post-concussion syndrome secondary to blast-related mTBI is really PTSD. No doubt there are grounds for controversy and many questions remain. Both PTSD and the post-concussion syndrome are clinical diagnoses made in disorders that have overlapping symptoms. Current definitions have lowered the threshold for diagnosing mTBI leading to questions as to whether we are now over diagnosing mTBI. Soldiers that have experienced blast-related mTBI have typically been exposed to psychological stressors as well and there are no biomarkers that can distinguish cognitive, affective, and somatic symptoms induced by a psychological stressor from those induced by physical trauma. PTSD is a well-established clinical syndrome while the effects of human low-level blast exposure including mTBI are still being established. Thus, there is much ground for legitimately questioning the importance of low-level blast exposure.
Animal models would seem ideal for determining the effects of primary blast free of many confounding variables present in natural human exposures. Indeed, a substantial literature now exists concerning the effects of blast in animal models. Yet, it must be admitted that this literature is also troubled by variability in experimental approach as well as at times lack of attention to modeling clinically relevant exposures or reliance on models that fail to isolate the effects of primary injury from tertiary injury. There has also been little attention given to defining what “mild” TBI is in an animal model and studies often describe models as “mild” TBI without any justification for why use of the term “mild” is appropriate.
Yet, despite these concerns, an abundance of evidence now supports the concept that low-level blast has significant long-term effects on the nervous system. While problems exist in applying definitions of human mTBI to animal models, conditions of low-level blast exposure can be defined that likely approximate human mTBI or subclinical exposure. In animals, blast exposures in these ranges exert a variety of biochemical, pathological, and physiological effects on the nervous system. Several studies in animals suggest that low-level blast exposure can induce PTSD-related behavioral traits in the absence of a psychological stressor. Indeed, if blast injury can induce PTSD like symptoms without a psychological stressor, then human cases that are presently being labeled PTSD may in fact be part of the spectrum of blast-related brain injury. Several observations in animal models also suggest that blast-related TBI is pathophysiologically distinct from nbTBI. A substantial body of neuroimaging data shows that blast-related mTBI in human beings is associated with chronic decreases in fractional anisotropy that are suggestive of chronic axonal injury and unlikely to be explained on the basis of co-morbid PTSD. In summary, while many questions remain concerning how blast overpressure waves affect the brain, there seems little doubt that low-level blast exposure should be of significant concern.
Author Contributions
All authors participated in the collection, review, and analysis of the relevant literature as well as the drafting and revising of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors have received research support from the Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research and Development Service Awards 1I01RX000179-01 and 1I01RX000996-01. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government. Stephen T. Ahlers: I am a military service member (or employee of the U.S. Government). This work was prepared as part of my official duties. Title 17 U.S.C. §105 provides that copyright protection under this title is not available for any work of the United States Government. Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
References
1. Bochicchio GV, Lumpkins K, O’Connor J, Simard M, Schaub S, Conway A, et al. Blast injury in a civilian trauma setting is associated with a delay in diagnosis of traumatic brain injury. Am Surg (2008) 74(3):267–70.
2. Jones E, Fear NT, Wessely S. Shell shock and mild traumatic brain injury: a historical review. Am J Psychiatry (2007) 164(11):1641–5. doi: 10.1176/appi.ajp.2007.07071180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Jones E, Thomas A, Ironside S. Shell shock: an outcome study of a First World War ‘PIE’ unit. Psychol Med (2007) 37(2):215–23. doi:10.1017/S0033291706009329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Rae R. An historical account of shell shock during the First World War and reforms in mental health in Australia 1914-1939. Int J Ment Health Nurs (2007) 16(4):266–73. doi:10.1111/j.1447-0349.2007.00476.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Anderson RJ. Shell shock: an old injury with new weapons. Mol Interv (2008) 8(5):204–18. doi:10.1124/mi.8.5.2
6. Jones E. Shell shock at Maghull and the Maudsley: models of psychological medicine in the UK. J Hist Med Allied Sci (2010) 65(3):368–95. doi:10.1093/jhmas/jrq006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Loughran T. Shell shock, trauma, and the First World War: the making of a diagnosis and its histories. J Hist Med Allied Sci (2012) 67(1):94–119. doi:10.1093/jhmas/jrq052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Shively SB, Perl DP. Traumatic brain injury, shell shock, and posttraumatic stress disorder in the military – past, present, and future. J Head Trauma Rehabil (2012) 27(3):234–9. doi:10.1097/HTR.0b013e318250e9dd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Linden SC, Jones E, Lees AJ. Shell shock at Queen Square: Lewis Yealland 100 years on. Brain (2013) 136(Pt 6):1976–88. doi:10.1093/brain/aws331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med (2008) 358(5):453–63. doi:10.1056/NEJMoa072972
11. Elder GA, Mitsis EM, Ahlers ST, Cristian A. Blast-induced mild traumatic brain injury. Psychiatr Clin North Am (2010) 33(4):757–81. doi:10.1016/j.psc.2010.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab (2010) 30(2):255–66. doi:10.1038/jcbfm.2009.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. DePalma RG, Burris DG, Champion HR, Hodgson MJ. Blast injuries. N Engl J Med (2005) 352(13):1335–42. doi:10.1056/NEJMra042083
14. Leung LY, VandeVord PJ, Dal Cengio AL, Bir C, Yang KH, King AI. Blast related neurotrauma: a review of cellular injury. Mol Cell Biomech (2008) 5(3):155–68.
15. Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. J Neurotrauma (2009) 26(6):815–25. doi:10.1089/neu.2007.0484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Okie S. Traumatic brain injury in the war zone. N Engl J Med (2005) 352(20):2043–7. doi:10.1056/NEJMp058102
17. Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known? J Neuropsychiatry Clin Neurosci (2006) 18(2):141–5. doi:10.1176/appi.neuropsych.18.2.141
18. Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil (2006) 21(5):398–402. doi:10.1097/00001199-200609000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Wolf SJ, Bebarta VS, Bonnett CJ, Pons PT, Cantrill SV. Blast injuries. Lancet (2009) 374(9687):405–15. doi:10.1016/S0140-6736(09)60257-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Akin FW, Murnane OD. Head injury and blast exposure: vestibular consequences. Otolaryngol Clin North Am (2011) 44(2):323–34,viii. doi:10.1016/j.otc.2011.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Bass CR, Panzer MB, Rafaels KA, Wood G, Shridharani J, Capehart B. Brain injuries from blast. Ann Biomed Eng (2012) 40(1):185–202. doi:10.1007/s10439-011-0424-0
22. Chen Y, Huang W. Non-impact, blast-induced mild TBI and PTSD: concepts and caveats. Brain Inj (2011) 25(7–8):641–50. doi:10.3109/02699052.2011.580313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Courtney MW, Courtney AC. Working toward exposure thresholds for blast-induced traumatic brain injury: thoracic and acceleration mechanisms. Neuroimage (2011) 54(Suppl 1):S55–61. doi:10.1016/j.neuroimage.2010.05.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. French LM. Military traumatic brain injury: an examination of important differences. Ann N Y Acad Sci (2010) 1208:38–45. doi:10.1111/j.1749-6632.2010.05696.x
25. Hicks RR, Fertig SJ, Desrocher RE, Koroshetz WJ, Pancrazio JJ. Neurological effects of blast injury. J Trauma (2010) 68(5):1257–63. doi:10.1097/TA.0b013e3181d8956d
26. Kobeissy F, Mondello S, Tumer N, Toklu HZ, Whidden MA, Kirichenko N, et al. Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front Neurol (2013) 4:186. doi:10.3389/fneur.2013.00186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Ling GS, Ecklund JM. Traumatic brain injury in modern war. Curr Opin Anaesthesiol (2011) 24(2):124–30. doi:10.1097/ACO.0b013e32834458da
28. Magnuson J, Leonessa F, Ling GS. Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep (2012) 12(5):570–9. doi:10.1007/s11910-012-0303-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rosenfeld JV, McFarlane AC, Bragge P, Armonda RA, Grimes JB, Ling GS. Blast-related traumatic brain injury. Lancet Neurol (2013) 12(9):882–93. doi:10.1016/S1474-4422(13)70161-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sayer NA. Traumatic brain injury and its neuropsychiatric sequelae in war veterans. Annu Rev Med (2012) 63:405–19. doi:10.1146/annurev-med-061610-154046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. DePalma R, Cross G, Beck L, Chandler D, editors. Epidemiology of mTBI (mild traumatic brain injury) due to blast: history, DOD/VA data bases: challenges and opportunities. Proceedings of the NATO RTO-MP-HFM-207 Symposium on a Survey of Blast Injury Across the Full Landscape of Military Science. Halifax (2011). p. 1–8.
32. DePalma R, Cross G, Buckley C, Gunnar W. Blast related traumatic brain injury: pathophysiology, comorbidities, and neurobehavioral outcomes. In: Levin H, Shum D, Chan R, editors. Understanding Traumatic Brain Injury: Current Research and Future Directions. Oxford: Oxford University Press (2014). p. 413–29.
33. Tanielian T, Jaycox LH, editors. Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. Santa Monica, CA: Rand Corporation (2008).
34. Warden DL, Ryan L, Helmick K, Schwab K, French L, Lu W, et al. War neurotrauma: the defense and veterans brain injury center (DVBIC) experience at the walter reed army medical center. J Neurotrauma (2005) 22:1178.
35. Bell RS, Vo AH, Neal CJ, Tigno J, Roberts R, Mossop C, et al. Military traumatic brain and spinal column injury: a 5-year study of the impact blast and other military grade weaponry on the central nervous system. J Trauma (2009) 66(4 Suppl):S104–11. doi:10.1097/TA.0b013e31819d88c8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Institute of Medicine. Gulf War and Health: Long-Term Effects of Blast Exposures. Vol. 9 Washington, DC: National Academies Press (2014).
37. Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci (2013) 14(2):128–42. doi:10.1038/nrn3407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Bigler ED, Maxwell WL. Neuropathology of mild traumatic brain injury: relationship to neuroimaging findings. Brain Imaging Behav (2012) 6(2):108–36. doi:10.1007/s11682-011-9145-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron (2012) 76(5):886–99. doi:10.1016/j.neuron.2012.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kucherov Y, Hubler G, DePalma R. Blast induced mild traumatic brain injury/concussion: a physical analysis. J Appl Phys (2012) 112:104701–104701. doi:10.1089/neu.2011.1895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Gupta RK, Przekwas A. Mathematical models of blast-induced TBI: current status, challenges, and prospects. Front Neurol (2013) 4:59. doi:10.3389/fneur.2013.00059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, et al. Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J Neurotrauma (2011) 28(6):1101–19. doi:10.1089/neu.2010.1442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Courtney AC, Courtney MW. A thoracic mechanism of mild traumatic brain injury due to blast pressure waves. Med Hypotheses (2009) 72(1):76–83. doi:10.1016/j.mehy.2008.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Benzinger TL, Brody D, Cardin S, Curley KC, Mintun MA, Mun SK, et al. Blast-related brain injury: imaging for clinical and research applications: report of the 2008 St. Louis workshop. J Neurotrauma (2009) 26(12):2127–44. doi:10.1089/neu.2009-0885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Zuckerman S. Experimental study of blast injuries to the lungs. Lancet (1940) 236:219–24. doi:10.1016/S0140-6736(01)08726-8
46. Clemedson CJ. Shock wave transmission to the central nervous system. Acta Physiol Scand (1956) 37(2–3):204–14. doi:10.1111/j.1748-1716.1956.tb01356.x
