 Juan M. Espinosa-Sanchez
Juan M. Espinosa-Sanchez Jose A. Lopez-Escamez
Jose A. Lopez-Escamez- 1Otology and Neurotology Group CTS495, Human DNA Variability Department, GENYO Centre for Genomics and Oncological Research Pfizer – University of Granada – Junta de Andalucia, Granada, Spain
- 2Department of Otolaryngology, Hospital San Agustin, Linares, Spain
- 3Department of Otolaryngology, Hospital de Poniente, El Ejido, Spain
Vestibular migraine (VM) is a common disorder in which genetic, epigenetic, and environmental factors probably contribute to its development. The pathophysiology of VM is unknown; nevertheless in the last few years, several studies are contributing to understand the neurophysiological pathways involved in VM. The current hypotheses are mostly based on the knowledge of migraine itself. The evidence of trigeminal innervation of the labyrinth vessels and the localization of vasoactive neuropeptides in the perivascular afferent terminals of these trigeminal fibers support the involvement of the trigemino-vascular system. The neurogenic inflammation triggered by activation of the trigeminal-vestibulocochlear reflex, with the subsequent inner ear plasma protein extravasation and the release of inflammatory mediators, can contribute to a sustained activation and sensitization of the trigeminal primary afferent neurons explaining VM symptoms. The reciprocal connections between brainstem vestibular nuclei and the structures that modulate trigeminal nociceptive inputs (rostral ventromedial medulla, ventrolateral periaqueductal gray, locus coeruleus, and nucleus raphe magnus) are critical to understand the pathophysiology of VM. Although cortical spreading depression can affect cortical areas involved in processing vestibular information, functional neuroimaging techniques suggest a dysmodulation in the multimodal sensory integration and processing of vestibular and nociceptive information, resulting from a vestibulo-thalamo-cortical dysfunction, as the pathogenic mechanism underlying VM. The elevated prevalence of VM suggests that multiple functional variants may confer a genetic susceptibility leading to a dysregulation of excitatory–inhibitory balance in brain structures involved in the processing of sensory information, vestibular inputs, and pain. The interactions among several functional and structural neural networks could explain the pathogenic mechanisms of VM.
Vestibular migraine (VM) is the most common cause of spontaneous episodic vestibular syndrome with a lifetime prevalence of about 1% (1). VM occurs in patients with current or previous history of migraine and recurrent episodes of vestibular symptoms accompanied by migraine features. In 2001, Neuhauser et al. proposed early diagnostic criteria for VM (2), and they have proven a high reliability in longitudinal studies (3); these criteria included:
1. Episodic vestibular symptoms of at least moderate intensity.
2. Migraine according to the International Headache Society (IHS).
3. At least one of the following migrainous symptoms in at least two vertiginous attacks: migrainous headaches, photophobia, phonophobia, and visual or other auras.
4. Other causes ruled out by appropriate investigations.
Currently, the diagnosis is based on the criteria developed by the Bárány Society and the IHS (4). The major differences between Neuhauser and Barany Society-IHS criteria are that the new definition sets out at least five episodes of vestibular symptoms with a duration of 5 min – 72 h. Furthermore, current diagnostic criteria use the Bárány Society’s Classification of Vestibular Symptoms (5).
The dissemination of these criteria and the inclusion in the beta version of the 3rd edition of the ICHD has renewed the interest in VM (6–9).
Neuro-otological examination between episodes is usually normal, although spontaneous and positional nystagmus can be found during the attacks. Vestibular function tests reveal normal findings in almost half of the patients, even though unilateral vestibular hypofunction and oculo-motor disturbances are also common.
The differential diagnosis includes benign paroxysmal positional vertigo (BPPV), Meniere’s disease (MD), labyrinth transient ischemic attacks, superior canal dehiscence syndrome, and chronic subjective dizziness, being the distinction between MD and VM the major diagnostic challenge, particularly in the first years of the disease.
The pathophysiology of VM is unknown. Several hypotheses have been proposed to explain the concurrence of episodic vestibular symptoms and migraine. Vestibular symptoms may appear in any phase of a migraine attack with a variable duration. In some episodes, there are only migraine symptoms, while in others migraine and vestibular symptoms are observed. Interestingly, within a single family, some members may suffer from migraine and other relatives have all the symptoms of VM. Moreover, it is unclear the relation between VM, MD, and BPPV (10).
We summarize the current knowledge on the pathophysiology of VM with a particular focus on genetic studies and multisensory integration network. Firstly, we will describe the mechanisms of migraine itself, since current hypotheses are mostly based on its pathophysiology.
Pathophysiology of Migraine
Migraine is a complex disorder with a controversial pathophysiology. In the last few years, several investigations have contributed to elucidate the causal mechanisms. It is widely accepted that the trigemino-vascular system (TVS) is the anatomical substrate and its activation and sensitization causes headache. The electrophysiological phenomenon of cortical spreading depression (CSD) could be the neurophysiological correlate of migraine aura, although CSD has not been linked with aura in humans (11).
