 Gregory A. Elder1,2,3,4*
Gregory A. Elder1,2,3,4* Miguel A. Gama Sosa2,4,5
Miguel A. Gama Sosa2,4,5 Rita De Gasperi2,4,5
Rita De Gasperi2,4,5 James Radford Stone6,7
James Radford Stone6,7 Dara L. Dickstein4,8,9
Dara L. Dickstein4,8,9 Fatemeh Haghighi2,4,5,8
Fatemeh Haghighi2,4,5,8 Patrick R. Hof4,8,9
Patrick R. Hof4,8,9 Stephen T. Ahlers10
Stephen T. Ahlers10
- 1Neurology Service, James J. Peters Department of Veterans Affairs Medical Center, Bronx, NY, USA
- 2Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 3Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 4Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 5Research and Development Service, James J. Peters Department of Veterans Affairs Medical Center, Bronx, NY, USA
- 6Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA
- 7Department of Neurosurgery, University of Virginia, Charlottesville, VA, USA
- 8Fishberg Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 9Department of Geriatrics and Palliative Care, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 10Department of Neurotrauma, Operational and Undersea Medicine Directorate, Naval Medical Research Center, Silver Spring, MD, USA
Blast-related traumatic brain injury (TBI) has received much recent attention because of its frequency in the conflicts in Iraq and Afghanistan. This renewed interest has led to a rapid expansion of clinical and animal studies related to blast. In humans, high-level blast exposure is associated with a prominent hemorrhagic component. In animal models, blast exerts a variety of effects on the nervous system including vascular and inflammatory effects that can be seen with even low-level blast exposures which produce minimal or no neuronal pathology. Acutely, blast exposure in animals causes prominent vasospasm and decreased cerebral blood flow along with blood-brain barrier breakdown and increased vascular permeability. Besides direct effects on the central nervous system, evidence supports a role for a thoracically mediated effect of blast; whereby, pressure waves transmitted through the systemic circulation damage the brain. Chronically, a vascular pathology has been observed that is associated with alterations of the vascular extracellular matrix. Sustained microglial and astroglial reactions occur after blast exposure. Markers of a central and peripheral inflammatory response are found for sustained periods after blast injury and include elevation of inflammatory cytokines and other inflammatory mediators. At low levels of blast exposure, a microvascular pathology has been observed in the presence of an otherwise normal brain parenchyma, suggesting that the vasculature may be selectively vulnerable to blast injury. Chronic immune activation in brain following vascular injury may lead to neurobehavioral changes in the absence of direct neuronal pathology. Strategies aimed at preventing or reversing vascular damage or modulating the immune response may improve the chronic neuropsychiatric symptoms associated with blast-related TBI.
Introduction
It was first recognized during world war (WW) I that blast exposure can be associated with psychological and neurological symptoms reminiscent of both the post-concussion syndrome and what would now be called post-traumatic stress disorder (PTSD) (1). While an uncommon cause of traumatic brain injury (TBI) in civilian life (2), blast-related TBI has been of longstanding interest in military medicine and has recently received wider attention because of its frequency in the military operations in Iraq and Afghanistan (3–7). Estimates are that 10–20% of veterans returning from these conflicts have suffered a TBI with the most frequent cause being blast exposure related to improvised explosive devices (IED) (8, 9).
Initially most attention focused on the moderate to severe end of the TBI spectrum (10), the type of injury that would be recognized in-theater, and the war in Iraq has resulted in the highest number of service-related severe TBIs since the Vietnam era (11). However, what became apparent was that many Iraq and Afghanistan veterans were presenting to veterans affairs (VA) hospitals and other facilities with symptoms suggestive of the residual effects of mild TBIs (mTBIs) that were never recognized prior to discharge. Indeed mTBIs vastly outnumber moderate to severe TBIs in these veterans (8, 9).
Interest in how blast affects the nervous system has led to a rapid expansion of clinical as well as animal studies. Animal studies show that blast pressure waves are transmitted through the brain and exert a variety of biochemical, pathological, and pathophysiological effects (12, 13). However, questions remain concerning how these pathologies lead to blast-related symptoms as well as how, or even if, the pathologies are related to one another. Blast-related vascular and inflammatory effects are well-established (14) and occur with even low-level exposures that produce minimal or no detectable neuronal pathology. Here, we review the evidence that blast exposure causes acute and chronic vascular as well as inflammatory effects and how these effects may play a role in the pathophysiology of chronic blast-related brain injury.
Vascular Pathology in Blast-Induced Brain Injury
Vascular pathology is a well-recognized component of non-blast TBI in humans and animals (15). Few human cases of acute blast exposure have come to autopsy and many that have sustained such severe injuries that they died within a few days of injury (16). The most prominent features in two cases from WWI studied by Mott (17) were punctate hemorrhages in subcortical gray and white matter regions. Cohen and Biskind (18) identified nine cases from WWII in the archives of the US Armed Forces Institute of Pathology, all of whom died within 5 days of injury. These cases also exhibited a prominent hemorrhagic component with diffuse leptomeningeal bleeding, intracerebral clots, and multifocal hemorrhages in white matter. More recent observations in-theater suggest that edema, intracranial hemorrhage, and vasospasm are prominent features of severe blast injury in the current conflicts (19, 20). In a review of neurosurgical consultations on soldiers transferred from Iraq to the National Navel Medical Center, Armonda et al. (19) found that vasospasm was present in nearly half of those undergoing angiography as part of a diagnostic evaluation for acute neurotrauma. Vasospasm was often prolonged averaging 14 days in duration with some cases persisting up to 30 days. Pseudoaneurysm formation as well as hemorrhage was often seen in association with vasospasm, which generally predicted a worse outcome (19). In a review of UK fatalities in Afghanistan, some form of hemorrhage was implicated as the proximal cause of death in most cases (21). Thus high-pressure blast waves can cause extensive CNS injury in the context of severe TBI in humans with a strong hemorrhagic component.
The role of vascular pathology in human cases of mTBI is less clear. Cases of mTBI rarely come to autopsy, and little is generally known about the pathology of mTBI especially in the acute phase (22). Only a few cases of blast-related TBI have been studied in the chronic phase (23–26). Several of these exhibited a chronic traumatic encephalopathy-like picture with a prominent tauopathy that included perivascular tau deposition (23–25). Ryu et al. (26) have also described amyloid precursor protein-positive axonal swellings typical of the diffuse axonal injury that can be seen long after non-blast TBI. In white matter, these abnormalities occurred primarily in a perivascular distribution (26).
Due to the lack of human data, most of our knowledge of how blast exposure affects the nervous system comes from animal studies. Animals have been exposed to various forms of blast ranging from direct exposure to live explosives to more commonly controlled blast waves produced by compressed-air generators with rats being most often used (3, 13). Approaches have varied. Some studies have subjected animals to whole body blast, while others attempting to isolate effects on the CNS have selectively exposed the head or utilized various forms of shielding to protect the body. Various pathological effects have followed blast overpressure injuries in these models (3, 13). These effects have in turn been associated with biochemical and pathophysiological changes as well as behavioral phenotypes. Issues concerning the use of these models have been discussed in several recent reviews (12, 27–29).
Blast-related vascular effects in animals are summarized in Tables 1 and 2, where studies are divided into those using whole body blast (Table 1) and those that have isolated exposure to the head or body (Table 2). As in humans, acute high-level blast exposure has a prominent hemorrhagic component in species including rats, mice, rabbits, ferrets, and goats (30–37). Varying degrees of subdural, subarachnoid, and intracerebral hemorrhage are visible grossly and microscopically along with edema and diffuse vascular congestion on the brain surface. Effects seem similar whether exposures under shock tube conditions or live explosives are used with a clear correlation between injury severity and blast overpressure levels or distance from the detonation in the case of live explosives.
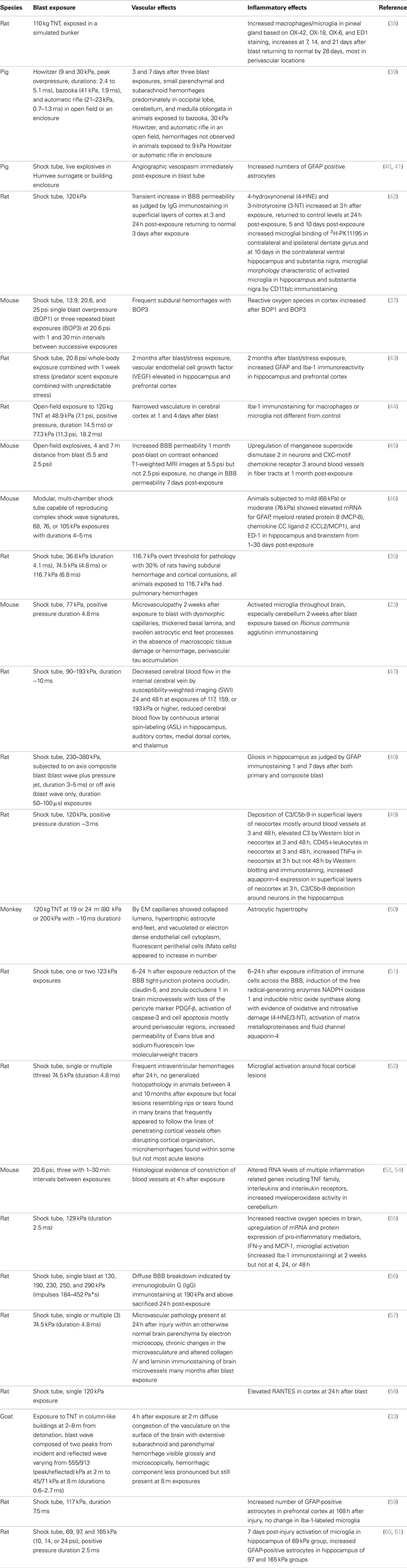
Table 1. Vascular and inflammatory changes in the nervous system following whole-body blast exposure in animal models.