47. Cohen H, Biskind G. Pathologic aspects of atmospheric blast injuries in man. Arch Pathol (1946) 42:12–34.
48. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet (1974) 2(7872):81–4. doi:10.1016/S0140-6736(74)91639-0
49. Jennett B, Teasdale G, Braakman R, Minderhoud J, Knill-Jones R. Predicting outcome in individual patients after severe head injury. Lancet (1976) 1(7968):1031–4. doi:10.1016/S0140-6736(76)92215-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Teasdale G, Knill-Jones R, van der Sande J. Observer variability in assessing impaired consciousness and coma. J Neurol Neurosurg Psychiatry (1978) 41(7):603–10. doi:10.1136/jnnp.41.7.603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Kay T, Adams R, Anderson T, Berrol S, Cicerone K, Dahlberg C, et al. Definition of mild traumatic brain injury. J Head Trauma Rehabil (1993) 8(3):86–7. doi:10.1097/00001199-199309000-00009
52. Centers for Disease Control and Prevention. Traumatic Brain Injury in the United States: A Report to Congress. (2003). Atlanta GA: CDCP. Available from: http://www.cdc.gov/traumaticbraininjury/tbi_report_to_congress.html
53. Carroll LJ, Cassidy JD, Holm L, Kraus J, Coronado VG. Methodological issues and research recommendations for mild traumatic brain injury: the WHO collaborating centre task force on mild traumatic brain injury. J Rehabil Med (2004) 36(43 Suppl):113–25. doi:10.1080/16501960410023877
54. Department of Defense/Department of Veterans Affairs. DoD/VA Common Definition of TBI. (2008). Available from: http://www.cdc.gov/nchs/data/icd/Sep08TBI.pdf
55. Williams DH, Levin HS, Eisenberg HM. Mild head injury classification. Neurosurgery (1990) 27(3):422–8. doi:10.1227/00006123-199009000-00014
56. Borgaro SR, Prigatano GP, Kwasnica C, Rexer JL. Cognitive and affective sequelae in complicated and uncomplicated mild traumatic brain injury. Brain Inj (2003) 17(3):189–98. doi:10.1080/0269905021000013183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Dagher JH, Richard-Denis A, Lamoureux J, de Guise E, Feyz M. Acute global outcome in patients with mild uncomplicated and complicated traumatic brain injury. Brain Inj (2013) 27(2):189–99. doi:10.3109/02699052.2012.729288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. de Guise E, Lepage JF, Tinawi S, LeBlanc J, Dagher J, Lamoureux J, et al. Comprehensive clinical picture of patients with complicated vs uncomplicated mild traumatic brain injury. Clin Neuropsychol (2010) 24(7):1113–30. doi:10.1080/13854046.2010.506199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Fay TB, Yeates KO, Taylor HG, Bangert B, Dietrich A, Nuss KE, et al. Cognitive reserve as a moderator of postconcussive symptoms in children with complicated and uncomplicated mild traumatic brain injury. J Int Neuropsychol Soc (2010) 16(1):94–105. doi:10.1017/S1355617709991007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Hessen E, Nestvold K. Indicators of complicated mild TBI predict MMPI-2 scores after 23 years. Brain Inj (2009) 23(3):234–42. doi:10.1080/02699050902748349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Iverson GL. Complicated vs uncomplicated mild traumatic brain injury: acute neuropsychological outcome. Brain Inj (2006) 20(13–14):1335–44. doi:10.1080/02699050601082156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Iverson GL, Lange RT, Waljas M, Liimatainen S, Dastidar P, Hartikainen KM, et al. Outcome from complicated versus uncomplicated mild traumatic brain injury. Rehabil Res Pract (2012) 2012:415740. doi:10.1155/2012/415740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kashluba S, Hanks RA, Casey JE, Millis SR. Neuropsychologic and functional outcome after complicated mild traumatic brain injury. Arch Phys Med Rehabil (2008) 89(5):904–11. doi:10.1016/j.apmr.2007.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Kennedy RE, Livingston L, Marwitz JH, Gueck S, Kreutzer JS, Sander AM. Complicated mild traumatic brain injury on the inpatient rehabilitation unit: a multicenter analysis. J Head Trauma Rehabil (2006) 21(3):260–71. doi:10.1097/00001199-200605000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Lange RT, Brickell TA, French LM, Merritt VC, Bhagwat A, Pancholi S, et al. Neuropsychological outcome from uncomplicated mild, complicated mild, and moderate traumatic brain injury in US military personnel. Arch Clin Neuropsychol (2012) 27(5):480–94. doi:10.1093/arclin/acs059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Lange RT, Iverson GL, Franzen MD. Neuropsychological functioning following complicated vs. uncomplicated mild traumatic brain injury. Brain Inj (2009) 23(2):83–91. doi:10.1080/02699050802635281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Strathmann FG, Schulte S, Goerl K, Petron DJ. Blood-based biomarkers for traumatic brain injury: evaluation of research approaches, available methods and potential utility from the clinician and clinical laboratory perspectives. Clin Biochem (2014) 47(10–11):876–88. doi:10.1016/j.clinbiochem.2014.01.028
68. Xiong KL, Zhu YS, Zhang WG. Diffusion tensor imaging and magnetic resonance spectroscopy in traumatic brain injury: a review of recent literature. Brain Imaging Behav (2014) 8(4):487–96. doi:10.1007/s11682-013-9288-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Harmon KG, Drezner JA, Gammons M, Guskiewicz KM, Halstead M, Herring SA, et al. American medical society for sports medicine position statement: concussion in sport. Br J Sports Med (2013) 47(1):15–26. doi:10.1136/bjsports-2012-091941
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Fear NT, Jones E, Groom M, Greenberg N, Hull L, Hodgetts TJ, et al. Symptoms of post-concussional syndrome are non-specifically related to mild traumatic brain injury in UK armed forces personnel on return from deployment in Iraq: an analysis of self-reported data. Psychol Med (2009) 39(8):1379–87. doi:10.1017/S0033291708004595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Terrio H, Brenner LA, Ivins BJ, Cho JM, Helmick K, Schwab K, et al. Traumatic brain injury screening: preliminary findings in a US Army Brigade Combat Team. J Head Trauma Rehabil (2009) 24(1):14–23. doi:10.1097/HTR.0b013e31819581d8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Verfaellie M, Lafleche G, Spiro A, Bousquet K. Neuropsychological outcomes in OEF/OIF veterans with self-report of blast exposure: associations with mental health, but not mTBI. Neuropsychology (2014) 28(3):337–46. doi:10.1037/neu0000027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Wilk JE, Thomas JL, McGurk DM, Riviere LA, Castro CA, Hoge CW. Mild traumatic brain injury (concussion) during combat: lack of association of blast mechanism with persistent postconcussive symptoms. J Head Trauma Rehabil (2010) 25(1):9–14. doi:10.1097/HTR.0b013e3181bd090f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Vasterling JJ, Verfaellie M, Sullivan KD. Mild traumatic brain injury and posttraumatic stress disorder in returning veterans: perspectives from cognitive neuroscience. Clin Psychol Rev (2009) 29(8):674–84. doi:10.1016/j.cpr.2009.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Southborough L. Report of the War Office Committee of Enquiry into “Shell-Shock.” London: His Majesty’s Stationery Office (1922).