The TVS involves the nociceptive pseudounipolar neurons of the trigeminal ganglion (TG), which innervate the pia, the dura mater, and the cranial blood vessels, mainly through the ophthalmic division of the trigeminal nerve (12). These first-order neurons project centrally to the trigemino-cervical complex (TCC), including the caudal portion of the trigeminal spinal nucleus (trigeminal nucleus caudalis) and the C1 and C2 dorsal horns of the spinal cord. The projections of the second-order neurons of TCC ascend through the quintothalamic tract and connect to the contralateral ventroposteromedial (VPM) and posterior thalamic nuclei, which in turn project to the primary and secondary somatosensory cortex, the insular cortex, and the anterior cingulate cortex. Moreover, the TCC makes reciprocal connections to brainstem centers involved in the processing of nociceptive information. These centers include the rostral ventromedial medulla (RVM) – particularly the nucleus raphe magnus – the ventrolateral periaqueductal gray (vlPAG), and hypothalamic areas. In fact, the modulation of the TCC depends upon descending pathways from the cortex, and posterior hypothalamus and these brainstem nuclei (13).
Experimental studies have demonstrated that the depolarization of the peripheral terminals of meningeal nociceptors leads to the activation of the TVS with the release of vasoactive neuropeptides from their perivascular endings. These substances, mainly substance P (SP), calcitonin gene-related peptide (CGRP), and neurokinin A (NK-A), cause vasodilation and an increase in cerebral blood flow, with plasma protein extravasation and mast cell degranulation on the dura with the release of pro-inflammatory factors that finally result in an inflammatory reaction known as neurogenic inflammation (11).
Pro-inflammatory mediators and the released glutamate increase the excitability of the first-order neurons in the TG (peripheral sensitization) producing throbbing pain. These mediators also sensitize second-order and third-order trigemino-vascular neurons (central sensitization) causing allodynia (14–16). Functional brain imaging studies show a participation of vlPAG, RVM, and nucleus cuneiformis in the maintenance of central sensitization.
Cortical spreading depression consists of a transient but massive neural depolarization, which generates a wave that slowly spread across cortex from occipital area, and it is followed by a prolonged suppression of cortical bioelectrical activity. CSD causes disruptions of transmembrane ionic gradients, increasing the intracellular concentrations of Ca2+ and the extracellular concentrations of H+, K+, glutamate, arachidonic acid, and nitric oxide (NO) in the synaptic cleft. These molecules are able to activate perivascular and meningeal nociceptors of TVS (17) and also central trigemino-vascular neurons in the spinal trigeminal nucleus (18).
The initiation of CSD is ignited by a local increase in the extracellular K+ concentration above a critical value and the release of glutamate from apical dendrites of cortical pyramidal cells and activation of NMDA receptors, probably due to the opening of voltage-gated Ca2+ channels (19). Recently, it has been experimentally demonstrated that CSD induces opening of neuronal pannexins channels and the release of pro-inflammatory substances that may activate the meningeal TVS (20).
Moreover, the concept of CSD as a migraine trigger is controversial since aura is not present in most patients with migraine. Some authors have postulated a silent aura mechanism, but the initial mechanism that causes activation of TVS is not known. A recent hypothesis proposes that migraine is a pathological brain state (21), in which multiple partially overlapping brain networks are involved. So, migraine headache arises from a dysfunction within diencephalic structures and brainstem nuclei that modulate trigeminal nociceptive inputs: RVM, vlPAG, posterior hypothalamus, and VPM thalamic nucleus (13). In this alternative hypothesis, direct activation of the TVS would occur instead of through activation of peripheral nociceptors and sequential sensitization of the first-, second-, and the third-order trigemino-vascular neurons.
Ischemic Events and VM
The hypotheses to explain the pathophysiology of VM have evolved over time from the vascular to the neurogenic theory. Reversible vasospasm of the internal auditory artery or its branches was one of the first proposed explanations (22). This mechanism could be responsible for the sudden onset of vestibular and/or auditory symptoms, spontaneous nystagmus or even could account for the link between VM and MD or BPPV. Moreover, a population-based study indicates that migraine could be associated with sudden sensorineural hearing loss (23).
Central spontaneous nystagmus and ocular-motor abnormalities cannot be elucidated by internal auditory artery ischemia, but it may be the manifestation of infarction in the anterior inferior cerebellar artery territory (AICA) (24). Meta-analyses indicate that migraine and stroke are not only comorbid conditions (25), but rather migraine is probably an independent cerebrovascular risk factor, especially in young people with migraine with aura (26, 27).
Several studies have reported reduced or absent vestibular evoked myogenic potential (VEMP) responses in patients with VM indicating a dysfunction of the vestibulo-collic reflex (28–31). These findings could be related with the hypoperfusion of the inner ear affecting the otolith organs, as well as ischemic lesions of the descending otolith pathways in the brainstem.
