 Nora Sandow1,2‡
Nora Sandow1,2‡ Simon Kim1†‡
Simon Kim1†‡ Claudia Raue1†
Claudia Raue1† Dennis Päsler1†
Dennis Päsler1† Zin-Juan Klaft1
Zin-Juan Klaft1 Leandro Leite Antonio1,3†
Leandro Leite Antonio1,3† Jan Oliver Hollnagel1
Jan Oliver Hollnagel1 Richard Kovacs1
Richard Kovacs1 Oliver Kann1,4
Oliver Kann1,4 Peter Horn2†Peter Vajkoczy2
Peter Horn2†Peter Vajkoczy2 Martin Holtkamp5
Martin Holtkamp5 Heinz-Joachim Meencke5
Heinz-Joachim Meencke5 Esper A. Cavalheiro3
Esper A. Cavalheiro3 Fritz Pragst6
Fritz Pragst6 Siegrun Gabriel1
Siegrun Gabriel1 Thomas-Nicolas Lehmann2†
Thomas-Nicolas Lehmann2† Uwe Heinemann1*
Uwe Heinemann1*- 1Institute of Neurophysiology, Charité Universitätsmedizin Berlin, Berlin, Germany
- 2Department of Neurosurgery, Charité Universitätsmedizin Berlin, Berlin, Germany
- 3Laboratorio de Neurologia Experimental, Universidade Federal de São Paulo-Escola Paulista de Medicina, São Paulo, Brazil
- 4Institute of Physiology and Pathophysiology, University of Heidelberg, Heidelberg, Germany
- 5Epilepsy Center of Berlin-Brandenburg, Ev. Krankenhaus Königin Elisabeth Herzberge, Berlin, Germany
- 6Institute of Forensic Medicine – Forensic Toxicology, Charité Universitätsmedizin Berlin, Berlin, Germany
Drug resistant patients undergoing epilepsy surgery have a good chance to become sensitive to anticonvulsant medication, suggesting that the resected brain tissue is responsible for drug resistance. Here, we address the question whether P-glycoprotein (Pgp) and multidrug resistance-associated proteins (MRPs) expressed in the resected tissue contribute to drug resistance in vitro. Effects of anti-epileptic drugs [carbamazepine (CBZ), sodium valproate, phenytoin] and two unspecific inhibitors of Pgp and MRPs [verapamil (VPM) and probenecid (PBN)] on seizure-like events (SLEs) induced in slices from 35 hippocampal and 35 temporal cortex specimens of altogether 51 patients (161 slices) were studied. Although in slice preparations the blood brain barrier is not functional, we found that SLEs predominantly persisted in the presence of anticonvulsant drugs (90%) and also in the presence of VPM and PBN (86%). Following subsequent co-administration of anti-epileptic drugs and drug transport inhibitors, SLEs continued in 63% of 143 slices. Drug sensitivity in slices was recognized either as transition to recurrent epileptiform transients (30%) or as suppression (7%), particularly by perfusion with CBZ in PBN containing solutions (43, 9%). Summarizing responses to co-administration from more than one slice per patient revealed that suppression of seizure-like activity in all slices was only observed in 7% of patients. Patients whose tissue was completely or partially sensitive (65%) presented with higher seizure frequencies than those with resistant tissue (35%). However, corresponding subgroups of patients do not differ with respect to expression rates of drug transporters. Our results imply that parenchymal MRPs and Pgp are not responsible for drug resistance in resected tissue.
Introduction
Patients suffering from partial epilepsies often present with therapeutic difficulties. Up to 80% of patients with temporal lobe epilepsy (TLE) are resistant to treatment with anti-epileptic drugs (AEDs) (1). Drug resistance may depend on the severity of disease (2), development of tolerance (3), changes in drug targets (4–6), network alterations (7, 8), and an open blood brain barrier (9). As drug resistance concerns drugs with different mechanisms of action, the “transporter hypothesis” received considerable attention (10–18). This hypothesis states that drug efflux pumps transport drug molecules out of the brain parenchyma and thereby impede the build-up of tissue AED-concentrations sufficient for seizure control. Multi drug transporter proteins (MDTs) include P-glycoprotein (Pgp) and the multidrug resistance-associated proteins (MRP1-5). Studies in human resected tissue of epilepsy patients show that such proteins are up-regulated or ectopically expressed at the blood brain barrier (19–22) and within the brain parenchyma (13, 18, 23–25).
The aim of our present study is to find out whether Pgp and multidrug resistance-associated proteins (MRPs) in acute hippocampal and temporal cortex slices contribute to drug resistance in the functionally blood brain barrier deprived tissue of drug resistant patients. Our previous study of the dentate gyrus (DG) (26) revealed that induced epileptiform activity in the DG from drug sensitive patients and patients with extra-hippocampal tumors was sensitive to CBZ, while similar activity of drug resistant patients was resistant. Here, we compare effects of drugs, which were “definitely” (phenytoin, PHT), “possibly” (metabolites of carbamazepine, CBZ), and “not” transported (valproate, VPA) by human Pgp (27). Additionally, verapamil (VPM) and probenecid (PBN) were chosen as unspecific inhibitors of Pgp and MRPs because clinical reports described that co-application of VPM with AEDs might restore pharmacosensitivity (28). Moreover, we already presented evidence that the efflux pump function of Pgp and MRPs expressed in slices from resected tissue was altered by VPM and PBN (29). Now, we extend our electrophysiological investigations to the subiculum (SUB) and the temporal neocortex (TCx), and focus on drug effects on induced seizure-like events (SLEs) resembling ictal discharges in intracranial recordings (30).
Materials and Methods
Surgery specimens were obtained from 72 patients suffering from TLE (years 2005–2009; 58 hippocampal and 52 cortical specimens) of which 65 patients were proven drug resistant, 4 patients were resistant but had adverse effects in at least 1 treatment strategy, and 3 patients were still not “proven” (1 year duration of epilepsy, high seizure frequency or tumor). Pre-surgical analysis including determination of drug resistance was performed in the Epilepsy Center of Berlin Brandenburg (Martin Holtkamp and Heinz-Joachim Meencke) as previously described (26, 31, 32).
All patients underwent a combined resection of the temporal pole with amygdalo-hippocampectomy (TPR+), using a cortical approach. Surgery was a two step procedure. Between 2–2.5 cm (dominant) and 3–3.5 cm (non-dominant) of the temporal pole were removed first, afterwards the mesial temporal structures were resected. The temporal pia mater covering the Sylvian vessels remained intact. After ablation of the temporal pole, the inferior horn was already open or could be readily opened (33). Operations were performed by Thomas-Nicolas Lehmann, Peter Vajkoczy, and Peter Horn in the Department of Neurosurgery, Charité.
For the purpose of the present study, 35 hippocampal and 35 temporal cortex specimens of altogether 51 patients (161 slices in which SLEs were induced) could be included.
The study was approved by the Ethics Committee at the Charité-Universitätsmedizin Berlin (EA125/2001, EA1/042/04) and performed in accordance with the declaration of Helsinki. A written informed consent was obtained from every patient before surgery. Neighboring sections of the tissue in this study were analyzed by the Department of Neuropathology, Charité for diagnostic purposes.
For some patients also the tissue concentrations of OXC, CBZ, and LTG were determined in the Department of Forensic Toxicology, Institute of Forensic Medicine, Charité. CBZ, CBZ-10,11-epoxide, oxcarbazepine, 10-hydroxy-oxcarbazepine, and lamotrigine were determined by high performance liquid chromatography with diode array detector (HPLC-DAD) described by Pragst and colleagues (34). Reference standards for calibration of the five compounds were obtained from LGC Promochem (Wesel, Germany). Between 200 and 500 mg brain tissue were exactly weighed and extracted with the threefold volume of acetonitrile by 2 h incubation and treatment in ultrasonic bath at room temperature. After centrifugation, the acetonitrile layer was completely separated. The residue was thoroughly washed with the same volume acetonitrile and centrifuged again. The extract and the washing were united and the acetonitrile evaporated in a nitrogen stream at 40°C to dryness. The residue was dissolved in 100 μl of the mobile phase (phosphate buffer pH 2.3/acetonitrile 63:37, v/v) and 50 μl were injected for HPLC.
Tissue Transport, Preparation, and Maintenance
One or two coronal sections of 5 mm thickness were cut from the resected temporal cortex and hippocampus, immediately transferred to cold (1–4°C) carbogenated transport solution containing (in mM) KCl 3, NaH2PO4 1.25, glucose 10, MgSO4 2, MgCl2 2, CaCl2 1.6, NaHCO3 21, sucrose 200, and (±) α-tocopherol 0.1 pre-dissolved in ethanol (pH 7.4, osmolality 303 mosmol/kg, 0.005 v% ethanol), and transported to the lab, as previously described (26, 31, 32). Subsequently, the tissue was coronally dissected into slices of 500 μm thicknesses using a vibratome (Campden Instruments Ltd., Leicester, UK). Slices subjected to electrophysiological recordings were immediately transferred to interface chambers, perfused at a rate of 1.7 ml/min with pre-warmed (34.5°C) carbogenated artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 129, KCl 3, NaH2PO4 1.25, glucose 10, MgCl2 2, CaCl2 1.6, NaHCO3 21, (±) α-tocopherol 0.03 (pH 7.4; osmolality 303 mosmol/kg, 0.002 v% ethanol). Recordings commenced 4 h after preparation of slices to permit optimal recovery after surgery and transport in solution with low sodium concentration.
Electrophysiological Recordings and Stimulation
Extracellular recordings were performed in the granule cell layer of the DG, in the pyramidal cell layer of the SUB, and in deep layers of the TCx (V–VI), 120–150 μm below the surface of the slice, using double-barreled K+-selective/reference microelectrodes as previously described (31). Signals were stored on a computer using Spike2 (version 4.01, CED; Cambridge, UK) with sampling rates of 10 kHz for field potentials (FPs, low pass filter cut-off 3 kHz) and 100 Hz for [K+]o (low pass filter cut-off 1.6 Hz). Paired electrical stimulation was performed with bipolar platinum electrodes (wire diameter 25 μm, tip distance about 50–100 μm, pulse duration 0.1 ms, pulse interval 50 ms). They were positioned in the hilus (26), in stratum radiatum of the (pro)SUB, and in the white matter below layer VI of the TCx.
Experimental Protocols
Viability of slices
To exclude that drug effects were mimicked by loss of slice viability, we routinely determined amplitudes of averaged responses (n = 5) to the first response of paired stimulus-evoked FPs (every 20 s, stimulus intensities eliciting 80 or 100% of the maximal FP amplitude) in the beginning and at the end of the experiment. Experiments were accepted when the evoked signals did not change more than 25% between the beginning and end of an experiment.
Regionally optimized induction of seizure-like events
In the DG, hilar stimulation and elevation of potassium concentration to 10–12 mM induced SLEs (26, 31) (Figure 1A). In the SUB elevation of potassium concentration to 10–12 mM could provoke SLEs (Figure 1B) while in the TCx 8 mM [K+] and 50 μM bicuculline–methiodide were sufficient. Bicuculline was added when [K+]o approached the plateau of equilibration (see Figure 1C). SLEs lasted for more than 5 s (Figures 2A,C). Epileptiform events shorter than 5 s were termed recurrent epileptiform transients (RETs; Figures 2B,C). Slices in which only RETs were observed were not included in the analysis. The differentiation between SLEs and RETs was based on the histogram of event durations (Figure 2C) and on the relation between event duration and event rate (not shown). The distribution of values displayed a first minimum around 5 s and a second minimum around 15 s. However, only the first minimum was associated with a change in the event rate. Therefore, we decided to categorize SLEs by a minimal event duration of 5.1 s.