76. Shepard B. ‘Pitiless psychology’: the role of prevention in British military psychiatry in the Second World War. Hist Psychiatry (1999) 10(40 Pt 4):491–524. doi:10.1177/0957154X9901004005
77. Gersons BP, Carlier IV. Post-traumatic stress disorder: the history of a recent concept. Br J Psychiatry (1992) 161:742–8. doi:10.1192/bjp.161.6.742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Bhattacharjee Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science (2008) 319(5862):406–8. doi:10.1126/science.319.5862.406
79. Joseph S, Masterson J. Posttraumatic stress disorder and traumatic brain injury: are they mutually exclusive? J Trauma Stress (1999) 12(3):437–53. doi:10.1023/A:1024762919372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Gil S, Caspi Y, Ben-Ari IZ, Koren D, Klein E. Does memory of a traumatic event increase the risk for posttraumatic stress disorder in patients with traumatic brain injury? A prospective study. Am J Psychiatry (2005) 162(5):963–9. doi:10.1176/appi.ajp.162.5.963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Harvey AG, Brewin CR, Jones C, Kopelman MD. Coexistence of posttraumatic stress disorder and traumatic brain injury: towards a resolution of the paradox. J Int Neuropsychol Soc (2003) 9(4):663–76. doi:10.1017/S1355617703940069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Hoge CW, Goldberg HM, Castro CA. Care of war veterans with mild traumatic brain injury – flawed perspectives. N Engl J Med (2009) 360(16):1588–91. doi:10.1056/NEJMp0810606
83. Kennedy JE, Leal FO, Lewis JD, Cullen MA, Amador RR. Posttraumatic stress symptoms in OIF/OEF service members with blast-related and non-blast-related mild TBI. NeuroRehabilitation (2010) 26(3):223–31. doi:10.3233/NRE-2010-0558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Nelson NW, Hoelzle JB, Doane BM, McGuire KA, Ferrier-Auerbach AG, Charlesworth MJ, et al. Neuropsychological outcomes of U.S. Veterans with report of remote blast-related concussion and current psychopathology. J Int Neuropsychol Soc (2012) 18(5):845–55. doi:10.1017/S1355617712000616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Verfaellie M, Lafleche G, Spiro A III, Tun C, Bousquet K. Chronic postconcussion symptoms and functional outcomes in OEF/OIF veterans with self-report of blast exposure. J Int Neuropsychol Soc (2013) 19(1):1–10. doi:10.1017/S1355617712000902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Schneiderman AI, Braver ER, Kang HK. Understanding sequelae of injury mechanisms and mild traumatic brain injury incurred during the conflicts in Iraq and Afghanistan: persistent postconcussive symptoms and posttraumatic stress disorder. Am J Epidemiol (2008) 167(12):1446–52. doi:10.1093/aje/kwn068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Belanger HG, Kretzmer T, Vanderploeg RD, French LM. Symptom complaints following combat-related traumatic brain injury: relationship to traumatic brain injury severity and posttraumatic stress disorder. J Int Neuropsychol Soc (2010) 16(1):194–9. doi:10.1017/S1355617709990841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Belanger HG, Kretzmer T, Yoash-Gantz R, Pickett T, Tupler LA. Cognitive sequelae of blast-related versus other mechanisms of brain trauma. J Int Neuropsychol Soc (2009) 15(1):1–8. doi:10.1017/S1355617708090036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Lange RT, Pancholi S, Brickell TA, Sakura S, Bhagwat A, Merritt V, et al. Neuropsychological outcome from blast versus non-blast: mild traumatic brain injury in U.S. military service members. J Int Neuropsychol Soc (2012) 18(3):595–605. doi:10.1017/S1355617712000239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lippa SM, Pastorek NJ, Benge JF, Thornton GM. Postconcussive symptoms after blast and nonblast-related mild traumatic brain injuries in Afghanistan and Iraq war veterans. J Int Neuropsychol Soc (2010) 16(5):856–66. doi:10.1017/S1355617710000743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Franke LM, Czarnota JN, Ketchum JM, Walker WC. Factor analysis of persistent postconcussive symptoms within a military sample with blast exposure. J Head Trauma Rehabil (2014). doi:10.1097/HTR.0000000000000042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Neipert L, Pastorek NJ, Troyanskaya M, Scheibel RS, Petersen NJ, Levin HS. Effect of clinical characteristics on cognitive performance in service members and veterans with histories of blast-related mild traumatic brain injury. Brain Inj (2014) 28(13–14):1667–74. doi:10.3109/02699052.2014.947623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Bazarian JJ, Donnelly K, Peterson DR, Warner GC, Zhu T, Zhong J. The relation between posttraumatic stress disorder and mild traumatic brain injury acquired during operations enduring freedom and Iraqi freedom. J Head Trauma Rehabil (2013) 28(1):1–12. doi:10.1097/HTR.0b013e318256d3d3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Kontos AP, Kotwal RS, Elbin RJ, Lutz RH, Forsten RD, Benson PJ, et al. Residual effects of combat-related mild traumatic brain injury. J Neurotrauma (2013) 30(8):680–6. doi:10.1089/neu.2012.2506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Mora AG, Ritenour AE, Wade CE, Holcomb JB, Blackbourne LH, Gaylord KM. Posttraumatic stress disorder in combat casualties with burns sustaining primary blast and concussive injuries. J Trauma (2009) 66(4 Suppl):S178–85. doi:10.1097/TA.0b013e31819ce2d6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Walilko T, North C, Young LA, Lux WE, Warden DL, Jaffee MS, et al. Head injury as a PTSD predictor among Oklahoma city bombing survivors. J Trauma (2009) 67(6):1311–9. doi:10.1097/TA.0b013e31819adc36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Walker WC, McDonald SD, Ketchum JM, Nichols M, Cifu DX. Identification of transient altered consciousness induced by military-related blast exposure and its relation to postconcussion symptoms. J Head Trauma Rehabil (2013) 28(1):68–76. doi:10.1097/HTR.0b013e318255dfd0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ruff RL, Riechers RG II, Wang XF, Piero T, Ruff SS. A case-control study examining whether neurological deficits and PTSD in combat veterans are related to episodes of mild TBI. BMJ Open (2012) 2(2):e000312. doi:10.1136/bmjopen-2011-000312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Macera CA, Aralis HJ, Macgregor AJ, Rauh MJ, Galarneau MR. Postdeployment symptom changes and traumatic brain injury and/or posttraumatic stress disorder in men. J Rehabil Res Dev (2012) 49(8):1197–208. doi:10.1682/JRRD.2011.07.0131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Reid MW, Miller KJ, Lange RT, Cooper DB, Tate DF, Bailie J, et al. A multisite study of the relationships between blast exposures and symptom reporting in a post-deployment active duty military population with mild traumatic brain injury. J Neurotrauma (2014) 31(23):1899–906. doi:10.1089/neu.2014.3455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Cernak I. Blast-induced neurotrauma models and their requirements. Front Neurol (2014) 5:128. doi:10.3389/fneur.2014.00128