Cortical Spreading Depression and VM
Although the vestibular symptoms in VM should not be consider as a type of sensory aura, it has been postulated that they could be explained by the mechanism of aura, i.e., CSD (32). The wave of CSD could reach the so-called vestibular cortex or even the brainstem vestibular nuclei causing vestibular symptoms. However, the duration of VM episodes (from seconds to days) does not fulfill the characteristics of an aura, and neither explains the unilateral canal paresis found in some patients. Aura can occur at any time during the attack, and aura without headache can also be observed. Furthermore, in the case of migraine with brainstem aura, previously called basilar-type, there are other brainstem symptoms such dysarthria, tinnitus, hypacusis, diplopia, ataxia, or decreased level of consciousness.
Involvement of TVS in VM
The inner ear receives innervation from the ophthalmic branch of the TG through the basilar artery and the AICA (33). The TG also innervates the cochlear nucleus and the superior olivary complex (34). In addition, experimental studies have shown that chemical and electrical stimulation of the TG cause a significant increase in the inner ear blood flow, and changes in vascular permeability with plasma protein extravasation into the inner ear (35–37). These findings open the doors to the participation of the TVS in the pathophysiology of VM. In this sense, painful trigeminal electric stimulation in patients with migraine without aura prompts a spontaneous nystagmus of probable peripheral origin, or modifies pre-existing spontaneous nystagmus, mostly increasing it (38). It has been identified the presence of vasoactive neuropeptides (e.g., SP, CGRP) in the trigeminal sensory fibers innervating the inner ear and in the vestibular nuclei (36, 39).
Genetic Factors
Recently, several genome-wide association studies (GWAS) have identified common genetic variants at several loci significantly associated with migraine susceptibility (40, 41). Three markers have shown a consistent association in independent cohorts of patients with migraine: rs2651899 in the PRDM16 gene, rs10166942 in theTRPM8 gene, and rs11172113 in the LRP1 gene. The common variant in the PRDM16 gene has been associated with migraine in European, Chinese, and North Indian populations, but the role of this gene in migraine is unknown.
Mutations in CACNA1A gene, which encodes the central pore-forming subunit of the voltage-gated CaV2.1 (P/Q-type) calcium channels, cause at least three neurological calcium channelopathy syndromes: episodic ataxia type 2, familial hemiplegic migraine type 1, and spinocerebellar ataxia type 6 (42). Accumulating evidence suggests that CaV2.1 channels could be involved in migraine and VM pathogenesis (6, 43). Thus, migraine may be conceived as a channelopathy in which CSD would be the result of different mutations that would increase the susceptibility to CSD by affecting several ionic channels, mostly involved in glutamate homeostasis, such that the common final result would be an increase of glutamate and K+ extracellular concentrations in the synaptic cleft, which could contribute to TVS activation, central sensitization, and CSD initiation (44).
Familial occurrence of VM has been reported suggesting a genetic component. Within the same family, there can be multiple-affected relatives, some individuals may have migraine and others VM or benign paroxysmal vertigo of childhood, suggesting a phenotypic heterogeneity. An autosomal dominant pattern with moderate to high penetrance is frequently appreciated (45, 46). However, Sanger sequencing did not identified mutations in patients with VM in genes causing familial hemiplegic migraine, such as CACNA1A, ATP1A2, SCN1A, or episodic ataxia type 5 (CACNB4) (45, 46).
Despite no causative mutation has been found in candidate gene studies, several loci have been identified. Lee et al. (47) studied 20 multicase families diagnosed with benign recurrent vertigo (BRV), demonstrating linkage to 22q12. BRV includes patients with recurrent episodes of spontaneous vertigo in the absence of cochlear signs (48). Nowadays, most of the patients previously diagnosed with BRV could be classified as VM, since 79% of the individuals diagnosed with BRV also fulfill criteria of migraine (47).
Subsequently, the same authors studied a large pedigree with VM (49). These investigators did not find a region of the genome shared by all affected family members, indicating a polygenic inheritance. Nevertheless, a region on 11q was shared by most affected females, suggesting that may contain a susceptibility allele that is penetrant predominantly in women.
Bahmad et al. studied a family with 10 members affected by VM demonstrating an autosomal dominant trait. Genome-wide linkage analysis and subsequent fine mapping revealed that the disease gene is located between markers rs244895 and D5S2073 in chromosome 5q35 (50).
According to prevalence of VM, it is unlikely to be a monogenic disease as familiar hemiplegic migraine or episodic ataxias, but it would be rather polygenic as MD. The involved functional genetic variants of multiple ionic channels and receptors, both in vestibular pathways and in the pain network, may determine the observed clinical heterogeneity in the VM phenotype. This would explain the concurrence of peripheral and central vestibular findings, and the migrainous features.
