 Julie A. Saugstad
Julie A. Saugstad- Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA
This review will focus on the current state of knowledge regarding non-coding RNAs (ncRNA) in stroke and neuroprotection. There will be a brief introduction to microRNAs (miRNA), long ncRNAs (lncRNA), and piwi-interacting RNAs (piRNA), followed by evidence for the regulation of ncRNAs in ischemia. This review will also discuss the effect of neuroprotection induced by a sublethal duration of ischemia or other stimuli given before a stroke (preconditioning) on miRNA expression and the role of miRNAs in preconditioning-induced neuroprotection. Experimental manipulation of miRNAs and/or their targets to induce pre- or post-stroke protection will also be presented, as well as discussion on miRNA responses to current post-stroke therapies. This review will conclude with a brief discussion of future directions for ncRNAs studies in stroke, such as new approaches to model complex ncRNA datasets, challenges in ncRNA studies, and the impact of extracellular RNAs on human diseases such as stroke.
Introduction
Despite advances in preclinical studies that have established mechanisms of cell death in brain ischemia and identified potential strategies to prevent or treat brain injury following stroke, few of these advances have successfully translated into clinical practice. Endogenous neuroprotective responses have been studied in experimental stroke largely using a model wherein a sublethal duration of ischemia is given before a stroke (preconditioning). However, additional methods of preconditioning are also effective in reducing stroke injury, i.e., hypoxia, anesthetic agents, and activation of Toll-like receptors, and natural states of neuroprotection in animals such as hibernation that lend important insight to the topic. More recent efforts have focused on strategies for neuroprotection after a stroke (post-conditioning), which is more relevant to the clinical presentation in human patients. This review will present the current state of knowledge regarding evidence for specific families of non-coding RNAs (ncRNA) in experimental stroke and neuroprotection: microRNAs (miRNA), long non-coding RNAs (lncRNA), and piwi-interacting RNAs (piRNA). The literature search was performed using PUBMED queried with a combination of terms listed in Table 1, and the number of references returned for each query.
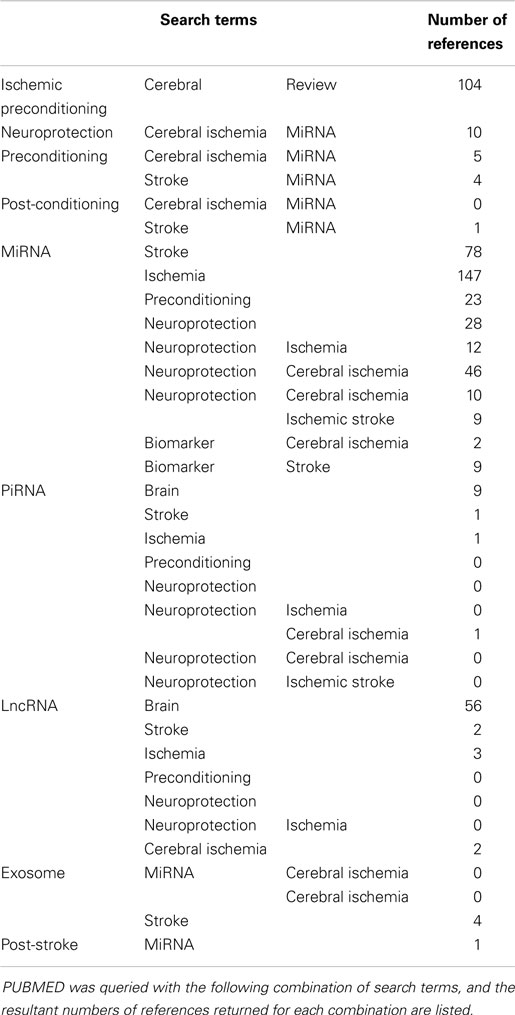
Table 1. Literature search strategy.
In recent years, there has been increasing focus on the role of ncRNAs as regulators of post-transcriptional gene expression, and of responses to ischemia and neuroprotection (1). Spinal cord injury and traumatic brain injury also lead to changes in miRNA expression that precedes injury (2). Further, dysregulation of ncRNA activities has been closely linked to the pathophysiology of cerebral vascular endothelium and neurologic functional disorders in the brain’s response to ischemia (3). Experimental stroke models have been used for global profiling of brain or blood miRNAs in animals, or to manipulate specific miRNAs or their targets to examine their potential as neuroprotective strategies for stroke. Given the current published reviews for the role of miRNAs in the occurrence and development of post-stroke depression (4), and in the regulation of miRNAs that lead to angiogenesis and remyelination (5), neurogenesis (6), and spontaneous recovery (7), they will not be discussed here. Also, outside the scope of this review are many miRNAs studies on cardiac ischemia and protection that may provide important insights on miRNAs in cerebral ischemia and neuroprotection (8).
Non-Coding RNAs
Early biochemistry studies identified three families of RNA that function cooperatively in the process of protein synthesis: messenger, transfer, and ribosomal RNA. Messenger RNAs (mRNA) carry genetic information copied from DNA in a series of three-base codes that specify a particular amino acid, specifying the polypeptide sequence. Transfer RNAs (tRNA) carry and bind a complementary amino acid to the growing end of a polypeptide chain. Ribosomal RNAs (rRNA) bind to protein complexes, which physically move along mRNAs and catalyze the assembly of amino acids into polypeptide chains (9). Additional families of ncRNAs that play essential roles in the processes of protein synthesis were subsequently discovered (10). For example, small nuclear RNAs (snRNA) bind to proteins to form spliceosomes that process pre-mRNA into mature mRNA in the nucleus (11), while small nucleolar RNAs (snoRNA) guide chemical modifications of other RNAs (12). This review will focus on three newer families of ncRNAs; (1) miRNAs that regulate post-transcriptional gene expression, largely by direct effects on mRNAs (13), (2) lncRNAs that can regulate transcription and translation, and act as epigenetic modifiers, and (3) piRNAs that largely exert effects on transcription and genomic maintenance. The reader is directed to a 2014 Frontiers in Cellular Neuroscience Research Topic entitled “Regulatory RNAs in the Nervous System” for comprehensive discussions regarding the biosynthesis and function of these ncRNAs, as well as a recent review entitled “Non-Coding RNAs as Potential Neuroprotectants against Ischemic Brain Injury,” which beautifully depicts the current understanding of the biogenesis pathways for each of these three ncRNAs discussed herein (14).
MicroRNAs
The miRNAs are the most well-characterized family of ncRNA to date. Mature, functional miRNA sequences are ~21–23 nucleotides (nt) in length, and are most recognized for their roles in the regulation of post-transcriptional gene expression via direct effects on the 3′-untranslated region (3′UTR) of mRNAs that lead to translational repression or mRNA degradation. MiRNA biogenesis has been previously described and will not be presented here, but the reader is guided to a recent comprehensive review of this topic (15). The miRNAs are particularly abundant in the nervous system where they serve as effectors of brain development (16–18) and maintenance of the neuronal phenotype (19, 20). MiRNAs also drive the maturation of dendrites and spines (21–23) and serve as effectors of synaptic plasticity and function (24–30). Emerging studies highlight roles for miRNAs in extracellular vesicles (EVs) where they contribute to cell–cell communication in the brain (31–33) and throughout the nervous system (34). As such, dysregulation of EVs may underlie diverse neurological disorders including brain tumors (35), neurodegeneration, and neuroinflammation (34), and EVs in biofluids are currently being evaluated for their potential as biomarkers or therapeutic strategies for the treatment or prevention of human diseases, including many brain disorders (36).