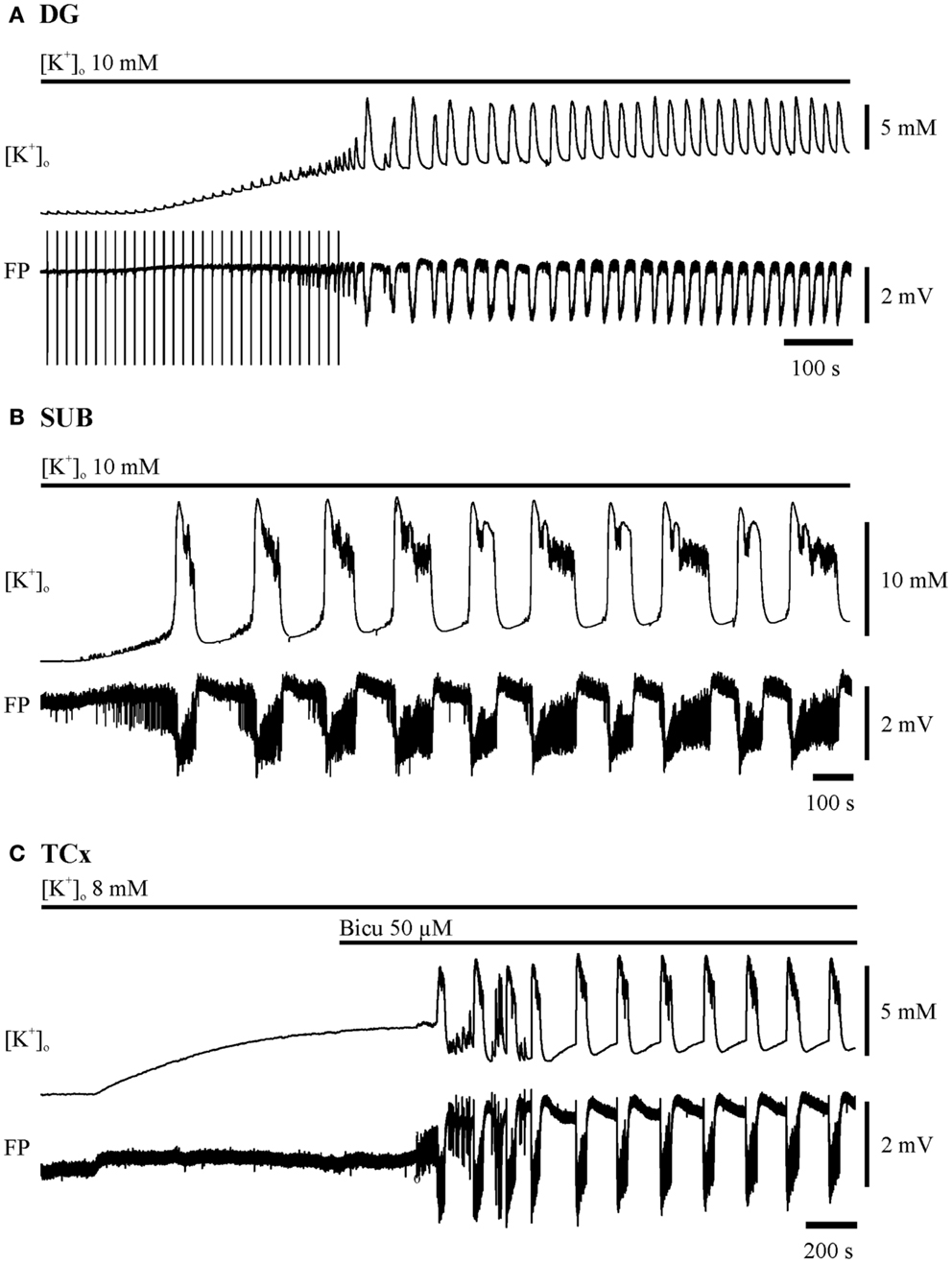
Figure 1. Induction of seizure-like events in the dentate gyrus (DG), subiculum (SUB), and in deep layers of the temporal neocortex (TCx). The two traces depicted for each region display recordings of [K+]o (top) and field potential (FP) bottom. Time and amplitude of signals are given by calibration bars on the right. Bars above each pair of traces mark the perfusion of the ictogenic buffer solution. (A) DG: hilar double pulse stimulation (pulse duration 0.1 ms, pulse interval 50 ms, stimulus intensity for pulses in the range of 80% of the maximal field potential amplitude, frequency 0.067 Hz). The stimulation was performed before and during elevation of [K+]o to 10 mM, and had been set off when epileptiform discharges appeared independent of electrical pulses. (B) SUB: elevation of [K+]o to 10 mM. (C) TCx: elevation of [K+]o to 8 mM and addition of 50 μM Bicuculline when [K+]o approximated the plateau of equilibration.
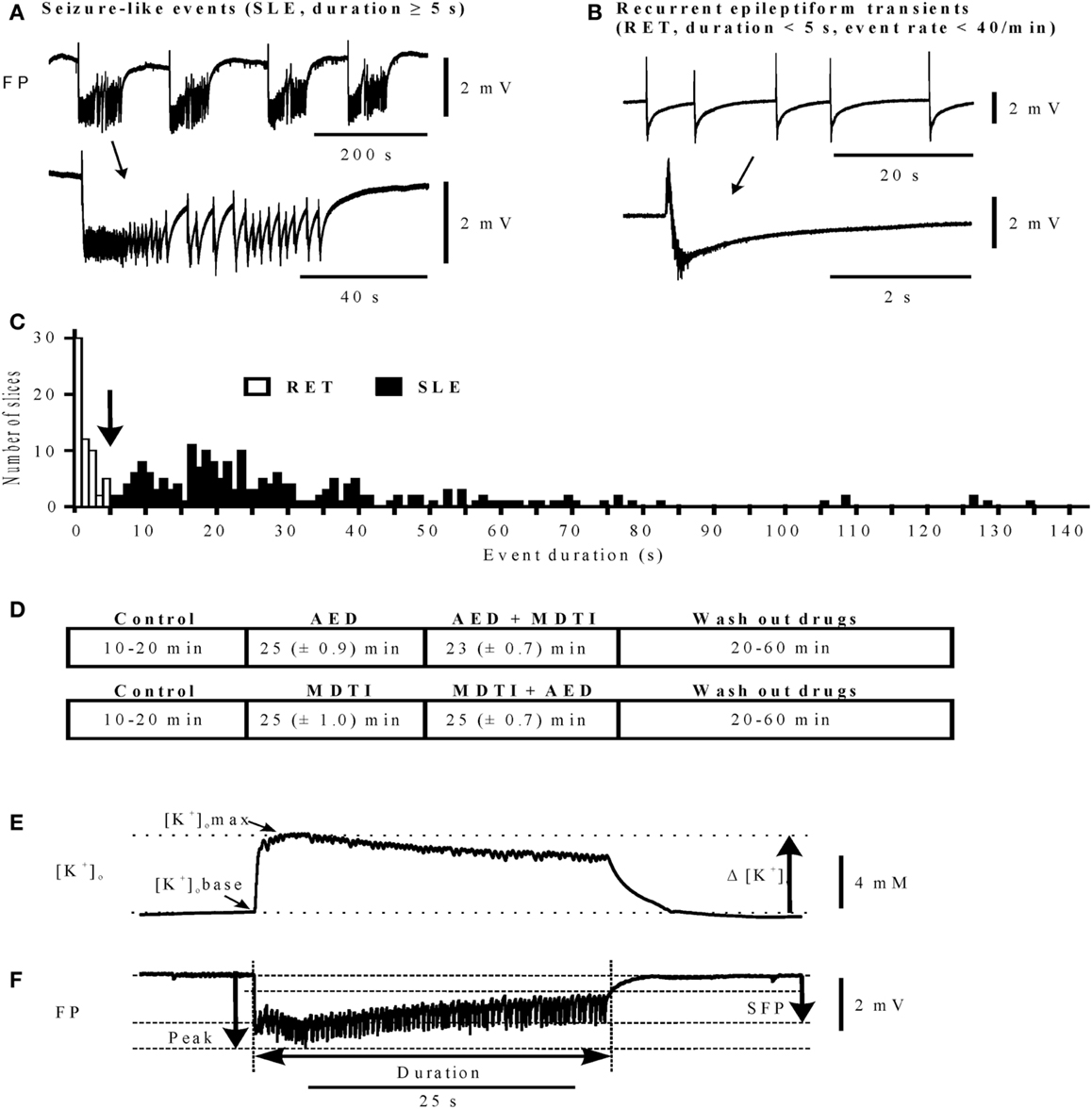
Figure 2. Differentiation of epileptiform activities and determination of characteristic parameters. (A,B) Traces of activity (top), single events marked by arrow are displayed on an expanded time scale (bottom), calibration bar for field potential amplitudes on the right. (A) Seizure-like events (SLEs); (B) recurrent epileptiform transients (RETs); (C) histogram of event durations for RETs and SLEs, bin width 1 s; (D) protocol sequences in main experiments: top (n = 120): control, AED, AED + MDTIs, and Wash out of drugs; bottom (n = 41): control, MDTIs, MDTIs + AED, and Wash out of drugs. For periods of Control and Wash out durations are given as Min and Max, and for the drug periods as Mean (±SEM). (E) Parameters determined for event-associated changes of extracellular potassium concentrations ([K+]o in mM): [K+]o base in the beginning of an event, [K+]o max at the maximum value, Δ [K+]o rise of [K+]o; (F) parameters determined for event-associated deviations of the field potential (FP): sfp (mV) maximum of the slow field potential amplitude, peak (mV) maximum of the peak amplitude, and duration (s) event duration (time from beginning of the event up to 2/3 recovery of the sfp deflection). (E,F) Calibration bar for [K+]o and FP on the right. Abscissa in (A,B,E,F): time in seconds (calibration bar below the FP traces).
Treatment protocols
After induction of SLEs, slices were subjected to two different sequential protocols denoted as “AED” followed by “AED + MDTI” and as “MDTIs” followed by “MDTIs + AED” (Figure 2D). Moreover, a small number of control experiments have been performed in which slices were not perfused with drugs, denoted as “No drug”-experiments (Figure 3).
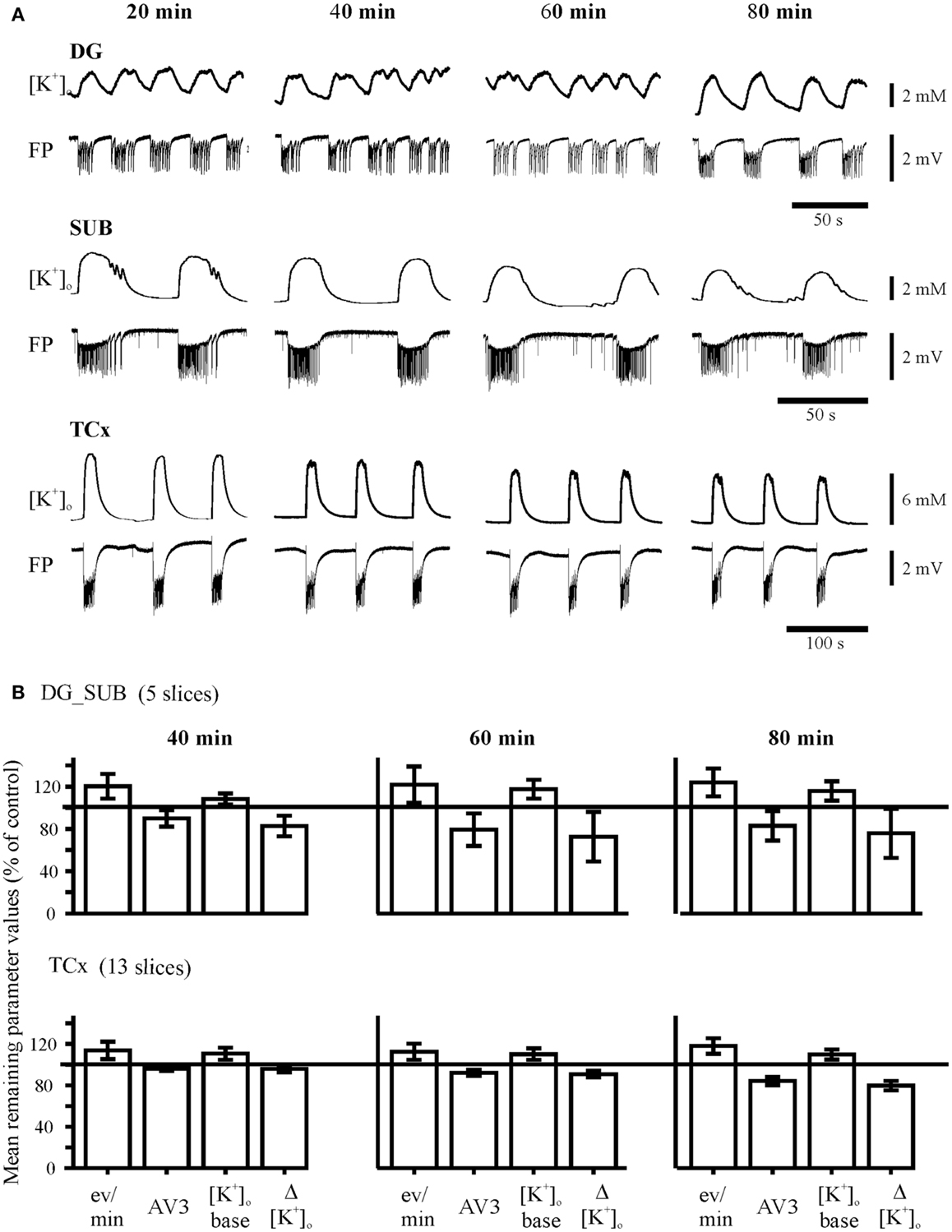
Figure 3. Control experiments. (A) Time-matched experiments without any drug perfusion in the dentate gyrus (DG), subiculum (SUB), and the temporal neocortex (TCx). The recordings of [K+]o and field potential (FP) were taken from the end of each time interval, marked above traces. Calibration bars for amplitude and time are given on the right. (B) Time matched remaining parameter values (percent of control values). The remaining parameter values for event rate (eV/min), AV3 (averaged remaining normalized parameter values for sfp, peak, and duration), [K+]o base, and ([K+]o show variability or slight run down but no pattern change throughout time.