Multisensory Integration and Vestibulo-Thalamic-Cortical Processing
The pathophysiology of VM has been explained by interactions between nociceptive and vestibular pathways (51). The reciprocal connections between the vestibular nuclei and other brainstem structures, such as the parabrachial nucleus, the raphe nuclei, and the locus coeruleus, may contribute to modulate the sensitivity of trigemino-vascular reflex and pain pathways (52–54). Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have revealed an activation of the dorsal brainstem during migraine attacks, significantly at the dorsal raphe nucleus and the vlPAG. In addition, reciprocal connections between the trigeminal nucleus caudalis and the vestibular nuclei could explain vestibular symptoms in migraneurs.
Some studies showed that patients with VM may have enhanced perceptual sensitivity for head motions that dynamically modulate canal and otolith inputs together (55), so it has been suggested that canal–otolith integration may be abnormal in these patients due to an altered signal processing in the caudal cerebellar vermis (56).
Electrophysiological and neuroanatomic studies in animals, along with functional neuroimaging techniques in humans by PET and fMRI, have identified the vestibular thalamus and the vestibular cortex as an essential part of the central vestibular system (57). Multiple thalamic nuclei are involved in vestibular processing, particularly the ventralposterolateral (VPL) and VPM nuclei. The thalamus is a major sensory relay station in the vestibular pathways, and it participates in the multisensory integration and processing of vestibular, visual, and proprioceptive inputs, so thalamic function could be a key event in multimodal sensorial sensitization. Thalamic activation has been demonstrated during a migraine attack using fMRI. VPL has been involved in controlling body orientation in space and the VPM receives trigeminothalamic input. We speculate that migraine attacks may transiently sensitize VPL and VPM nuclei and enhance the sensory perception, including vestibular information. This hypothesis would also explain the susceptibility to motion sickness described in migraine and VM patients (58, 59).
Although a specific vestibular cortex has not been clearly identified in humans, several cortical areas are involved in the processing of vestibular information, particularly the insula, the parietal operculum, and the temporo-parietal junction, as well as the posterior parietal cortex, the cingulate cortex, the somatosensory cortex, and the lateral and medial frontal cortices (60–63). These regions are multisensory areas where several sensory modalities converge. Equally, a nociceptive-specific area neither has been found, but it is worth noting that the posterior insular–opercular region has been postulated as the primary nociceptive cortex (64) and it also may provide a site for interaction between vestibular and nociceptive processing (61, 65). The vestibular cortical projections strongly overlap with the somatosensory cortical projections, probably due to intrainsular connections, giving rise to vestibular–somatosensory interactions as proven by both caloric and galvanic vestibular stimulation (66, 67). So, it has been suggested that vestibular input could influence processing in other sensory modalities.
This thalamo-cortical involvement has also been demonstrated in PET studies in VM (68). Subtraction images comparing ictal with interictal images showed an increased metabolism in the bilateral cerebellum, frontal cortices, temporal cortex, posterior insula, and thalami. It was also observed hypometabolism in the bilateral occipitotemporal and posterior parietal cortices. The cerebellar hypermetabolism is interpreted by an adaptive mechanism to suppress the hyperactive vestibular system, whereas the occipitotemporal deactivation is explained by reciprocal inhibition between visual and vestibular systems (69). This study indicates activation of the vestibulo-thalamo-cortical pathway in VM.
Neuroimaging in migraineurs with blood oxygenation level-dependent (BOLD) fMRI, have revealed the participation of several cortical areas others than primary visual cortex. These include anterior cingulate, prefrontal, and orbitofrontal cortices. Using BOLD fMRI, caloric vestibular stimulation elicited a significant activation in insular cortex, parietal cortex, thalamus, brainstem (including PAG), and cerebellum, and a significantly decreased response in anterior cingulate cortex. This pattern is observed in both healthy volunteers and patients with migraine without aura or VM during an interictal phase (70). Nevertheless, patients with VM showed also an increased mediodorsal thalamic activation ipsilateral to the vestibular stimulation as compared with both, healthy controls and patients with migraine without aura. Moreover, the degree of thalamic activation was correlated with frequency of migraine attacks in patients with VM.
Recently, patients with VM have been studied using MRI-based voxel-based morphometry to analyze gray matter (GM) volume differences between VM patients and healthy controls (71). VM patients showed a decrease of GM volume bilaterally in the inferior temporal gyrus, the cingulate cortex, and the posterior insula. GM decrease was also observed in the left side of superior temporal gyrus, middle temporal gyrus, supramarginal gyrus, and superior parietal lobules, and on the right sides of the dorsolateral prefrontal cortex and the inferior occipital gyrus. These findings are consistent with above-mentioned studies that suggested that these areas are involved in cortical processing of vestibular and nociceptive information.