Long non-coding RNAs
The lncRNAs are >200 nt in size, and they differ greatly from each other with respect to their size, interacting partners, and modes of action (37). The lncRNAs are processed through pathways similar to those of protein-coding genes, and have similar histone-modification profiles, splicing signals, and exon/intron lengths. The complex intergenic, overlapping, and antisense patterns of lncRNAs relative to adjacent protein-coding genes suggests that many lncRNAs regulate the expression of adjacent protein-coding genes. LncRNAs are also implicated in the regulation of transcription and post-transcriptional gene expression, as comprehensively reviewed (38). LncRNAs can stabilize or promote the translation of target mRNAs through extended base pairing, or they can inhibit target mRNA translation or facilitate mRNA decay via partial base pairing. Even in the absence of any base pair complementarity, lncRNAs can suppress precursor mRNA splicing and translation by acting as decoys for RNA-binding proteins or miRNAs, or compete for miRNA-mediated inhibition of mRNAs resulting in increased expression of corresponding proteins. Over one-half of all lncRNAs are expressed in the brain (39) and they regulate many CNS processes (40). Hence, dysregulation of lncRNAs can also contribute to CNS pathologies such as neurodegeneration, neuroimmunological disorders, primary brain tumors, and psychiatric diseases (41). As such, lncRNAs present new targets for the understanding, diagnosis, and treatment of CNS disorders (42).
Piwi-interacting RNAs
The piRNAs are ~25–33 nt in length, depending on the PIWI protein group they bind to, and they lack sequence conservation between organisms. Recent reviews highlight the complexity of piRNA biogenesis pathways, which have just begun to be elucidated (43, 44). PiRNA biogenesis pathways are complicated as they are distinct in individual organisms, and they are distinct from miRNA pathways in that there is no evidence for a double-stranded RNA precursor and biogenesis is independent of Dicer (45–47). There are two proposed pathways for generating piRNAs: a primary processing pathway and a “ping-pong” amplification loop, as recently reviewed (14). PiRNA clusters are transcribed in the sense or antisense direction, and the long single-stranded RNA serves as the basis for piRNA production. The piRNAs protect germline cells from transposons in organisms as they guide PIWI proteins to complementary RNAs derived from transposable elements, and then the PIWI proteins cleave transposon RNA. Transposons are genetic elements that copy themselves to RNA, then back to DNA, and then integrate back into the genome where they may induce mutations that cause disease by inserting near or within other genes to disrupt their function. The piRNAs have been dubbed as the guardians of the genome because of their ability to silence deleterious retrotransposons (48). Very recent studies also show piRNAs as effectors in the epigenetic control of long-term memory storage in the brain (49).
Together, these three newer families of ncRNAs serve to regulate many aspects of cellular biology that ultimately control phenotype, function, and genomic integrity. As such, dysregulation of any or all of these ncRNAs will likely have profound impact on CNS disorders.
Ischemia
Overall Effect of Ischemia on miRNA Expression
The overall effect of stroke on miRNA expression in brain and blood has been examined in experimental rodent models and in human subjects. Most rodent studies have used transient middle cerebral artery occlusion (MCAO) to induce focal cerebral ischemia. The first such study used microarray analysis to examine overall expression of miRNAs in rat brain and blood at 24 and 48 h after ischemia and showed that miRNAs are regulated by focal ischemia and may potentially serve as blood biomarkers for ischemia (50). Focal ischemia studies in spontaneously hypertensive rats also showed altered miRNA profiles at all reperfusion times examined (3 h to 3 days), with several miRNAs altered at multiple time points (51). Bioinformatic analysis predicted mRNA targets of the ischemia-regulated miRNAs as proteins known to mediate inflammation, transcription, neuroprotection, receptor function, and ionic homeostasis (51). Further, in silico analysis of 8 ischemia-induced miRNAs revealed sequence complementarity to 877 gene promoters, suggesting that ischemia-regulated miRNAs directly modulate gene expression. Importantly, these studies also showed that focal ischemia did not alter mRNA levels for Drosha, Dicer, Pasha, and Exportin-5, proteins involved in the miRNA biogenesis pathway (51). Studies focused on humans revealed that circulating blood miRNA profiles were altered in young ischemic stroke patients (18–49 years) and that specific miRNAs that could classify subtypes of stroke (large-vessel atherosclerosis vs. small-vessel disease vs. cardioembolism) were still detectable in blood several months after injury (52). The first RNAseq studies identified significant changes in 78 known and 24 novel miRNAs in rat brain following ischemia, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis predicted that protein targets of these miRNAs function in signaling transduction, MAPK signaling, NF-kappaB signaling, and neurotrophin signaling (53).
Together, these studies show that miRNAs are regulated in brain and blood where they likely regulate translation, and transcription, in post-ischemic brain. These studies also established the potential for blood miRNAs to be developed as clinical biomarkers for the diagnosis and prognosis of ischemic stroke in humans.
Sex-Specific miRNA Responses to Ischemic Injury
Females are more resistant to stroke injury than males until menopause (54), and responses to experimental stroke and cell death are sexually dimorphic (55). In males, stroke initiates mitochondrial release of apoptosis-inducing factors that result in caspase-independent cell death, while in females stroke primarily triggers mitochondrial cytochrome c release and subsequent caspase activation. At baseline, females have higher levels than males of brain mRNA for X-linked inhibitor of apoptosis (XIAP), the primary endogenous inhibitor of caspases, and stroke significantly decreases XIAP mRNA in females but not in males. However, XIAP protein levels are decreased in both sexes after stroke, likely due to different levels of miR-23a, which binds to the XIAP 3′UTR, in male and female ischemic brain (55). Subsequent studies also revealed distinct differences in miRNA responses to ischemia in male and female brain, as well as a signature miRNA response to ischemia common to males and females (56). The finding that there are sex differences in miRNA expression during development in rat cortex may bear on differential responses to ischemia in adults (57). Accordingly, a recent study investigated the impact of age and sex on miRNA expression in adult (6 months) and middle-aged (11–12 months) female and male rats subjected to the endothelin-1 model of MCAO (58). Consistent with MCAO injury, infarct volume and sensory-motor deficits were significantly reduced in adult females compared with middle-aged females, adult males, or middle-aged males. Analysis of blood at 2 days post-stroke revealed 21 differentially regulated circulating miRNAs, and principal component analysis demonstrated that most of the variance was due to age. At 5 days post-stroke, analysis of blood revealed 78 differentially regulated circulating miRNAs, and principal component analysis confirmed that most of the variance was associated with sex. Brain miRNAs analyzed at 5 days post-stroke revealed a small cohort of miRNAs (miR-15a, miR-19b, miR-32, miR-136, and miR-199a-3p) highly expressed exclusively in adult females. These patterns of circulating miRNA expression suggest that age is the primary influence on the initial severity of stroke pathology, but sex is the primary influence on recovery from stroke. Together, these studies support that miRNA expression can be different between males and females, which may contribute to sexually dimorphic responses to ischemia.
Effect of Ischemia on Specific miRNAs and Targets
Subsequent studies began to investigate the functional significance of individual ischemia-regulated miRNAs on the molecular mechanisms that lead to brain cell death. In addition to those described above, several of these specific miRNAs are discussed below.
miR-497
The expression of miR-497 was significantly induced in mouse brain 24 h after MCAO, and in mouse N2A neuroblastoma (N2A) cells after in vitro ischemia induced by oxygen-glucose deprivation (OGD) (59). Yin and colleagues showed a direct correlation between miR-497 and cell death: gain or loss of miR-497 promoted or decreased OGD-induced neuronal cell loss, respectively. Luciferase assays showed direct binding of miR-497 to the 3′UTR of B-cell lymphoma 2 (Bcl-2) and Bcl-w mRNAs, whose proteins function as key regulators in attenuating stroke-induced apoptotic cell death (60–63). Inhibition of miR-497 in mouse brain enhanced Bcl-2/-w protein expression in the ischemic region, attenuated ischemic brain infarction, and improved neurological outcomes in response to focal ischemia.
miR-29b
Subsequent studies showed that miR-29b promoted ischemic cell death by targeting Bcl-2. MiR-29b expression was significantly increased in rat brain by focal ischemia, and in primary cultured neurons by OGD (64). Cell death was also directly correlated with miR-29b expression: gain or loss of miR-29b promoted or decreased OGD-induced neuronal cell loss, respectively. Additionally, miR-29b directly bound to the 3′UTR of Bcl-2 mRNA, and overexpression of the bcl-2 gene rescued neuronal cell death induced by miR-29b.