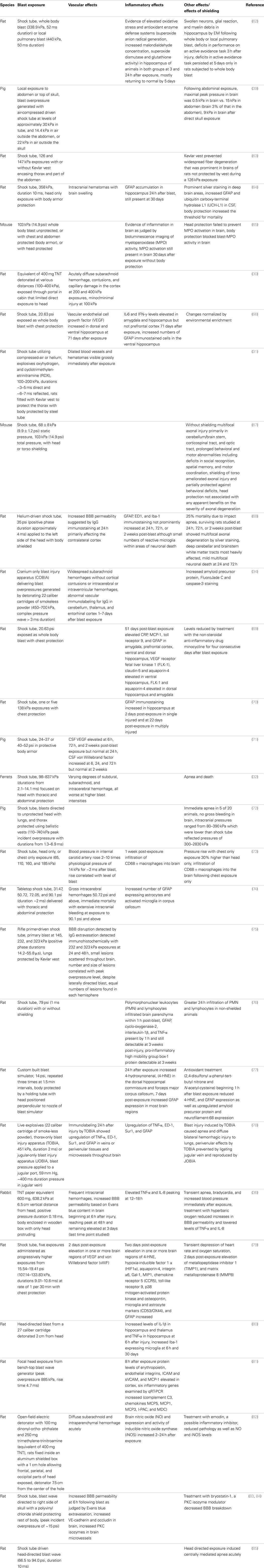
Table 2. Vascular and inflammatory effects on the nervous system following restricted cranial or thoracic exposure to blast in experimental animals.
While the hemorrhagic component is more apparent at high-level blast, pathologically hemorrhage also seems to be a feature of lower-level exposure. For example, Saljo et al. (39) observed small parenchymal and subarachnoid hemorrhages, predominately in the occipital lobe, cerebellum, and medulla oblongata in pigs at 3 and 7 days after relatively low-pressure exposures from a bazooka, Howitzer, or an automatic rifle in an open field. Gama Sosa et al. (52), studying rats at 24 h after single or multiple 74.5 kPa blast exposures while observing no generalized histopathology, observed frequent intraventricular hemorrhages.
Histologically, blast exposure can also induce a microvascular pathology in the absence of hemorrhage. Goldstein et al. (23), studying mice exposed to 77 kPa overpressures, observed a microvasculopathy 2 weeks after exposure with dysmorphic capillaries, thickened basal lamina, and swollen astrocytic end-feet in the absence of macroscopic tissue damage or hemorrhage. In rats exposed to single or multiple 74.5 kPa blast exposures, Gama Sosa et al. (57) observed a microvascular pathology 24 h after injury within an otherwise normal brain parenchyma. In these studies, chronic microvascular changes in the form of altered collagen IV and laminin immunostaining of brain microvessels was observed many months after blast exposure (57). Other studies have described vascular narrowing or constriction acutely after blast (44, 53).
Physiologically acute blast exposure causes prominent vasospasm (40) and decreased cerebral blood flow (47). Bauman et al. (40), studying pigs, demonstrated vasospasm angiographically immediately after exposure in a shock tube. Bir et al. (47), studying rats exposed to 90–193 kPa overpressures, found decreased cerebral blood flow in the internal cerebral vein by susceptibility-weighted imaging (SWI) at 24 and 48 h following exposures of 117 kPa or higher, as well as reduced cerebral blood flow by continuous arterial spin-labeling (ASL) in multiple brain regions for up to 72 h across the entire range of pressures.
At the functional level, acutely blast exposure has been associated with increased vascular permeability and blood-brain barrier (BBB) breakdown. Multiple studies have described at least transient increases in BBB permeability acutely as judged by IgG immunostaining (34, 42, 56, 68, 75). This staining, which has mostly been observed at higher blast exposures, typically returned to normal by 72 h, although Kuehn et al. (34) observed abnormal vascular immunolabeling for IgG in cerebellum, thalamus, and entorhinal cortex for as long as 7 days after blast exposure in rats. Several studies have documented increased BBB permeability to Evans blue (36, 51, 83, 84). Abdul-Muneer et al. (51) observed increased permeability of Evans blue and sodium-fluorescein low molecular-weight tracers 6–24 h after rats had been exposed to one or two 123 kPa exposures. Thus, a variety of studies have documented that functional breakdown of the BBB occurs acutely at least with relatively higher-level blast exposures. Whether chronic effects on BBB integrity exist has been less studied although one study (45) examining mice exposed to relatively low-level 5.5 psi overpressures generated by live explosives found increased BBB permeability 1 month post-blast on contrast enhanced T1-weighted MRI. Interestingly, these studies found no change in BBB permeability 7 days post-exposure, suggesting an evolving process that was not apparent acutely.
The Pathophysiological Basis of Blast-Related Vascular Pathology
Morphological and functional data indicate that both large and small brain vessels are affected by blast injury. Little is known about the molecular changes associated with these abnormalities. Reductions of the BBB tight-junction proteins occludin, claudin-5, and zonula occludens 1 (ZO-1) in brain microvessels have been observed 6–24 h after exposure (51), although others have found increased occludin and VE-cadherin following blast (83, 84). Altered protein kinase C (PKC) isozymes have been observed in brain microvessels acutely following blast (84). Goldstein et al. (23) found perivascular accumulation of tau 2 weeks after blast exposure.
The long-term effects of blast on the vasculature are likewise poorly understood, although one study observed altered immunostaining of the microvasculature with collagen IV and laminin several months after blast exposure (57) (Figures 1–3). This immunostaining, which occurred without the partial proteolytic digestion needed to unmask such antigens in perfusion-fixed tissue of adult rat brain (86), suggests a loss of the normal tightness of the gliovascular junctions and ongoing vascular remodeling (57). Why vascular remodeling would continue many months after blast exposure is unknown but suggests a chronic pathology. Beyond these findings, the molecular basis for blast-related vascular pathology remains poorly understood. Further examination of the molecular changes occurring particularly in the chronic phase is needed to understand the pathophysiological basis of blast-related vascular injury.
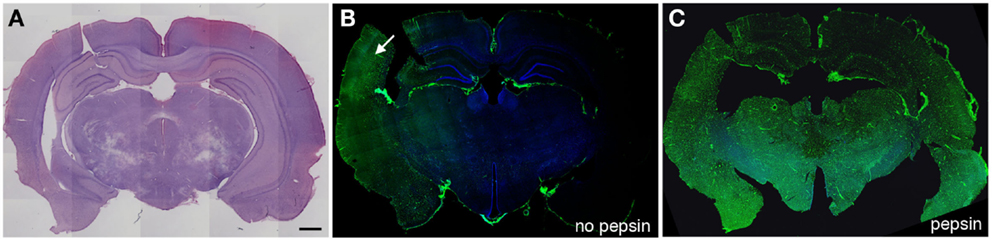
Figure 1. Altered collagen IV immunostaining in the microvasculature of blast-exposed rats. Shown are sections from a rat sacrificed 10 months after receiving three 74.5 kPa blast exposures delivered on consecutive days. (A) shows a coronal section stained with hematoxylin and eosin. (B,C) show adjacent sections immunostained with collagen IV without (B) or with pepsin (C) treatment. Immunostaining was performed as described in Gama Sosa et al. (57). Pepsin treatment (C) unmasks widespread collagen IV immunostaining. Arrow in (B) indicates a region that shows collagen IV immunostaining without pepsin pretreatment. Scale bar: 1 mm.
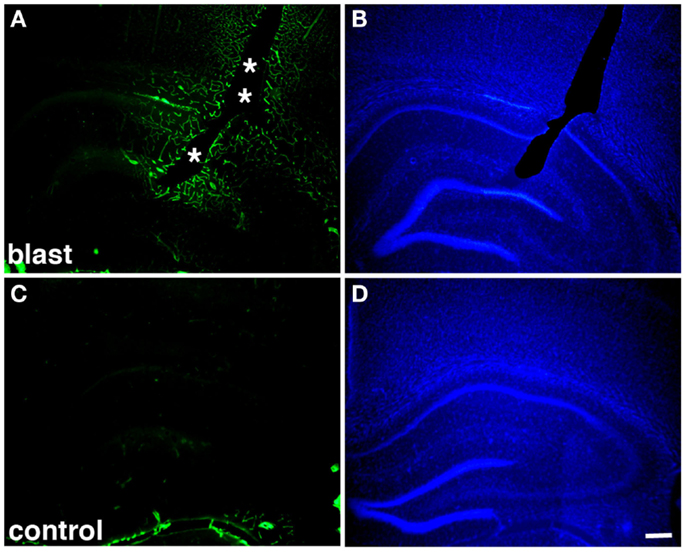
Figure 2. Altered collagen IV immunostaining around blast-induced shear-related lesion. Shown are sections from a rat sacrificed 10 months after receiving three 74.5 kPa blast exposures (A–B) or a non-blast exposed control (C–D). Sections were immunostained for collagen IV without pepsin pretreatment (A,C) and counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (B,D) as described in Gama Sosa et al. (57). A focal blast-induced lesion (indicted by asterisks) is apparent in (A,B). Note the vascular staining with collagen IV in the blast-exposed animal despite the lack of pepsin treatment (A) in comparison to the unstained control (C). Scale bar: 250 μm.
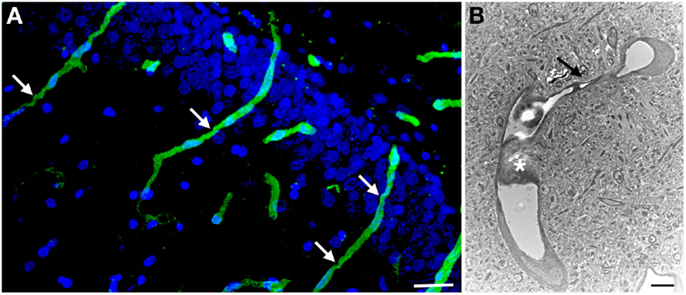
Figure 3. Chronic microvascular pathology following blast exposure. In (A) a section of the hippocampal dentate gyrus is shown from a rat sacrificed 6 months after receiving three 74.5 kPa blast exposures. Sections were immunostained for collagen IV without pepsin pretreatment and counterstained with DAPI as in Figure 2. Note the prominent vascular staining despite the lack of pepsin treatment. Arrows indicate strictures in the vessels. In (B) an electron micrograph is shown taken from the frontal cortex of a rat that received three 74.5 kPa blast exposures and was sacrificed 6 months after the last exposure. Note the amorphous material in the lumen creating a near complete occlusion (asterisk). The vessel also becomes narrowed (arrow). The brain parenchyma surrounding the vessel appears normal. Electron microscopy was performed as described in Gama Sosa et al. (57). Scale bar: 25 μm (A); 2 μm (B).
As noted above, acute blast exposure is associated with prominent vasospasm (40). Vasospasm would be expected to lead to decreased blood flow, which has been documented acutely in vivo (47). Sato et al. (87) have also suggested that a blast wave applied directly to the brain can induce acute physiological changes. In these studies, a laser-induced shock wave was applied directly to the rat brain through the skull under conditions where no changes in systemic physiological parameters occurred. Blood vessels at the brain surface showed vasodilatation for 3–4 min followed by long-lasting vasoconstriction. These changes were associated with electroencephalographic evidence of spreading depression, long-lasting hypoxemia, and signal changes indicating mitochondrial energy impairment.