An intrinsic hypersensitivity to different sensory stimuli has been described in migraineurs. Exposure to a specific stimulus results not only in hypersensitivity to that stimulus, but it may also result in further enhancement of hypersensitivity to other stimuli. It has been appreciated an altered central processing of sensory stimuli in patients with migraine including trigeminal inputs (72), in such a way that some authors interpret migraine as a disorder of multisensory integration (73). As we have seen, multisensory integration has been described in all vestibular relays, including vestibular nuclei, thalamus, and cerebral cortex. These multimodal sensory areas are responsible for the integration and processing of somatosensory, visual, and vestibular information. An intrinsic enhanced vestibular sensory perception and these cross-modal interactions at (vestibular) brainstem-thalamo-cortical level could explain, at least partly, the link between migraine and some vestibular disorders including VM.
We hypothesized that VM responds to an abnormal brain sensitization leading to a dysmodulation of multimodal sensory integration and processing at vestibulo-thalamo-cortical level (Figure 1). At the molecular level, this may result from functional variants affecting several ionic channels and receptors, and that can confer susceptibility to VM. Multiple brain networks are probably involved not only in the pain matrix and the vestibular pathways, but also in other structures such as the limbic system.
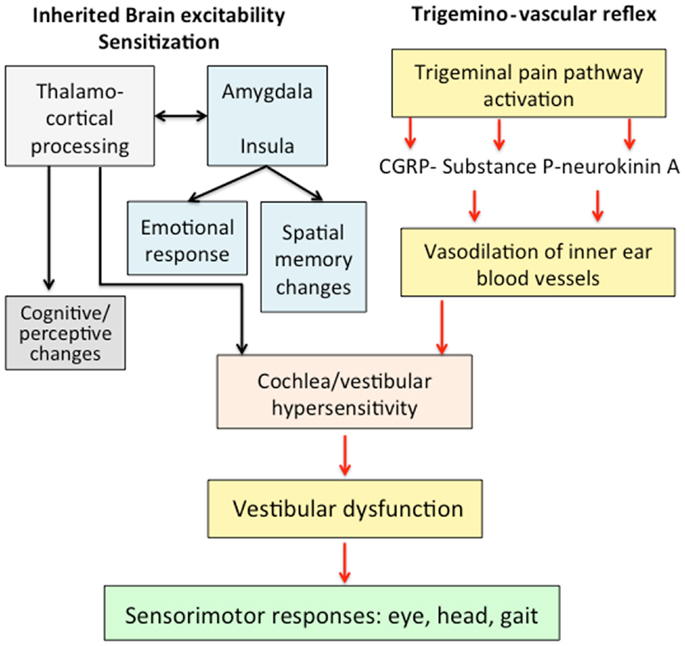
Figure 1. Mechanisms involved in the pathophysiology of vestibular migraine. An abnormal brain sensitization leading to a dysmodulation of multimodal sensory integration in thalamo-cortical processing could interact with the trigemino-vascular reflex. The abnormal processing of vestibular and nociceptive information could determine a transient vestibular dysfunction associated with migraine features.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Neuhauser HK, Radtke A, von Brevern M, Feldmann M, Lezius F, Ziese T, et al. Migrainous vertigo: prevalence and impact on quality of life. Neurology (2006) 67:1028–33. doi: 10.1212/01.wnl.0000237539.09942.06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Neuhauser H, Leopold M, von Brevern M, Arnold G, Lempert T. The interrelations of migraine, vertigo, and migrainous vertigo. Neurology (2001) 27(56):436–41. doi:10.1212/WNL.56.4.436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Radtke A, Neuhauser H, von Brevern M, Hottenrott T, Lempert T. Vestibular migraine – validity of clinical diagnostic criteria. Cephalalgia (2011) 31:906–13. doi:10.1177/0333102411405228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Lempert T, Olesen J, Furman J, Waterston J, Seemungal B, Carey J, et al. Vestibular migraine: diagnostic criteria. J Vestib Res (2012) 22:167–72. doi:10.3233/VES-2012-0453
5. Bisdorff A, Von Brevern M, Lempert T, Newman-Toker DE. Classification of vestibular symptoms: towards an international classification of vestibular disorders. J Vestib Res (2009) 19:1–13. doi:10.3233/VES-2009-0343
6. Furman JM, Marcus DA, Balaban CD. Vestibular migraine: clinical aspects and pathophysiology. Lancet Neurol (2013) 12:706–15. doi:10.1016/S1474-4422(13)70107-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Lempert T. Vestibular migraine. Semin Neurol (2013) 33:212–8. doi:10.1055/s-0033-1354596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Stolte B, Holle D, Naegel S, Diener HC, Obermann M. Vestibular migraine. Cephalalgia (2014). doi:10.1177/0333102414535113
9. Bisdorff A. Migraine and dizziness. Curr Opin Neurol (2014) 27:105–10. doi:10.1097/WCO.0000000000000061
10. Espinosa-Sanchez JM, Martin-Sierra C, Lopez-Escamez JA. Meniere’s syndrome and migraine. In: Colombo B, Teggi R, editors. Vestibular Migraine and Related Syndromes. Heidelberg: Springer International Publishing (2014). p. 129–41.
11. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol (2013) 75:365–91. doi:10.1146/annurev-physiol-030212-183717
12. Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience (2009) 161(2):327–41. doi:10.1016/j.neuroscience.2009.03.019
13. Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci (2011) 12:570–84. doi:10.1038/nrn3057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol (2009) 8:679–90. doi:10.1016/S1474-4422(09)70090-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Burstein R, Jakubowski M, Rauch SD. The science of migraine. J Vestib Res (2011) 21:305–14. doi:10.3233/VES-2012-0433
16. Bernstein C, Burstein R. Sensitization of the trigeminovascular pathway: perspective and implications to migraine pathophysiology. J Clin Neurol (2012) 8:89–99. doi:10.3988/jcn.2012.8.2.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci (2010) 30:8807–14. doi:10.1523/JNEUROSCI.0511-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol (2011) 69:855–65. doi:10.1002/ana.22329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci (2014) 15:379–93. doi:10.1038/nrn3770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science (2013) 339:1092–5. doi:10.1126/science.1231897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Charles A. Migraine: a brain state. Curr Opin Neurol (2013) 26:235–9. doi:10.1097/WCO.0b013e32836085f4
23. Chu CH, Liu CJ, Fuh JL, Shiao AS, Chen TJ, Wang SJ. Migraine is a risk factor for sudden sensorineural hearing loss: a nationwide population-based study. Cephalalgia (2013) 33:80–6. doi:10.1177/0333102412468671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Lee H. Isolated vascular vertigo. J Stroke (2014) 16:124–30. doi:10.5853/jos.2014.16.3.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wang SJ, Chen PK, Fuh JL. Comorbidities of migraine. Front Neurol (2010) 1:16. doi:10.3389/fneur.2010.00016
26. Kruit MC, van Buchem MA, Launer LJ, Terwindt GM, Ferrari MD. Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: the population-based MRI CAMERA study. Cephalalgia (2010) 30:129–36. doi:10.1111/j.1468-2982.2009.01904.x
27. Guidetti D, Rota E, Morelli N, Immovilli P. Migraine and stroke: “vascular” comorbidity. Front Neurol (2014) 8(5):193. doi:10.3389/fneur.2014.00193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Baier B, Stieber N, Dieterich M. Vestibular-evoked myogenic potentials in vestibular migraine. J Neurol (2009) 256:1447–54. doi:10.1007/s00415-009-5132-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Boldingh MI, Ljøstad U, Mygland A, Monstad P. Vestibular sensitivity in vestibular migraine: VEMPs and motion sickness susceptibility. Cephalalgia (2011) 31:1211–9. doi:10.1177/0333102411409074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Hong SM, Kim SK, Park CH, Lee JH. Vestibular-evoked myogenic potentials in migrainous vertigo. Otolaryngol Head Neck Surg (2011) 144:284–7. doi:10.1177/0194599810391755
31. Zuniga MG, Janky KL, Schubert MC, Carey JP. Can vestibular-evoked myogenic potentials help differentiate Ménière disease from vestibular migraine? Otolaryngol Head Neck Surg (2012) 146:788–96. doi:10.1177/0194599811434073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Dieterich M, Brandt T. Episodic vertigo related to migraine (90 cases): vestibular migraine? J Neurol (1999) 246:883–92. doi:10.1007/s004150050478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Vass Z, Shore SE, Nuttall AL, Miller JM. Direct evidence of trigeminal innervation of the cochlear blood vessels. Neuroscience (1998) 84:559–67. doi:10.1016/S0306-4522(97)00503-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Shore SE, Vass Z, Wys NL, Altschuler RA. Trigeminal ganglion innervates the auditory brainstem. J Comp Neurol (2000) 419:271–85. doi:10.1002/(SICI)1096-9861(20000410)419:3<271::AID-CNE1>3.0.CO;2-M
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Vass Z, Steyger PS, Hordichok AJ, Trune DR, Jancsó G, Nuttall AL. Capsaicin stimulation of the cochlea and electric stimulation of the trigeminal ganglion mediate vascular permeability in cochlear and vertebro-basilar arteries: a potential cause of inner ear dysfunction in headache. Neuroscience (2001) 103:189–201. doi:10.1016/S0306-4522(00)00521-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Vass Z, Dai CF, Steyger PS, Jancsó G, Trune DR, Nuttall AL. Co-localization of the vanilloid capsaicin receptor and substance P in sensory nerve fibers innervating cochlear and vertebro-basilar arteries. Neuroscience (2004) 124:919–27. doi:10.1016/j.neuroscience.2003.12.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Koo JW, Balaban CD. Serotonin-induced plasma extravasation in the murine inner ear: possible mechanism of migraine-associated inner ear dysfunction. Cephalalgia (2006) 26:1310–9. doi:10.1111/j.1468-2982.2006.01208.