 Pankaj Kumar Singh*
Pankaj Kumar Singh* Sweta Singh
Sweta Singh
- Department of Translational Medicine and Neurogenetics, Institut de Génétique et de Biologie Moléculaire et Cellulare (IGBMC), Illkirch, France
In brain, glycogen metabolism is predominantly restricted to astrocytes but it also indirectly supports neuronal functions. Increased accumulation of glycogen in neurons is mysteriously pathogenic triggering neurodegeneration as seen in “Lafora disease” (LD) and in other transgenic animal models of neuronal glycogen accumulation. LD is a fatal neurodegenerative disorder with excessive glycogen inclusions in neurons. Autophagy, a pathway for bulk degradation of obsolete cellular constituents also degrades metabolites like lipid and glycogen. Recently, defects in this pathway emerged as a plausible reason for glycogen accumulation in neurons in LD, although some contradictions prevail. Albeit surprising, a reciprocal regulation of autophagy by glycogen in neurons has also just been proposed. Notably, increasing evidences of interaction between proteins of autophagy and glycogen metabolism from diverse model systems indicate a conserved, dynamic, and regulatory cross-talk between these two pathways. Concerning these findings, we herein provide certain models for the molecular basis of this cross-talk and discuss its potential implication in the pathophysiology of LD.
Introduction
A common feature of many neurodegenerative disorders is the defect in protein quality control mechanisms including ubiquitin proteasome system (UPS) and macroautophagy (hereafter referred as autophagy) leading to biogenesis and accumulation of protein aggregates or inclusions (1). During the last decade, autophagy has been increasingly recognized as the primary reason behind pathogenesis of several neurodegenerative disorders. Defects in autophagy perturb neuronal functions, progressively leading to neurodegeneration (2). Beyond proteolysis, autophagy also plays a pivotal role in nutrient recycling and metabolic homeostasis by degrading lipids and glycogen (3). Surprisingly, while proteolytic dysfunction of autophagy is well recognized to underlie pathogenesis of neurodegenerative disorder, influence of its metabolic aspect is comparatively less explored.
In animals, glycogen has evolved as an efficient means of energy storage. In addition, the metabolism of this carbohydrate in the liver helps to maintain the blood and cerebral glucose level within the physiological limits during hypoglycemia and starvation. Intriguingly, despite glucose being the preferential energy source for the neurons and the presence of molecular machinery to synthesize glycogen, neurons synthesize very low glycogen under physiological conditions compared to most other cell types in animals (4). Glycogen or glycogen like inclusions called polyglucosan bodies, nevertheless do accumulate in neurons in (a) pathologies like Pompe disease (5), Lafora disease (LD) (6), Alzheimer’s disease (AD) (7) amyotrophic lateral sclerosis (8), adult polyglucosan body disease (APBD) (9), (b) under pathophysiological conditions like diabetes (10), hypoxia (11), and during aging (9). Surprisingly, this accumulation correlates with neurodegeneration in Pompe disease (5), in LD (6), in fly/mouse models expressing constitutive active glycogen synthase (GS) in neurons (12), and with reduced neuronal functions during aging (13). These findings, therefore, suggest that glycogen or related inclusion bodies are a pathogenic entity in the brain.
Remarkably, a direct impact of glycogen or its metabolic/regulatory proteins in control of neuronal functions is slowly growing. Thus, while induced glycogen accumulation in neurons promotes neurodegeneration, the prevention of polyglucosan accumulation in neurons by knocking down GS improves neurological functions and increases life span in aged fly (13). Notably, accumulation of carbohydrate inclusion (9) and reduced autophagy activity (14) in neurons, are two independent hypotheses proposed for the decline of neuronal functions with aging. Autophagy degrades glycogen and a recent indication of converse regulation of autophagy by glycogen or its constituent proteins, has suggested a direct functional link between glycogen and this proteolytic pathway. Mounting evidences for interaction between proteins of these two pathways has further compelled us to uncover the molecular mechanism linking autophagy activity, glycogen accumulation, and neuronal survival. This review summarizes all the recent findings of evolving autophagy–glycogen connection and its contribution to pathogenesis of neurodegenerative disorders, particularly LD.