102. Ho J, Kleiven S. Can sulci protect the brain from traumatic injury? J Biomech (2009) 42(13):2074–80. doi:10.1016/j.jbiomech.2009.06.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Panzer MB, Bass CR, Rafaels KA, Shridharani J, Capehart BP. Primary blast survival and injury risk assessment for repeated blast exposures. J Trauma Acute Care Surg (2012) 72(2):454–66. doi:10.1097/TA.0b013e31821e8270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Sundaramurthy A, Alai A, Ganpule S, Holmberg A, Plougonven E, Chandra N. Blast-induced biomechanical loading of the rat: an experimental and anatomically accurate computational blast injury model. J Neurotrauma (2012) 29(13):2352–64. doi:10.1089/neu.2012.2413
105. Panzer MB, Wood GW, Bass CR. Scaling in neurotrauma: how do we apply animal experiments to people? Exp Neurol (2014) 261C:120–6. doi:10.1016/j.expneurol.2014.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Cernak I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front Neurol (2010) 1:151. doi:10.3389/fneur.2010.00151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Ahlers ST, Vasserman-Stokes E, Shaughness MC, Hall AA, Shear DA, Chavko M, et al. Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front Neurol (2012) 3:32. doi:10.3389/fneur.2012.00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Xie K, Kuang H, Tsien JZ. Mild blast events alter anxiety, memory, and neural activity patterns in the anterior cingulate cortex. PLoS One (2013) 8(5):e64907. doi:10.1371/journal.pone.0064907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Elder GA, Dorr NP, De Gasperi R, Gama Sosa MA, Shaughness MC, Maudlin-Jeronimo E, et al. Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J Neurotrauma (2012) 29(16):2564–75. doi:10.1089/neu.2012.2510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Gama Sosa MA, De Gasperi R, Paulino AJ, Pricop PE, Shaughness MC, Maudlin-Jeronimo E, et al. Blast overpressure induces shear-related injuries in the brain of rats exposed to a mild traumatic brain injury. Acta Neuropathol Commun (2013) 1(1):51. doi:10.1186/2051-5960-1-51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Gama Sosa MA, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun (2014) 2:67. doi:10.1186/2051-5960-2-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma (2009) 26(6):827–40. doi:10.1089/neu.2008.0748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Risling M, Plantman S, Angeria M, Rostami E, Bellander BM, Kirkegaard M, et al. Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage (2011) 54(Suppl 1):S89–97. doi:10.1016/j.neuroimage.2010.05.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Svetlov SI, Prima V, Kirk DR, Gutierrez H, Curley KC, Hayes RL, et al. Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J Trauma (2010) 69(4):795–804. doi:10.1097/TA.0b013e3181bbd885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Garman RH, Jenkins LW, Switzer RC III, Bauman RA, Tong LC, Swauger PV, et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma (2011) 28(6):947–59. doi:10.1089/neu.2010.1540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Budde MD, Shah A, McCrea M, Cullinan WE, Pintar FA, Stemper BD. Primary blast traumatic brain injury in the rat: relating diffusion tensor imaging and behavior. Front Neurol (2013) 4:154. doi:10.3389/fneur.2013.00154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Park E, Gottlieb JJ, Cheung B, Shek PN, Baker AJ. A model of low-level primary blast brain trauma results in cytoskeletal proteolysis and chronic functional impairment in the absence of lung barotrauma. J Neurotrauma (2011) 28(3):343–57. doi:10.1089/neu.2009.1050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Pun PB, Kan EM, Salim A, Li Z, Ng KC, Moochhala SM, et al. Low level primary blast injury in rodent brain. Front Neurol (2011) 2:19. doi:10.3389/fneur.2011.00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Saljo A, Bolouri H, Mayorga M, Svensson B, Hamberger A. Low-level blast raises intracranial pressure and impairs cognitive function in rats: prophylaxis with processed cereal feed. J Neurotrauma (2010) 27(2):383–9. doi:10.1089/neu.2009.1053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Saljo A, Mayorga M, Bolouri H, Svensson B, Hamberger A. Mechanisms and pathophysiology of the low-level blast brain injury in animal models. Neuroimage (2011) 54(Suppl 1):S83–8. doi:10.1016/j.neuroimage.2010.05.050
121. Baalman KL, Cotton RJ, Rasband SN, Rasband MN. Blast wave exposure impairs memory and decreases axon initial segment length. J Neurotrauma (2013) 30(9):741–51. doi:10.1089/neu.2012.2478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med (2012) 4(134):134ra60. doi:10.1126/scitranslmed.3003716
123. Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis (2013) 37(2):309–23. doi:10.3233/JAD-130182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT, Mahota TM, et al. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis (2011) 41(2):538–51. doi:10.1016/j.nbd.2010.10.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol (2011) 70(5):399–416. doi:10.1097/NEN.0b013e3182189f06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Heldt SA, Elberger AJ, Deng Y, Guley NH, Del Mar N, Rogers J, et al. A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front Neurol (2014) 5:2. doi:10.3389/fneur.2014.00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol (2011) 232(2):280–9. doi:10.1016/j.expneurol.2011.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, et al. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis (2013) 54:1–11. doi:10.1016/j.nbd.2013.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Woods AS, Colsch B, Jackson SN, Post J, Baldwin K, Roux A, et al. Gangliosides and ceramides change in a mouse model of blast induced traumatic brain injury. ACS Chem Neurosci (2013) 4(4):594–600. doi:10.1021/cn300216h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Chavko M, Watanabe T, Adeeb S, Lankasky J, Ahlers ST, McCarron RM. Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J Neurosci Methods (2011) 195(1):61–6. doi:10.1016/j.jneumeth.2010.11.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Chavko M, Koller WA, Prusaczyk WK, McCarron RM. Measurement of blast wave by a miniature fiber optic pressure transducer in the rat brain. J Neurosci Methods (2007) 159(2):277–81. doi:10.1016/j.jneumeth.2006.07.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Mott D, Young T, Schwer D. Blast Loading on the Head Under a Military Helmet: Effect of Face Shield and Mandible Protection. 52nd Aerospace Sciences Meeting. American Institute of Aeronautics and Astronautics (2014). Available from: http://dx.doi.org/10.2514/6.2014-0948