In considering potential transcranial effects of blast, focused ultrasound (FUS) serves as an interesting comparison. Similar to blast, FUS involves generation of a compression-rarefaction longitudinal wave. However, in contrast to blast, a FUS longitudinal wave has a higher frequency content than a blast wave, and is applied focally in a typically pulsed fashion leading to generally higher peak pressures than a blast wave (56, 88). Yet, similar to blast, Raymond et al. (89) demonstrated BBB disruption and acute vasospasm following direct transcranial application of 10 ms bursts of ultrasound at 1 MHz. Hynynen et al. (88) similarly showed BBB disruption following direct transcranial application of 10 ms bursts of ultrasound at 260 kHz. Sheikov et al. (90) demonstrated loss of tight junction proteins occludin, claudin-5, and ZO-1 by immunoelectron microscopy following application of 10 ms bursts of ultrasound at 1.5 MHz. Thus despite differences in the physical characteristics of the two exposures, the biological effects have interesting similarities.
Alford et al. (91, 92) have suggested that blast-induced vasospasm may play another role by initiating a phenotypic switch in vascular smooth muscle cells that has long-term consequences. They employed high-velocity stretching of engineered arterial lamellae to simulate the mechanical forces of a blast pulse on the vasculature. One hour after a simulated blast, injured tissue displayed altered intracellular calcium dynamics leading to hypersensitivity to a contractile stimulus with endothelin-1. One day after simulated blast, tissues exhibited a prolonged hypercontraction and changes in vascular smooth muscle cells suggestive of a phenotypic switch that was blast force-dependent. They argued that blast-induced cerebral vasospasm causes a mechanically transduced genetic switch in vascular smooth muscle cells that potentiates vascular remodeling leading to further vasospasm and lumen occlusion. Although this phenomenon has yet to be demonstrated in vivo, blast effects on large cerebral blood vessels acutely are well established (40). Another mechanism would need to be invoked to explain effects on the microvasculature where a smooth muscle layer is lacking.
Thoracic and Systemic Effects of Blast on the Nervous System
The primary blast wave may affect the brain as it is transmitted through the tissue. Blast waves also cause acceleration/rotation of the head imparting mechanical energy to the brain that may cause injuries similar to those seen in non-blast TBI. A third mechanism that has been considered is that the blast wave striking the body may cause indirect CNS injury through what has been referred to as a thoracic or systemic mechanism (65, 93). Support for this mechanism came initially from observations in animals suggesting that peripheral injuries by conventional ballistics could cause indirect CNS damage through pressure waves transmitted from the peripheral impact site to the CNS. Studies in pigs, for example, showed that high-frequency pressure waves could be recorded in the brain of an anesthetized pig that was shot in the thigh with a projectile (94–98). Along with pressure changes, microscopic damage was detected in the hippocampus and cerebellum (97). Studies in dogs produced similar results (99). In rats exposed to 12 low-level blast exposures (34.5 kPa), we (unpublished data) have noted a stripping away of the intra-vascular glycocalyx, evidence consistent with increased hydrostatic pressure resulting from the blast event.
Cernak and collaborators first brought attention to a possible systemic mechanism in blast (62, 65, 100). In rats subjected to a local pulmonary blast, they found electron microscopic evidence of swollen neurons, a glial reaction, and myelin debris in the hippocampus following either whole body or local pulmonary exposure (62). With both exposures, deficits were noted in an active avoidance task along with evidence of oxidative stress and activation of antioxidant enzyme defense systems in hippocampus immediately following the insult. Initially, thoracic effects were thought to be vagally mediated, as bilateral transections of the vagus, glossopharyngeal, and hypoglossal nerves in rabbits were found to prevent the apnea, hypotension and bradycardia, as well as mitigate some of the metabolic changes in brain (100). Irwin et al. (101) observed similar effects of vagotomy in rats.
More recently, the mechanism of the thoracic/systemic effect has been modified to emphasize blast wave transmission through the vasculature (65, 102, 103). Specifically, it was proposed that a high-pressure blast wave hitting the body compresses the abdomen and chest inducing oscillating high-pressure waves that can be transmitted through the systemic circulation to the brain. In brain, preferential damage to cellular elements close to cerebral vessels was suggested to damage the cerebral vasculature and disrupt the BBB. The key element of this proposal is that the kinetic energy of the shock wave is transferred into hydraulic energy that is carried through the cardiovascular system causing rapid blood displacement into the lower pressure intracranial compartment. Clearly, if correct, this injury mechanism has implications not only for understanding blast-related pathophysiology but also for design of protective body armor.
Subsequently, a number of studies have addressed the relative importance of body vs. head exposure using selective shielding or a variety of designs for selective exposure. These studies are summarized in Table 2. Among them, multiple studies support the notion that shielding the body can block some blast effects on the CNS (63, 65, 67, 78). For example, Long et al. (63) showed that fitting rats with a Kevlar vest encasing the thorax and part of the abdomen reduced mortality as well as prevented widespread axonal degeneration seen in brains of unprotected rats exposed to a whole body blast. Koliatsos et al. (67), studying mice exposed to a whole body blast, found that without shielding there was multifocal axonal injury as well as deficits in social recognition, spatial memory, and motor coordination. Shielding of the torso reduced axonal injury and partially protected against behavioral deficits, while head protection was not associated with any apparent benefits on the severity of axonal degeneration (67). Studies in mice have found that head protection fails to prevent an inflammatory response in brain as judged by bioluminescence imaging of myeloperoxidase (MPO) activity, while body protection blocked blast-induced MPO activity in brain (65).
Simard et al. (78) have provided support for the systemic effect occurring through a vascular mechanism. These authors constructed devices that allow selective blast exposures to the thorax or the jugular veins of rats. A thorax-only device delivers a collimated blast wave to the right lateral thorax of the rat, precluding direct impact on the cranium. The jugular device delivers a blast wave to the fluid-filled port of an extracorporeal intravenous infusion device whose catheter is inserted retrogradely into the jugular vein, precluding lung injury. Thorax-only exposure caused apnea with diffuse and bilateral pulmonary hemorrhage. At 24 h after thoracic exposure, immunolabeling found that around veins, perivenular tissues, and microvessels in brain there was upregulation of tumor necrosis factor-α (TNF-α), ED-1, sulfonylurea receptor 1 (Sur1), and glial fibrillary acidic protein (GFAP). Perivenular inflammatory effects induced by thorax-only exposure were prevented by ligating the jugular vein and were reproduced by the jugular exposure, all consistent with blast injury to the thorax leading to perivenular inflammation and a reactive gliosis on the basis of a hydrodynamic pulse transmitted through the vasculature (78). Consistent with the systemic effect being cardiovascular, studies in rats have shown that blood pressure in the internal carotid arteries rises 30% more following a selective chest exposure than a brain exposure (73).
In contrast, other studies clearly document that head exposure alone without body exposure induces significant injury to the nervous system (30–32, 34, 36, 63, 64, 66, 68–72, 74, 75, 79–85, 104–106). Indeed, it seems that use of some form of body shielding or selective head exposure has become the norm for blast exposure experiments in many laboratories. Examples include that cranium-only blast exposure in rats leads to widespread subarachnoid hemorrhage with abnormal vascular immunolabeling for IgG, increased amyloid precursor protein staining, and scattered cell death (34) (Table 2). Garmen et al. (68) found that following a blast applied to the left side of the head with the body shielded, 25% of rats died due to impact apnea. Surviving rats studied at 24 h to 2 weeks post-blast showed multifocal axonal degeneration along with scattered neuronal death and increased BBB permeability that primarily affected the contralateral cortex. Studies in rats and ferrets across a range of pressures found that exposures focused on the head with thoracic and abdominal protection caused varying degrees of subdural, subarachnoid, and intracerebral hemorrhage, as well as apnea and death, especially at higher level exposures (32, 74). Pigs and rats experience frequent apnea after blast exposure to the unprotected head (72, 85). Head-directed blast may also induce systemic effects as Prima et al. (105) found that in rats fitted with body armor protection, thrombin generation in blood increased along with changes in other peripheral markers.
Therefore multiple studies show that body shielding can reduce or eliminate the adverse effects of blast on the brain (63, 65, 67, 78) with the studies of Simard et al. (78), perhaps providing the most direct evidence for transmission of the pressure wave through the vasculature. Thus, support exists for a thoracic mechanism playing a substantial role at least under some experimental conditions. In contrast, multiple other studies document that blast exposure of the head alone can reproduce a range of CNS pathology (30–32, 34, 36, 63, 64, 66, 68–72, 74, 75, 79–85, 104–106).
Unfortunately, many technical factors related to the characteristics of the blast wave produced by different shock tubes as well as variations in specimen mounting and degree of head restraint among other factors limit comparisons between studies even within the same species (12). Yet, in looking for differences that may explain the seemingly divergent findings summarized in Table 2, it is noteworthy that the studies using head exposure alone have typically reported adverse effects at higher blast pressures than those used in studies involving whole body or systemic exposures. For example, Turner et al. (74) subjected rats to 31.47, 50.72, 72.05, and 90.1 psi exposures, (duration ~2 ms, ~216.98–621.22 kPa) delivered with thoracic and abdominal protection using a tabletop shock tube. They found gross intracerebral hemorrhages with the 50.72 psi exposure and above but none at the 31.47 psi exposure, a pressure within a range that commonly leads to gross cerebral hemorrhages with whole body exposures (35). It is therefore tempting to speculate that a threshold may exist above which isolated cranial exposures are sufficient to cause significant CNS injury but below which the effects of systemic/thoracic mechanisms are critical. Whether thoracic effects play a role in the low-level blast range including mTBI and subclinical blast is unclear. Answering this question in part depends on how mTBI is defined in animal models, a subject that has recently been discussed elsewhere (12). It depends as well on assessing outcomes more subtle than intracerebral hemorrhage.