Neuronal Glycogen Metabolism: Growing Understanding from a Rare Epilepsy – “Lafora Disease”
During the last decade, understanding the pathophysiological mechanism of a rare progressive myoclonus epilepsy “LD” has uncovered many interesting aspects of neuronal glycogen metabolism. LD is a fatal neurodegenerative disorder characterized by accumulation of insoluble, hyperphosphorylated, and less branched form of glycogen called Lafora bodies (LBs or polyglucosan more commonly) in several tissues including neurons of patients and mouse models (6, 15–17). Laforin and malin, two proteins pathologically linked to LD, were found to promote neuronal survival by restricting glycogen synthesis (18). A complex of these proteins was reported to keep neuronal glycogen synthetic machinery constitutively silent by enforcing GS inactivation and degradation of protein targeting to glycogen (PTG, an adaptor subunit of protein phosphatase 1 and activator of glycogen synthesis), through proteasome (18). This report is, however, challenged by subsequent findings demonstrating no alteration in the level of PTG (19, 20) and conflicting reports about GS activity in the brain of LD mouse models (17, 19, 20). Increased phosphorylation of glycogen in the absence of glucan phosphatase laforin is hypothesized as another reason behind neuronal glycogen accumulation (19). This hypothesis has also been recently challenged by Gayarre et al. (21), who show that the phosphatase activity of laforin is dispensable for prevention of LB formation and rescuing LD pathogenesis. An inadequate understanding of the precise regulatory role of LD proteins in glycogen metabolism therefore makes it inconceivable, how polyglucosan biogenesis is elicited in LD.
Inhibition of glycogen synthesis prevents polyglucosan accumulation and neurodegeneration in double knockout mice of PTG/laforin (22), GS/laforin (23), and PTG/malin (24). Thus, it appears that glycogen is regulated primarily at the level of its synthesis in neurons. Interestingly, potential significance of glycogen degradation in buildup of neuronal glycogen is also now gaining considerable attention. Thus, subsiding a previous report about absence of glycogen and its degradative enzyme glycogen phosphorylase in neurons, Saez et al. have demonstrated the presence of glycogen and its rapid turnover by glycogen phosphorylase (GP) (4). Furthermore, identification of autophagy defect in LD particularly in the presence of functional glycogen phosphorylase suggests this pathway as an alternative route of neuronal glycogen degradation (25–27). Although the relative contribution of glycogen phosphorylase and autophagy in glycogen degradation is undetermined, autophagy axis is steadily gaining more attention.
Involvement of Autophagy in Neuronal Glycogen Degradation: Current Understanding and Emerging Controversies
Several indirect evidences now exist that as in many other tissues, autophagy may degrade the glycogen in neurons. For example, defects in lysosomal enzyme “acid alpha-glucosidase (GAA)” are linked to glycogen accumulation in many tissues including neurons (5). Recently, autophagy defects at the level of compromised autophagosome formation, defective lysosomal structure, activity, and endosomal–lysosomal trafficking have been found in the brain of LD mice models (25–27). Further, as a rather direct evidence, Gayarre et al. have reported that forestalling autophagy defects in EPM2A−/− mice correlates with the absence of LB in the brain (21). These authors by expressing transgene encoding either active laforin (wild type, LAFWT) or its phosphatase inactive mutant (LAFC265S) in laforin-deficient mouse (Epm2a−/−) observed that both proteins are equally efficient in forestalling autophagy defects and prevent LBs formation in Epm2a−/− mouse. Still at molecular level, a direct role of laforin in both these processes and involvement of autophagy in neuronal glycogen degradation is not clear from this study. Nonetheless, together these findings convincingly indicate possible involvement of autophagy in neuronal glycogen degradation.