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Marano E, Marcelli V, Di Stasio E, Bonuso S, Vacca G, Manganelli F, et al. Trigeminal stimulation elicits a peripheral vestibular imbalance in migraine patients. Headache (2005) 45:325–31. doi:10.1111/j.1526-4610.2005.05069.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ahn SK, Khalmuratova R, Jeon SY, Kim JP, Park JJ, Hur DG, et al. Colocalization of 5-HT1F receptor and calcitonin gene-related peptide in rat vestibular nuclei. Neurosci Lett (2009) 465:151–6. doi:10.1016/j.neulet.2009.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Gasparini CF, Sutherland HG, Griffiths LR. Studies on the pathophysiology and genetic basis of migraine. Curr Genomics (2013) 14(5):300–15. doi:10.2174/13892029113149990007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Anttila V, Winsvold BS, Gormley P, Kurth T, Bettella F, McMahon G, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet (2013) 45:912–7. doi:10.1038/ng.2676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Requena T, Espinosa-Sanchez JM, Lopez-Escamez JA. Genetics of dizziness: cerebellar and vestibular disorders. Curr Opin Neurol (2014) 27:98–104. doi:10.1097/WCO.0000000000000053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Pietrobon D. Calcium channels and migraine. Biochim Biophys Acta (2013) 1828:1655–65. doi:10.1016/j.bbamem.2012.11.012
44. Gasparini CF, Griffiths LR. The biology of the glutamatergic system and potential role in migraine. Int J Biomed Sci (2013) 9:1–8.
45. Kim JS, Yue Q, Jen JC, Nelson SF, Baloh RW. Familial migraine with vertigo: no mutations found in CACNA1A. Am J Med Genet (1998) 79:148–51. doi:10.1002/(SICI)1096-8628(19980901)79:2<148::AID-AJMG11>3.0.CO;2-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. von Brevern M, Ta N, Shankar A, Wiste A, Siegel A, Radtke A, et al. Migrainous vertigo: mutation analysis of the candidate genes CACNA1A, ATP1A2, SCN1A, and CACNB4. Headache (2006) 46:1136–41. doi:10.1111/j.1526-4610.2006.00504.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lee H, Jen JC, Wang H, Chen Z, Mamsa H, Sabatti C, et al. A genome-wide linkage scan of familial benign recurrent vertigo: linkage to 22q12 with evidence of heterogeneity. Hum Mol Genet (2006) 15:251–8. doi:10.1093/hmg/ddi441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Slater R. Benign recurrent vertigo. J Neurol Neurosurg Psychiatry (1979) 42:363–7. doi:10.1136/jnnp.42.4.363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lee H, Jen JC, Cha YH, Nelson SF, Baloh RW. Phenotypic and genetic analysis of a large family with migraine-associated vertigo. Headache (2008) 48:1460–7. doi:10.1111/j.1526-4610.2007.01002.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Bahmad F Jr, DePalma SR, Merchant SN, Bezerra RL, Oliveira CA, Seidman CE, et al. Locus for familial migrainous vertigo disease maps to chromosome 5q35. Ann Otol Rhinol Laryngol (2009) 118:670–6. doi:10.1177/000348940911800912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Balaban CD. Migraine, vertigo and migrainous vertigo: links between vestibular and pain mechanisms. J Vestib Res (2011) 21:315–21. doi:10.3233/VES-2011-0428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Halberstadt AL, Balaban CD. Organization of projections from the raphe nuclei to the vestibular nuclei in rats. Neuroscience (2003) 120:573–94. doi:10.1016/S0306-4522(02)00952-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Halberstadt AL, Balaban CD. Anterograde tracing of projections from the dorsal raphe nucleus to the vestibular nuclei. Neuroscience (2006) 143:641–54. doi:10.1016/j.neuroscience.2006.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Balaban CD, Jacob RG, Furman JM. Neurologic bases for comorbidity of balance disorders, anxiety disorders and migraine: neurotherapeutic implications. Expert Rev Neurother (2011) 11:379–94. doi:10.1586/ern.11.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Lewis RF, Priesol AJ, Nicoucar K, Lim K, Merfeld DM. Dynamic tilt thresholds are reduced in vestibular migraine. J Vestib Res (2011) 21(6):323–30. doi:10.3233/VES-2011-0422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. King S, Wang J, Priesol A, Lewis R. Central integration of canal and otolith signals is abnormal in vestibular migraine. Front Neurol (2014) 5:233. doi:10.3389/fneur.2014.00233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Lopez C, Blanke O. The thalamocortical vestibular system in animals and humans. Brain Res Rev (2011) 67:119–46. doi:10.1016/j.brainresrev.2010.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Murdin L, Chamberlain F, Cheema S, Arshad Q, Gresty MA, Golding JF, et al. Motion sickness in migraine and vestibular disorders. J Neurol Neurosurg Psychiatry (2014). doi:10.1136/jnnp-2014-308331