During the protocol “AED” (120 slices, 37 patients, Figure 2D, top), one of three AEDs CBZ, 50 μM, VPA, 1 mM, and PHT, 50 μM was applied for at least 20 min. Subsequently, during the protocol “AED + MDTIs,” the MDTIs PBN (400 μM) and/or VPM (40 μM) were added to the perfusion solution for another 20 min.
In order to test whether VPM and/or PBN may alter seizure-like activity independently of their interaction with potential AED transport, we also studied their effects on SLEs before the respective AEDs were added to the perfusate (Figure 2D, bottom). During this protocol, referred to as “MDTIs,” PBN, and/or VPM were applied first (41 slices, 15 patients) and then, in the protocol “MDTIs + AED,” one of the AEDs was added to the perfusion solution. The time schedule “MDTI” and “MDTI + AED” was similar to that for “AED” and “AED + MDTI.” During both protocols, the MDTIs VPM and PBN were simultaneously administered to 115 slices, PBN alone to 22 slices, and VPM alone to 22 slices.
The “No drug”-protocol (17 slices, 9 specimens, 7 patients) lasted 80 min without drug administration (Figure 3A). This permits to test for stability of induced activity, and to compare drug effects to a time-matched “No drug”-group (Figure S2 in Supplementary Material).
Drug effects were considered if at least a partial recovery of epileptiform activity was achieved. All substances were administered through the ictogenic perfusion medium.
Immunohistochemistry of Multidrug Transport Proteins
Expression patterns of multidrug transporters in specimens of drug resistant patients included in the present study have been already published (29). In brief, we used a modified glucose oxidase-diaminobenzidine (DAB) method (35). Tissue samples were fixed overnight (4% PFA) and 10 μm thin sections were cut in a cryostat (Leica, Jung CM 1800) and then incubated (24 h at 4°C) with diluted primary antibody [monoclonal antibodies: Pgp, JSB-1 antibody (1:50); MRP1, MRPr1 antibody (1:20); MRP2, M2III-6 antibody (1:50), Alexis Biochemicals; MRP5, M51-1 antibody (1:20) DCS/Signet, Hamburg, Germany]. The antibodies were diluted in normal goat serum (10%), Triton X-100 (0.3%), BSA, and 0.1 M PB (pH 7.4). Subsequently, slices were incubated for 1 h in biotinylated secondary antibody (1:100), washed in AB complex for 1 h, followed by DAB oxidation (ABC kit, Vector Labs Burlingame, CA, USA), and then counterstained with Vector Hematoxylin Nuclear Counterstain (Vector Labs).
Cell counting and quantification of immunohistochemistry data were carried out semi-automatically by using the software Kappa Image (Metreo Software, Kappa Optoelectronics) based on the method of West and Gundersen (36). The ratio of multidrug transporter expressing cells refers to the total cell number, determined in counterstained slices, and corrected following the method of Abercrombie (37). The ratio values were given in percent of the corresponding total cell number, averaged with respect to each transporter type, region, and cell type per patient.
Data Analysis and Statistics
Initial analysis
Initial analysis was based on the categorization of effects in a given slice (persistence of SLE, transition of SLE to RET, or suppression of SLE). Quantification of drug effects followed previous protocols (26). Changes of [K+]o were described (i) for the onset of events ([K+]obase), (ii) for the event-associated maximum of [K+]o ([K+]omax), and (iii) for the event-associated rise of [K+]o (Δ [K+]o = [K+]omax − [K+]obase, Figure 2E). Deflections of the FP (Figure 2F) were characterized by their event rate (n/min), maximal slow FP amplitude [sfp (mV)], maximal peak amplitude [peak (mV)], considering the largest negative transient, and event duration [duration (s)]. Fluctuations with a size of <0.3 mV and a time interval of <3 ms were excluded from the analysis.
Moreover, all activity parameters were normalized with reference to the control value, determined after a stabilization period lasting 10–20 min, and given as remaining parameter values in a percentage of the control value. Three normalized parameter values (sfp, peak, and duration) were averaged as AV3 [see also Ref. (26)]. In order to prevent statistical overweight of event rate, normalized changes >150% were set to 150%.
In addition, a patient oriented analysis was performed, allowing correlation with clinical data. All pharmacological responses from more than one slice of the same specimen/patient were again categorized with respect to quality and heterogeneity. In order to relate some of our data to serum concentrations of clinically employed drugs with different pharmacokinetic properties and ranges of effectiveness, serum concentrations were normalized to the maximal therapeutic serum level for each AED [set by the clinical analysis, following available literature, i.e., Ref. (38)] and given as a percentage of the maximum level.
Statistical analysis
Group data of ratio variables are displayed as mean ± SEM throughout the manuscript. Data of nominal and ordinal variables are given as proportions of group members assigned to the response categories. As the Shapiro–Wilk tests indicated deviation from the normal distribution of values for some of the variables, comparisons within groups and between groups were performed using non-parametric tests (Wilcoxon, Friedman; Mann–Whitney U, Kruskal–Wallis-H). Proportional differences within and between groups were evaluated applying the McNemar and the Chi2-homogeneity test (Fisher’s exact test). All statistics were calculated using the PASW statistics 18 (SPSS, Chicago, IL, USA).
Results
The present pharmacological study is based on experiments in 161 slices (35 hippocampal and 35 cortical specimens of 51 patients) in which SLEs could be induced (2038 with Table 1). First, we investigated pharmacosensitivity of SLEs to AEDs or MDTIs, and then to co-administration of AEDs and MDTIs.
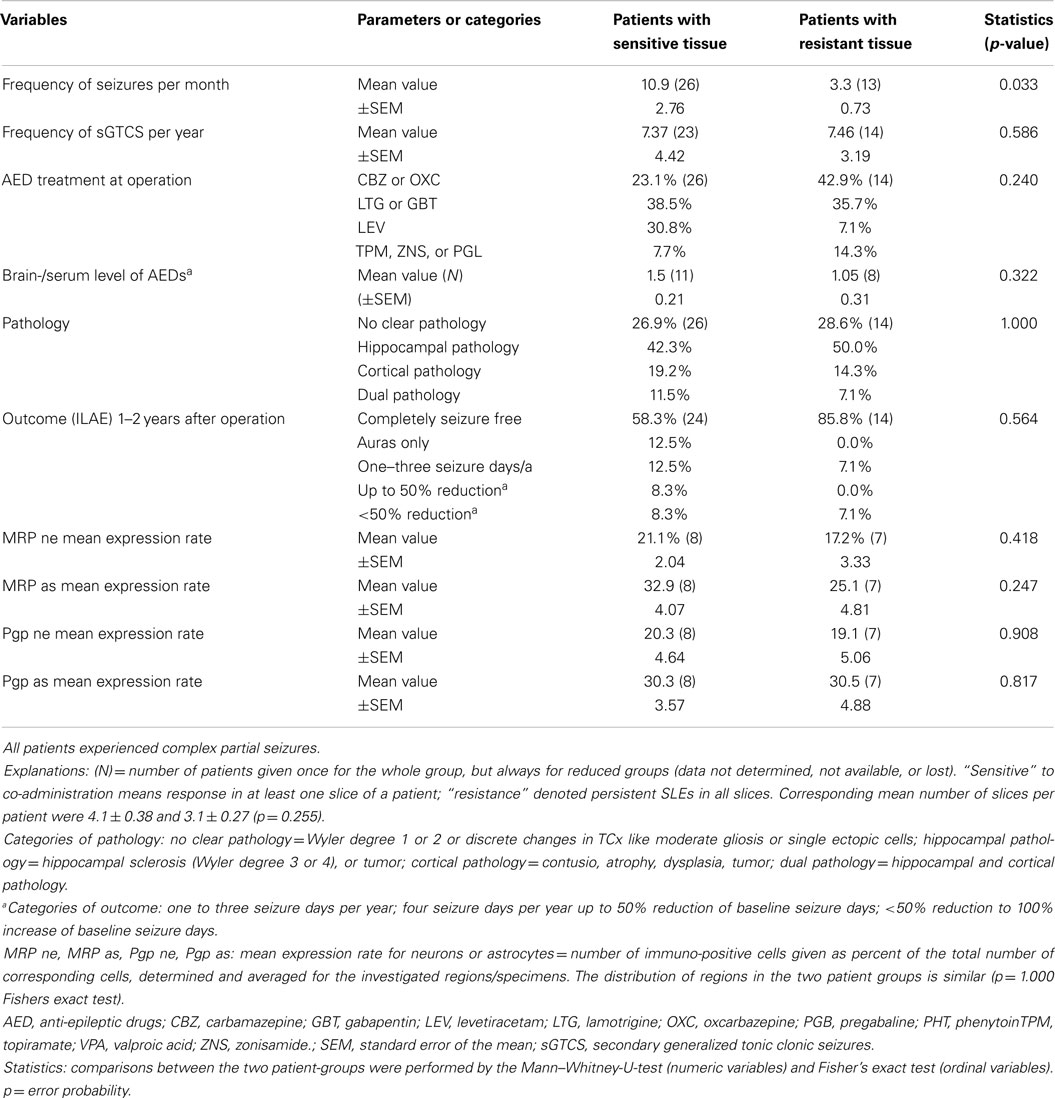
Table 1. Comparison of patient data with respect to in vitro sensitivity or resistance of SLEs to co-administration of one AED and probenecid or/and verapamil for 40 patients providing more than one slice to the analysis.
As shown in Figures 1 and 2A, SLEs were usually characterized by a large negative FP-shift of more than 5 s duration, associated with a rise in [K+]o. Event durations varied considerably (Figure 2C). In a given slice, SLEs recurred regularly after induction. In No drug experiments, the incidence of SLEs increased with time while duration and rises in [K+]o declined with time (Figure 3B). Also, the value of AV3 became reduced. However, these changes were <20% in hippocampal slices and <10% in cortical slices. Spontaneous transition of SLEs to RETs was not observed in any of the 17 slices. SLEs were not a priori resistant (26) (here Figure S1 in Supplementary Material).
Resistance of SLEs Against Carbamazepine, Valproic Acid, and Phenytoin Extends to Cortical Tissue Slices
Seizure-like events predominantly persisted in slices from hippocampal and temporal neocortical specimens. Figures 4 and 5 give examples of drug effects on induced SLEs in the DG, SUB, and TCx in sister slices from the same hippocampal or cortical specimen and show that SLEs persisted in presence of CBZ, VPA, and PHT.
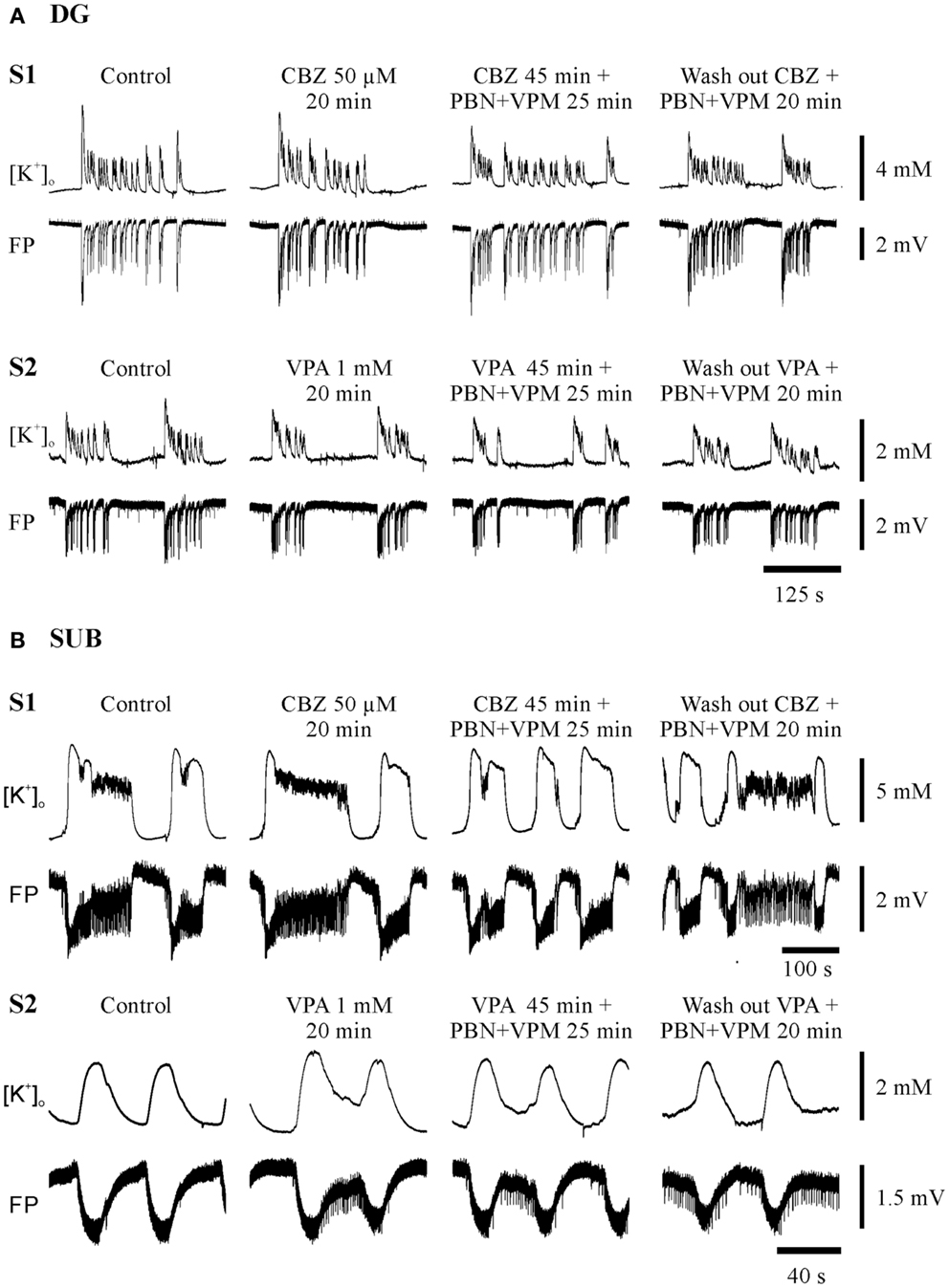
Figure 4. Typical experiments in sister-slices from the same hippocampal specimen show persistence of SLE at the end of each protocol sequences (control, AED, AED + MDTIs, washout). (A) In the dentate gyrus (DG), (B) in the subiculum (SUB), S1 slice 1 with application of CBZ, S2 slice 2 with application of VPA for both regions. The drugs applied are described above the pairs of traces, which display [K+]o (top), and field potential (FP) bottom. Amplitudes and time are given by calibration bars on the right.