In contrast to this, Kakhlon et al. (28) have demonstrated failure of autophagy to degrade polyglucosan in a primary neuronal model of polyglucosan accumulation created by knockdown of glycogen branching enzyme (Gbe1). These authors failed to detect polyglucosan within any of the vesicular compartments of autophagy even after inducing autophagy by rapamycin (mammalian target of rapamycin, mTOR, inhibitor) treatment and starvation. Moreover, they noticed that rapamycin decreases polyglucosan accumulation by inhibiting GS activity and that this decrease is not prevented by autophagy blockade.
In the middle of this controversy, a completely new prospect of autophagy–glycogen relation emerged when Duran et al. (29) showed that in brain, autophagy activity corresponds to cellular glycogen level. The authors using malin-deficient mice (malinKO, an LD model) and double knockout mice with either partial (malinKO + MGSHet) or complete (malinKO + MGSKO) disability to store glycogen owing to partial (MGSHet) or complete (MGSKO) lack of GS in the brain showed that, reduction in glycogen level significantly rescued autophagy impairments and neurodegeneration. Conversely, they found that transgenic mice overexpressing, PTG (PTGOE) or non-inactivatable form of GS 9A-MGS (9A-MGSOE) show increased glycogen accumulation and autophagy impairments in the brain. Based on these observations, the authors claim to have resolved the question whether glycogen accumulation is a cause or consequence of autophagy defect, favoring the former possibility. This could be possible that in neurons, polyglucosan buildup hamper vesicular trafficking important for proper autophagy function. The enigma will, however, continue until it is ascertained that altered activity/level of GS/PTG protein does not modulate autophagy function intrinsically and has no direct involvement in this process. The reason for this concern is made apparent in the following sections.
Although autophagy-mediated glycogen degradation is well established, evidences for its existence in neurons are so far still in the preliminary stage. In this regard, conflicting reports like autophagy does not degrade neuronal polyglucosan (28), but at the same time, autophagy might prevent LBs accumulation (21), has created confusion as to whether, and under what context, autophagy could degrade neuronal glycogen. The discrepancy is further compounded by the finding that glycogen level determines autophagy activity (29). We believe that all these findings could have distinct physiological reasons and in a larger context, determining mutual regulation of autophagy and glycogen metabolism could help us to understand these discrepancies. Therefore, an in-depth discussion of these findings is prerequisite to evoke future research interest in the field to suggest the possible directions of investigation.
Herein, going against our own belief, we have first discussed how autophagy might fail to degrade neuronal glycogen. Afterwards, we have provided logical assumptions to contradict models rejecting the role of autophagy in neuronal glycogen degradation. Finally, envisaging certain models showing how proteins of these two cellular pathways may interact and regulate each other, we support the role of autophagy in neuronal glycogen degradation and insist a fresh revisit to investigate this metabolic nexus.
Possible Prospects of Polyglucosan Degradation by Autophagy
Glycogen is seemingly pathogenic to neurons, yet it is poorly understood why or how the neuronal machinery fails to clear it. It is noteworthy that neurons preferentially synthesize polyglucosan over glycogen. The term “Polyglucosan or Polyglucosan bodies” is attributed to structure, which represents less branched form of glycogen. These are hyperphosphorylated, poorly branched, amylase insensitive, and insoluble entities in contrast to glycogen (19). Perhaps, due to these structural and biochemical differences, the protein/s recruiting glycogen for autophagic degradation might fail to recognize polyglucosan. This opinion is based on finding that LD protein laforin shows more affinity for polyglucosan than glycogen (30). Therefore, altered affinity of proteins indispensable for glycogen degradation toward polyglucosan would obviously affect its metabolism. An interesting quest to this end would thus be to find out proteins mediating glycogen–autophagy interaction and identify their affinity for polyglucosan.