133. Saljo A, Arrhen F, Bolouri H, Mayorga M, Hamberger A. Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J Neurotrauma (2008) 25(12):1397–406. doi:10.1089/neu.2008.0602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Moochhala SM, Md S, Lu J, Teng CH, Greengrass C. Neuroprotective role of aminoguanidine in behavioral changes after blast injury. J Trauma (2004) 56(2):393–403. doi:10.1097/01.TA.0000066181.50879.7A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. De Gasperi R, Gama Sosa MA, Kim SH, Steele JW, Shaughness MC, Maudlin-Jeronimo E, et al. Acute blast injury reduces brain abeta in two rodent species. Front Neurol (2012) 3:177. doi:10.3389/fneur.2012.00177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Genovese RF, Simmons LP, Ahlers ST, Maudlin-Jeronimo E, Dave JR, Boutte AM. Effects of mild TBI from repeated blast overpressure on the expression and extinction of conditioned fear in rats. Neuroscience (2013) 254:120–9. doi:10.1016/j.neuroscience.2013.09.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res (2008) 167:151–69. doi:10.1016/S0079-6123(07)67011-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci (2012) 35:24–35. doi:10.1016/j.tins.2011.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Kwon SK, Kovesdi E, Gyorgy AB, Wingo D, Kamnaksh A, Walker J, et al. Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front Neurol (2011) 2:12. doi:10.3389/fneur.2011.00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Kamnaksh A, Kovesdi E, Kwon SK, Wingo D, Ahmed F, Grunberg NE, et al. Factors affecting blast traumatic brain injury. J Neurotrauma (2011) 28(10):2145–53. doi:10.1089/neu.2011.1983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Kovesdi E, Gyorgy AB, Kwon SK, Wingo DL, Kamnaksh A, Long JB, et al. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front Neurosci (2011) 5:42. doi:10.3389/fnins.2011.00042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Mao JC, Pace E, Pierozynski P, Kou Z, Shen Y, VandeVord P, et al. Blast-induced tinnitus and hearing loss in rats: behavioral and imaging assays. J Neurotrauma (2012) 29(2):430–44. doi:10.1089/neu.2011.1934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Meyer DL, Davies DR, Barr JL, Manzerra P, Forster GL. Mild traumatic brain injury in the rat alters neuronal number in the limbic system and increases conditioned fear and anxiety-like behaviors. Exp Neurol (2012) 235(2):574–87. doi:10.1016/j.expneurol.2012.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Reger ML, Poulos AM, Buen F, Giza CC, Hovda DA, Fanselow MS. Concussive brain injury enhances fear learning and excitatory processes in the amygdala. Biol Psychiatry (2012) 71(4):335–43. doi:10.1016/j.biopsych.2011.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, et al. Stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell (2005) 123(4):697–709. doi:10.1016/j.cell.2005.08.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Brocke B, Lesch KP, Armbruster D, Moser DA, Muller A, Strobel A, et al. Stathmin, a gene regulating neural plasticity, affects fear and anxiety processing in humans. Am J Med Genet B Neuropsychiatr Genet (2009) 153B(1):243–51. doi:10.1002/ajmg.b.30989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Ehlis AC, Bauernschmitt K, Dresler T, Hahn T, Herrmann MJ, Roser C, et al. Influence of a genetic variant of the neuronal growth associated protein Stathmin 1 on cognitive and affective control processes: an event-related potential study. Am J Med Genet B Neuropsychiatr Genet (2011) 156B(3):291–302. doi:10.1002/ajmg.b.31161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury: football, warfare, and long-term effects. N Engl J Med (2010) 363:1293–6. doi:10.1056/NEJMp1007051
149. Gandy S, Ikonomovic MD, Mitsis E, Elder G, Ahlers ST, Barth J, et al. Chronic traumatic encephalopathy: clinical-biomarker correlations and current concepts in pathogenesis. Mol Neurodegener (2014) 9(1):37. doi:10.1186/1750-1326-9-37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Gavett BE, Cantu RC, Shenton M, Lin AP, Nowinski CJ, McKee AC, et al. Clinical appraisal of chronic traumatic encephalopathy: current perspectives and future directions. Curr Opin Neurol (2011) 24(6):525–31. doi:10.1097/WCO.0b013e32834cd477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med (2011) 30(1):179–88,xi. doi:10.1016/j.csm.2010.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol (2009) 68(7):709–35. doi:10.1097/NEN.0b013e3181a9d503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol (2010) 69(9):918–29. doi:10.1097/NEN.0b013e3181ee7d85
154. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery (2005) 57(1):128–34. doi:10.1227/01.NEU.0000163407.92769.ED