Whether all vascular pathology associated with low-level blast exposure in animal models can be explained by a thoracic/systemic mechanism is also unclear. Relatively low-level blast pressure waves (~35 kPa) are transmitted to brain even when the body is protected (107). Other studies have found that rats exposed to whole body 74.5 kPa exposures, while developing no general histopathology, exhibited focal cortical lesions that likely represent shear-related effects due to pressure differentials transmitted through the perivascular spaces in the brain (52). This conclusion was drawn based on observations that the focal lesions typically followed the course of penetrating cortical vessels but were seldom associated with hemorrhage as would be expected if the pressure wave was transmitted through the vasculature. In contrast, hemorrhages might not occur if the blast pressure wave was transmitted through the vascular compartment but not through the blood vessel lumen, an effect that could occur if the main pressure wave was transmitted through the Virchow–Robin compartments. Many studies have documented increases in intracranial pressure acutely following blast exposure (23, 39, 40, 72, 107–114). Increased CSF pressure transmitted through the Virchow–Robin compartments could generate local pressure differentials at the interface between the vascular basal lamina and the surrounding tissues. Shearing along this plane would conceptually leave the blood vessel wall intact preventing hemorrhages. Disruption of the Virchow–Robin compartments could have another effect in that recently a brain-wide network of paravascular channels has been identified, termed the “glymphatic” pathway, along which CSF moves through the brain parenchyma (115). This pathway which facilitates clearance of interstitial solutes (115) would likely be disrupted by blast driven pressures transmitted through the Virchow–Robin compartments.
Inflammation in Blast-Induced Brain Injury
Inflammatory changes in brain after blast exposure in animals are well documented (Tables 1 and 2). Blast exposure induces a number of cytokines/inflammatory mediators including interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-1β (IL-1β), interferon-γ (IFN- γ), C-reactive protein (CRP), monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 1 (MIP1), galectin-1 (Gal-1), TNF-α, and toll receptor 9 (36, 49, 66, 69, 76, 78–80). cDNA microarray studies find altered RNA levels of multiple inflammation related genes including TNF family-related genes, interleukins, and interleukin receptors (54).
At the cellular level, microglial activation has been reported in many studies (23, 38, 42, 52, 55, 60, 61, 74, 79). Increased numbers of GFAP-positive astrocytes (48, 59–61, 66, 70, 74, 76, 77) or elevated GFAP levels in brain (41, 64, 69, 78, 79) have also been reported. Polymorphonuclear leukocytes and lymphocytes can infiltrate brain parenchyma within 1 h of a blast exposure (76) and CD45+ leukocytes have been found increased in cortex 3 and 48 h after blast (49). Elevated cortical C3 levels and perivascular deposition of complement C3/C5b-9 has been observed in superficial cortical layers (49). Evidence of oxidative stress which is commonly found in association with inflammation is also seen following blast (37, 42, 51, 55, 62, 77, 79, 82).
Inflammatory changes in brain have been studied mostly following acute blast exposure with some of the changes being short-lived. For example, TNF-α while increased in cortex at 3 h was not increased at 48 h (49). However, Kovesdi et al. (66) found that IL-6 and IFN-γ were elevated in amygdala and hippocampus for at least 71 days after blast exposure and another study (69) found elevated toll receptor 9, MCP-1, and CRP, 51 days post-exposure, suggesting that some changes persist. At 1 month after exposure, Rubovitch et al. (45) also observed increased perivascular expression of the CXC-motif chemokine receptor 3 which regulates leukocyte trafficking across the vasculature. Consistent with chronic cellular infiltration in brain, MPO bioluminescence imaging suggests polymorphonuclear leukocyte accumulation for at least 30 days after blast exposure (65).
In animal models, blast is also associated with markers of inflammation in blood and plasma (Table 3). Elevations in CRP, IL-1 and IL-10, plasma complement levels, blood and plasma MPO, and TNF-α have all been observed (48, 49, 53, 58, 69, 79, 106). Kamnaksh et al. (79) recently identified a range of elevated inflammatory, extracellular matrix, vascular and oxidative stress related proteins in plasma after blast exposure. As in brain, many of the changes have been short lived. For example in one study, elevations of IL-1β, TNF-α, and IL-10 while detectible at 3 h post-exposure peaked at 24 h and returned to normal by 48 h (58), although as in brain, elevations in CRP and MCP-1 have been reported in blood 51 days after blast exposure (69).
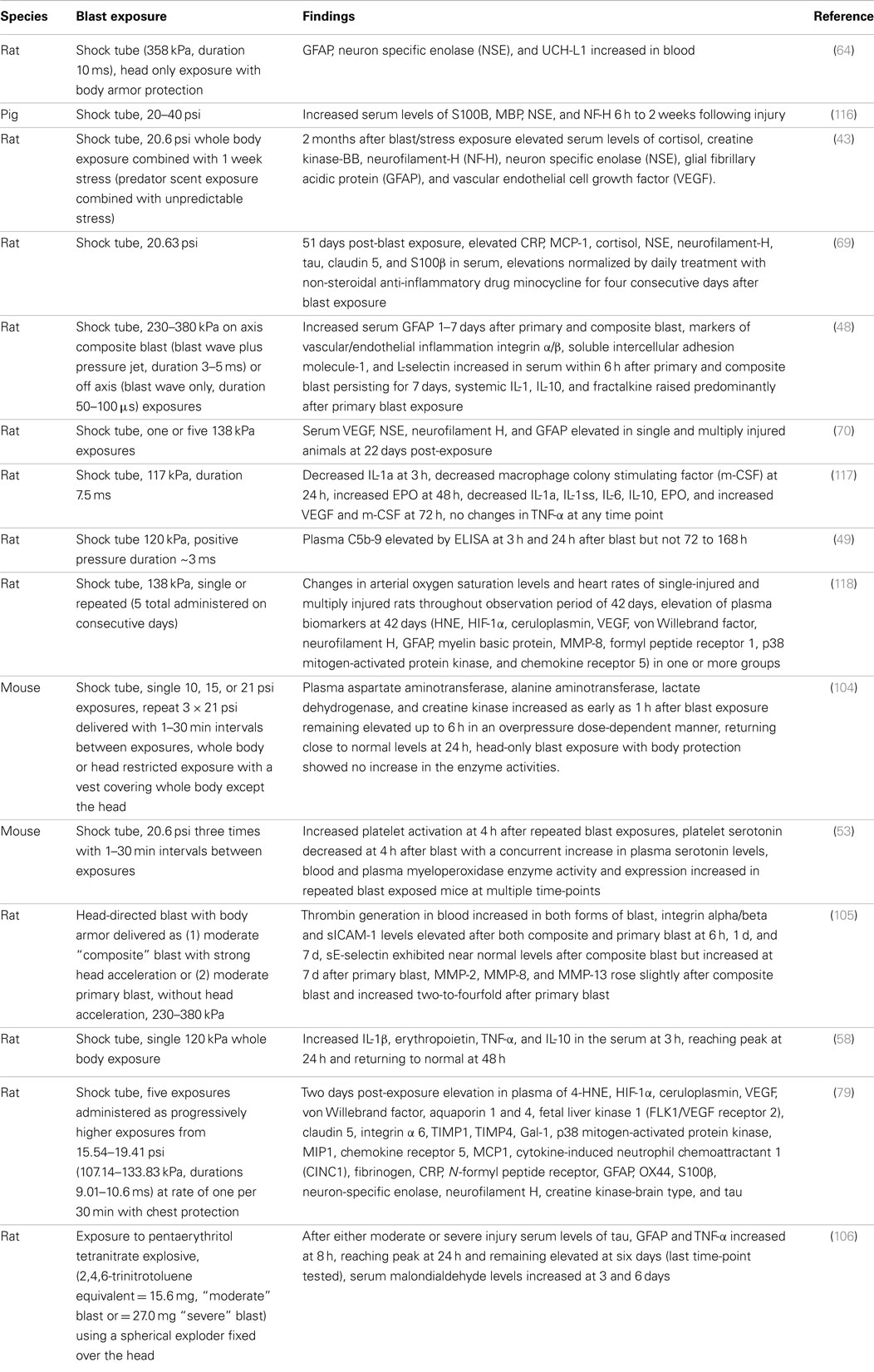
Table 3. Changes in blood or plasma in experimental animals after blast.
Linking Vascular and Inflammatory Factors to the Neuropsychiatric Features of Blast-Related TBI
One of the striking features of the mTBI cases in veterans returning from Iraq and Afghanistan is the frequent presence of PTSD. PTSD or depression is present in over one-third of Iraq veterans who suffered mTBI (8, 119). The presence of PTSD has complicated diagnosis as the clinical distinction between a post-concussion syndrome and PTSD is often difficult (3). While the frequent overlap could represent dual exposures to TBI as well as PSTD-related psychological stressors, other studies have suggested that many of the adverse physical or neuropsychological outcomes following blast-related mTBI are only non-specifically related to blast or better explained by PTSD (12). However, several studies now show that blast exposure in animals can induce PTSD-related traits in the absence of a psychological stressor (120–122). Blast exposure in animals has been found to induce anxiety, increased acoustic startle, increased prepulse inhibition, altered fear conditioning, and enhanced responses to a predator scent challenge (12, 120–122), with one study finding that these traits were still present many months after blast exposure (120). Human neuroimaging studies also show that blast-related mTBI is associated with chronic effects that are unlikely to be explained by comorbid PTSD (12). Thus, multiple lines of evidence support the notion that blast exposure is associated with chronic neurobehavioral effects independent of PTSD.
Whether vascular and inflammatory factors play a role in neurobehavioral effects associated with blast injury is unknown. Supporting a potential role is a substantial literature suggesting that chronic low-grade inflammation is a consistent feature of many neuropsychiatric disorders including major depression and PTSD (123, 124). Epidemiological studies suggest inflammation as a risk factor for major depression (123). Multiple studies have reported increased circulating lymphocytes and phagocytic cells in major depression along with elevated plasma levels of acute phase reactants such as CRP and increased proinflammatory cytokines, including interleukins IL-1β, IL-2, and IL-6, and TNF-α (123). Levels of oxidative stress are also linked to inflammation and major depression (125). Relatively similar changes occur in PTSD where greater total lymphocytes, increased number and activity of natural killer (NK) cells, and activated T lymphocytes have been noted along with increased plasma levels of CRP and proinflammatory cytokines including IL-1β, IL-6, and TNF-α (124, 126). Inflammatory changes may in addition be linked to alterations in the hypothalamic pituitary axis (HPA) found in PTSD and recently epigenetic changes in genes related to the HPA axis have been found that may affect peripheral immune reactions (124).
How inflammation is related to the clinical features of major depression and PTSD is not clear. Some of the most compelling evidence for cytokine production playing an etiological role comes from patients treated with interferons which have documented high rates of depression with either IFN-α or IFN-γ (127). Proinflammatory cytokines also influence pathways affected in major depression including serotonin, noradrenalin, and kynurenine metabolism, all of which have links to neurotransmitter systems implicated in depression (123).