miR-134
Overexpression of miR-134 promoted hippocampal cell death in cultured neurons subjected to OGD, while inhibition of miR-134 was protective. Expression of cyclic AMP (cAMP) response element-binding protein (CREB), a putative target of miR-134, was inversely regulated by miR-134 (65).
miR-21
In situ hybridization showed increased expression of miR-21 in rat neurons in the ischemic boundary zone at 2 and 7 days after embolic MCAO, and RT-qPCR studies verified a quantitative increase in mature miR-21 in neurons isolated from the ischemic boundary zone (66). Overexpression of miR-21 protected cortical neurons from cell death induced by OGD, while inhibition of miR-21 promoted OGD-induced cell death. Overexpression of miR-21 in neurons significantly reduced levels of Fas Ligand (FASLG), a cell death-inducing ligand, and luciferase assays showed that miR-21 binds to the 3′UTR of FASLG mRNA.
miR-347
Permanent focal cerebral ischemia in rat brain also leads to regulation of miRNAs between 3 h and 14 days following MCAO (67). Of particular interest was miR-347 that was upregulated at both acute and late phases following ischemia. Overexpression of miR-347 increased neuronal apoptosis and induced the expression of novel mRNA targets including acyl-CoA synthetase long-chain family member 4 (Acsl4), Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3-like (Bnip3l), and phytanoyl-CoA 2-hydroxylase interacting protein (Phyhip) (67).
These independent studies demonstrate that although there are diverse miRNAs regulated by ischemia, multiple miRNAs act directly and indirectly on cell-death proteins and can influence both pro- and anti-apoptotic pathways.
Temporal and Regional Expression of Specific miRNAs in Ischemia
MiRNAs can induce distinct outcomes in a temporal- and cell-specific manner. Cultured rat cortical neurons and astrocytes subjected to OGD exhibit unique miRNA expression patterns. OGD increased miR-29b expression in neurons by twofold after 6 h and by fourfold after 24 h compared to miR-21 that remained unchanged until after 24 h of OGD (68). However, OGD increased miR-29b and miR-21 in astrocytes only after 12 h, and further, altered expression of miR-30b, miR-107, and miR-137 was specific to astrocytes. Responses to ischemia in mouse brain also showed increased expression of miR-181 in the core, where cells die, and decreased expression of miR-181 in the penumbra, where cells survive (69). Reduction in miR-181a was correlated with increased levels of glucose-regulated protein, 78 kDa (GRP78), a heat shock protein that functions within the chaperone network to increase resistance to and improve recovery from ischemic brain injury (69, 70). Further studies compared miRNA expression induced by OGD in rat cortical neurons to in vivo miRNA expression in mice exposed to the three-vessel occlusion model of ischemia (71). These studies showed that while multiple miRNAs were differentially regulated by ischemia in each model, miR-19b, miR-29b-2*, and miR-339-5p were all significantly upregulated in common to both models (71). These studies suggest that there are signature miRNA responses to ischemia in salvageable brain regions, information that may lend new insights into cell- and temporal-specific protective strategies for the treatment of ischemia.
lncRNAs in Ischemia
The effect of ischemia on lncRNA expression was first examined in adult male spontaneously hypertensive rats at 3, 6, and 12 h after MCAO (72). The results showed that of 8314 lncRNAs analyzed, 443 were significantly changed at all time points (359 upregulated and 84 downregulated). Of these, 61 stroke-responsive lncRNAs showed >90% sequence homology with exons of protein-coding genes, but in vitro translation of these lncRNAs did not form any protein products (72). Based on the roles of lncRNAs in cellular homeostasis during development and disease via interactions with chromatin-modifying proteins (CMPs), the effect of ischemia on the association of lncRNAs with CMPs, Sin3A, and corepressors of the RE1 (Repressor Element 1)-silencing transcription factor (coREST) was examined (73). These studies revealed that 177 of the 2497 lncRNAs in rat cerebral cortex showed increased binding to either Sin3A or coREST following ischemia, and that of these, 26 lncRNAs enriched with Sin3A and 11 lncRNAs enriched with coREST were upregulated by ischemia. Thus, these studies suggest that ischemia-induced lncRNAs might associate with CMPs to modulate the post-ischemic epigenetic landscape.
piRNAs in Ischemia
The first evidence for expression of piRNAs in the nervous system was revealed by RNAseq analysis of mouse hippocampal RNA (74). PiRNA expression in adult male spontaneously hypertensive rats examined 24 h after focal ischemia revealed that of ~40,000 piRNAs analyzed, 105 piRNAs were significantly altered in cerebral cortex with 54 upregulated and 51 downregulated (75). Bioinformatic analysis of four of the top stroke-responsive piRNAs (two up, two down) revealed that their transposon targets belong exclusively to the class of retrotransposons. Further, analysis of the promoter region of 10 stroke-responsive piRNAs revealed binding sites for 159 transcription factors, including the zinc finger, Kruppel, and E2F transcription factor-like families, supporting that transcription factors control the expression of stroke-responsive piRNAs.
Neuroprotection
Preconditioning
The studies discussed here will focus only on miRNA responses to preconditioning models that lead to neuroprotection against stroke as there are no published studies regarding the impact of post-conditioning on miRNA, lncRNA, or piRNA in cerebral ischemia to date. Preconditioning is a phenomenon whereby a brief, non-lethal duration of a stimulus induces a transient state of resistance (tolerance) to a subsequent lethal duration of a stimulus. Ischemic preconditioning (IPC) is an experimental technique induced by a short duration of ischemia that protects the brain from a subsequent injurious duration of ischemia (76, 77). New protein synthesis is required for IPC-induced tolerance (78), and the signature of ischemic tolerance is global repression of gene expression (79–81). However, additional preconditioning stimuli such as hypoxia, anesthesia, and activation of the Toll-like receptors can induce tolerance and protection against ischemia, as discussed below.
Ischemic preconditioning
Ischemic tolerance was first examined in a gerbil model of global cerebral ischemia (82, 83). The first studies to examine miRNA responses to ischemia in mice evaluated their expression in response to IPC (15 min MCAO), ischemia (60 min MCAO), and tolerance (15 min MCAO, 72 h reperfusion, 60 min MCAO). Microarray analysis of cortical tissue isolated from adult male mice 24 h after each treatment revealed that miRNAs were regulated by each treatment with 192 upregulated and 95 downregulated by IPC, and that there was very little overlap in the miRNAs regulated between all three treatment groups (84). Bioinformatic analysis focused on those miRNAs downregulated by IPC that could result in increased protein synthesis revealed that predicted targets included transcriptional regulators such as methyl cytosine-phosphate-guanine (CpG) binding protein 2 (MeCP2) (84). MecCP2 is a global transcriptional regulator that largely serves to repress gene expression but can also activate gene expression (85). MeCP2 protein, but not mRNA expression, increased in mouse cortex by 8 h after IPC, and MeCP2 knockout mice showed increased sensitivity to the preconditioning stimulus and were not tolerant to a subsequent ischemic event (84). Recent studies in a cardiac ischemia model showed that mesenchymal stem cells, which protect ischemic cardiomyocytes by secretion of paracrine factors, secreted exosomes enriched with miR-22 following IPC, and uptake of these exosomes by cardiomyocytes reduced ischemia-induced apoptosis via direct targeting of MeCP2 (86). These studies support a protective role for MeCP2 in both cerebral and cardiac ischemia, although the specific gene targets in each model need to be identified.
Analogous studies to examine miRNA expression in a rat model of IPC induced in adult male spontaneously hypertensive rats by 10 min MCAO revealed significant changes in 51 miRNAs (26 upregulated and 25 downregulated) at 6 h of reperfusion, and 20 of these miRNAs remained altered 3 days after IPC (87). Bioinformatic analysis of the miRNAs and KEGG Pathway analysis of their predicted targets showed MAP-kinase and mammalian target of rapamycin (mTOR) signaling as the top two pathways targeted by the upregulated miRNAs, and Wnt and gonadotropin releasing hormone (GnRH) signaling as the top two pathways targeted by the downregulated miRNAs. Interestingly, these studies also showed that multiple miRNAs downregulated by IPC were predicted to target MeCP2 in rat, consistent with previous studies on IPC in mouse (84).