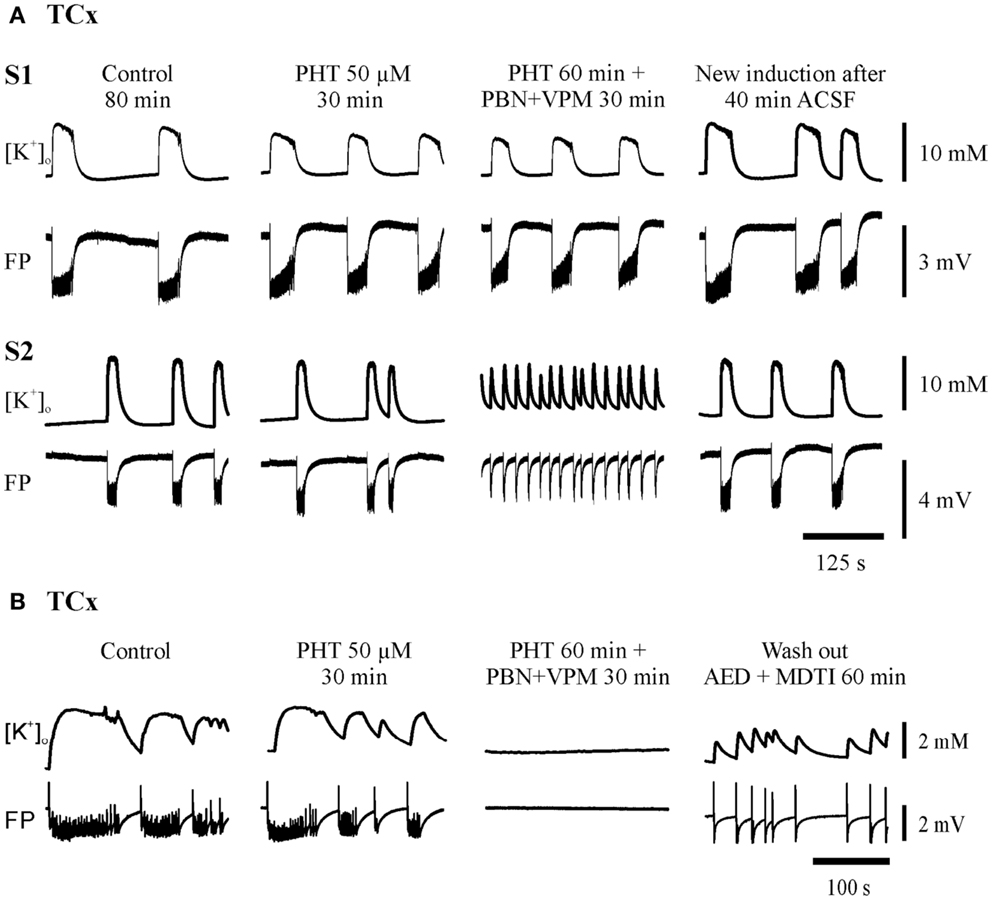
Figure 5. Typical experiments in slices from TCx-specimens. The traces were taken at the end of all subsequent protocol sequences (control, AED, AED + MDTIs, washout). (A) In sister-slices from the same specimen: S1 displaying resistance of SLEs against PHT and PHT + PBN + VPM; S2 illustrates resistance against PHT but transition of SLE to RET during co-administration (PHT + PBN + VPM); (B) in another specimen also displaying resistance of SLE against PHT but suppression of SLE during co-administration (PHT + PBN + VPM). The drugs applied are described above each pair of traces, which display [K+]o (top) and field potential (FP) (bottom). Calibration bars for amplitude and time are given on the right.
The synopsis of all AED drug experiments (Figure 6) revealed that initial SLEs (120 slices) persisted in 90% of slices, were replaced by RETs in 8.4%, and suppressed in only 1.6%, in spite of high AED-concentrations in the perfusion solution. Regarding the different specimens, AEDs were similarly effective in modifying SLEs in hippocampal and TCx slices (Figure 6A).
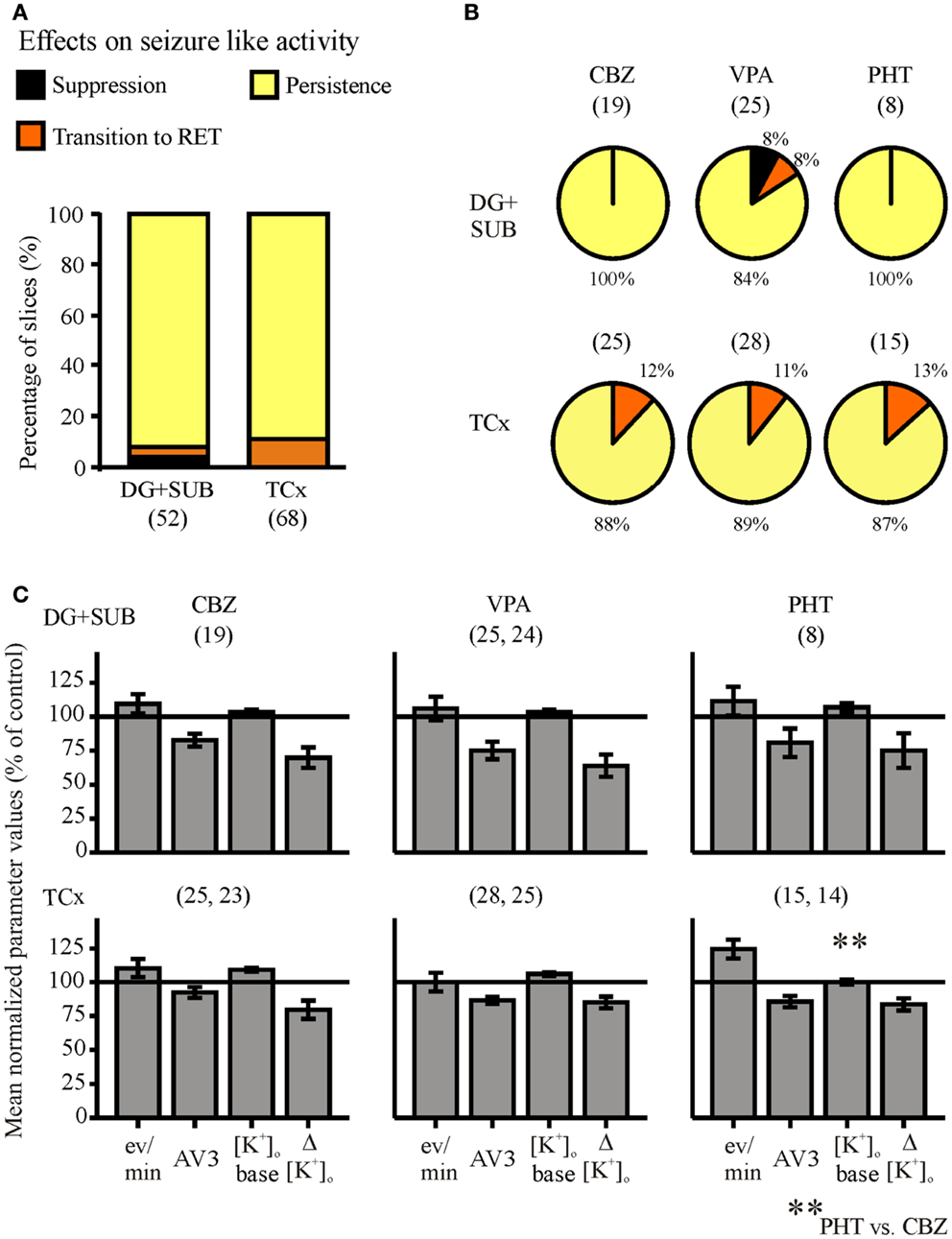
Figure 6. Summarized effects of anti-epileptic drugs (AEDs) on seizure-like events. Categories of drug effects, marked by different colors, are shown on top of (A). (A) Proportional distribution of AED-effects in hippocampal and cortical slice-groups. (B) Effects of carbamazepine (CBZ), valproate (VPA), and phenytoin (PHT) in hippocampal (top) and neocortical slices (bottom). (C) Corresponding averaged remaining normalized parameter values for event rate (eV/min), AV3 (average of normalized values for sfp, peak, and duration), [K+]o base, and ([K+]o. Ordinates: (A) percentage of slices; (C) means ± SEM for remaining percentages of control values. Signs for significant differences between ranges of values for PHT vs. CBZ **p ≤ 0.01. Numbers in parenthesis give number of slices in sub-samples (A,B), or number of slices analyzed (C). The second numbers in parenthesis of (C) display the number of slices with reliable measurements of [K+]o.
With respect to the three AEDs tested (Figure 6B), we found in slices from hippocampal and cortical specimens that effects of CBZ, VPA, and PHT were not significantly different, suggesting that the efficiency of the three AEDs in the chosen concentration range is similar. Studies of drug effects on seizure parameters (Figure 6C) also revealed that there were no significant differences between the different drugs, except for the baseline [K+]o in slices from cortical specimens, which differed between CBZ and PHT.
Seizure-Like Activity also Persists in the Presence of Drug Transport Inhibitors
When the MDTIs VPM and/or PBN were applied first (Figure 7, second column of traces; Figure 8, summary of 41 slices), the proportion of slices with continuing SLEs amounted to 85.4%. SLEs were never suppressed but could be transformed to RETs. Regional differences in sensitivity to MDTIs were observed. In hippocampal slices (n = 19) SLEs persisted in 100% of slices while in cortical slices (n = 22) SLEs persisted only in 73% (p = 0.023, Fisher’s exact test), indicating that induced activity in cortical slices was more sensitive to MDTIs than in hippocampal slices (Figure 8A). Some slices were investigated for singular effects of PBN (n = 11) or VPM (n = 7). There were no proportional differences between effects of PBN, VPM, and PBN plus VPM in hippocampal as well as cortical slices (Figure 8B). Nevertheless, it should be noted that in four temporal cortex slices in which VPM was applied alone (Figure 8B, bottom), this drug led to transition of SLE into RETs in two slices, an effect that was not seen with PBN. Similarly, seizure parameters in temporal cortex slices (Figure 8C) showed that effects of VPM were conspicuously larger than for PBN. However, the difference could not be proven significant, due to the small number of slices tested.