155. Lange RT, Brickell TA, Ivins B, Vanderploeg RD, French LM. Variable, not always persistent, postconcussion symptoms after mild TBI in U.S. military service members: a five-year cross-sectional outcome study. J Neurotrauma (2013) 30(11):958–69. doi:10.1089/neu.2012.2743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus (2011) 31(5):E3. doi:10.3171/2011.9.FOCUS11178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement (2014) 10(3 Suppl):S242–53. doi:10.1016/j.jalz.2014.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol (2007) 208(2):185–92. doi:10.1016/j.expneurol.2007.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest (2005) 115(5):1121–9. doi:10.1172/JCI25100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci (2010) 11(5):361–70. doi:10.1038/nrn2808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol (2004) 190(1):192–203. doi:10.1016/j.expneurol.2004.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS, et al. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol (2007) 64(4):541–4. doi:10.1001/archneur.64.4.541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid beta peptide in traumatic axonal injury. J Neurotrauma (2002) 19(5):601–14. doi:10.1089/089771502753754073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One (2012) 7(4):e34504. doi:10.1371/journal.pone.0034504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med (2009) 15(4):377–9. doi:10.1038/nm.1940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Loane DJ, Washington PM, Vardanian L, Pocivavsek A, Hoe HS, Duff KE, et al. Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J Neurotrauma (2011) 28(2):225–36. doi:10.1089/neu.2010.1595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Yu F, Zhang Y, Chuang DM. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J Neurotrauma (2012) 29(13):2342–51. doi:10.1089/neu.2012.2449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Tian L, Guo R, Yue X, Lv Q, Ye X, Wang Z, et al. Intranasal administration of nerve growth factor ameliorate beta-amyloid deposition after traumatic brain injury in rats. Brain Res (2012) 1440:47–55. doi:10.1016/j.brainres.2011.12.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST. Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann Neurol (2009) 66(3):407–14. doi:10.1002/ana.21731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Tran HT, LaFerla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci (2011) 31(26):9513–25. doi:10.1523/JNEUROSCI.0858-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Smith DH, Nakamura M, McIntosh TK, Wang J, Rodriguez A, Chen XH, et al. Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol (1998) 153(3):1005–10. doi:10.1016/S0002-9440(10)65643-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Schwetye KE, Cirrito JR, Esparza TJ, Mac Donald CL, Holtzman DM, Brody DL. Traumatic brain injury reduces soluble extracellular amyloid-beta in mice: a methodologically novel combined microdialysis-controlled cortical impact study. Neurobiol Dis (2010) 40(3):555–64. doi:10.1016/j.nbd.2010.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Zohar O, Lavy R, Zi X, Nelson TJ, Hongpaisan J, Pick CG, et al. PKC activator therapeutic for mild traumatic brain injury in mice. Neurobiol Dis (2011) 41(2):329–37. doi:10.1016/j.nbd.2010.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, et al. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J Neural Transm (2004) 111(4):523–36. doi:10.1007/s00702-003-0095-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KS, Shohami E, et al. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia (2008) 56(5):552–67. doi:10.1002/glia.20638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol (2004) 165(2):357–71. doi:10.1016/S0002-9440(10)63303-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Murakami N, Yamaki T, Iwamoto Y, Sakakibara T, Kobori N, Fushiki S, et al. Experimental brain injury induces expression of amyloid precursor protein, which may be related to neuronal loss in the hippocampus. J Neurotrauma (1998) 15(11):993–1003. doi:10.1089/neu.1998.15.993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Van Den Heuvel C, Blumbergs P, Finnie J, Manavis J, Lewis S, Jones N, et al. Upregulation of amyloid precursor protein and its mRNA in an experimental model of paediatric head injury. J Clin Neurosci (2000) 7(2):140–5. doi:10.1054/jocn.1999.0168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Ciallella JR, Ikonomovic MD, Paljug WR, Wilbur YI, Dixon CE, Kochanek PM, et al. Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J Neurotrauma (2002) 19(12):1555–67. doi:10.1089/089771502762300229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Wang Y, Wei Y, Oguntayo S, Wilkins W, Arun P, Valiyaveettil M, et al. Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J Neurotrauma (2011) 28(10):2171–83. doi:10.1089/neu.2011.1990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett (1993) 160(2):139–44. doi:10.1016/0304-3940(93)90398-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Lewen A, Li GL, Nilsson P, Olsson Y, Hillered L. Traumatic brain injury in rat produces changes of beta-amyloid precursor protein immunoreactivity. Neuroreport (1995) 6(2):357–60. doi:10.1097/00001756-199501000-00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J Neurosci (1996) 16(3):1083–90.
184. Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol (1994) 87(1):55–62. doi:10.1007/BF00386254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res (2000) 871(2):288–302. doi:10.1016/S0006-8993(00)02485-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Kuehn R, Simard PF, Driscoll I, Keledjian K, Ivanova S, Tosun C, et al. Rodent model of direct cranial blast injury. J Neurotrauma (2011) 28(10):2155–69. doi:10.1089/neu.2010.1532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Arun P, Abu-Taleb R, Oguntayo S, Tanaka M, Wang Y, Valiyaveettil M, et al. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: role of compromised cell membrane integrity. Neurosci Lett (2013) 552:87–91. doi:10.1016/j.neulet.2013.07.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Vanderploeg RD, Belanger HG, Curtiss G. Mild traumatic brain injury and posttraumatic stress disorder and their associations with health symptoms. Arch Phys Med Rehabil (2009) 90(7):1084–93. doi:10.1016/j.apmr.2009.01.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Graner J, Oakes TR, French LM, Riedy G. Functional MRI in the investigation of blast-related traumatic brain injury. Front Neurol (2013) 4:16. doi:10.3389/fneur.2013.00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Warden DL, French LM, Shupenko L, Fargus J, Riedy G, Erickson ME, et al. Case report of a soldier with primary blast brain injury. Neuroimage (2009) 47(Suppl 2):T152–3. doi:10.1016/j.neuroimage.2009.01.060