Glycogen degradation in tissues like liver and heart particularly under starvation or hypoglycemic conditions primarily serves the purpose of instant energy supply by providing glucose. The neuronal energy demand under such conditions is, however, compensated by surrounding glial cells (31) and therefore even under glycogenolytic signals, polyglucosan degradation in neurons will prove futile with no metabolic/physiological advantage to them. The basal autophagy operates at low level in neurons and it is quite resilient to induction under glycogenolytic condition like starvation that otherwise induces autophagy in the liver and heart tissues (31). Thus, with limited autophagy induction, neurons seem more prone to accumulate polyglucosan inclusions when the rate of synthesis of these inclusions exceeds the degradative capacity of autophagy. An interesting exploration would thus be to see if compensatory autophagy induction clears extra-neuronal polyglucosan. This will also uncover whether there exists any fundamental difference in metabolisms of this entity between neuronal and non-neuronal tissues.
These hypotheses about failure of autophagy to degrade polygucosan are, however, unjustified in realms of reports that, a structurally similar storage carbohydrate of plant; “starch” is degraded by autophagy (32) and autophagy might inhibits polyglucosan accumulation (21). How then Kakhlon et al. might have failed to observe autophagic clearance of polyglucosan? Autophagy is a selective process and specific adaptor proteins determine its substrate specificity in signal-dependent manner (33). Therefore, one reason could be that rapamycin treatment and starvation are not specific signals for glycogen degradation through autophagy in neurons. In fact, the extent of in vivo induction of autophagy in brain by both these manipulations is quite debatable. Interestingly, as discussed in the following section, these signals might rather inhibit glycogen recruitment to autophagosome. Thus, specific pharmacological or genetic manipulations that induce autophagy in brain must be tested before abandoning autophagy’s role in neuronal polyglucosan clearance. Additionally, it would be interesting to investigate whether such manipulations also manage to clear extra-neuronal polyglucosan.
With emergence of glycogenic proteins at different steps of autophagy (recruitment, and autophagosome synthesis), as discussed in the following section, glycogen metabolism seems a relatively more complex process than believed to be previously. Thus, it might be possible that despite active autophagy, loss of a specific glycogenic protein hampers glycogen degradation by perturbing its recruitment to autophagosome. Exploration of the role of glycogen branching enzyme (Gbe1) in this aspect would not only assert this hypothesis but would also settle the controversy stirred by Kakhlon et al. study.
Another intriguing exploration would be the impact of phosphorylation–dephosphosphorylation of glucosyl unit of glycogen over its autophagic degradation. Polyglucosan is hyperphosphorylated compared to glycogen and this biochemical modification of glucosyl unit in starch assists its degradation (34). Understanding the effect of this modification in autophagic degradation of starch might therefore uncover whether and how polyglucosan could be degraded by autophagy machinery in animal. Together, autophagy-mediated polyglucosan degradation in neurons is possibly provided, the process of autophagy induction, polyglucosan recruitment to autophagosome and lysosomal activity are intact, and the burden of polyglucosan remains within the physiological limit of autophagy capacity.
Autophagy–Glycogen Cross-Talk – A New Paradigm
The metabolic fate of glycogen (synthesis vs. degradation) is a function of active communication among its constituent proteins, which are perhaps also involved in other cellular processes. For example, GS has recently been found to interact with Atg8 (35), its human ortholog GABARAPL1 (36), and to regulate autophagy. Moreover, proteins like laforin, malin are supposed to play a role in autophagosome synthesis (25, 26). In addition, there are proteins like starch-binding domain-containing protein 1 (Stbd1) (37) and receptor of activated protein kinase C 1 (Rack1) (38), with poorly defined functions but, both interact with players of glycogen metabolism and autophagy (Table 1). The studies performed under loss or gain of function of these proteins, therefore would obviously tell the eventual metabolic fate of glycogen resulting from number of molecular changes happening “within, around, and outside the domain of glycogen granules” and consequent cellular adaptation. The autophagy–glycogen relationship derived from such studies therefore, should be analyzed with extreme caution.

Table 1. The so far identified potential proteins with dual roles in glycogen metabolism and autophagy.