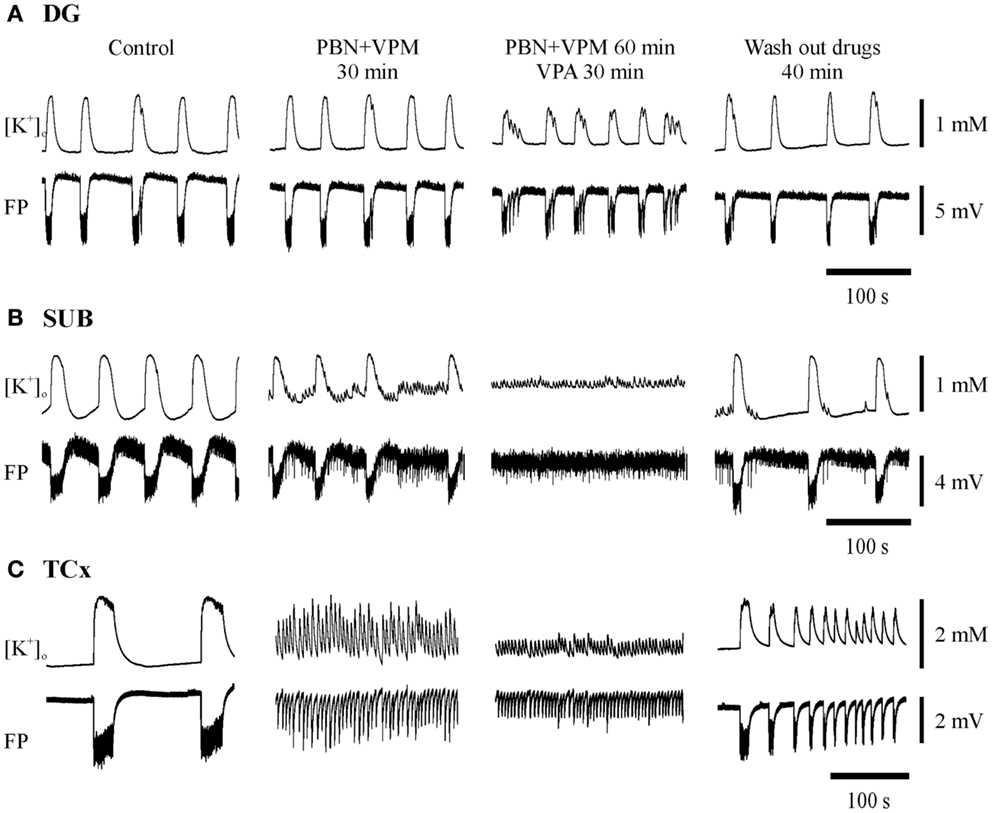
Figure 7. Typical experiments with administration of probenecid (PBN) and verapamil (VPM) followed by addition of an AED in the three regions investigated. Substances applied are described above the pairs of traces displaying [K+]o (top) and FP (bottom). Traces were taken at the end of each protocol-sequence. (A,B) Synchronous recordings from one hippocampal specimen in DG and SUB display resistance of SLE against PBN + VPM in both regions while co-administration with VPA resulted in persistence of SLEs in the DG and in replacement of SLEs by RETs in the SUB; (C) recordings from a cortical specimen of another patient show transition to RET already after application of PBN + VPM, thereby indicating seizure modifying effects of MDTIs, which become more pronounced after co-administration with VPA. (A,C) Amplitudes and time are given by calibration bars on the right.
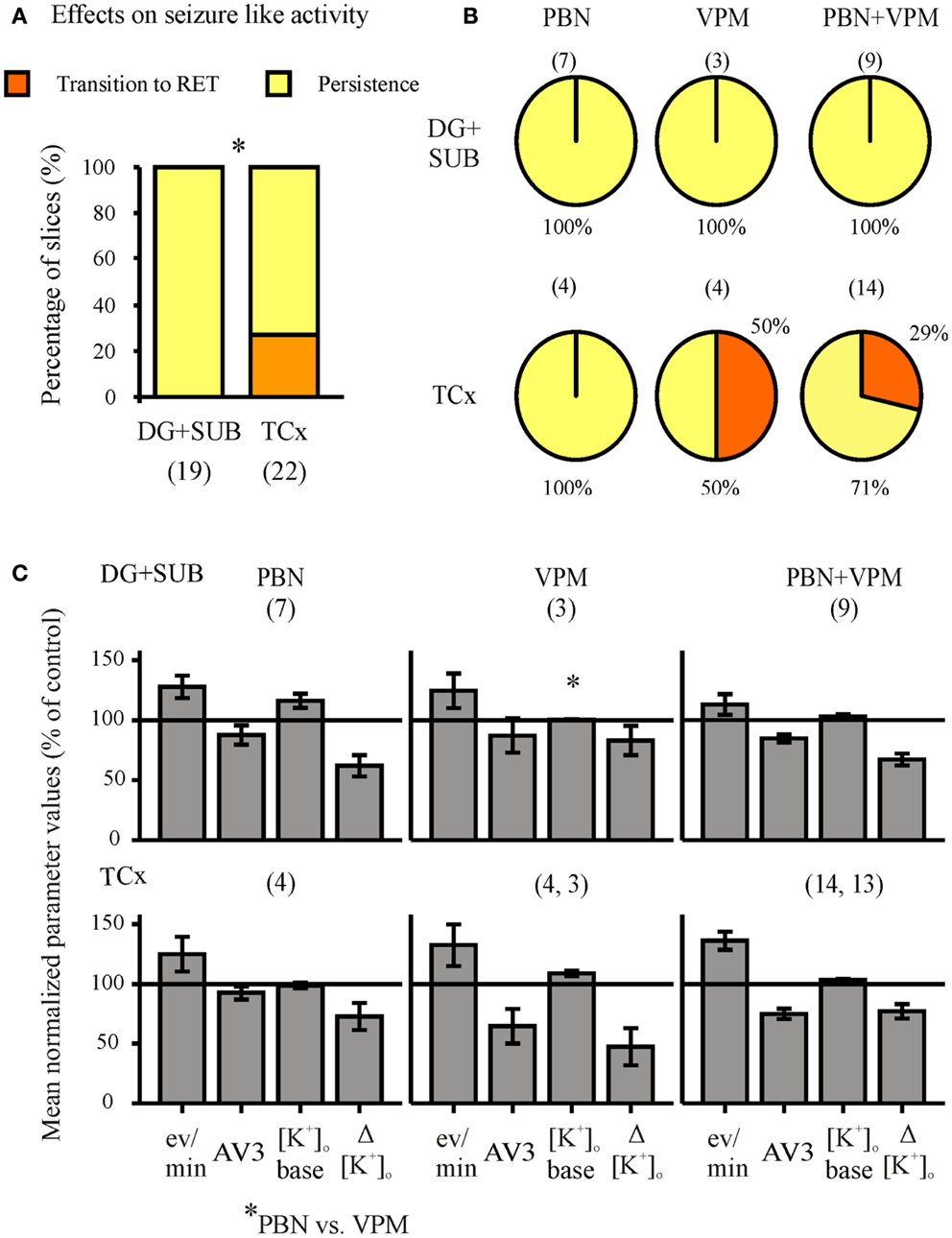
Figure 8. Summarized effects of unspecific inhibitors of P-glycoprotein (Pgp) and multi-drug resistance-associated proteins (MRPs). Categories of drug effects, marked by different colors, are shown on top of (A). (A) Proportional distribution of effects after administration of MDTIs in hippocampal and cortical slice-groups. (B) Circles representing the distribution of effects by probenecid (PBN), verapamil (VPM), and both PBN and VPM in hippocampal (top) and cortical slice-groups (bottom). (C) Corresponding averaged remaining normalized parameter values for event rate, AV3 (average of normalized values for sfp, peak, and duration), [K+]o base, and ([K+]o. Ordinates: (A) percentage of slices; (C) means ± SEM for percentages of control values. Signs for significant differences between proportional distributions *p ≤ 0.05, or ranges of values for PBN vs. PBN + VPM *p ≤ 0.05. Numbers in parenthesis give number of slices in sub-samples (A,B). The numbers in (B) also apply to (C).
Co-Administration of AEDs and MDTIs Increases Responses of Slices
In order to find out whether co-administration of anti-seizure drugs and MDT-inhibitors is able to reverse resistance to standard AEDs, we added PBN and/or VPM to the perfusion solution already containing one AED (AED + MDTIs) (Figures 4 and 5, third column of traces). Alternatively, one AED was added to the perfusion solution already containing MDTIs (MDTIs + AED) (Figure 7, third column of traces). As there were no differences of co-administration effects on SLEs resistant against AEDs or MDTIs (n = 143, p = 0.234, Fisher’s exact test), the subsamples AED + MDTI and MDTI + AED were pooled. Together, during combined treatment with an AED and MDTIs, SLEs persisted in 63% of slices, became converted into RETs in 30%, and suppressed in 7%. Thus, co-application of an AED and MDTIs resulted more frequently in transition to RET compared to slices treated with AEDs or MDTIs alone (p < 0.001, McNemar–Bowker test). Different from slices of cortical specimens, SLEs in hippocampal slices continued more often (69 vs. 58%), were more often suppressed (10.4 vs. 3.9%), and less often transformed to RET (21 vs. 38%; Figure 9A, p = 0.042).
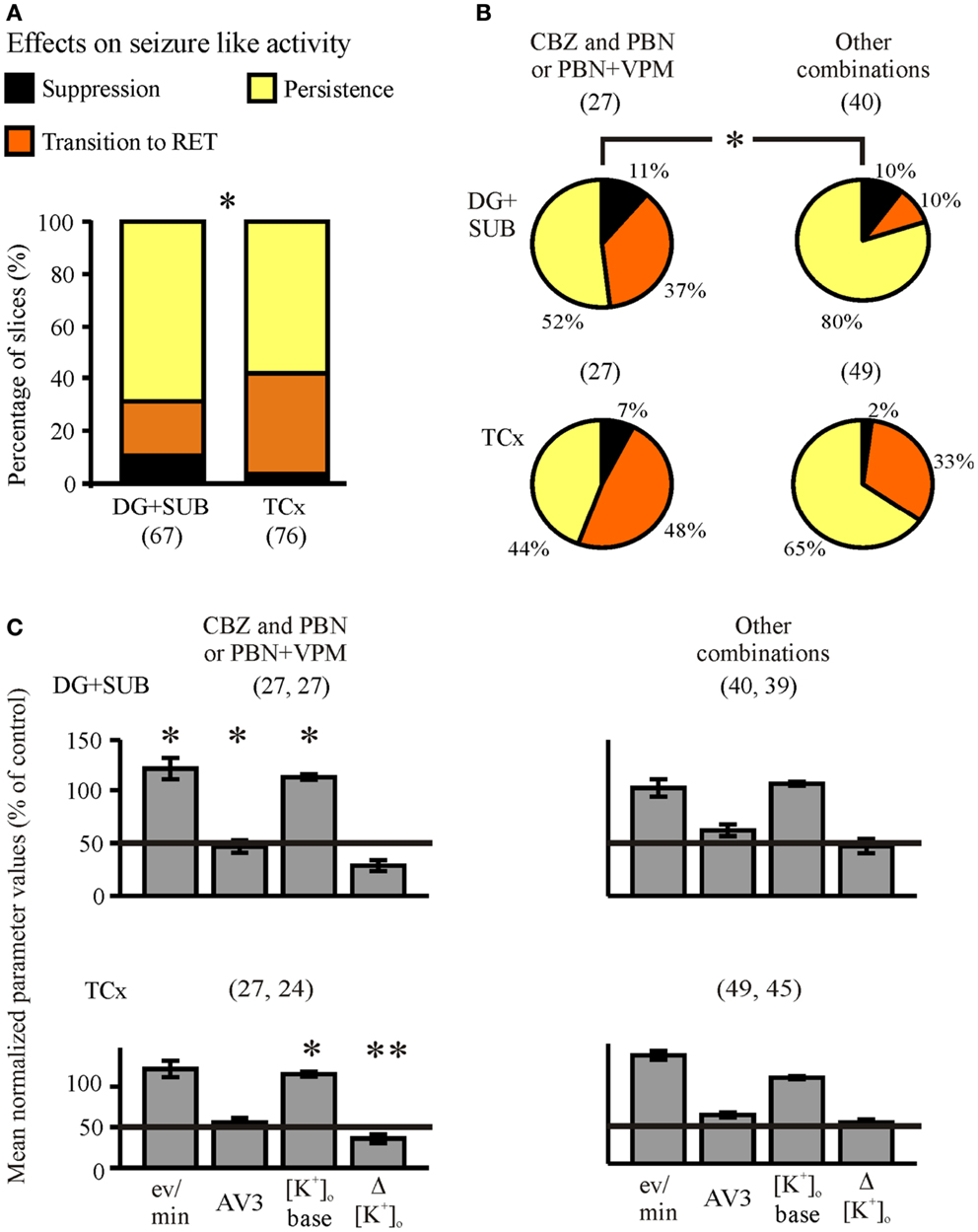
Figure 9. Summarized effects of one AED and MDTIs (co-administration). Categories of drug effects, marked by different colors, are shown on top of (A). (A) Proportions of slices showing different effects of co-administration in hippocampal and cortical slice-groups. (B) Proportional distributions of effects by CBZ and PBN-containing solutions (CBZ + PBN, CBZ + PBN + VPM) and by other combinations (CBZ + VPM, VPA + PBN, VPA + VPM, VPA + PBN + VPM, and PHT + PBN + VPM), given in circles for hippocampal (top) and cortical slice-groups (bottom). (C) Corresponding averaged remaining normalized parameter values for event rate (eV/min), AV3 (average of normalized values for sfp, peak, and duration), [K+]o base, and ([K+]o. Ordinates: (A) percentage of slices; (C) means ± SEM for remaining percentages of control values. Signs for significant differences between proportional distributions of effects or between ranges of values in separate groups (treatment or regions) *p ≤ 0.05, **p ≤ 0.01. Numbers in parenthesis give number of slices in sub-samples (A,B) or number of slices analyzed (C). The second number in parenthesis of (C) displays the corresponding number of slices with reliable measurements of [K+]o.