191. Levin HS, Wilde E, Troyanskaya M, Petersen NJ, Scheibel R, Newsome M, et al. Diffusion tensor imaging of mild to moderate blast-related traumatic brain injury and its sequelae. J Neurotrauma (2010) 27(4):683–94. doi:10.1089/neu.2009.1073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Peskind ER, Petrie EC, Cross DJ, Pagulayan K, McCraw K, Hoff D, et al. Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war Veterans with persistent post-concussive symptoms. Neuroimage (2011) 54(Suppl 1):S76–82. doi:10.1016/j.neuroimage.2010.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Mac Donald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med (2011) 364(22):2091–100. doi:10.1056/NEJMoa1008069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Davenport ND, Lim KO, Armstrong MT, Sponheim SR. Diffuse and spatially variable white matter disruptions are associated with blast-related mild traumatic brain injury. Neuroimage (2012) 59(3):2017–24. doi:10.1016/j.neuroimage.2011.10.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Hayes JP, Morey RA, Tupler LA. A case of frontal neuropsychological and neuroimaging signs following multiple primary-blast exposure. Neurocase (2012) 18(3):258–69. doi:10.1080/13554794.2011.588181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Matthews SC, Spadoni AD, Lohr JB, Strigo IA, Simmons AN. Diffusion tensor imaging evidence of white matter disruption associated with loss versus alteration of consciousness in warfighters exposed to combat in operations enduring and Iraqi freedom. Psychiatry Res (2012) 204(2–3):149–54. doi:10.1016/j.pscychresns.2012.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Morey RA, Haswell CC, Selgrade ES, Massoglia D, Liu C, Weiner J, et al. Effects of chronic mild traumatic brain injury on white matter integrity in Iraq and Afghanistan war veterans. Hum Brain Mapp (2013) 34(11):2986–99. doi:10.1002/hbm.22117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Fischer BL, Parsons M, Durgerian S, Reece C, Mourany L, Lowe MJ, et al. Neural activation during response inhibition differentiates blast from mechanical causes of mild to moderate traumatic brain injury. J Neurotrauma (2014) 31(2):169–79. doi:10.1089/neu.2013.2877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Mac Donald C, Johnson A, Cooper D, Malone T, Sorrell J, Shimony J, et al. Cerebellar white matter abnormalities following primary blast injury in US military personnel. PLoS One (2013) 8(2):e55823. doi:10.1371/journal.pone.0055823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Vakhtin AA, Calhoun VD, Jung RE, Prestopnik JL, Taylor PA, Ford CC. Changes in intrinsic functional brain networks following blast-induced mild traumatic brain injury. Brain Inj (2013) 27(11):1304–10. doi:10.3109/02699052.2013.823561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Han K, Mac Donald CL, Johnson AM, Barnes Y, Wierzechowski L, Zonies D, et al. Disrupted modular organization of resting-state cortical functional connectivity in U.S. military personnel following concussive ‘mild’ blast-related traumatic brain injury. Neuroimage (2014) 84:76–96. doi:10.1016/j.neuroimage.2013.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Petrie EC, Cross DJ, Yarnykh VL, Richards T, Martin NM, Pagulayan K, et al. Neuroimaging, behavioral, and psychological sequelae of repetitive combined blast/impact mild traumatic brain injury in Iraq and Afghanistan war veterans. J Neurotrauma (2014) 31(5):425–36. doi:10.1089/neu.2013.2952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Taber KH, Hurley RA, Haswell CC, Rowland JA, Hurt SD, Lamar CD, et al. White matter compromise in veterans exposed to primary blast forces. J Head Trauma Rehabil (2014). doi:10.1097/HTR.0000000000000030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Hetherington HP, Hamid H, Kulas J, Ling G, Bandak F, de Lanerolle NC, et al. MRSI of the medial temporal lobe at 7 T in explosive blast mild traumatic brain injury. Magn Reson Med (2014) 71(4):1358–67. doi:10.1002/mrm.24814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Yeh PH, Wang B, Oakes TR, French LM, Pan H, Graner J, et al. Postconcussional disorder and PTSD symptoms of military-related traumatic brain injury associated with compromised neurocircuitry. Hum Brain Mapp (2014) 35(6):2652–73. doi:10.1002/hbm.22358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Stocker RP, Cieply MA, Paul B, Khan H, Henry L, Kontos AP, et al. Combat-related blast exposure and traumatic brain injury influence brain glucose metabolism during REM sleep in military veterans. Neuroimage (2014) 99C:207–14. doi:10.1016/j.neuroimage.2014.05.067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Strigo IA, Spadoni AD, Lohr J, Simmons AN. Too hard to control: compromised pain anticipation and modulation in mild traumatic brain injury. Transl Psychiatry (2014) 4:e340. doi:10.1038/tp.2013.116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Tate DF, York GE, Reid MW, Cooper DB, Jones L, Robin DA, et al. Preliminary findings of cortical thickness abnormalities in blast injured service members and their relationship to clinical findings. Brain Imaging Behav (2014) 8(1):102–9. doi:10.1007/s11682-013-9257-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Daniels JK, Lamke JP, Gaebler M, Walter H, Scheel M. White matter integrity and its relationship to PTSD and childhood trauma – a systematic review and meta-analysis. Depress Anxiety (2013) 30(3):207–16. doi:10.1002/da.22044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Mac Donald CL, Johnson AM, Wierzechowski L, Kassner E, Stewart T, Nelson EC, et al. Prospectively assessed clinical outcomes in concussive blast vs nonblast traumatic brain injury among evacuated US military personnel. JAMA Neurol (2014) 71(8):994–1002. doi:10.1001/jamaneurol.2014.1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Tate CM, Wang KK, Eonta S, Zhang Y, Carr W, Tortella FC, et al. Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study. J Neurotrauma (2013) 30(19):1620–30. doi:10.1089/neu.2012.2683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: animal models, blast, human studies, post-traumatic stress disorder, traumatic brain injury
Citation: Elder GA, Stone JR and Ahlers ST (2014) Effects of low-level blast exposure on the nervous system: is there really a controversy? Front. Neurol. 5:269. doi: 10.3389/fneur.2014.00269
Received: 12 June 2014; Accepted: 29 November 2014;
Published online: 19 December 2014.
Edited by:
Ibolja Cernak, University of Alberta, CanadaReviewed by:
David Ritzel, Dyn-FX Consulting Ltd, CanadaRalph George Depalma, Office of Research and Development, USA
Copyright: © 2014 Elder, Stone and Ahlers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory A. Elder, Neurology Service (3E16), James J. Peters Department of Veterans Affairs Medical Center, 130 West Kingsbridge Road, Bronx, NY 10468, USA e-mail:Z3JlZ29yeS5lbGRlckB2YS5nb3Y=