With unfolding of interaction between protein of glycogen metabolism and autophagy, it seems that these two pathways are in continuous cross-talk and depending upon specific metabolic signals and/or, active/inactive state of specific proteins, they regulate each other in a manner beneficial to the cell. For instance, GS–Atg8 interaction depends upon GS activity as mutation affecting GS ability to bind with its allosteric activator glucose-6-phospate not only inhibits its activity but also perturbs its interaction with Atg8 (35). Conversely, GS insensitive for starvation-induced activity suppression by GS kinase 3 beta (GSK3β), still interact with Atg8 during starvation (35). Therefore, a dynamic balance between the relative ratio of free GS (for glycogen synthesis) and GS–Atg8 (determining autophagy activity) is essential for both these process to take place under basal condition. An activity-dependent change in GS–Atg8 interaction therefore would perturb the cellular availability of free Atg8 for autophagosome formation and autophagy activity as such, or recruitment of a specific substrate like glycogen to the autophagosome. Going by this hypothesis, reduced GS–Atg8 interaction and perturbed recruitment of polyglucosan in autophagosome upon GS inhibition by rapamycin/starvation could be a reason behind Kakhlon et al. failure to see polyglucosan in autophagic compartment despite autophagy induction.
Relevance of Glycogen–Autophagy Cross-Talk in LD
In neurons, change in the sub-cellular localization of GS and not just its increased level or activity determines glycogen synthesis (18). Therefore, signals that induce movement of GS from nucleus to the cytoplasm particularly in the brain might promote glycogen synthesis with concomitant suppression of autophagosome synthesis due to increased GS–Atg8 interaction. Laforin and GS both show nuclear–cytoplasmic translocation in response to glycogen availability (39, 40) and laforin interacts with GS (41). Therefore, loss of laforin-induced cytoplasmic translocation of GS triggering glycogen synthesis and perturbing autophagy could be a plausible reason of LB biogenesis. This functional model of GS–Atg8 also explains the molecular basis of autophagy defect seen in malin knockout mice (malinKO) with increased GS level (29) and unaltered mammalian target of rapamycin activity (26). This also unequivocally explains how the loss of GS in malin-GS double knockout mice (malinKO + MGSHet and malinKO + MGSKO) could rescue autophagy defect and prevent glycogen accumulation (29).
Future Directions
The discussed GS–Atg8 interaction is just one example of many such possible interactions between the proteins of these two cellular pathways. As both these pathways encompass several proteins, this cross-talk could supposedly be more complex involving several axis, with each composed of a specific set of proteins functional in one or other physiological conditions. For example, both GS and Stbd1 bind with glycogen and contain Atg8-interacting motif (AIM) predicted to be required for selective degradation of cargo (perhaps glycogen). Moreover, Stbd1 interacts with GS and other glycogenic proteins like laforin and debranching enzyme (42). In the light of such a complex interaction network (Figure 1), it would be difficult to predict the mechanism of Stbd1- or GS-mediated glycogen degradation by autophagy at this stage. Nevertheless, it can be speculated that, perhaps several independent and redundant mechanisms operate for dynamic regulation of autophagy–glycogen axis to meet metabolic demands under different physiological conditions.