The brain vasculature is a central target of blast-induced effects (Tables 1 and 2) with recent studies suggesting that the cerebral vasculature is selectively vulnerable (57). Blast exposure is associated with chronic inflammatory changes in brain and blood in experimental animals (Tables 1–3) with chronic changes in the microvasculature being apparent many months after blast exposure (57) (Figure 3), at a time when persistent behavioral traits are present (120). Figure 4 summarizes how an initial vascular insult might lead to chronic neurobehavioral effects. Mechanical injury to blood vessels might induce local production of inflammatory mediators including cytokines, chemokines, and cell adhesion molecules, effects that could occur through cell mediated mechanisms activated by physical trauma (128). Oxidative stress induced by mechanical injury and other factors is also associated with induction of an inflammatory response (125). Changes in BBB permeability could have various functional consequences including potentiating an inflammatory reaction. Blast also damages the choroid plexus (52, 129), which has been associated with inflammatory responses due to altered blood-CSF barrier function (130).
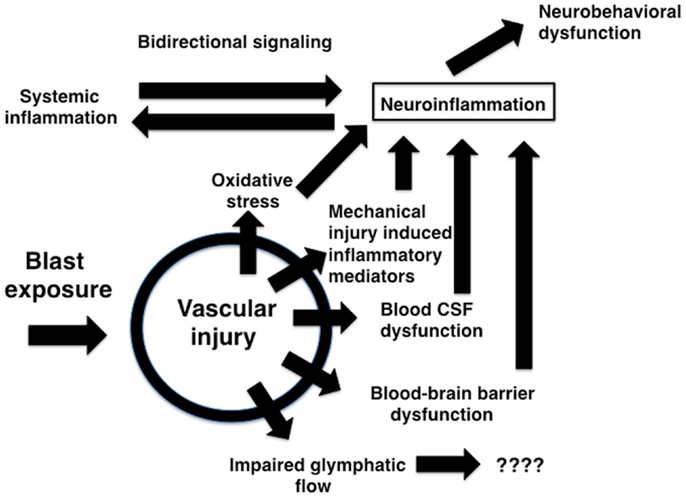
Figure 4. Potential mechanisms relating blast-induced vascular injury to neuroinflammation and neurobehavioral dysfunction. Mechanical injury to blood vessels induces local production of inflammatory mediators including cytokines, chemokines, and cell adhesion molecules through largely cell-mediated mechanisms. Mechanical injury also induces oxidative stress which can be associated with induction of an inflammatory response. Changes in BBB permeability could support initiation of an inflammatory reaction acutely and help sustain a response chronically. Blast-induced damage to the choroid plexus may alter blood-CSF barrier function which has been linked to induction of an inflammatory response. Impaired BBB function and bidirectional signaling between CNS and systemic inflammatory responses could amplify both reactions. Chronic immune activation could lead to neurobehavioral changes in the absence of direct neuronal pathology. Vascular pathology could also disrupt recently described glymphatic pathways that move CSF through the brain parenchyma.
Chronic vascular pathology could have another effect in that recently a brain-wide network of paravascular channels has been identified, termed the “glymphatic” pathway along which CSF moves through the brain parenchyma (115). Through this pathway substances including the amyloid β and tau proteins are transported out of the CNS (115). Interestingly transport through this pathway is impaired in mice with a null mutation of the astroglial water channel aquaporin-4 and multiple studies have identified changes in aquaporin-4 expression following blast injury (49–51, 131). Multiple studies have also identified accumulation of tau (23, 58, 66, 132, 133) following blast exposure although β-amyloid levels are decreased acutely following blast injury (134). In non-blast TBI models, it has recently been shown that one route, whereby serum markers such as S100β, GFAP, and neuron-specific enolase reach the systemic circulation is through the brain glymphatics and the cervical lymph nodes (135).
The cellular response following blast includes a microglial and astroglial reaction. Microglia play central roles in brain inflammatory responses and could therefore play critical roles in activating a brain inflammatory response following blast (136, 137). Astrocytes also regulate BBB permeability and influence microglial activation (123). Chemokines released by astrocytes recruit monocytes and macrophages as well as other immune cells into the CNS (123), and astrocytes are activated by TNF-α, IFN-γ, IL-1, and IL-6 (123). Activated microglia produce proinflammatory cytokines including IL-1β, IL-6, and TNF-α (138, 139). A microglial response often precedes the activation of astrocytes and is thought to be involved in maintaining an astroglial response (140). Inflammatory signaling through microglia has been linked to serotonin metabolism through effects on the kynurenine pathway leading to altered 5-hydroxytryptophan availability and lower levels of serotonin (127). Microglial activation is also a feature of non-blast TBI (141).
An additional cellular element that likely plays a role in this process is the pericyte, which is damaged acutely in blast-associated vascular injury (57). Pericytes have a close structural relationship with endothelial cells and regulate capillary permeability through mechanical means, as well as secretion of cytokines and production of nitric oxide and matrix metalloproteinases (142). Pericytes are increasingly being recognized as having a role in normal physiology as well as disease (143). Like astrocytes, pericytes regulate the BBB and control leukocyte migration into brain through production of soluble factors as well as effects on adhesion molecule expression (142). Under physiological conditions, pericytes appear to function as immune-suppressors, supporting the notion that their loss or damage secondary to blast increases immune activation in brain.
Markers of both a central and a peripheral inflammatory response are found after blast injury (48). While the relationships between the central and peripheral inflammatory responses after blast exposure are not known, one possibility is that the systemic inflammatory response serves at least in part as a driver of the CNS response, an effect that could be accentuated in the presence of an impaired CNS vasculature following blast exposure. Cytokines cross the BBB either through active transport or damaged endothelium (144). Thus the CNS can be affected not only by inflammatory mediators produced within the brain, but as well through actions of mediators originating from the periphery. Interestingly, increased BBB permeability along with elevated intrathecal production of IgG is found in a subgroup of patients with major depression (123). These changes seem most prominent in patients with the most treatment-resistant symptoms, supporting the notion that a peripheral inflammatory response might drive a chronic CNS inflammatory response. Chronic immune activation in the brain following vascular injury could lead to neurobehavioral changes in the absence of direct neuronal pathology.
Thus, evidence collectively points to the need to develop strategies to prevent or repair vascular damage following blast injury. It also points to targeting the immune response. Inflammation is a recognized component of non-blast TBI (145, 146) and reversal of established anxiety-like behavior in rats after a lateral fluid percussion injury has been reported following immunomodulatory therapy (147). While studies following blast exposure are limited, one has reported that treatment with minocycline, a drug whose effects include anti-inflammatory properties, can reverse behavioral deficits and neurochemical changes in a rat model of blast-related TBI (69). Another study found that a complement inhibitor could attenuate many of the effects of acute blast injury including BBB breakdown (58), while Du et al. (77) found that treatment with a combination of antioxidants could reduce blast-induced brain injury. In addition, microglial activation is becoming increasingly recognized as a factor in many neurological diseases and might be another therapeutic target (136, 138, 139).
Conclusion
The possible effects of blast exposure on the nervous system have been discussed since WWI. Public interest in this topic as well as interest in the scientific community has expanded rapidly in recent years because of the prominence of blast-related TBI in the conflicts in Iraq and Afghanistan. In humans, high-pressure blast exposure can cause extensive CNS injury with a prominent hemorrhagic component. Less is known about the pathology of human blast-related mTBI, and animal studies are the source of most knowledge about low-level blast exposure. As in humans, acute high-level blast exposure in experimental animals is associated with a prominent hemorrhagic component. At the functional level, high-level blast exposure acutely disrupts the BBB. Low levels of blast exposure are also associated with a microvascular pathology that can be seen in the presence of an otherwise normal brain parenchyma, suggesting that the vasculature may be selectively vulnerable to blast injury. Multiple studies have shown that shielding the body can reduce or eliminate the adverse effects of blast on the brain supporting the role for thoracically mediated effects whereby pressure waves transmitted through the systemic circulation cause damage in brain.
Inflammatory changes after blast exposure in animals are also well known. In brain, blast exposure induces a number of inflammatory mediators including many interleukins, interferons and cytokines. At the cellular level, microglial and astroglial reactions occur. In animal models, blast is associated with increases in inflammatory markers in blood and plasma including many of the factors elevated in brain. Evidence of oxidative stress which is commonly found in association with inflammation has also been documented in brain and blood.
Thus much evidence supports a role for vascular and inflammatory factors in the pathophysiology of blast-related brain injury, although most animal studies have utilized relatively high-level blast exposures that likely more approximate human moderate to severe TBI than mTBI. Where information is limited is on the effects of lower-level exposures that more approximate mTBI by far the most common clinical exposure in the most recent conflicts. There is also little information on whether vascular and inflammatory effects persist chronically. Many of the vascular and inflammatory changes have been documented to occur only transiently. Few studies in animals have addressed changes weeks or months after blast exposure, the phase most relevant to the chronic persistent symptoms that are the most troublesome following human blast-related mTBI.
Yet, despite the paucity of long-term studies, a chronic vascular pathology has been observed in animals. In addition, some blast-related inflammatory effects have been documented to be present weeks or months following exposure. Chronic low-grade inflammation is a consistent feature of many neuropsychiatric disorders including major depression and PTSD. Clearly, a need exists to better understand the nature of vascular damage following blast exposure and the relationship of vascular changes to neuroinflammatory effects. Understanding this relationship will be critical to determining whether chronic immune activation in brain following vascular injury may underlie blast-associated neurobehavioral changes.
Author Contributions
All authors participated in the collection, review, and analysis of the relevant literature as well as the drafting and revising of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors have received research support from the Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research and Development Service Awards 1I01RX000179-01 and 1I01RX000996-01. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government. STA is a military service member (or employe of the U.S. Government). This work was prepared as part of his official duties. Title 17 U.S.C. §105 provides that copyright protection under this title is not available for any work of the United States Government. Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employe of the U.S. Government as part of that person’s official duties.
References
1. Jones E, Fear NT, Wessely S. Shell shock and mild traumatic brain injury: a historical review. Am J Psychiatry (2007) 164(11):1641–5. doi: 10.1176/appi.ajp.2007.07071180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Bochicchio GV, Lumpkins K, O’Connor J, Simard M, Schaub S, Conway A, et al. Blast injury in a civilian trauma setting is associated with a delay in diagnosis of traumatic brain injury. Am Surg (2008) 74(3):267–70.