Concerning the combinations of AEDs and MDTIs administered (Figure 9B), we found that SLEs were more often sensitive to CBZ + PBN or CBZ + PBN + VPM (n = 54) than to all other combined applications (n = 89, p = 0.017, Fishers exact test). Although this was numerically true in slices from hippocampal and cortical specimens, the error probability reached significance (p = 0.022) in hippocampal specimens only. Out of the remaining normalized parameter values event rate, AV3 and [K+]o base significantly differed between the combinations in the hippocampus, while [K+]o base and Δ[K+]o differed in the cortex (Figure 9C). Taken together, co-administration of one AED and MDTIs modifies appearance of SLEs by transition to RETs but rarely by suppression.
Co-Administration Experiments Reveal Homogeneous and Heterogeneous Responses in Slices of the Same Patient
When studying the effects of CBZ in the DG of hippocampal slices from the same patient (26), we noted that the substance failed to suppress SLEs in one slice but was effective in another. This may indicate differences of drug sensitivity throughout the resected tissue, prompting investigation of multiple slices from each patient.
One to seven slices from one specimen could be investigated for responses of SLEs to co-administration of AEDs and MDTIs (Figure 10A). In specimens with more than one slice (Figure S4 in Supplementary Material), we observed different effects in 52.6% of 19 hippocampal specimens and in 42.9% of 28 cortical specimens. Consequently, categorization of the patient’s in vitro drug response also needs consideration of heterogeneity of effects in two or more slices of the same patient.
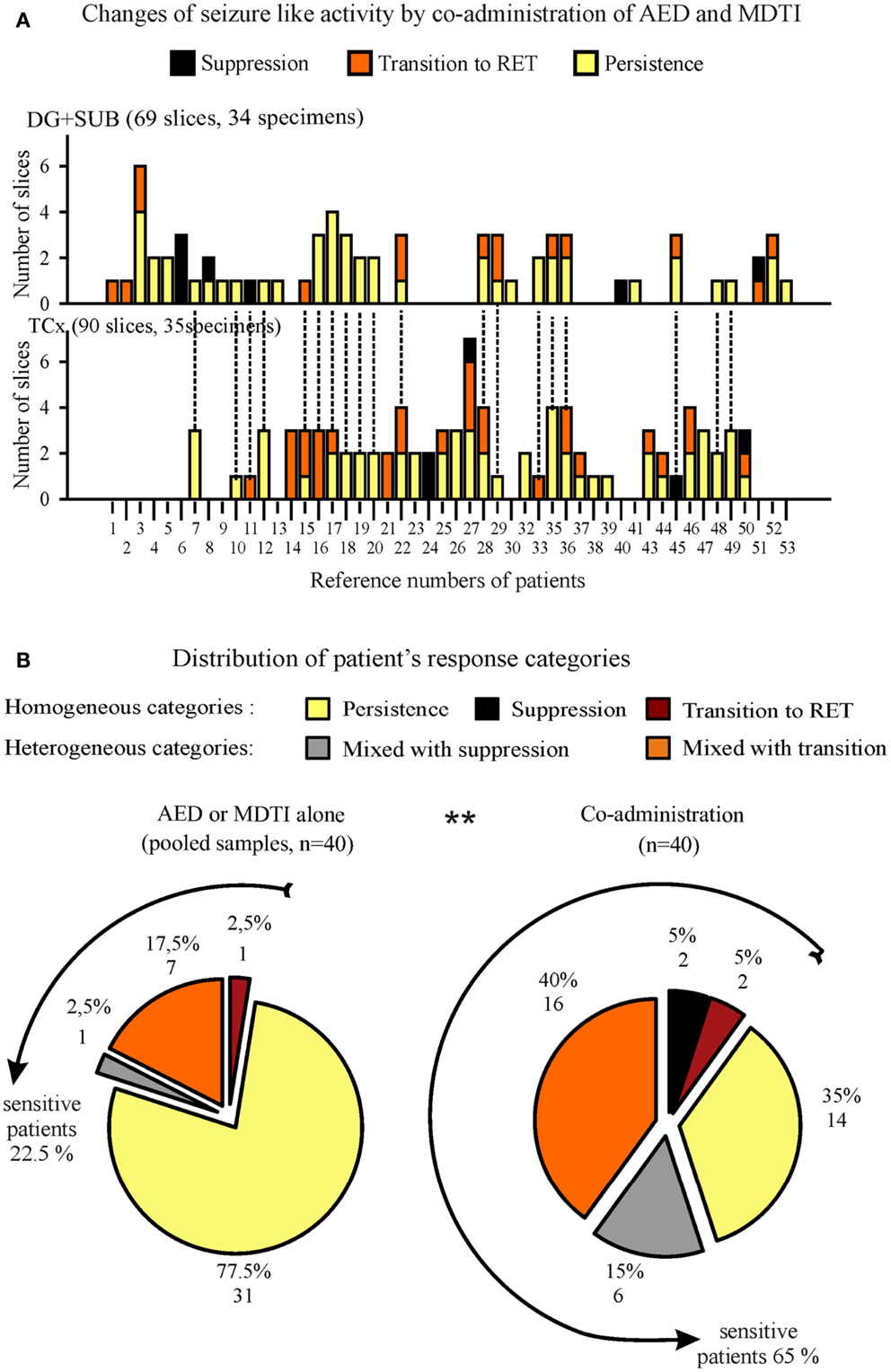
Figure 10. Summarized responses to co-administration of AED and MDTIs in specimens and patients. (A) Effects in slices from hippocampal (top) and cortical (bottom) specimens, ordered by patient number for determination of the patient’s response category. Patients providing both, hippocampal and cortical specimens are marked by dotted lines on top of columns in (B). (B) Proportional distribution of different response categories for patients with at least two slices in the analysis. In order to evaluate the changes by co-administration, we present two circles, one for responses to AED or MDTIs alone (pooled sample, left) and another one for responses to co-administration (right). The lines around circles indicate subgroups of patients who were “sensitive” to the respective treatment in at least one slice. Numbers in parenthesis denote the number of slices/specimens (A) or the number of patients (B).
In summary of both specimens (Figure 10B), 40 patients provided between 2 and 7 slices to the categorization procedure. In Figure 10B (left), responses to AED or MDTIs alone were summarized. Pharmacoresistance was homogenous in 77.5% of patients. In the remaining 22.5% of patients, sensitivity to AED or MDTIs was found in at least one slice. In Figure 10B (right), the combined effects of AEDs and MDTIs were summarized. The graph shows a significantly larger proportion of patient material in which we observed sensitivity (65%). Compared to treatment with either AEDs or MDTIs alone, we noted a strong increase in the sensitivity to co-administration of one AED and MDTIs (p = 0.001, McNemar).
Relations of in vitro Response Categories to Patient Data
Patients’ with at least two slices in the analysis were compared between in vitro “sensitive” and “resistant” groups concerning co-administration of an AED and MDTIs as displayed in Table 1.
We also tested for age at resection, onset of epilepsy, gender, and site of resection and found no significant differences between the two groups (p = 0.314; 0.100, 0.739, 0.510). The two patient groups only differ with respect to the distribution of in vivo seizure frequencies (high for sensitive patients, low for resistant patients) but not between their quotients of brain tissue- to serum-concentrations of AEDs. Besides, we recognized that local tissue concentrations of AEDs or their metabolites (in case of CBZ or OXC) are positively correlated to corresponding serum concentrations in the hippocampus (Pearson’s r = 0.764, n = 18, p < 0,001) but not in the neocortex (r = 0.253, N = 15, p = 0.362). Also, the local expression rates of MRPs and Pgp determined for neurons and astrocytes in corresponding subgroups do not differ (Table 1). These findings put doubt on a relevant role of Pgp and MRPs in parenchymal drug resistance.
To find out, whether specimen-specific variations of patient data may differ, we performed a separate analysis for both specimens and display a corresponding table for TCx-specimens in the Supplementary Material (Table S1 in Supplementary Material). The error probability of 0.046 for the difference between brain/serum concentrations of AEDs between resistant and sensitive tissue in TCx-specimens was not accepted as “significant” (SPSS 18) because of the small sample sizes and the large spread of values in both groups.
Compared with the patient groups providing TCx-specimens (sensitive vs. resistant) corresponding subgroups of hippocampal specimens differ concerning age of onset (8.5 ± 2.92 years, n = 6 vs. 22.0 ± 3.61 years, n = 11) and gender [sensitive patients are mainly male (75%) resistant patients mainly female (82%, p = 0.024 Fisher’s exact test)]. For subgroups in both specimens (TCx, Hippocampus), there is no difference with respect to pathologies (TCx p = 0.124, Hippocampus p = 0.500 for Wyler graduation), resistance, or AED-treatment at operation.
Discussion
In the present study, we investigated effects of AEDs and inhibitors of Pgp and MRPs on artificially induced recurrent SLEs in slices from hippocampal and cortical specimens of drug resistant patients.
AED-Resistance of Patients is Preserved in the Resected Tissue
The vast majority of slices retained SLEs when CBZ, VPA, or PHT was applied. In a previous study of the DG, we described that epileptiform activity induced in slices of drug resistant patients remained predominantly resistant to CBZ (26). Our present findings on seizure-like activity support the previous statement and we now show that this applies also to VPA or PHT. Additionally, the results indicate that not only the DG but also the SUB and TCx of surgically resected tissue are frequently characterized by pharmacoresistance of SLEs. Considering that in certain patients the blood brain barrier is leaky (39–41), and that drug concentrations in the tissue are close to plasma levels or even exceed those (42), we used high AED-concentrations in the tissue experiments. Despite such high concentrations, SLEs were resistant.
The present results are in line with reports on effects of AEDs on epileptiform activity in human cortical slices, focused on short recurrent discharges induced by lowering [Mg2+] in the perfusion medium. In a study by Oby and colleagues, application of 50 μM CBZ or PHT resulted in a slight modification of the repetition rate and a decline of the integrated amplitude of short recurrent epileptiform discharges (43). Levetiracetam in human temporal cortex slices did not affect spontaneous sharp waves or Mg2+-free induced epileptiform FPs (44). The agreement of results from several studies using different induction protocols suggests that pharmacological investigations aiming at detection of new suitable drugs for drug resistant patients are also possible in centers where only neocortical tissue is available.
Verapamil and Probenecid Partially Modify Induced Epileptiform Activity in Cortical Slices
Verapamil and PBN were used as broad spectrum drug transport inhibitors. However, these agents also have unspecific anticonvulsant effects. Indeed, VPM and PBN, administered in the absence of AEDs, led to transformation of SLEs into RETs in 30% of TCx slices, which was never observed in the DG or SUB. This effect might be related to direct anticonvulsant effects.
Verapamil, known as an organic L-type calcium channel antagonist, was shown to exert anticonvulsant effects in a number of experimental models of epilepsy (45–49). Even in human tissue, VPM reduced spontaneous FP transients and low Mg2+ and bicuculline-induced epileptiform activity (50, 51).
Less is known about anticonvulsant effects of PBN. PBN might affect release of ATP and (conversion to) adenosine (52) as well as regulatory processes within a complex metabolic chain providing for neuroprotective effects of the NMDA-receptor inhibitor kynurenic acid and also the neurotoxic quinolinic acid (53, 54). Substances such as adenosine (55–59), taurine (60, 61), and kynurenic acid (54) are known to exert anticonvulsant effects. More recently, it was suggested that PBN blocks pannexin hemi channels (62), which might contribute to seizure activity when opened (63). Thus, our finding that PBN and VPM administered before application of CBZ, VPA, or PHT can convert SLEs into RETs suggests that any of those agents or their combination support the effect of AEDs and therefore might indicate a rational basis for multi drug treatment in some instances.
Is There Evidence for a Contribution of Pgp or MRPs to Drug Resistance?
Clinical data exist, which show that VPM can sometimes be used as an additive drug in the treatment of refractory epilepsy (28), in severe myoclonic epilepsy in infancy (64), and in the treatment of pharmacoresistant status epilepticus (65–67).