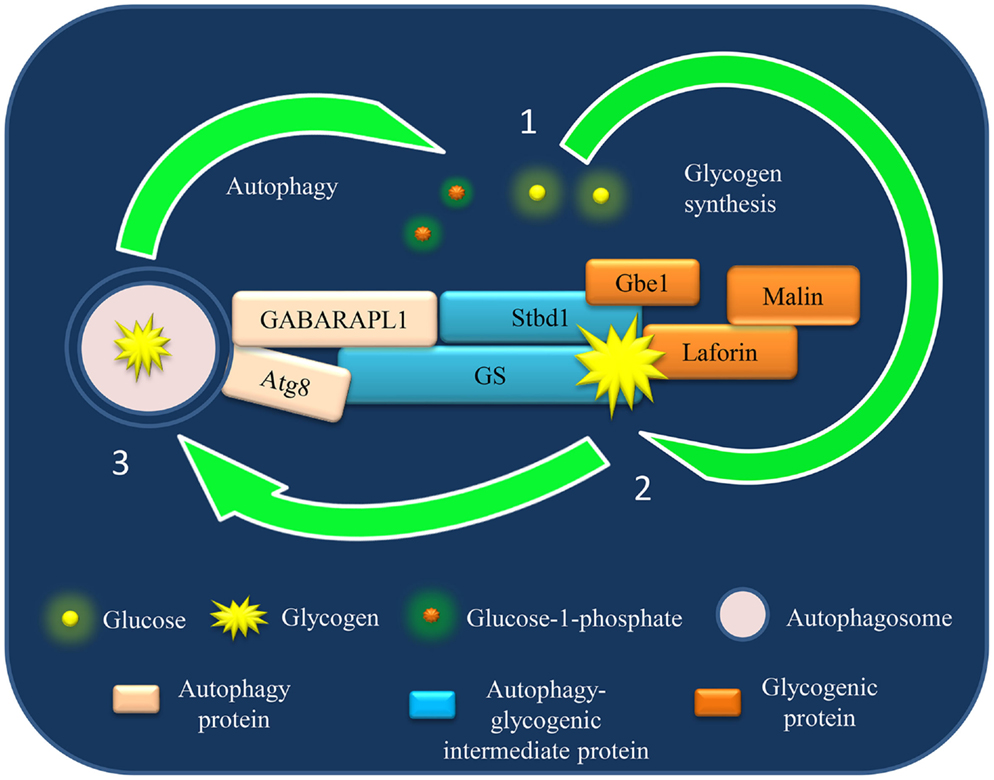
Figure 1. A model of autophagy–glycogen proteins interaction based on current understanding and its potential implication in Lafora disease (LD). The schematic representation shows the identified interaction until now between the proteins of glycogen metabolism and autophagy network. The physiological implication of these interactions in context of Lafora disease (LD) is discussed in this review.
Importantly, since these interactions are identified in cells of diverse origins, this cross-talk appears to be a conserved mechanism. This though could have cell/tissues specificity in context of the proteins involved particularly considering the tissue-specific isoforms of several glycogen metabolism proteins (43) and the fact that, glycogen proteome itself shows differences in its constituent proteins among various tissues (44). Cumulatively, these evidences postulate the existence of an intricate functional relationship between glycogen metabolism and autophagy and are intriguingly encouraging to explore the autophagy–glycogen communication with a fresher perspective, particularly in neurons. For this, animal models of glycogen storage (GS, PTG overexpression mice) and deficiency (GS knockout mice) can be utilized to study autophagy activity in brain. Likewise, autophagy-related gene (Atg)-deficient mice could be beneficial in investigating the status of neuronal glycogen metabolism.
Conclusion
The hypotheses proposed herein based on current evidences fully support autophagy-mediated glycogen degradation in neurons. In this direction, therefore, we urge to identify additional components of glycogen metabolism and autophagy machinery that interact and mutually regulate each other perhaps, under varied physiological conditions. Furthermore, in order to identify a direct link between these two pathways, an immediate challenge is to generate an appropriate cellular/animal model for dynamic monitoring of both these pathways simultaneously. As autophagy defect is seen in a number of neurodegenerative disorders and glycogen accumulation induces neurodegeneration, understanding a causal relationship between the two should be one of the prime focuses of future investigations in order to enhance the therapeutic potential of the diseases like LD, and to target proteins of glycogen metabolism as novel therapeutic interventions.