3. Elder GA, Mitsis EM, Ahlers ST, Cristian A. Blast-induced mild traumatic brain injury. Psychiatr Clin North Am (2010) 33(4):757–81. doi:10.1016/j.psc.2010.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab (2010) 30(2):255–66. doi:10.1038/jcbfm.2009.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Wolf SJ, Bebarta VS, Bonnett CJ, Pons PT, Cantrill SV. Blast injuries. Lancet (2009) 374(9687):405–15. doi:10.1016/S0140-6736(09)60257-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Rosenfeld JV, McFarlane AC, Bragge P, Armonda RA, Grimes JB, Ling GS. Blast-related traumatic brain injury. Lancet Neurol (2013) 12(9):882–93. doi:10.1016/S1474-4422(13)70161-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. DePalma RG, Buckley CJ, Ecklund JM, Cross GM, Gunnar W. Blast related traumatic brain injury: pathophysiology, comorbidities, and neurobehavioral outcomes. In: Levin H, Shum D, Chan R, editors. Understanding Traumatic Brain Injury: Current Research and Future Directions. Oxford: Oxford University Press (2014). p. 413–29.
8. Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med (2008) 358(5):453–63. doi:10.1056/NEJMoa072972
9. Tanielian T, Jaycox LH, editors. Invisible Wounds of War: Psychological and Cognitive Injuries, their Consequences, and Services to Assist Recovery. Santa Monica, CA: Rand Corporation (2008).
10. Warden DL, Ryan L, Helmick K, Schwab K, French L, Lu W, et al. War neurotrauma: the defense and veterans brain injury center (DVBIC) experience at the Walter Reed Army Medical Center. J Neurotrauma (2005) 22:1178. doi:10.1089/neu.2005.22.1163
11. Bell RS, Vo AH, Neal CJ, Tigno J, Roberts R, Mossop C, et al. Military traumatic brain and spinal column injury: a 5-year study of the impact blast and other military grade weaponry on the central nervous system. J Trauma (2009) 66(4 Suppl):S104–11. doi:10.1097/TA.0b013e31819d88c8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Elder GA, Stone JR, Ahlers ST. Effects of low-level blast exposure on the nervous system: is there really a controversy? Front Neurol (2014) 5:269. doi:10.3389/fneur.2014.00269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Kobeissy F, Mondello S, Tumer N, Toklu HZ, Whidden MA, Kirichenko N, et al. Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front Neurol (2013) 4:186. doi:10.3389/fneur.2013.00186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Shetty AK, Mishra V, Kodali M, Hattiangady B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front Cell Neurosci (2014) 8:232. doi:10.3389/fncel.2014.00232
15. Alves JL. Blood-brain barrier and traumatic brain injury. J Neurosci Res (2014) 92(2):141–7. doi:10.1002/jnr.23300
16. Kocsis JD, Tessler A. Pathology of blast-related brain injury. J Rehabil Res Dev (2009) 46(6):667–72. doi:10.1682/JRRD.2008.08.0100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Cohen H, Biskind G. Pathologic aspects of atmospheric blast injuries in man. Arch Pathol (1946) 42:12–34.
19. Armonda RA, Bell RS, Vo AH, Ling G, DeGraba TJ, Crandall B, et al. Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery (2006) 59(6):1215–25. doi:10.1227/01.NEU.0000249190.46033.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. J Neurotrauma (2009) 26(6):815–25. doi:10.1089/neu.2007.0484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Singleton JA, Gibb IE, Hunt NC, Bull AM, Clasper JC. Identifying future ‘unexpected’ survivors: a retrospective cohort study of fatal injury patterns in victims of improvised explosive devices. BMJ Open (2013) 3(8):e003130. doi:10.1136/bmjopen-2013-003130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Bigler ED, Maxwell WL. Neuropathology of mild traumatic brain injury: relationship to neuroimaging findings. Brain Imaging Behav (2012) 6(2):108–36. doi:10.1007/s11682-011-9145-0
23. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med (2012) 4(134):134ra60. doi:10.1126/scitranslmed.3003716
24. Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus (2011) 31(5):E3. doi:10.3171/2011.9.FOCUS11178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement (2014) 10(3 Suppl):S242–53. doi:10.1016/j.jalz.2014.04.003
26. Ryu J, Horkayne-Szakaly I, Xu L, Pletnikova O, Leri F, Eberhart C, et al. The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol Commun (2014) 2(1):153. doi:10.1186/s40478-014-0153-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Cernak I. Blast-induced neurotrauma models and their requirements. Front Neurol (2014) 5:128. doi:10.3389/fneur.2014.00128
28. Panzer MB, Wood GW, Bass CR. Scaling in neurotrauma: how do we apply animal experiments to people? Exp Neurol (2014) 261C:120–6. doi:10.1016/j.expneurol.2014.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Goldstein LE, McKee AC, Stanton PK. Considerations for animal models of blast-related traumatic brain injury and chronic traumatic encephalopathy. Alzheimers Res Ther (2014) 6(5):64. doi:10.1186/s13195-014-0064-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Cheng J, Gu J, Ma Y, Yang T, Kuang Y, Li B, et al. Development of a rat model for studying blast-induced traumatic brain injury. J Neurol Sci (2010) 294(1–2):23–8. doi:10.1016/j.jns.2010.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Reneer DV, Hisel RD, Hoffman JM, Kryscio RJ, Lusk BT, Geddes JW. A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J Neurotrauma (2011) 28(1):95–104. doi:10.1089/neu.2010.1513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Rafaels KA, Bass CR, Panzer MB, Salzar RS, Woods WA, Feldman SH, et al. Brain injury risk from primary blast. J Trauma Acute Care Surg (2012) 73(4):895–901. doi:10.1097/TA.0b013e31825a760e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Li BC, Li Y, Xu C, Wang J, Chen Z, Li G, et al. Blast-induced traumatic brain injury of goats in confined space. Neurol Res (2014) 36:974–82. doi:10.1179/1743132813Y.0000000314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kuehn R, Simard PF, Driscoll I, Keledjian K, Ivanova S, Tosun C, et al. Rodent model of direct cranial blast injury. J Neurotrauma (2011) 28(10):2155–69. doi:10.1089/neu.2010.1532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ahlers ST, Vasserman-Stokes E, Shaughness MC, Hall AA, Shear DA, Chavko M, et al. Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front Neurol (2012) 3:32. doi:10.3389/fneur.2012.00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Zhang Y, Yang Y, Tang H, Sun W, Xiong X, Smerin D, et al. Hyperbaric oxygen therapy ameliorates local brain metabolism, brain edema and inflammatory response in a blast-induced traumatic brain injury model in rabbits. Neurochem Res (2014) 39:950–60. doi:10.1007/s11064-014-1292-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Wang Y, Wei Y, Oguntayo S, Wilkins W, Arun P, Valiyaveettil M, et al. Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J Neurotrauma (2011) 28(10):2171–83. doi:10.1089/neu.2011.1990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kaur C, Singh J, Lim MK, Ng BL, Ling EA. Macrophages/microglia as ‘sensors’ of injury in the pineal gland of rats following a non-penetrative blast. Neurosci Res (1997) 27(4):317–22. doi:10.1016/S0168-0102(97)01164-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Saljo A, Arrhen F, Bolouri H, Mayorga M, Hamberger A. Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J Neurotrauma (2008) 25(12):1397–406. doi:10.1089/neu.2008.0602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Bauman RA, Ling G, Tong L, Januszkiewicz A, Agoston D, Delanerolle N, et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma (2009) 26(6):841–60. doi:10.1089/neu.2009-0898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. de Lanerolle NC, Bandak F, Kang D, Li AY, Du F, Swauger P, et al. Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol (2011) 70(11):1046–57. doi:10.1097/NEN.0b013e318235bef2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM, et al. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res (2010) 88(16):3530–9. doi:10.1002/jnr.22510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kwon SK, Kovesdi E, Gyorgy AB, Wingo D, Kamnaksh A, Walker J, et al. Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front Neurol (2011) 2:12. doi:10.3389/fneur.2011.00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Pun PB, Kan EM, Salim A, Li Z, Ng KC, Moochhala SM, et al. Low level primary blast injury in rodent brain. Front Neurol (2011) 2:19. doi:10.3389/fneur.2011.00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol (2011) 232(2):280–9. doi:10.1016/j.expneurol.2011.09.018
46. Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT, Mahota TM, et al. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis (2011) 41(2):538–51. doi:10.1016/j.nbd.2010.10.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bir C, Vandevord P, Shen Y, Raza W, Haacke EM. Effects of variable blast pressures on blood flow and oxygen saturation in rat brain as evidenced using MRI. Magn Reson Imaging (2012) 30(4):527–34. doi:10.1016/j.mri.2011.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Svetlov SI, Prima V, Glushakova O, Svetlov A, Kirk DR, Gutierrez H, et al. Neuro-glial and systemic mechanisms of pathological responses in rat models of primary blast overpressure compared to “composite” blast. Front Neurol (2012) 3:15. doi:10.3389/fneur.2012.00015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dalle Lucca JJ, Chavko M, Dubick MA, Adeeb S, Falabella MJ, Slack JL, et al. Blast-induced moderate neurotrauma (BINT) elicits early complement activation and tumor necrosis factor alpha (TNFalpha) release in a rat brain. J Neurol Sci (2012) 318(1–2):146–54. doi:10.1016/j.jns.2012.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Lu J, Ng KC, Ling G, Wu J, Poon DJ, Kan EM, et al. Effect of blast exposure on the brain structure and cognition in Macaca fascicularis. J Neurotrauma (2012) 29(7):1434–54. doi:10.1089/neu.2010.1591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med (2013) 60:282–91. doi:10.1016/j.freeradbiomed.2013.02.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Gama Sosa MA, De Gasperi R, Paulino AJ, Pricop PE, Shaughness MC, Maudlin-Jeronimo E, et al. Blast overpressure induces shear-related injuries in the brain of rats exposed to a mild traumatic brain injury. Acta Neuropathol Commun (2013) 1(1):51. doi:10.1186/2051-5960-1-51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Valiyaveettil M, Alamneh Y, Wang Y, Arun P, Oguntayo S, Wei Y, et al. Contribution of systemic factors in the pathophysiology of repeated blast-induced neurotrauma. Neurosci Lett (2013) 539:1–6. doi:10.1016/j.neulet.2013.01.