In the present study, co-administration of one AED and MDTIs in slices resulted more often in a transition of SLEs to RETs than administration of AEDs or MDTIs alone. However, full suppression of SLEs suggesting reversal of drug resistance was rarely observed. This would be in agreement with a previous report of Rivers and colleagues who could not assess reversion of the drug resistance phenotype and possible interactions with ABC-drug transporters for any of the AEDs used in our study (68).
We showed that CBZ co-applied with PBN− or PBN + VPM containing solutions was more effective than other combinations, particularly in the hippocampus. The finding that in the hippocampus weak or no effects were noted in presence of AEDs or MDTIs alone but strong effects during co-administration of CBZ and MDTIs might be a sign for interaction of metabolites of CBZ with MRPs in the hippocampus. However, our previous results on slices of a subgroup of similar patients showed that efflux pump function is preserved in both specimens (57% of temporal cortex slices and 60% of hippocampal slices). Moreover, corresponding sub-samples of slices did not differ with respect to expression of MDTs in the TCx and hippocampus (29).
We further calculated tissue-serum quotients of AED-levels in a subset of patients and found that they were only rarely and not homogeneously low. Although there are no data about the unbound fraction of AEDs in the present study, the value for total levels of AEDs in the tissue told us that AEDs reached the brain tissue in both subgroups, patients with in vitro sensitive as well as resistant tissue, at least in the hippocampus.
Provided that PHT is a substrate of Pgp but not VPA (69–71), blocking the transport might augment effects of PHT. However, a selective augmentation for PHT and MDTIs was not observed. Metabolites of CBZ with anticonvulsant or convulsant properties [Ref. (72) for a convulsant metabolite of CBZ] may be subject to drug efflux pump transport (73, 74). Therefore, it is of interest that we detected more frequently and stronger effects of CBZ when co-administered with PBN or PBN and VPM than for VPA and PHT in corresponding experimental conditions. In contrast, Luna-Tortos and colleagues reported that there was no PBN- or MK571-inhibitable transport of CBZ, VPA, and PHT in experiments on cell lines transfected with human MRPs (75).
Taken together, it seems to be less likely that our results on modification of SLEs can be ascribed to increased drug concentration at cellular targets of AEDs but rather are the consequence of combined drug effects at several anti-seizure targets.
Heterogeneity of in vitro Drug Effects
Here, we show for the first time that slices of the same patient display different responses of SLEs to co-administration of one AED and MDTIs. This might be caused by different consequences of surgical handling and slicing in the laboratory as well as by different pathological changes in individual slices of the same specimen. To clarify the reasons for heterogeneity of pharmacological responses in slices of the same specimen a new study of differential morphological, biochemical, and immunological properties in “responding” and “non-responding” slices is ongoing. When responses of hippocampal and cortical specimens were categorized for treatment with AED or MDTIs, we noted mixed effects (resistance and pattern change) in 19% of patients. Such mixed effects increased to 54% during co-administration of one AED and MDTIs, and homogeneous suppression also increased from 0 to 7% of 41 patients. Nevertheless, the finding that there were no differences between the response categories regarding MRP- and Pgp local expression rates or tissue/serum quotients of AED-concentrations suggest a rather marginal contribution of MDTs to drug resistance in tissue without a functional blood brain barrier.
Limitations
1. We did not observe spontaneous SLEs in human tissue slices and needed induction of seizure-like activity by modification of the electrolyte supplying the slices. The fact that spontaneous seizures were never detected presumably results from interruption of essential connectivity by surgery and preparation of slices. Even if the tissue would be capable to generate SLEs spontaneously, the low incidence of seizures in TLE patients would make it unlikely that we observed spontaneous seizures during the possible observation time.
2. Our study does not address the mechanisms underlying different vulnerability of hippocampal and neocortical tissue to modifications in potassium concentration. With respect to the requirement of bicuculline for induction of SLEs in slices of cortical specimens it might be of advantage to synchronously investigate changes of inter-neurons concerning morphology and molecular biology of their afferent connections and synaptic transmission to target cells.
3. It is presently not known whether resistance of induced SLEs in slices reflects resistance of spontaneous seizures of patients and if potential mechanisms of multidrug resistance found in this model system can be translated to the clinical setting of refractory epilepsy. None of the available models of pharmacoresistance so far is clinically validated. There are no drugs available, which are more effective in such models than the clinically employed AEDs. This is also true for the induced SLEs in this study. However, when potential seizure controlling drugs are effective ex vivo in human tissue, this might encourage further development and eventually testing in pharmacoresistant patients.
4. The non-selectivity of PBN and VPM with respect to inhibition of drug transport provides no clear argument against the design of our co-administration experiments. We found similar effects of adding MK 571 or PBN to VPA and VPM in the same slice (Figure S3 in Supplementary Material). Additionally, our findings that VPM and PBN altered drug efflux pump function in slices (29) support the use of VPM and PBN as drug transport inhibitors.
Conclusion
The results of the present study indicate that SLEs in resected human temporal cortex and hippocampus are predominantly resistant to CBZ, VPA, and PHT as well as to drug transport inhibitors like VPM and PBN.
Complete suppression of seizure-like activity was rarely observed after co-administration of an AED with PBN and VPM, suggesting that drug resistance in slices is not reversed by inhibition of drug transport with PBN and VPM. This suggestion is supported by the fact that patients with in vitro sensitive tissue do not differ from patients with in vitro resistant tissue concerning expression rates of Pgp and MRPs or quotients of tissue/serum levels of anticonvulsants.
Co-administration of an AED with PBN and VPM in vitro increased the efficiency in modifying induced seizures (SLEs), rather by adding several anticonvulsant effects than by interaction with drug transport.
The heterogeneity of drug effects in different slices of the same patient remains to be elucidated.
Provided that the resistance of induced SLEs as well as the heterogeneity of drug effects reflect rather disease dependent than methodological consequences, our findings suggest that human tissue slices can be used for the evaluation of new drugs for patients with drug refractory epilepsy, i.e., by performing experimental multi-center studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. H. Siegmund, Dr. H-J. Gabriel, and B. Schacht for technical support, to D. Meincken-Jäggi, S. Bartsch, and particularly to N. Wieczorek for documentation, to S. Frosinski and Dr. K. Schulze for secretarial assistance, and to M. L. Anderson for language correction and critical comments on the manuscript. Additionally, the authors owe thanks to the Department of Neuropathology at the Charité for cooperation. Funding: This research was supported by a DFG grant (SFB TR3 C7, EXC 257 NeuroCure, the EU-grants FP6, LSAM-CT-206-037315, FP7 Desire Project Nr. 602531), and the Hertie foundation.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fneur.2015.00030/abstract
Abbreviations
AEDs, anti-epileptic drugs; CBZ, carbamazepine; DG, dentate gyrus; FP, extracellular field potential; [K+]o, extracellular potassium concentration; MDTs, multidrug transporters, MDTIs, multidrug transporter inhibitors; MRP, multidrug resistance-associated protein; PBN, probenecid; Pgp, P-glycoprotein; PHT, phenytoin; RETs, recurrent epileptiform transients; SLEs, seizure-like events; SUB, subiculum; TCx, temporal neocortex; TLE, temporal lobe epilepsy; VPA, valproate; VPM, verapamil.
References
1. Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology (1998) 51(5):1256–62. doi: 10.1212/WNL.51.5.1256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Schmidt D, Loscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr (2009) 9(2):47–52. doi:10.1111/j.1535-7511.2008.01289.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Loscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia (2006) 47(8):1253–84. doi:10.1111/j.1528-1167.2006.00607.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Remy S, Gabriel S, Urban BW, Dietrich D, Lehmann TN, Elger CE, et al. A novel mechanism underlying drug resistance in chronic epilepsy. Ann Neurol (2003) 53(4):469–79. doi:10.1002/ana.10473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain (2006) 129(Pt 1):18–35. doi:10.1093/brain/awh682
6. Beck H. Plasticity of antiepileptic drug targets. Epilepsia (2007) 48(Suppl 1):14–8. doi:10.1111/j.1528-1167.2007.00994.x
7. Lehmann TN, Gabriel S, Eilers A, Njunting M, Kovacs R, Schulze K, et al. Fluorescent tracer in pilocarpine-treated rats shows widespread aberrant hippocampal neuronal connectivity. Eur J Neurosci (2001) 14(1):83–95. doi:10.1046/j.0953-816x.2001.01632.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Lehmann TN, Gabriel S, Kovacs R, Eilers A, Kivi A, Schulze K, et al. Alterations of neuronal connectivity in area CA1 of hippocampal slices from temporal lobe epilepsy patients and from pilocarpine-treated epileptic rats. Epilepsia (2000) 41(Suppl 6):S190–4. doi:10.1111/j.1528-1157.2000.tb01580.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Salar S, Maslarova A, Lippmann K, Nichtweiss J, Weissberg I, Sheintuch L, et al. Blood-brain barrier dysfunction can contribute to pharmacoresistance of seizures. Epilepsia (2014) 55(8):1255–63. doi:10.1111/epi.12713
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Tishler DM, Weinberg KI, Hinton DR, Barbaro N, Annett GM, Raffel C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia (1995) 36(1):1–6. doi:10.1111/j.1528-1157.1995.tb01657.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Lazarowski A, Sevlever G, Taratuto A, Massaro M, Rabinowicz A. Tuberous sclerosis associated with MDR1 gene expression and drug-resistant epilepsy. Pediatr Neurol (1999) 21(4):731–4. doi:10.1016/S0887-8994(99)00074-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Lee G, Dallas S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev (2001) 53(4):569–96. doi:10.1146/annurev.pharmtox.41.1.569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sisodiya SM, Lin WR, Squier MV, Thom M. Multidrug-resistance protein 1 in focal cortical dysplasia. Lancet (2001) 357(9249):42–3. doi:10.1016/S0140-6736(00)03573-X
14. Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci (2005) 6(8):591–602. doi:10.1038/nrn1728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev (2006) 58(2):140–61. doi:10.1124/pr.58.2.3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lazarowski A, Czornyj L, Lubienieki F, Girardi E, Vazquez S, D’Giano C. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia (2007) 48(Suppl 5):140–9. doi:10.1111/j.1528-1167.2007.01302.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Loscher W. Mechanisms of drug resistance in status epilepticus. Epilepsia (2007) 48(Suppl 8):74–7. doi:10.1111/j.1528-1167.2007.01357.x
18. Aronica E, Sisodiya SM, Gorter JA. Cerebral expression of drug transporters in epilepsy. Adv Drug Deliv Rev (2012) 64(10):919–29. doi:10.1016/j.addr.2011.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Marchi N, Hallene KL, Kight KM, Cucullo L, Moddel G, Bingaman W, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med (2004) 2:37. doi:10.1186/1741-7015-2-37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Sisodiya SM, Martinian L, Scheffer GL, van der Valk P, Scheper RJ, Harding BN, et al. Vascular colocalization of P-glycoprotein, multidrug-resistance associated protein 1, breast cancer resistance protein and major vault protein in human epileptogenic pathologies. Neuropathol Appl Neurobiol (2006) 32(1):51–63. doi:10.1111/j.1365-2990.2005.00699.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Begley DJ, Lechardeur D, Chen ZD, Rollinson C, Bardoul M, Roux F, et al. Functional expression of P-glycoprotein in an immortalised cell line of rat brain endothelial cells, RBE4. J Neurochem (1996) 67(3):988–95. doi:10.1046/j.1471-4159.1996.67030988.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Liu JY, Thom M, Catarino CB, Martinian L, Figarella-Branger D, Bartolomei F, et al. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain (2012) 135(Pt 10):3115–33. doi:10.1093/brain/aws147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain (2002) 125(Pt 1):22–31. doi:10.1093/brain/awf002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Aronica E, Gorter JA, Ramkema M, Redeker S, Ozbas-Gerceker F, van Vliet EA, et al. Expression and cellular distribution of multidrug resistance-related proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Epilepsia (2004) 45(5):441–51. doi:10.1111/j.0013-9580.2004.4510erratum.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Nies AT, Jedlitschky G, Konig J, Herold-Mende C, Steiner HH, Schmitt HP, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience (2004) 129(2):349–60. doi:10.1016/j.neuroscience.2004.07.