59. Sharon JD, Hullar TE. Motion sensitivity and caloric responsiveness in vestibular migraine and Meniere’s disease. Laryngoscope (2014) 124(4):969–73. doi:10.1002/lary.24285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Lopez C, Blanke O, Mast FW. The human vestibular cortex revealed by coordinate-based activation likelihood estimation meta-analysis. Neuroscience (2012) 212:159–79. doi:10.1016/j.neuroscience.2012.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Baier B, Zu Eulenburg P, Best C, Geber C, Müller-Forell W, Birklein F, et al. Posterior insular cortex – site of vestibular-somato sensory interaction? Brain Behav (2013) 3:519–24. doi:10.1002/brb3.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Klingner CM, Volk GF, Flatz C, Brodoehl S, Dieterich M, Witte OW, et al. Components of vestibular cortical function. Behav Brain Res (2013) 236:194–9. doi:10.1016/j.bbr.2012.08.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Mazzola L, Lopez C, Faillenot I, Chouchou F, Mauguière F, Isnard J. Vestibular responses to direct stimulation of the human insular cortex. Ann Neurol (2014) 76:609–19. doi:10.1002/ana.24252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Garcia-Larrea L. The posterior insular-opercular region and the search of a primary cortex for pain. Neurophysiol Clin (2012) 42:299–313. doi:10.1016/j.neucli.2012.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. zu Eulenburg P, Baumgärtner U, Treede RD, Dieterich M. Interoceptive and multimodal functions of the operculo-insular cortex: tactile, nociceptive and vestibular representations. Neuroimage (2013) 83:75–86. doi:10.1016/j.neuroimage.2013.06.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Bottini G, Gandola M, Sedda A, Ferrè ER. Caloric vestibular stimulation: interaction between somatosensory system and vestibular apparatus. Front Integr Neurosci (2013) 7:66. doi:10.3389/fnint.2013.00066
67. Ferrè ER, Day BL, Bottini G, Haggard P. How the vestibular system interacts with somatosensory perception: a sham-controlled study with galvanic vestibular stimulation. Neurosci Lett (2013) 550:35–40. doi:10.1016/j.neulet.2013.06.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Shin JH, Kim YK, Kim HJ, Kim JS. Altered brain metabolism in vestibular migraine: comparison of interictal and ictal findings. Cephalalgia (2014) 34:58–67. doi:10.1177/0333102413498940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Brandt T, Bartenstein P, Janek A, Dieterich M. Reciprocal inhibitory visual-vestibular interaction. Visual motion stimulation deactivates the parieto-insular vestibular cortex. Brain (1998) 121:1749–58. doi:10.1093/brain/121.9.1749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Russo A, Marcelli V, Esposito F, Corvino V, Marcuccio L, Giannone A, et al. Abnormal thalamic function in patients with vestibular migraine. Neurology (2014) 82:2120–6. doi:10.1212/WNL.0000000000000496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Obermann M, Wurthmann S, Steinberg BS, Theysohn N, Diener HC, Naegel S. Central vestibular system modulation in vestibular migraine. Cephalalgia (2014) 34:1053–61. doi:10.1177/0333102414527650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. de Tommaso M, Ambrosini A, Brighina F, Coppola G, Perrotta A, Pierelli F, et al. Altered processing of sensory stimuli in patients with migraine. Nat Rev Neurol (2014) 10:144–55. doi:10.1038/nrneurol.2014.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Schwedt TJ, Chong CD, Chiang CC, Baxter L, Schlaggar BL, Dodick DW. Enhanced pain-induced activity of pain-processing regions in a case-control study of episodic migraine. Cephalalgia (2014) 34:947–58. doi:10.1177/0333102414526069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: migraine, aura, vertigo, multisensory integration, vestibulo-thalamo-cortical system, Meniere’s disease, vestibular system
Citation: Espinosa-Sanchez JM and Lopez-Escamez JA (2015) New insights into pathophysiology of vestibular migraine. Front. Neurol. 6:12. doi: 10.3389/fneur.2015.00012
Received: 30 October 2014; Accepted: 19 January 2015;
Published online: 06 February 2015.
Edited by:
Alexandre Bisdorff, Centre Hospitalier Emile Mayrisch, LuxembourgReviewed by:
Stefan Evers, University of Muenster, GermanyStephanie J. Nahas, Thomas Jefferson University, USA
Copyright: © 2015 Espinosa-Sanchez and Lopez-Escamez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose A. Lopez-Escamez, Otology and Neurotology Group CTS495, GENYO Centre for Genomics and Oncological Research Pfizer – Universidad de Granada – Junta de Andalucía, PTS, Avda. de la Ilustración, 114 18016 Granada, Spain e-mail:YW50b25pby5sb3BlemVzY2FtZXpAZ2VueW8uZXM=