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Valiyaveettil M, Alamneh YA, Miller SA, Hammamieh R, Arun P, Wang Y, et al. Modulation of cholinergic pathways and inflammatory mediators in blast-induced traumatic brain injury. Chem Biol Interact (2013) 203(1):371–5. doi:10.1016/j.cbi.2012.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Cho HJ, Sajja VS, Vandevord PJ, Lee YW. Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience (2013) 253:9–20. doi:10.1016/j.neuroscience.2013.08.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Skotak M, Wang F, Alai A, Holmberg A, Harris S, Switzer RC, et al. Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma (2013) 30(13):1147–60. doi:10.1089/neu.2012.2652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Gama Sosa MA, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun (2014) 2:67. doi:10.1186/2051-5960-2-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Li Y, Chavko M, Slack JL, Liu B, McCarron RM, Ross JD, et al. Protective effects of decay-accelerating factor on blast-induced neurotrauma in rats. Acta Neuropathol Commun (2013) 1(1):52. doi:10.1186/2051-5960-1-52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sajja VS, Perrine SA, Ghoddoussi F, Hall CS, Galloway MP, VandeVord PJ. Blast neurotrauma impairs working memory and disrupts prefrontal myo-inositol levels in rats. Mol Cell Neurosci (2014) 59:119–26. doi:10.1016/j.mcn.2014.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Sajja VS, Ereifej ES, VandeVord PJ. Hippocampal vulnerability and subacute response following varied blast magnitudes. Neurosci Lett (2014) 570:33–7. doi:10.1016/j.neulet.2014.03.072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Sajja VS, Hubbard WB, VandeVord PJ. Subacute oxidative stress and glial reactivity in the amygdala are associated with increased anxiety following blast neurotrauma. Shock (2014). doi:10.1097/SHK.0000000000000311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Cernak I, Wang Z, Jiang J, Bian X, Savic J. Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma (2001) 50(4):695–706. doi:10.1097/00005373-200104000-00017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma (2009) 26(6):827–40. doi:10.1089/neu.2008.0748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Svetlov SI, Prima V, Kirk DR, Gutierrez H, Curley KC, Hayes RL, et al. Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J Trauma (2010) 69(4):795–804. doi:10.1097/TA.0b013e3181bbd885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Cernak I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front Neurol (2010) 1:151. doi:10.3389/fneur.2010.00151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kovesdi E, Gyorgy AB, Kwon SK, Wingo DL, Kamnaksh A, Long JB, et al. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front Neurosci (2011) 5:42. doi:10.3389/fnins.2011.00042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol (2011) 70(5):399–416. doi:10.1097/NEN.0b013e3182189f06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Garman RH, Jenkins LW, Switzer RC III, Bauman RA, Tong LC, Swauger PV, et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma (2011) 28(6):947–59. doi:10.1089/neu.2010.1540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol (2012) 3:111. doi:10.3389/fneur.2012.00111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kamnaksh A, Kwon SK, Kovesdi E, Ahmed F, Barry ES, Grunberg NE, et al. Neurobehavioral, cellular, and molecular consequences of single and multiple mild blast exposure. Electrophoresis (2012) 33(24):3680–92. doi:10.1002/elps.201200319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Ahmed F, Gyorgy A, Kamnaksh A, Ling G, Tong L, Parks S, et al. Time-dependent changes of protein biomarker levels in the cerebrospinal fluid after blast traumatic brain injury. Electrophoresis (2012) 33(24):3705–11. doi:10.1002/elps.201200299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Shridharani JK, Wood GW, Panzer MB, Capehart BP, Nyein MK, Radovitzky RA, et al. Porcine head response to blast. Front Neurol (2012) 3:70. doi:10.3389/fneur.2012.00070
73. Assari S, Laksari K, Barbe M, Darvish K. Cerebral blood pressure rise during blast exposure in a rat model of blast-induced traumatic brain injury. From the ASME 2013 International Mechanical Engineering Congress and Exposition; 2013 Nov 15–21; San Diego, CA. Published in Proceedings Biomedical and Biotechnology Engineering, Volume 3A. American Society of Mechanical Engineers (ASME) (2013). doi:10.1115/IMECE2013-64992
74. Turner RC, Naser ZJ, Logsdon AF, DiPasquale KH, Jackson GJ, Robson MJ, et al. Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp Neurol (2013) 248:520–9. doi:10.1016/j.expneurol.2013.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Ann Biomed Eng (2013) 41(10):2206–14. doi:10.1007/s10439-013-0805-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Tompkins P, Tesiram Y, Lerner M, Gonzalez LP, Lightfoot S, Rabb CH, et al. Brain injury: neuro-inflammation, cognitive deficit, and magnetic resonance imaging in a model of blast induced traumatic brain injury. J Neurotrauma (2013) 30(22):1888–97. doi:10.1089/neu.2012.2674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Du X, Ewert DL, Cheng W, West MB, Lu J, Li W, et al. Effects of antioxidant treatment on blast-induced brain injury. PLoS One (2013) 8(11):e80138. doi:10.1371/journal.pone.0080138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Simard JM, Pampori A, Keledjian K, Tosun C, Schwartzbauer G, Ivanova S, et al. Exposure of the thorax to a sublethal blast wave causes a hydrodynamic pulse that leads to perivenular inflammation in the brain. J Neurotrauma (2014) 31(14):1292–304. doi:10.1089/neu.2013.3016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kamnaksh A, Ahmed F, Kovedsi E, Barry E, Grunberg N, Long J, et al. Molecular mechanisms of increased cerebral vulnerability after repeated mild blast-induced traumatic brain injury. Trans Proteomics (2014) 3:22–37. doi:10.1016/j.trprot.2013.11.001
80. Perez-Polo JR, Rea HC, Johnson KM, Parsley MA, Unabia GC, Xu GY, et al. A rodent model of mild traumatic brain blast injury. J Neurosci Res (2015) 93(4):549–61. doi:10.1002/jnr.23513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Risdall JE, Carter AJ, Kirkman E, Watts SA, Taylor C, Menon DK. Endothelial activation and chemoattractant expression are early processes in isolated blast brain injury. Neuromolecular Med (2014) 16(3):606–19. doi:10.1007/s12017-014-8313-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Ma Y, Xia X, Cheng JM, Kuang YQ, Yang T, Yang LB, et al. Emodin inhibits inducible nitric oxide synthase in a rat model of craniocerebral explosive injury. Neurochem Res (2014) 39(9):1809–16. doi:10.1007/s11064-014-1395-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci (2014) 8:421. doi:10.3389/fncel.2014.00421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, et al. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol (2014). doi:10.1007/s12035-014-8902-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Adams S, Condrey JA, Tsai HW, Svetlov SI, Davenport PW. Respiratory responses following blast-induced traumatic brain injury in rats. Respir Physiol Neurobiol (2014) 204:112–9. doi:10.1016/j.resp.2014.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Franciosi S, De Gasperi R, Dickstein DL, English DF, Rocher AB, Janssen WG, et al. Pepsin pretreatment allows collagen IV immunostaining of blood vessels in adult mouse brain. J Neurosci Methods (2007) 163(1):76–82. doi:10.1016/j.jneumeth.2007.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Sato S, Kawauchi S, Okuda W, Nishidate I, Nawashiro H, Tsumatori G. Real-time optical diagnosis of the rat brain exposed to a laser-induced shock wave: observation of spreading depolarization, vasoconstriction and hypoxemia-oligemia. PLoS One (2014) 9(1):e82891. doi:10.1371/journal.pone.0082891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Hynynen K, McDannold N, Vykhodtseva N, Raymond S, Weissleder R, Jolesz FA, et al. Focal disruption of the blood-brain barrier due to 260-kHz ultrasound bursts: a method for molecular imaging and targeted drug delivery. J Neurosurg (2006) 105(3):445–54. doi:10.3171/jns.2006.105.3.445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Raymond SB, Skoch J, Hynynen K, Bacskai BJ. Multiphoton imaging of ultrasound/Optison mediated cerebrovascular effects in vivo. J Cereb Blood Flow Metab (2007) 27(2):393–403. doi:10.1038/sj.jcbfm.9600336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Sheikov N, McDannold N, Sharma S, Hynynen K. Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. Ultrasound Med Biol (2008) 34(7):1093–104. doi:10.1016/j.ultrasmedbio.2007.12.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Alford PW, Dabiri BE, Goss JA, Hemphill MA, Brigham MD, Parker KK. Blast-induced phenotypic switching in cerebral vasospasm. Proc Natl Acad Sci U S A (2011) 108(31):12705–10. doi:10.1073/pnas.1105860108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Hald ES, Alford PW. Smooth muscle phenotype switching in blast traumatic brain injury-induced cerebral vasospasm. Transl Stroke Res (2014) 5(3):385–93. doi:10.1007/s12975-013-0300-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Courtney AC, Courtney MW. A thoracic mechanism of mild traumatic brain injury due to blast pressure waves. Med Hypotheses (2009) 72(1):76–83. doi:10.1016/j.mehy.2008.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Goransson AM, Ingvar DH, Kutyna F. Remote cerebral effects on EEG in high-energy missile trauma. J Trauma (1988) 28(1 Suppl):S204–5. doi:10.1097/00005373-198801001-00042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Suneson A, Hansson HA, Seeman T. Peripheral high-energy missile hits cause pressure changes and damage to the nervous system: experimental studies on pigs. J Trauma (1987) 27(7):782–9. doi:10.1097/00005373-198707000-00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Suneson A, Hansson HA, Seeman T. Central and peripheral nervous damage following high-energy missile wounds in the thigh. J Trauma (1988) 28(1 Suppl):S197–203. doi:10.1097/00005373-198801001-00041
97. Suneson A, Hansson HA, Seeman T. Pressure wave injuries to the nervous system caused by high-energy missile extremity impact: part II. Distant effects on the central nervous system-a light and electron microscopic study on pigs. J Trauma (1990) 30(3):295–306.
98. Suneson A, Hansson HA, Seeman T. Pressure wave injuries to the nervous system caused by high-energy missile extremity impact: part I. Local and distant effects on the peripheral nervous system-a light and electron microscopic study on pigs. J Trauma (1990) 30(3):281–94.