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Jandova K, Pasler D, Antonio LL, Raue C, Ji S, Njunting M, et al. Carbamazepine-resistance in the epileptic dentate gyrus of human hippocampal slices. Brain (2006) 129(Pt 12):3290–306. doi:10.1093/brain/awl218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Zhang C, Kwan P, Zuo Z, Baum L. The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv Rev (2012) 64(10):930–42. doi:10.1016/j.addr.2011.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Summers MA, Moore JL, McAuley JW. Use of verapamil as a potential P-glycoprotein inhibitor in a patient with refractory epilepsy. Ann Pharmacother (2004) 38(10):1631–4. doi:10.1345/aph.1E068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kovacs R, Raue C, Gabriel S, Heinemann U. Functional test of multidrug transporter activity in hippocampal-neocortical brain slices from epileptic patients. J Neurosci Methods (2011) 200(2):164–72. doi:10.1016/j.jneumeth.2011.06.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, et al. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci (2011) 14(5):627–34. doi:10.1038/nn.2790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, et al. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci (2004) 24(46):10416–30. doi:10.1523/JNEUROSCI.2074-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Kann O, Kovacs R, Njunting M, Behrens CJ, Otahal J, Lehmann TN, et al. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain (2005) 128(Pt 10):2396–407. doi:10.1093/brain/awh568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Helmstaedter C, Richter S, Roske S, Oltmanns F, Schramm J, Lehmann TN. Differential effects of temporal pole resection with amygdalohippocampectomy versus selective amygdalohippocampectomy on material-specific memory in patients with mesial temporal lobe epilepsy. Epilepsia (2008) 49(1):88–97. doi:10.1111/j.1528-1167.2007.01386.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Pragst F, Herzler M, Erxleben BT. Systematic toxicological analysis by high-performance liquid chromatography with diode array detection (HPLC-DAD). Clin Chem Lab Med (2004) 42(11):1325–40. doi:10.1515/CCLM.2004.251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Shu SY, Ju G, Fan LZ. The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neurosci Lett (1988) 85(2):169–71. doi:10.1016/0304-3940(88)90346-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol (1990) 296(1):1–22. doi:10.1002/cne.902960102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec (1946) 94:239–47. doi:10.1002/ar.1090940210
38. Neels HM, Sierens AC, Naelaerts K, Scharpe SL, Hatfield GM, Lambert WE. Therapeutic drug monitoring of old and newer anti-epileptic drugs. Clin Chem Lab Med (2004) 42(11):1228–55. doi:10.1515/CCLM.2004.245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Rigau V, Morin M, Rousset MC, de Bock F, Lebrun A, Coubes P, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain (2007) 130(Pt 7):1942–56. doi:10.1093/brain/awm118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. van Vliet EA, da Costa Araujo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain (2007) 130(Pt 2):521–34. doi:10.1093/brain/awl318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Friedman A, Kaufer D, Heinemann U. Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res (2009) 85(2–3):142–9. doi:10.1016/j.eplepsyres.2009.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Rambeck B, Jurgens UH, May TW, Pannek HW, Behne F, Ebner A, et al. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia (2006) 47(4):681–94. doi:10.1111/j.1528-1167.2006.00504.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Oby E, Caccia S, Vezzani A, Moeddel G, Hallene K, Guiso G, et al. In vitro responsiveness of human-drug-resistant tissue to antiepileptic drugs: insights into the mechanisms of pharmacoresistance. Brain Res (2006) 1086(1):201–13. doi:10.1016/j.brainres.2006.02.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Gorji A, Hohling JM, Madeja M, Straub H, Kohling R, Tuxhorn I, et al. Effect of levetiracetam on epileptiform discharges in human neocortical slices. Epilepsia (2002) 43(12):1480–7. doi:10.1046/j.1528-1157.2002.23702.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Aicardi G, Schwartzkroin PA. Suppression of epileptiform burst discharges in CA3 neurons of rat hippocampal slices by the organic calcium channel blocker, verapamil. Exp Brain Res (1990) 81(2):288–96. doi:10.1007/BF00228118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Vezzani A, Wu HQ, Stasi MA, Angelico P, Samanin R. Effect of various calcium channel blockers on three different models of limbic seizures in rats. Neuropharmacology (1988) 27(5):451–8. doi:10.1016/0028-3908(88)90126-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bingmann D, Speckmann EJ. Specific suppression of pentylenetetrazol-induced epileptiform discharges in CA3 neurons (hippocampal slice, guinea pig) by the organic calcium antagonists flunarizine and verapamil. Exp Brain Res (1989) 74(2):239–48. doi:10.1007/BF00248856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Link MC, Wiemann M, Bingmann D. Organic and inorganic calcium antagonists inhibit veratridine-induced epileptiform activity in CA3 neurons of the guinea pig. Epilepsy Res (2008) 78(2–3):147–54. doi:10.1016/j.eplepsyres.2007.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Witte OW, Speckmann EJ, Walden J. Motor cortical epileptic foci in vivo: actions of a calcium channel blocker on paroxysmal neuronal depolarizations. Electroencephalogr Clin Neurophysiol (1987) 66(1):43–55. doi:10.1016/0013-4694(87)90137-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Kohling R, Lucke A, Straub H, Speckmann EJ, Tuxhorn I, Wolf P, et al. Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain (1998) 121(Pt 6):1073–87. doi:10.1093/brain/121.6.1073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Straub H, Kohling R, Luke A, Fauteck JD, Speckmann EJ, Moskopp D, et al. The effects of verapamil and flunarizine on epileptiform activity induced by bicuculline and low Mg2+ in neocortical tissue of epileptic and primary non-epileptic patients. Brain Res (1996) 733(2):307–11. doi:10.1016/0006-8993(96)00802-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Heinrich A, Ando RD, Turi G, Rozsa B, Sperlagh B. K+ depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study. Br J Pharmacol (2012) 167(5):1003–20. doi:10.1111/j.1476-5381.2012.01932.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Vecsei L, Miller J, MacGarvey U, Beal MF. Kynurenine and probenecid inhibit pentylenetetrazol- and NMDLA-induced seizures and increase kynurenic acid concentrations in the brain. Brain Res Bull (1992) 28(2):233–8. doi:10.1016/0361-9230(92)90184-Y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Nemeth H, Robotka H, Kis Z, Rozsa E, Janaky T, Somlai C, et al. Kynurenine administered together with probenecid markedly inhibits pentylenetetrazol-induced seizures. An electrophysiological and behavioural study. Neuropharmacology (2004) 47(6):916–25. doi:10.1016/j.neuropharm.2004.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Kostopoulos G, Drapeau C, Avoli M, Olivier A, Villemeure JG. Endogenous adenosine can reduce epileptiform activity in the human epileptogenic cortex maintained in vitro. Neurosci Lett (1989) 106(1–2):119–24. doi:10.1016/0304-3940(89)90212-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol (1992) 32(5):618–24. doi:10.1002/ana.410320504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Kovac S, Sirin Y, Speckmann EJ, Gorji A. Different regional neuroinhibitory effects of adenosine on stimulus-induced patterns of bioelectric activity of rat hippocampal and neocortical tissues. Neuroscience (2008) 152(2):547–57. doi:10.1016/j.neuroscience.2007.11.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Lee KS, Schubert P, Heinemann U. The anticonvulsive action of adenosine: a postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Res (1984) 321(1):160–4. doi:10.1016/0006-8993(84)90694-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Klaft ZJ, Schulz SB, Maslarova A, Gabriel S, Heinemann U, Gerevich Z. Extracellular ATP differentially affects epileptiform activity via purinergic P2X7 and adenosine A1 receptors in naive and chronic epileptic rats. Epilepsia (2012) 53(11):1978–86. doi:10.1111/j.1528-1167.2012.03724.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kirchner A, Breustedt J, Rosche B, Heinemann UF, Schmieden V. Effects of taurine and glycine on epileptiform activity induced by removal of Mg2+ in combined rat entorhinal cortex-hippocampal slices. Epilepsia (2003) 44(9):1145–52. doi:10.1046/j.1528-1157.2003.01603.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Wu JY, Prentice H. Role of taurine in the central nervous system. J Biomed Sci (2010) 17(Suppl 1):S1. doi:10.1186/1423-0127-17-S1-S1
62. Silverman W, Locovei S, Dahl G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol (2008) 295(3):C761–7. doi:10.1152/ajpcell.00227.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, et al. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science (2008) 322(5907):1555–9. doi:10.1126/science.1165209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Iannetti P, Parisi P, Spalice A, Ruggieri M, Zara F. Addition of verapamil in the treatment of severe myoclonic epilepsy in infancy. Epilepsy Res (2009) 85(1):89–95. doi:10.1016/j.eplepsyres.2009.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Iannetti P, Spalice A, Parisi P. Calcium-channel blocker verapamil administration in prolonged and refractory status epilepticus. Epilepsia (2005) 46(6):967–9. doi:10.1111/j.1528-1167.2005.59204.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Pirker S, Baumgartner C. Termination of refractory focal status epilepticus by the P-glycoprotein inhibitor verapamil. Eur J Neurol (2011) 18(12):e151. doi:10.1111/j.1468-1331.2011.03513.x
67. Schmitt FC, Dehnicke C, Merschhemke M, Meencke HJ. Verapamil attenuates the malignant treatment course in recurrent status epilepticus. Epilepsy Behav (2010) 17(4):565–8. doi:10.1016/j.yebeh.2010.01.166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Rivers F, O’Brien TJ, Callaghan R. Exploring the possible interaction between anti-epilepsy drugs and multidrug efflux pumps; in vitro observations. Eur J Pharmacol (2008) 598(1–3):1–8. doi:10.1016/j.ejphar.2008.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Baltes S, Fedrowitz M, Tortos CL, Potschka H, Loscher W. Valproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J Pharmacol Exp Ther (2007) 320(1):331–43. doi:10.1124/jpet.106.102491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Loscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology (2007) 52(2):333–46. doi:10.1016/j.neuropharm.2006.07.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Luna-Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology (2008) 55(8):1364–75. doi:10.1016/j.neuropharm.2008.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Ghosh C, Marchi N, Hossain M, Rasmussen P, Alexopoulos AV, Gonzalez-Martinez J, et al. A pro-convulsive carbamazepine metabolite: quinolinic acid in drug resistant epileptic human brain. Neurobiol Dis (2012) 46(3):692–700. doi:10.1016/j.nbd.2012.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Zhang C, Zuo Z, Kwan P, Baum L. In vitro transport profile of carbamazepine, oxcarbazepine, eslicarbazepine acetate, and their active metabolites by human P-glycoprotein. Epilepsia (2011) 52(10):1894–904. doi:10.1111/j.1528-1167.2011.03140.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Zhang C, Kwan P, Zuo Z, Baum L. In vitro concentration dependent transport of phenytoin and phenobarbital, but not ethosuximide, by human P-glycoprotein. Life Sci (2010) 86(23–24):899–905. doi:10.1016/j.lfs.2010.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Luna-Tortos C, Fedrowitz M, Loscher W. Evaluation of transport of common antiepileptic drugs by human multidrug resistance-associated proteins (MRP1, 2 and 5) that are overexpressed in pharmacoresistant epilepsy. Neuropharmacology (2010) 58(7):1019–32. doi:10.1016/j.neuropharm.2010.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: TLE, surgically resected tissue, seizure-like events, carbamazepine, sodium valproate, phenytoin, verapamil, probenecid
Citation: Sandow N, Kim S, Raue C, Päsler D, Klaft Z-J, Antonio LL, Hollnagel JO, Kovacs R, Kann O, Horn P, Vajkoczy P, Holtkamp M, Meencke H-J, Cavalheiro EA, Pragst F, Gabriel S, Lehmann T-N and Heinemann U (2015) Drug resistance in cortical and hippocampal slices from resected tissue of epilepsy patients: no significant impact of P-glycoprotein and multidrug resistance-associated proteins. Front. Neurol. 6:30. doi: 10.3389/fneur.2015.00030
Received: 14 October 2014; Accepted: 03 February 2015;
Published online: 18 February 2015.
Edited by:
Jeremy Daniel Slater, University of Texas Medical School at Houston, USAReviewed by:
Mario Alonso, Instituto Nacional de Neurologia y Neurocirugia, MexicoDave F. Clarke, Dell Children’s Medical Center of Central Texas, USA
Copyright: © 2015 Sandow, Kim, Raue, Päsler, Klaft, Antonio, Hollnagel, Kovacs, Kann, Horn, Vajkoczy, Holtkamp, Meencke, Cavalheiro, Pragst, Gabriel, Lehmann and Heinemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uwe Heinemann, Neuroscience Research Center, Institute of Neurophysiology, Charité Universitätsmedizin Berlin, Garystr. 5, Berlin D 14195, Germany e-mail:dXdlLmhlaW5lbWFubkBjaGFyaXRlLmRl
†Present address: Simon Kim, Klinik und Poliklinik für Chirurgie, Abteilung für Unfall- und Wiederherstellungschirurgie, Greifswald, Germany;
Claudia Raue, Department of Neurosurgery, Uniklinikum Dresden, Dresden, Germany;
Dennis Päsler, Department of Neurosurgery, Universitätsmedizin Greifswald, Greifswald, Germany;
Leandro Leite Antonio, University of Texas at Brownsville, Brownsville, TX, USA;
Peter Horn, Department of Neurosurgery, HSK Wiesbaden, Wiesbaden, Germany;
Thomas-Nicolas Lehmann, Department of Neurosurgery, Helios-Klinikum Bad Saarow, Bad Saarow, Germany
‡Nora Sandow and Simon Kim have contributed equally to this work.