Author Contributions
PS; conception and design. PS, SS; writing and discussion.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med (2004) 10(Suppl):S10–7. doi:10.1038/nm1066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441:885–9. doi:10.1038/nature04724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330:1344–8. doi:10.1126/science.1193497
4. Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, et al. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab (2014) 34:945–55. doi:10.1038/jcbfm.2014.33
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Sidman RL, Taksir T, Fidler J, Zhao M, Dodge JC, Passini MA. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol (2008) 67:803–18. doi:10.1097/NEN.0b013e3181815994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ganesh S, Puri R, Singh S, Mittal S, Dubey D. Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J Hum Genet (2006) 51:1–8. doi:10.1007/s10038-005-0321-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Inoue M, Yagishita S, Itoh Y, Amano N, Matsushita M. Coexistence of paired helical filaments and polyglucosan bodies in the same neuron in an autopsy case of Alzheimer’s disease. Acta Neuropathol (1996) 92:511–4. doi:10.1007/s004010050553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Dodge JC, Treleaven CM, Fidler JA, Tamsett TJ, Bao C, Searles M, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci U S A (2013) 110:10812–7. doi:10.1073/pnas.1308421110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Cavanagh JB. Corpora-amylacea and the family of polyglucosan diseases. Brain Res Rev (1999) 29:265–95. doi:10.1016/S0165-0173(99)00003-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Bestetti G, Rossi GL. Hypothalamic lesions in rats with long-term streptozotocin-induced diabetes mellitus. A semiquantitative light- and electron-microscopic study. Acta Neuropathol (1980) 52:119–27. doi:10.1007/BF00688009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Ibrahim MZ. The response of the brain to hypoxia and ischaemia. J Neurol Sci (1972) 17:271–9. doi:10.1016/0022-510X(72)90032-9
12. Duran J, Tevy MF, Garcia-Rocha M, Calbó J, Milán M, Guinovart JJ. Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol Med (2012) 4:719–29. doi:10.1002/emmm.201200241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sinadinos C, Valles-Ortega J, Boulan L, Solsona E, Tevy MF, Marquez M, et al. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell (2014) 13:935–45. doi:10.1111/acel.12254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet (2008) 24:604–12. doi:10.1016/j.tig.2008.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Yokoi S, Austin J, Witmer F, Sakai M. Studies in myoclonus epilepsy (Lafora body form). I. Isolation and preliminary characterization of Lafora bodies in two cases. Arch Neurol (1968) 19:15–33. doi:10.1001/archneur.1968.00480010033002
16. Ganesh S, Delgado-Escueta AV, Sakamotom T, Avila MR, Machado-Salas J, Hoshii Y, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet (2002) 11:1251–62. doi:10.1093/hmg/11.11.1251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann Neurol (2010) 68:925–33. doi:10.1002/ana.22156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García-Fojeda B, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci (2007) 10:1407–13. doi:10.1038/nn1998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, et al. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem (2008) 283:33816–25. doi:10.1074/jbc.M807428200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J Biol Chem (2010) 285:25372–81. doi:10.1074/jbc.M110.148668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gayarre J, Duran-Trío L, Criado Garcia O, Aguado C, Juana-López L, Crespo I, et al. The phosphatase activity of laforin is dispensable to rescue Epm2a−/− mice from Lafora disease. Brain (2014) 137:806–18. doi:10.1093/brain/awt353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Turnbull J, DePaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet (2011) 7:e1002037. doi:10.1371/journal.pgen.1002037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Pederson BA, Turnbull J, Epp JR, Weaver SA, Zhao X, Pencea N, et al. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol (2013) 74:297–300. doi:10.1002/ana.23899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N, Wang P, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol (2014) 75:442–6. doi:10.1002/ana.24104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Aguado C, Sarkar S, Korolchuk VI, Criado O, Vernia S, Boya P, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet (2010) 19:2867–76. doi:10.1093/hmg/ddq190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Criado O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, Vernia S, et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet (2012) 21:1521–33. doi:10.1093/hmg/ddr590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Puri R, Suzuki T, Yamakawa K, Ganesh S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum Mol Genet (2012) 21:175–84. doi:10.1093/hmg/ddr452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kakhlon O, Glickstein H, Feinstein N, Liu Y, Baba O, Terashima T, et al. Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. J Neurochem (2013) 127:101–13. doi:10.1111/jnc.12277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Duran J, Gruart A, García-Rocha M, Delgado-García JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet (2014) 23:3147–56. doi:10.1093/hmg/ddu024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P, et al. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet (2004) 13:1117–29. doi:10.1093/hmg/ddh130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta (2009) 1793:1496–507. doi:10.1016/j.bbamcr.2009.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Wang Y, Yu B, Zhao J, Guo J, Li Y, Han S, et al. Autophagy contributes to leaf starch degradation. Plant Cell (2013) 25:1383–99. doi:10.1105/tpc.112.108993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif – crucial for selective autophagy. J Cell Sci (2013) 126:3237–47. doi:10.1242/jcs.126128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Silver DM, Kötting O, Moorhead GB. Phosphoglucan phosphatase function sheds light on starch degradation. Trends Plant Sci (2014) 19:471–8. doi:10.1016/j.tplants.2014.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Zirin J, Nieuwenhuis J, Perrimon N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol (2013) 11:e1001708. doi:10.1371/journal.pbio.1001708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature (2010) 466:68–76. doi:10.1038/nature09204
37. Jiang S, Wells CD, Roach PJ. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun (2011) 413:420–5. doi:10.1016/j.bbrc.2011.08.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Erdi B, Nagy P, Zvara A, Varga A, Pircs K, Ménesi D, et al. Loss of the starvation-induced gene Rack1 leads to glycogen deficiency and impaired autophagic responses in Drosophila. Autophagy (2012) 8:1124–35. doi:10.4161/auto.20069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Singh PK, Singh S, Ganesh S. The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol Cell Biol (2012) 32:652–63. doi:10.1128/MCB.06353-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Cid E, Cifuentes D, Baqué S, Ferrer JC, Guinovart JJ. Determinants of the nucleocytoplasmic shuttling of muscle glycogen synthase. FEBS J (2005) 272:3197–213. doi:10.1111/j.1742-4658.2005.04738.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem (2006) 281:30412–8. doi:10.1074/jbc.M606117200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Zhu Y, Zhang M, Kelly AR, Cheng A. The carbohydrate-binding domain of overexpressed STBD1 is important for its stability and protein-protein interactions. Biosci Rep (2014) 1:34. doi:10.1042/BSR20140053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J (2012) 441:763–87. doi:10.1042/BJ20111416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Stapleton D, Nelson C, Parsawar K, Flores-Opazo M, McClain D, Parker G. The 3T3-L1 adipocyte glycogen proteome. Proteome Sci (2013) 11:11. doi:10.1186/1477-5956-11-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Zeng L, Wang Y, Baba O, Zheng P, Liu Y, Liu Y. Laforin is required for the functional activation of malin in endoplasmic reticulum stress resistance in neuronal cells. FEBS J (2012) 279:2467–78. doi:10.1111/j.1742-4658.2012.08627.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Pederson BA, Schroeder JM, Parker GE, Smith MW, DePaoli-Roach AA, Roach PJ. Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes (2005) 54:3466–73. doi:10.2337/diabetes.54.12.3466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: autophagy, glycogen, polyglucosan, neurodegeneration, Lafora disease
Citation: Singh PK and Singh S (2015) Changing shapes of glycogen–autophagy nexus in neurons: perspective from a rare epilepsy. Front. Neurol. 6:14. doi: 10.3389/fneur.2015.00014
Received: 16 December 2014; Accepted: 20 January 2015;
Published online: 04 February 2015.
Edited by:
Tibor Hortobágyi, University of Debrecen, HungaryReviewed by:
Tibor Hortobágyi, University of Debrecen, HungaryKrisztina Deak-Pocsai, University of Debrecen, Hungary
Pascual Sanz, Consejo Superior de Investigaciones Científicas, Spain
Copyright: © 2015 Singh and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj Kumar Singh, Department of Translational Medicine and Neurogenetics, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), CNRS UMR 7104, INSERM U 964, 1 rue Laurent Fries, BP 10142, Illkirch 67404, France e-mail:c3BhbmthajA4MUBnbWFpbC5jb20=