99. Wang Q, Wang Z, Zhu P, Jiang J. Alterations of myelin basic protein and ultrastructure in the limbic system at the early stage of trauma-related stress disorder in dogs. J Trauma (2004) 56(3):604–10. doi:10.1097/01.TA.0000058122.57737.0E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Cernak I, Savic J, Malicevic Z, Zunic G, Radosevic P, Ivanovic I, et al. Involvement of the central nervous system in the general response to pulmonary blast injury. J Trauma (1996) 40(3 Suppl):S100–4. doi:10.1097/00005373-199603001-00023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Irwin RJ, Lerner MR, Bealer JF, Mantor PC, Brackett DJ, Tuggle DW. Shock after blast wave injury is caused by a vagally mediated reflex. J Trauma (1999) 47(1):105–10. doi:10.1097/00005373-199907000-00023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Bhattacharjee Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science (2008) 319(5862):406–8. doi:10.1126/science.319.5862.406
103. Chen Y, Huang W, Constantini S. Concepts and strategies for clinical management of blast-induced traumatic brain injury and posttraumatic stress disorder. J Neuropsychiatry Clin Neurosci (2013) 25(2):103–10. doi:10.1176/appi.neuropsych.12030058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Arun P, Oguntayo S, Alamneh Y, Honnold C, Wang Y, Valiyaveettil M, et al. Rapid release of tissue enzymes into blood after blast exposure: potential use as biological dosimeters. PLoS One (2012) 7(4):e33798. doi:10.1371/journal.pone.0033798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Prima V, Serebruany VL, Svetlov A, Hayes RL, Svetlov SI. Impact of moderate blast exposures on thrombin biomarkers assessed by calibrated automated thrombography in rats. J Neurotrauma (2013) 30(22):1881–7. doi:10.1089/neu.2012.2758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Liu MD, Luo P, Wang ZJ, Fei Z. Changes of serum Tau, GFAP, TNF-alpha and malonaldehyde after blast-related traumatic brain injury. Chin J Traumatol (2014) 17(6):317–22.
107. Chavko M, Watanabe T, Adeeb S, Lankasky J, Ahlers ST, McCarron RM. Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J Neurosci Methods (2011) 195(1):61–6. doi:10.1016/j.jneumeth.2010.11.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Bolander R, Mathie B, Bir C, Ritzel D, VandeVord P. Skull flexure as a contributing factor in the mechanism of injury in the rat when exposed to a shock wave. Ann Biomed Eng (2011) 39(10):2550–9. doi:10.1007/s10439-011-0343-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Chavko M, Koller WA, Prusaczyk WK, McCarron RM. Measurement of blast wave by a miniature fiber optic pressure transducer in the rat brain. J Neurosci Methods (2007) 159(2):277–81. doi:10.1016/j.jneumeth.2006.07.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Clemedson CJ, Pettersson H. Propagation of a high explosive air shock wave through different parts of an animal body. Am J Physiol (1956) 184(1):119–26.
111. Dal Cengio Leonardi A, Keane NJ, Bir CA, Ryan AG, Xu L, Vandevord PJ. Head orientation affects the intracranial pressure response resulting from shock wave loading in the rat. J Biomech (2012) 45(15):2595–602. doi:10.1016/j.jbiomech.2012.08.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Leonardi AD, Bir CA, Ritzel DV, VandeVord PJ. Intracranial pressure increases during exposure to a shock wave. J Neurotrauma (2011) 28(1):85–94. doi:10.1089/neu.2010.1324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Saljo A, Mayorga M, Bolouri H, Svensson B, Hamberger A. Mechanisms and pathophysiology of the low-level blast brain injury in animal models. Neuroimage (2011) 54(Suppl 1):S83–8. doi:10.1016/j.neuroimage.2010.05.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Kawoos U, Meng X, Huang SM, Rosen A, McCarron RM, Chavko M. Telemetric intracranial pressure monitoring in blast-induced traumatic brain injury. IEEE Trans Biomed Eng (2014) 61(3):841–7. doi:10.1109/TBME.2013.2291239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci (2014) 34(49):16180–93. doi:10.1523/JNEUROSCI.3020-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Gyorgy A, Ling G, Wingo D, Walker J, Tong L, Parks S, et al. Time-dependent changes in serum biomarker levels after blast traumatic brain injury. J Neurotrauma (2011) 28(6):1121–6. doi:10.1089/neu.2010.1561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Sajja VS, Tenn C, McLaws LJ, Vandevord PJ. A temporal evaluation of cytokines in rats after blast exposure. Biomed Sci Instrum (2012) 48:374–9.
118. Ahmed FA, Kamnaksh A, Kovesdi E, Long JB, Agoston DV. Long-term consequences of single and multiple mild blast exposure on select physiological parameters and blood-based biomarkers. Electrophoresis (2013) 34(15):2229–33. doi:10.1002/elps.201300077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Vasterling JJ, Verfaellie M, Sullivan KD. Mild traumatic brain injury and posttraumatic stress disorder in returning veterans: perspectives from cognitive neuroscience. Clin Psychol Rev (2009) 29(8):674–84. doi:10.1016/j.cpr.2009.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Elder GA, Dorr NP, De Gasperi R, Gama Sosa MA, Shaughness MC, Maudlin-Jeronimo E, et al. Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J Neurotrauma (2012) 29(16):2564–75. doi:10.1089/neu.2012.2510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Heldt SA, Elberger AJ, Deng Y, Guley NH, Del Mar N, Rogers J, et al. A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front Neurol (2014) 5:2. doi:10.3389/fneur.2014.00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Xie K, Kuang H, Tsien JZ. Mild blast events alter anxiety, memory, and neural activity patterns in the anterior cingulate cortex. PLoS One (2013) 8(5):e64907. doi:10.1371/journal.pone.0064907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Muller N. Immunology of major depression. Neuroimmunomodulation (2014) 21(2–3):123–30. doi:10.1159/000356540
124. Wieck A, Grassi-Oliveira R, Hartmann do Prado C, Teixeira AL, Bauer ME. Neuroimmunoendocrine interactions in post-traumatic stress disorder: focus on long-term implications of childhood maltreatment. Neuroimmunomodulation (2014) 21(2–3):145–51. doi:10.1159/000356552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Bakunina N, Pariante CM, Zunszain PA. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology (2015) 144(3):365–73. doi:10.1111/imm.12443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Gill JM, Saligan L, Woods S, Page G. PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr Care (2009) 45(4):262–77. doi:10.1111/j.1744-6163.2009.00229.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Watkins CC, Sawa A, Pomper MG. Glia and immune cell signaling in bipolar disorder: insights from neuropharmacology and molecular imaging to clinical application. Transl Psychiatry (2014) 4:e350. doi:10.1038/tp.2013.119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Davis C, Fischer J, Ley K, Sarembock IJ. The role of inflammation in vascular injury and repair. J Thromb Haemost (2003) 1(8):1699–709. doi:10.1046/j.1538-7836.2003.00292.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Kaur C, Singh J, Lim MK, Ng BL, Yap EP, Ling EA. Studies of the choroid plexus and its associated epiplexus cells in the lateral ventricles of rats following an exposure to a single non-penetrative blast. Arch Histol Cytol (1996) 59(3):239–48. doi:10.1679/aohc.59.239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol (2014) 128(2):267–77. doi:10.1007/s00401-013-1227-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Zou YY, Kan EM, Lu J, Ng KC, Tan MH, Yao L, et al. Primary blast injury-induced lesions in the retina of adult rats. J Neuroinflammation (2013) 10:79. doi:10.1186/1742-2094-10-79
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis (2013) 37(2):309–23. doi:10.3233/JAD-130182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Arun P, Abu-Taleb R, Oguntayo S, Tanaka M, Wang Y, Valiyaveettil M, et al. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: role of compromised cell membrane integrity. Neurosci Lett (2013) 552:87–91. doi:10.1016/j.neulet.2013.07.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. De Gasperi R, Gama Sosa MA, Kim SH, Steele JW, Shaughness MC, Maudlin-Jeronimo E, et al. Acute blast injury reduces brain abeta in two rodent species. Front Neurol (2012) 3:177. doi:10.3389/fneur.2012.00177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci (2015) 35(2):518–26. doi:10.1523/JNEUROSCI.3742-14.2015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci (2014) 15(5):300–12. doi:10.1038/nrn3722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol (2013) 39(1):3–18. doi:10.1111/nan.12011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Gertig U, Hanisch UK. Microglial diversity by responses and responders. Front Cell Neurosci (2014) 8:101. doi:10.3389/fncel.2014.00101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Giunti D, Parodi B, Cordano C, Uccelli A, Kerlero de Rosbo N. Can we switch microglia phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology (2013) 141:328–38. doi:10.1111/imm.12177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Rodgers KM, Bercum FM, McCallum DL, Rudy JW, Frey LC, Johnson KW, et al. Acute neuroimmune modulation attenuates the development of anxiety-like freezing behavior in an animal model of traumatic brain injury. J Neurotrauma (2012) 29(10):1886–97. doi:10.1089/neu.2011.2273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Front Neurol (2013) 4:30. doi:10.3389/fneur.2013.00030
142. Hurtado-Alvarado G, Cabanas-Morales AM, Gomez-Gonzalez B. Pericytes: brain-immune interface modulators. Front Integr Neurosci (2014) 7:80. doi:10.3389/fnint.2013.00080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol (2011) 122(1):1–9. doi:10.1007/s00401-011-0847-6
144. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol (2006) 147(Suppl 1):S232–40. doi:10.1038/sj.bjp.0706400
145. Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WS, Graham DI, et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol (2013) 39(6):654–66. doi:10.1111/nan.12008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol (2013) 4:18. doi:10.3389/fneur.2013.00018
147. Rodgers KM, Deming YK, Bercum FM, Chumachenko SY, Wieseler JL, Johnson KW, et al. Reversal of established traumatic brain injury-induced, anxiety-like behavior in rats after delayed, post-injury neuroimmune suppression. J Neurotrauma (2014) 31(5):487–97. doi:10.1089/neu.2013.3090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: animal models, blast, inflammation, traumatic brain injury, vascular pathology
Citation: Elder GA, Gama Sosa MA, De Gasperi R, Stone JR, Dickstein DL, Haghighi F, Hof PR and Ahlers ST (2015) Vascular and inflammatory factors in the pathophysiology of blast-induced brain injury. Front. Neurol. 6:48. doi: 10.3389/fneur.2015.00048
Received: 27 January 2015; Paper pending published: 12 February 2015;
Accepted: 23 February 2015; Published online: 16 March 2015.
Edited by:
Firas H. Kobeissy, University of Florida, USAReviewed by:
Deborah Shear, Walter Reed Army Institute of Research, USARalph George Depalma, Department of Veterans Affairs Office of Research and Development, USA
Copyright: © 2015 Elder, Gama Sosa, De Gasperi, Stone, Dickstein, Haghighi, Hof and Ahlers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory A. Elder, James J. Peters Department of Veterans Affairs Medical Center, Neurology Service (3E16), 130 West Kingsbridge Road, Bronx, NY 10468, USA e-mail:Z3JlZ29yeS5lbGRlckB2YS5nb3Y=