Additional studies in adult male mice subjected to IPC or ischemia revealed 8 miRNAs that together comprise the miR-200 and miR-182 families of miRNAs that were selectively upregulated 3 h after IPC, and 2 miRNAs (miR-681 and miR-197) that were selectively upregulated 24 h after IPC (88). No miRNAs were downregulated at 3 h after IPC, and only 1 miRNA (miR-468) was downregulated at 24 h after IPC. Transfection of the 8 IPC upregulated miRNAs into N2A cells revealed 5 miRNAs that increased cell survival in response to OGD, and decreased expression of prolyl hydroxylase 2 (PHD2), a predicted target of the miR-200 family involved in the ubiquitin/proteosomal pathway. Reduced levels of PHD2 protein were also detected 24 h after IPC, but not after ischemia.
Hypoxic preconditioning
Hypoxic preconditioning (HPC) induced by autohypoxia (acute and repetitive exposure to progressive hypoxia) in adult male mice significantly decreased ischemic injury by mechanisms involving multiple conventional protein kinase C βII (cPKCβII)-interacting proteins (89). Bioinformatic analysis of 17 miRNAs (4 upregulated and 13 downregulated) in mouse cortex between HPC and ischemia predicted that cPKCβII, γ, and novel protein kinase Cε (nPKCε)-interacting proteins as targets of the HPC-specific miRNAs (90). Together, these studies support that HPC-induced protection against ischemia involves regulation of cPKC proteins and modulation of calcium signaling in mouse brain.
Anesthetic preconditioning
Several experimental studies show that inhalational anesthetics serve as preconditioning agents that lead to neuroprotection against stroke. Administration of isoflurane, halothane, sevoflurane, desflurane, nitrous oxide, and other agents is protective in the setting of stroke, and potential mechanisms of anesthetic preconditioning include antagonism of AMPA and NMDA receptors and reduced excitotoxicity, modulation of cerebral catecholamine release, direct anti-oxidant effects of anesthesia, and reduction of the cerebral metabolic rate (91). Recent studies to examine the contribution of miRNAs to anesthetic preconditioning revealed that isoflurane significantly increased expression of miR-203 in rat B35 neuron-like cells, and there was a trend toward increased miR-203 expression in rat cerebral cortex after isoflurane exposure (92). Overexpression of miR-203 in B35 cells increased both tolerance to ischemia and expression of Phospho-Akt, a protein kinase that promotes cell survival. The effect of sevoflurane preconditioning on rat brain miRNA expression following transient cerebral ischemia revealed 3 upregulated and 9 downregulated miRNAs (93). Among these, miR-15b expression was increased after ischemia, while sevoflurane preconditioning downregulated miR-15b expression and increased expression of Bcl-2, a target of miR-15b. These studies suggest that sevoflurane may exert anti-apoptotic effects by inhibiting miR-15b expression leading to increased expression of Bcl-2.
Toll-like receptor preconditioning
Preconditioning can be induced by activation of the Toll-like receptors (94, 95). CpG oligodeoxynucleotide is a Toll-like receptor 9 agonist that when administered 72 h prior to ischemia can reduce injury in rodent and non-human primate models of experimental stroke (96, 97). MiRNA expression studies in adult male mice revealed that miRNAs are not regulated at 3, 24, or 72 h after CpG preconditioning (98), suggesting that alterations in miRNA expression in the brain are not necessary for the neuroprotection induced by systemic CpG preconditioning. Interestingly, CpG-preconditioned mice had fewer total miRNAs regulated in response to MCAO compared with saline-treated mice (57 vs. 73 miRNAs), suggesting that CpG preconditioning alters the miRNA response to stroke (98). Subsequent analysis of the miRNAs differentially expressed in CpG-preconditioned and saline-treated mice at 24 h after stroke revealed that miRNA suppression correlated with the upregulation of genes involved in neuroprotection, suggesting that suppression of differential miRNAs in CpG preconditioning may reprogram the genomic response to stroke to favor a protective outcome.
Hibernation torpor
Torpor is a form of sleep that helps animals conserve valuable resources in times of stress, such as in cold or very hot, dry weather. Hibernation torpor (HT) is used to model endogenous mechanisms of tolerance, as metabolic rates decrease in order to conserve energy in both conditions. HT induces massive global SUMOylation in ground squirrels, and overexpression of Ubc9, SUMO-1, or SUMO-2/3 is protective against ischemic injury in cortical neurons subjected to OGD, and in mouse brain (99, 100). SUMO conjugation and global protein conjugation by ubiquitin-like modifiers (ULMs) including NEDD8, ISG15, UFM1, and FUB1 significantly increased in ground squirrel brain during HT. MiRNA analysis of ground squirrel brain during HT revealed that the miR-200 and miR-182 families were among the most consistently depressed miRNAs relative to active animals (101). Inhibition of these miRNA families in SHSY5Y cells resulted in increased global protein conjugation and tolerance to OGD-induced cell death (101). These studies link endogenous tolerance to brain ischemia in hibernating animals to the regulation of a broad range of ULMs via repression of miRNAs.
Neuroprotection via Manipulation of miRNAs and/or Their Targets
The previous studies support that manipulation of specific miRNAs or their targets altered by IPC or ischemia may be translated into viable neuroprotective strategies for stroke. Identified targets thus far include epigenetic modifiers, heat shock proteins, ubiquitin modifiers, DNA repair and cell cycle proteins, autophagosome assembly proteins, and mediators of inflammatory responses to injury in astrocytes.
miR-29
Previous studies showed that miR-29 is highly expressed in adult rat brain, and is significantly downregulated by focal ischemia (51). Bioinformatic analysis revealed DNA (cytosine-5)-methyltransferase 3a (DNMT3a) as a target of miR-29c, suggesting that increased DNMT3a expression in ischemic brain may contribute to stroke injury (102). Indeed, treatment with premiR-29c was protective against ischemia both in vitro (PC12 cells) and in vivo, suggesting that strategies to replace miR-29c may decrease cell death following ischemia. In addition, the miR-29c gene promoter region contains binding sites for REST, and treatment of PC12 cells with REST siRNA prevented ischemia-induced downregulation of miR-29c, which maintained repression of DNMT3a resulting in significantly reduced ischemic cell death (102).
miR-132
Studies show that miR-132 is a neuron-specific miRNA that contains a RE1 site within its promoter regions in mouse and is a validated target of REST in mammalian cells (19). Transient global ischemia markedly decreased miR-132 expression in rat hippocampal CA1 cells at 24 and 48 h after ischemia, due to ischemia-induced enrichment of REST at the miR-132 promoter and repressed expression of miR-132 (103). REST-dependent silencing was specific to miR-132, as REST did not alter expression of miR-9 or miR-124a that also has RE1 sites in their promoter regions. In addition, the repression of miR-132 was selectively induced in vulnerable CA1 neurons, but not in CA3 neurons, supporting that REST-dependent repression of miR-132 is critical to ischemia-induced neuronal death (103). Depletion of REST in vivo blocked ischemia-induced loss of miR-132 in hippocampal neurons, while overexpression of miR-132 in vitro and in vivo afforded robust protection against ischemia-induced neuronal death. These studies support that REST-dependent repression of miR-132 induces neuronal death in global ischemia.
miR-181b and miR-134
The expression of miR-181b and miR-134 is decreased by HPC and ischemia in adult mouse cortex, but increased in tolerant brain (90). Targets for miR-181b include heat shock protein A5 (HSPA5) and ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1), and expression of miR-181b in N2A cells repressed HSPA5 and UCHL1 and protected cells against OGD-induced cell death. Further, miR-181b antagomirs reduced caspase-3 cleavage and neuronal cell loss in ischemic cortex and improved neurological deficits in mice after ischemia (104). A subsequent study showed that downregulation of miR-134 in mouse N2A cells is protective against OGD by upregulating heat shock protein A12B (HSPA12B) (105). HSPA12B was validated as a target of miR-134, and increased expression of HSPA12B correlated with attenuation of neural cell damage in OGD-treated N2A cells, and reduced infarct size and improved neurological outcomes in mice with ischemic stroke (4).
miR-30a
Downregulation of miR-30a following ischemia in N2A cells and in mouse brain correlated with an increased conversion ratio of LC3 (microtubule-associated protein 1 light chain 3)-II/LC3-I and increased expression of Beclin 1, which are required for formation of the autophagosome (106).
miR-124
Ischemia leads to reduced expression of miR-124 in rat brain (107, 108) and downregulation of miR-124 in rats subjected to focal ischemia correlated with increased expression of Ku70, a DNA repair protein. In vivo administration of a miR-124 antagomir into rat brain increased expression of Ku70 mRNA and protein in the ischemic region, resulting in reduced cell death and infarct size, and improved neurological outcomes in rat (109).
miR-424
Expression of miR-424 is decreased in the plasma of patients with acute ischemic stroke, and in mouse plasma and brain after ischemia (110). Overexpression of miR-424 decreased infarct size and brain edema due to MCAO when administered either pre- or post-treatment. MiR-424 mimics reduced mRNA and protein levels of cell division cycle 25 homolog A (CDC25A), cyclin D1, and cell division protein kinase 6 (CDK6) in BV2 microglial cells, all of which are upregulated in ischemic brain (110).
miR-103-1
Recent studies examined miR-103-1 as a treatment strategy for stroke, based on its potential to regulate the expression of the Na+/Ca2+ exchanger (NCX1), which mediates bidirectional flux of calcium and sodium across the synaptic membrane and increases ischemic damage in rat brain (111). NCX1 was validated as a target of miR-103-1 in cortical neurons, and intracerebroventricular infusion of anti-miR-103-1 not only prevented NCX1 reduction induced by stroke but also significantly reduced the extent of brain ischemia when infused 24 h before stroke onset (112).
Astrocyte miRNAs
Ischemic brain extract contains factors that elicit increased expression of stromal cell-derived factor-1 (SDF-1), a chemokine that is induced by proinflammatory stimuli and that activates leukocytes, in C6 cells and primary astrocytes (113). SDF-1 expression increased in preconditioned astrocytes contributes to brain tolerance via the regulation of the miR-223/miR-27b pathways (113). Neuroprotection by manipulation of miRNA-related pathways are also found in astrocyte-rich miRNAs, including miR-181 and miR-29 families, and miR-146a, and their validated targets, GRP78, and Bcl-2 family members (114).
Effects of Post-Stroke Neuroprotective Therapies on miRNAs
Recent studies show that post-stroke therapies can effectively regulate the expression of miRNAs and their targets, and these miRNAs may serve as direct targets for clinical intervention in stroke. For example, post-stroke treatment of rats with valproic acid, a histone deacetylase inhibitor and a mood stabilizer, upregulated the expression of several miRNAs including miR-331 and miR-885-3p, and improved neurological deficits and motor performance following ischemia (115). In addition, studies show that using Velcade (a proteasome inhibitor that may also suppress TLR signaling) administered in combination with tissue plasminogen activator (tPA) is neuroprotective against ischemia in aged rats (116). The combination of Velcade plus tPA resulted in increased levels of miR-l46a on cerebral endothelial cells, which express TLRs, and outcomes were associated with reduced expression of vascular interleukin-1 receptor-activated kinases 1 (116). Further, it is well known that ischemic injury is more severe in older animals as compared to younger animals, and this difference is associated with reduced availability of insulin-like growth factor-1 (IGF-1) whose levels decrease with age. IGF-1 infusion following stroke, which prevents estrogen neurotoxicity in middle-aged female rats, altered the expression of two conserved IGF pathway regulatory miRNAs, Let-7f, and miR-1. Adult female rats treated with anti-miR-1 as late as 4 h after ischemia had significantly reduced cortical infarct volume, and anti-Let-7 robustly reduced both cortical and striatal infarcts and preserved sensorimotor function and inter-hemispheric neural integration (117). However, anti-Let-7 was only effective in intact females and had no effect on males or ovariectomized females, indicating that the gonadal steroid environment critically modifies miRNA action (117). Further studies examined the effect of post-stroke IGF-1 treatment on miRNA expression in middle-aged female rats (118). IGF-1 administered immediately after ischemia significantly reduced infarct volume in 9- to 11-month female rats 24 h after stroke. Post-stroke IGF-1 treatment significantly downregulated 8 out of 168 disease-related miRNAs in tissue isolated 4 h after ischemia. KEGG pathway analysis predicted that targets of these miRNAs include proteins involved in PI3K–Akt signaling, cell adhesion/ECM receptor pathways, and T-and B-cell signaling. Further, Phospho-Akt was reduced by 4 h after ischemia, but was elevated by IGF-1 treatment at 24 h after ischemia. IGF-1 also induced Akt activation, an effect that was preceded by a reduction of BBB permeability and global suppression of cytokines at 4 h post-stroke (118).
Conclusion
The goal of this review was to present the current state of knowledge regarding ncRNAs and their role(s) in stroke and neuroprotection. However, of these three relatively new families of ncRNAs, miRNAs are the most well studied to date. Yet the lncRNAs and piRNAs are certain to contribute to the setting of ischemia and neuroprotection, and there are several important future lines of investigation for all ncRNA studies. The foregoing studies highlight the difficulty of analyzing miRNA expression data, due to the diversity of ischemia and neuroprotection models, the time points examined, and the fact that since miRNAs can act in concert to exert effects on target mRNAs, focus on a single miRNA is unlikely to realistically capture endogenous responses. Thus, the first such studies must focus on ways to meaningfully translate the outcome of multiple expression profiling studies from diverse labs in order to identify global signaling pathways or targets that have been underappreciated to date. To address this issue, Xiao and colleagues sought to model complex interactions in order to identify synergistic effects within miRNA responses to brain ischemia (119). miRNA array expression data submitted to the Gene Expression Omnibus database from multiple studies in different labs were examined to identify mouse brain miRNAs differentially expressed between ischemia and control. Common predicted targets of the miRNAs were then used as a candidate subset to identify functional modules. An miRNA functional synergistic network, constructed by assembling all miRNA synergistic pairs, revealed 51 differentially expressed miRNAs identified between ischemia and control (32 upregulated and 19 downregulated). Among them, 24 miRNAs commonly regulating at least one target gene were used to construct a network. Subsequent analyses revealed 16 miRNAs forming 20 miRNA interaction pairs that participated in inflammatory responses, suggesting these 16 miRNAs as underlying targets for prevention and/or treatment of stroke (119). These analyses highlight that the power of global miRNA and ncRNA studies lies in their potential to identify proteins or pathways involved in responses to ischemia. The second such studies must address the need to identify the effects of ncRNAs acting in concert to regulate cellular phenotypes or responses to stress. To date, each family of ncRNA has largely been examined individually, due to technical barriers to combinatorial analysis of all ncRNAs. RNAseq studies can be used to identify all RNA species in a given sample set, but even this method is limited in the ability to analyze distinct sizes of RNA species. Thus, lncRNAs (>200 nt) are not yet typically sequenced together with small ncRNAs such as the miRNAs and piRNAs (~20–36 nt). A third line of exciting studies will come from the not-so-recent finding that extracellular RNAs exist in biofluids, mostly contained within EVs such as microvesicles, and can serve as biomarkers of neurological disorders (120). EVs and their molecular components are increasingly shown to be effectors of cell–cell communication in the brain and their uptake can alter the phenotype of a recipient cell (32). Thus, future lines of investigation should include examining the consequences of disrupted blood flow and metabolism, such as during a stroke, on communication mediated by EVs. Further, given that EVs mediate communication between distinct cell types in the brain, future therapeutic strategies for the treatment or prevention of stroke and other brain disorders might include focused delivery of molecular or pharmaceutical agents by EVs, potentially using vesicles isolated from ones own biofluids. The importance and excitement of such studies is highlighted by the recent formation of the Extracellular RNA Communication program (http://commonfund.nih.gov/Exrna/index) supported by the National Institutes of Health Common Fund that aims to discover fundamental biological principles about the mechanisms of exRNA generation, secretion, and transport; to identify and develop a catalog of exRNA in normal human body fluids; and to investigate the potential for using exRNAs as therapeutic molecules or biomarkers of disease. Thus, there are many new and exciting future opportunities for research and development in the field of ncRNAs, and in particular for their role in, contribution to, and regulation of the treatment of human diseases, including stroke.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I sincerely thank Dr. Michael Roberts and Dr. Theresa Lusardi for their excellent comments and editorial contributions to this manuscript. While all efforts were made to provide a comprehensive review on this topic, the author sincerely apologizes for any studies that may have been inadvertently overlooked during creation of the manuscript. Supported by National Institutes of Health R01NS064270 and R21NS082832.
References
1. Vemuganti R. All’s well that transcribes well: non-coding RNAs and post- stroke brain damage. Neurochem Int (2013) 63:438–49. doi: 10.1016/j.neuint.2013.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Bhalala OG, Srikanth M, Kessler JA. The emerging roles of microRNAs in CNS injuries. Nat Rev Neurol (2013) 9:328–39. doi:10.1038/nrneurol.2013.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Yin KJ, Hamblin M, Chen YE. Non-coding RNAs in cerebral endothelial pathophysiology: emerging roles in stroke. Neurochem Int (2014) 77:9–16. doi:10.1016/j.neuint.2014.03.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Yan H, Fang M, Liu XY. Role of microRNAs in stroke and poststroke depression. ScientificWorldJournal (2013) 2013:459692. doi:10.1155/2013/459692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Wang Y, Wang Y, Yang GY. MicroRNAs in cerebral ischemia. Stroke Res Treat (2013) 2013:276540. doi:10.1155/2013/276540
6. Liu XS, Chopp M, Zhang RL, Zhang ZG. MicroRNAs in cerebral ischemia-induced neurogenesis. J Neuropathol Exp Neurol (2013) 72:718–22. doi:10.1097/NEN.0b013e31829e4963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Liu FJ, Lim KY, Kaur P, Sepramaniam S, Armugam A, Wong PT, et al. microRNAs involved in regulating spontaneous recovery in embolic stroke model. PLoS One (2013) 8:e66393. doi:10.1371/journal.pone.0066393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kukreja RC, Yin C, Salloum FN. MicroRNAs: new players in cardiac injury and protection. Mol Pharmacol (2011) 80:558–64. doi:10.1124/mol.111.073528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Lodish HF. Translational control of protein synthesis. Annu Rev Biochem (1976) 45:39–72. doi:10.1146/annurev.bi.45.070176.000351
10. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell (2014) 157:77–94. doi:10.1016/j.cell.2014.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol (2014) 15:108–21. doi:10.1038/nrm3778
12. Bratkovic T, Rogelj B. The many faces of small nucleolar RNAs. Biochim Biophys Acta (2014) 1839:438–43. doi:10.1016/j.bbagrm.2014.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr Opin Struct Biol (2005) 15:331–41. doi:10.1016/j.sbi.2005.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Kaur P, Liu F, Tan JR, Lim KY, Sepramaniam S, Karolina DS, et al. Non-coding RNAs as potential neuroprotectants against ischemic brain injury. Brain Sci (2013) 3:360–95. doi:10.3390/brainsci3010360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15:509–24. doi:10.1038/nrm3838
16. Iyengar BR, Choudhary A, Sarangdhar MA, Venkatesh KV, Gadgil CJ, Pillai B. Non-coding RNA interact to regulate neuronal development and function. Front Cell Neurosci (2014) 8:47. doi:10.3389/fncel.2014.00047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Petri R, Malmevik J, Fasching L, Akerblom M, Jakobsson J. miRNAs in brain development. Exp Cell Res (2014) 321:84–9. doi:10.1016/j.yexcr.2013.09.022
18. Sun E, Shi Y. MicroRNAs: small molecules with big roles in neurodevelopment and diseases. Exp Neurol (2014). doi:10.1016/j.expneurol.2014.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A (2006) 103:2422–7. doi:10.1073/pnas.0511041103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Jovicic A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, et al. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci (2013) 33:5127–37. doi:10.1523/JNEUROSCI.0600-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Fiore R, Khudayberdiev S, Christensen M, Siegel G, Flavell SW, Kim TK, et al. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J (2009) 28:697–710. doi:10.1038/emboj.2009.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A (2010) 107:20382–7. doi:10.1073/pnas.1015691107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature (2006) 439:283–9. doi:10.1038/nature04367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Aksoy-Aksel A, Zampa F, Schratt G. MicroRNAs and synaptic plasticity – a mutual relationship. Philos Trans R Soc Lond B Biol Sci (2014) 369:20130515. doi:10.1098/rstb.2013.0515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Bredy TW, Lin Q, Wei W, Baker-Andresen D, Mattick JS. MicroRNA regulation of neural plasticity and memory. Neurobiol Learn Mem (2011) 96:89–94. doi:10.1016/j.nlm.2011.04.004
26. Olde Loohuis NF, Kos A, Martens GJ, Van Bokhoven H, Nadif Kasri N, Aschrafi A. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci (2012) 69:89–102. doi:10.1007/s00018-011-0788-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Paschou M, Paraskevopoulou MD, Vlachos IS, Koukouraki P, Hatzigeorgiou AG, Doxakis E. miRNA regulons associated with synaptic function. PLoS One (2012) 7:e46189. doi:10.1371/journal.pone.0046189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Siegel G, Saba R, Schratt G. microRNAs in neurons: manifold regulatory roles at the synapse. Curr Opin Genet Dev (2011) 21:491–7. doi:10.1016/j.gde.2011.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Tognini P, Pizzorusso T. MicroRNA212/132 family: molecular transducer of neuronal function and plasticity. Int J Biochem Cell Biol (2012) 44:6–10. doi:10.1016/j.biocel.2011.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Wibrand K, Pai B, Siripornmongcolchai T, Bittins M, Berentsen B, Ofte ML, et al. MicroRNA regulation of the synaptic plasticity-related gene Arc. PLoS One (2012) 7:e41688. doi:10.1371/journal.pone.0041688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Chivet M, Javalet C, Hemming F, Pernet-Gallay K, Laulagnier K, Fraboulet S, et al. Exosomes as a novel way of interneuronal communication. Biochem Soc Trans (2013) 41:241–4. doi:10.1042/BST20120266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Fruhbeis C, Frohlich D, Kuo WP, Kramer-Albers EM. Extracellular vesicles as mediators of neuron-glia communication. Front Cell Neurosci (2013) 7:182. doi:10.3389/fncel.2013.00182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Sharma P, Schiapparelli L, Cline HT. Exosomes function in cell-cell communication during brain circuit development. Curr Opin Neurobiol (2013) 23:997–1004. doi:10.1016/j.conb.2013.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Rajendran L, Bali J, Barr MM, Court FA, Kramer-Albers EM, Picou F, et al. Emerging roles of extracellular vesicles in the nervous system. J Neurosci (2014) 34:15482–9. doi:10.1523/JNEUROSCI.3258-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Garnier D, Jabado N, Rak J. Extracellular vesicles as prospective carriers of oncogenic protein signatures in adult and paediatric brain tumours. Proteomics (2013) 13:1595–607. doi:10.1002/pmic.201200360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. El Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov (2013) 12:347–57. doi:10.1038/nrd3978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet (2009) 10:155–9. doi:10.1038/nrg2521
38. Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol (2013) 425(19):3723–30. doi:10.1016/j.jmb.2012.11.024
39. Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A (2008) 105:716–21. doi:10.1073/pnas.0706729105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci (2012) 13:528–41. doi:10.1038/nrn3234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Qureshi IA, Mattick JS, Mehler MF. Long non-coding RNAs in nervous system function and disease. Brain Res (2010) 1338:20–35. doi:10.1016/j.brainres.2010.03.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Qureshi IA, Mehler MF. Long non-coding RNAs: novel targets for nervous system disease diagnosis and therapy. Neurotherapeutics (2013) 10:632–46. doi:10.1007/s13311-013-0199-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Weick EM, Miska EA. piRNAs: from biogenesis to function. Development (2014) 141:3458–71. doi:10.1242/dev.094037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet (2013) 14:447–59. doi:10.1038/nrg3462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell (2008) 31:79–90. doi:10.1016/j.molcel.2008.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell (2007) 129:69–82. doi:10.1016/j.cell.2007.03.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science (2006) 313:320–4. doi:10.1126/science.1129333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell (2009) 136:656–68. doi:10.1016/j.cell.2009.01.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Landry CD, Kandel ER, Rajasethupathy P. New mechanisms in memory storage: piRNAs and epigenetics. Trends Neurosci (2013) 36:535–42. doi:10.1016/j.tins.2013.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Jeyaseelan K, Lim KY, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke (2008) 39:959–66. doi:10.1161/STROKEAHA.107.500736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab (2009) 29:675–87. doi:10.1038/jcbfm.2008.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Tan KS, Armugam A, Sepramaniam S, Lim KY, Setyowati KD, Wang CW, et al. Expression profile of MicroRNAs in young stroke patients. PLoS One (2009) 4:e7689. doi:10.1371/journal.pone.0007689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wang C, Pan Y, Cheng B, Chen J, Bai B. Identification of conserved and novel micrornas in cerebral ischemia-reperfusion injury of rat using deep sequencing. J Mol Neurosci (2014) 54(4):671–83. doi:10.1007/s12031-014-0383-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J (2004) 45:147–59. doi:10.1093/ilar.45.2.147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Siegel C, Li J, Liu F, Benashski SE, McCullough LD. miR-23a regulation of X-linked inhibitor of apoptosis (XIAP) contributes to sex differences in the response to cerebral ischemia. Proc Natl Acad Sci U S A (2011) 108:11662–7. doi:10.1073/pnas.1102635108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Lusardi TA, Murphy SJ, Phillips JI, Chen Y, Davis CM, Young JM, et al. MicroRNA responses to focal cerebral ischemia in male and female mouse brain. Front Mol Neurosci (2014) 7:11. doi:10.3389/fnmol.2014.00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Murphy SJ, Lusardi TA, Phillips JI, Saugstad JA. Sex differences in microRNA expression during development in rat cortex. Neurochem Int (2014) 77:24–32. doi:10.1016/j.neuint.2014.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Selvamani A, Williams MH, Miranda RC, Sohrabji F. Circulating miRNA profiles provide a biomarker for severity of stroke outcomes associated with age and sex in a rat model. Clin Sci (Lond) (2014) 127:77–89. doi:10.1042/CS20130565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, et al. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis (2010) 38(1):17–26. doi:10.1016/j.nbd.2009.12.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Chen J, Simon RP, Nagayama T, Zhu R, Loeffert JE, Watkins SC, et al. Suppression of endogenous bcl-2 expression by antisense treatment exacerbates ischemic neuronal death. J Cereb Blood Flow Metab (2000) 20:1033–9. doi:10.1097/00004647-200007000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Linnik MD, Zahos P, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke (1995) 26:1670–4. doi:10.1161/01.STR.26.9.1670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron (1994) 13:1017–30. doi:10.1016/0896-6273(94)90266-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Sun Y, Jin K, Clark KR, Peel A, Mao XO, Chang Q, et al. Adeno-associated virus-mediated delivery of BCL-w gene improves outcome after transient focal cerebral ischemia. Gene Ther (2003) 10:115–22. doi:10.1038/sj.gt.3301868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y, et al. Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res (2012) 216:225–30. doi:10.1007/s00221-011-2925-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Huang W, Liu X, Cao J, Meng F, Li M, Chen B, et al. miR-134 regulates ischemia/reperfusion injury-induced neuronal cell death by regulating CREB signaling. J Mol Neurosci (2014) 2:1–9. doi:10.1007/s12031-014-0434-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Buller B, Liu X, Wang X, Zhang RL, Zhang L, Hozeska-Solgot A, et al. MicroRNA-21 protects neurons from ischemic death. FEBS J (2010) 277:4299–307. doi:10.1111/j.1742-4658.2010.07818.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Gubern C, Camos S, Ballesteros I, Rodriguez R, Romera VG, Canadas R, et al. miRNA expression is modulated over time after focal ischaemia: up-regulation of miR-347 promotes neuronal apoptosis. FEBS J (2013) 280:6233–46. doi:10.1111/febs.12546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Ziu M, Fletcher L, Rana S, Jimenez DF, Digicaylioglu M. Temporal differences in microRNA expression patterns in astrocytes and neurons after ischemic injury. PLoS One (2011) 6:e14724. doi:10.1371/journal.pone.0014724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE, et al. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis (2012) 45:555–63. doi:10.1016/j.nbd.2011.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Ouyang YB, Giffard RG. MicroRNAs regulate the chaperone network in cerebral ischemia. Transl Stroke Res (2013) 4:693–703. doi:10.1007/s12975-013-0280-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Dhiraj DK, Chrysanthou E, Mallucci GR, Bushell M. miRNAs-19b, -29b-2* and -339-5p show an early and sustained up-regulation in ischemic models of stroke. PLoS One (2013) 8:e83717. doi:10.1371/journal.pone.0083717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Dharap A, Nakka VP, Vemuganti R. Effect of focal ischemia on long noncoding RNAs. Stroke (2012) 43:2800–2. doi:10.1161/STROKEAHA.112.669465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Dharap A, Pokrzywa C, Vemuganti R. Increased binding of stroke-induced long non-coding RNAs to the transcriptional corepressors Sin3A and coREST. ASN Neuro (2013) 5:283–9. doi:10.1042/AN20130029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Lee EJ, Banerjee S, Zhou H, Jammalamadaka A, Arcila M, Manjunath BS, et al. Identification of piRNAs in the central nervous system. RNA (2011) 17:1090–9. doi:10.1261/rna.2565011
75. Dharap A, Nakka VP, Vemuganti R. Altered expression of PIWI RNA in the rat brain after transient focal ischemia. Stroke (2011) 42:1105–9. doi:10.1161/STROKEAHA.110.598391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Rejdak R, Rejdak K, Sieklucka-Dziuba M, Stelmasiak Z, Grieb P. Brain tolerance and preconditioning. Pol J Pharmacol (2001) 53:73–9.
77. Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, et al. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol (2014) 114:58–83. doi:10.1016/j.pneurobio.2013.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke (1998) 29:1937–50. doi:10.1161/01.STR.29.9.1937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke (2007) 38:680–5. doi:10.1161/01.STR.0000251444.56487.4c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Stenzel-Poore MP, Stevens SL, Simon RP. Genomics of preconditioning. Stroke (2004) 35:2683–6. doi:10.1161/01.STR.0000143735.89281.bb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet (2003) 362:1028–37. doi:10.1016/S0140-6736(03)14412-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, et al. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res (1991) 561:203–11. doi:10.1016/0006-8993(91)91596-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res (1990) 528:21–4. doi:10.1016/0006-8993(90)90189-I
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Lusardi TA, Farr CD, Faulkner CL, Pignataro G, Yang T, Lan J, et al. Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J Cereb Blood Flow Metab (2010) 30:744–56. doi:10.1038/jcbfm.2009.253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science (2008) 320:1224–9. doi:10.1126/science.1153252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Feng Y, Huang W, Wani M, Yu X, Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS One (2014) 9:e88685. doi:10.1371/journal.pone.0088685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Dharap A, Vemuganti R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J Neurochem (2010) 113:1685–91. doi:10.1111/j.1471-4159.2010.06735.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Lee ST, Chu K, Jung KH, Yoon HJ, Jeon D, Kang KM, et al. MicroRNAs induced during ischemic preconditioning. Stroke (2010) 41:1646–51. doi:10.1161/STROKEAHA.110.579649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, et al. Proteomic analysis of cPKCbetaII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem (2011) 117:346–56. doi:10.1111/j.1471-4159.2011.07209.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Liu C, Peng Z, Zhang N, Yu L, Han S, Li D, et al. Identification of differentially expressed microRNAs and their PKC-isoform specific gene network prediction during hypoxic pre-conditioning and focal cerebral ischemia of mice. J Neurochem (2012) 120:830–41. doi:10.1111/j.1471-4159.2011.07624.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kitano H, Kirsch JR, Hurn PD, Murphy SJ. Inhalational anesthetics as neuroprotectants or chemical preconditioning agents in ischemic brain. J Cereb Blood Flow Metab (2007) 27:1108–28. doi:10.1038/sj.jcbfm.9600410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Cao L, Feng C, Li L, Zuo Z. Contribution of microRNA-203 to the isoflurane preconditioning-induced neuroprotection. Brain Res Bull (2012) 88:525–8. doi:10.1016/j.brainresbull.2012.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Shi H, Sun BL, Zhang J, Lu S, Zhang P, Wang H, et al. miR-15b suppression of Bcl-2 contributes to cerebral ischemic injury and is reversed by sevoflurane preconditioning. CNS Neurol Disord Drug Targets (2013) 12:381–91. doi:10.2174/1871527311312030011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience (2009) 158:1007–20. doi:10.1016/j.neuroscience.2008.07.067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Vartanian K, Stenzel-Poore M. Toll-like receptor tolerance as a mechanism for neuroprotection. Transl Stroke Res (2010) 1:252–60. doi:10.1007/s12975-010-0033-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Bahjat FR, Williams-Karnesky RL, Kohama SG, West GA, Doyle KP, Spector MD, et al. Proof of concept: pharmacological preconditioning with a toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab (2011) 31:1229–42. doi:10.1038/jcbfm.2011.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab (2008) 28:1040–7. doi:10.1038/sj.jcbfm.9600606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Vartanian KB, Mitchell HD, Stevens SL, Conrad VK, McDermott JE, Stenzel-Poore MP. CpG preconditioning regulates miRNA expression that modulates genomic reprogramming associated with neuroprotection against ischemic injury. J Cereb Blood Flow Metab (2014) 35(2):257–66. doi:10.1038/jcbfm.2014.193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Lee YJ, Castri P, Bembry J, Maric D, Auh S, Hallenbeck JM. SUMOylation participates in induction of ischemic tolerance. J Neurochem (2009) 109:257–67. doi:10.1111/j.1471-4159.2009.05957.x
100. Lee YJ, Miyake S, Wakita H, McMullen DC, Azuma Y, Auh S, et al. Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. J Cereb Blood Flow Metab (2007) 27:950–62. doi:10.1038/sj.jcbfm.9600395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Lee YJ, Johnson KR, Hallenbeck JM. Global protein conjugation by ubiquitin-like-modifiers during ischemic stress is regulated by microRNAs and confers robust tolerance to ischemia. PLoS One (2012) 7:e47787. doi:10.1371/journal.pone.0047787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Pandi G, Nakka VP, Dharap A, Roopra A, Vemuganti R. MicroRNA miR-29c down-regulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS One (2013) 8:e58039. doi:10.1371/journal.pone.0058039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Hwang JY, Kaneko N, Noh KM, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol (2014) 426:3454–66. doi:10.1016/j.jmb.2014.07.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Peng Z, Li J, Li Y, Yang X, Feng S, Han S, et al. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J Neurosci Res (2013) 91:1349–62. doi:10.1002/jnr.23255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Chi W, Meng F, Li Y, Li P, Wang G, Cheng H, et al. Impact of microRNA-134 on neural cell survival against ischemic injury in primary cultured neuronal cells and mouse brain with ischemic stroke by targeting HSPA12B. Brain Res (2014) 1592:22–33. doi:10.1016/j.brainres.2014.09.072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, et al. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res (2014) 39:1279–91. doi:10.1007/s11064-014-1310-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs expression and function in cerebral ischemia reperfusion injury. J Mol Neurosci (2014) 53:242–50. doi:10.1007/s12031-014-0293-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Peng T, Jia YJ, Wen QQ, Guan WJ, Zhao EY, Zhang BA. [Expression of microRNA in neonatal rats with hypoxic-ischemic brain damage]. Zhongguo Dang Dai Er Ke Za Zhi (2010) 12:373–6.
109. Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci (2014) 52:148–55. doi:10.1007/s12031-013-0155-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Zhao H, Wang J, Gao L, Wang R, Liu X, Gao Z, et al. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke (2013) 44:1706–13. doi:10.1161/STROKEAHA.111.000504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Pignataro G, Gala R, Cuomo O, Tortiglione A, Giaccio L, Castaldo P, et al. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke (2004) 35:2566–70. doi:10.1161/01.STR.0000143730.29964.93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Vinciguerra A, Formisano L, Cerullo P, Guida N, Cuomo O, Esposito A, et al. MicroRNA-103-1 selectively downregulates brain NCX1 and its inhibition by anti-miRNA ameliorates stroke damage and neurological deficits. Mol Ther (2014) 22:1829–38. doi:10.1038/mt.2014.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Shin JH, Park YM, Kim DH, Moon GJ, Bang OY, Ohn T, et al. Ischemic brain extract increases SDF-1 expression in astrocytes through the CXCR2/miR-223/miR-27b pathway. Biochim Biophys Acta (2014) 1839:826–36. doi:10.1016/j.bbagrm.2014.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Ouyang YB, Xu L, Yue S, Liu S, Giffard RG. Neuroprotection by astrocytes in brain ischemia: importance of microRNAs. Neurosci Lett (2014) 565:53–8. doi:10.1016/j.neulet.2013.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Hunsberger JG, Fessler EB, Wang Z, Elkahloun AG, Chuang DM. Post-insult valproic acid-regulated microRNAs: potential targets for cerebral ischemia. Am J Transl Res (2012) 4:316–32.
116. Zhang L, Chopp M, Liu X, Teng H, Tang T, Kassis H, et al. Combination therapy with VELCADE and tissue plasminogen activator is neuroprotective in aged rats after stroke and targets microRNA-146a and the toll-like receptor signaling pathway. Arterioscler Thromb Vasc Biol (2012) 32:1856–64. doi:10.1161/ATVBAHA.112.252619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Selvamani A, Sathyan P, Miranda RC, Sohrabji F. An antagomir to microRNA Let7f promotes neuroprotection in an ischemic stroke model. PLoS One (2012) 7:e32662. doi:10.1371/journal.pone.0032662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Bake S, Selvamani A, Cherry J, Sohrabji F. Blood brain barrier and neuroinflammation are critical targets of IGF-1-mediated neuroprotection in stroke for middle-aged female rats. PLoS One (2014) 9:e91427. doi:10.1371/journal.pone.0091427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Xiao S, Ma Y, Zhu H, Sun H, Yin Y, Feng G. miRNA functional synergistic network analysis of mice with ischemic stroke. Neurol Sci (2014) 36(1):143–8. doi:10.1007/s10072-014-1904-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Colombo E, Borgiani B, Verderio C, Furlan R. Microvesicles: novel biomarkers for neurological disorders. Front Physiol (2012) 3:63. doi:10.3389/fphys.2012.00063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cerebral ischemia, neuroprotection, preconditioning, non-coding RNA, microRNA
Citation: Saugstad JA (2015) Non-coding RNAs in stroke and neuroprotection. Front. Neurol. 6:50. doi: 10.3389/fneur.2015.00050
Received: 30 January 2015; Paper pending published: 10 February 2015;
Accepted: 23 February 2015; Published online: 13 March 2015.
Edited by:
Giuseppe Pignataro, Federico II University of Naples, ItalyReviewed by:
Sebastien S. Hebert, Université Laval, CanadaAshutosh Dharap, Los Alamos National Laboratory, USA
Copyright: © 2015 Saugstad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie A. Saugstad, Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, HRC5N, Portland, OR 97239-3098, USA e-mail:c2F1Z3N0YWRAb2hzdS5lZHU=