 Dilek Inekci
Dilek Inekci Ditte Svendsen Jonesco
Ditte Svendsen Jonesco Sophie Kennard
Sophie Kennard Morten Asser Karsdal1
Morten Asser Karsdal1 Kim Henriksen
Kim Henriksen- 1Nordic Bioscience, Biomarkers and Research, Herlev, Denmark
- 2Systems Biology, Technical University of Denmark, Lyngby, Denmark
The diagnosis of dementia is challenging and early stages are rarely detected limiting the possibilities for early intervention. Another challenge is the overlap in the clinical features across the different dementia types leading to difficulties in the differential diagnosis. Identifying biomarkers that can detect the pre-dementia stage and allow differential diagnosis could provide an opportunity for timely and optimal intervention strategies. Also, such biomarkers could help in selection and inclusion of the right patients in clinical trials of both Alzheimer’s disease and other dementia treatment candidates. The cerebrospinal fluid (CSF) has been the most investigated source of biomarkers and several candidate proteins have been identified. However, looking solely at protein levels is too simplistic to provide enough detailed information to differentiate between dementias, as there is a significant crossover between the proteins involved in the different types of dementia. Additionally, CSF sampling makes these biomarkers challenging for presymptomatic identification. We need to focus on disease-specific protein fragmentation to find a fragment pattern unique for each separate dementia type – a form of protein fragmentology. Targeting protein fragments generated by disease-specific combinations of proteins and proteases opposed to detecting the intact protein could reduce the overlap between diagnostic groups as the extent of processing as well as which proteins and proteases constitute the major hallmark of each dementia type differ. In addition, the fragments could be detectable in blood as they may be able to cross the blood–brain barrier due to their smaller size. In this review, the potential of the fragment-based biomarker discovery for dementia diagnosis and prognosis is discussed, especially highlighting how the knowledge from CSF protein biomarkers can be used to guide blood-based biomarker development.
Introduction
Dementias are brain disorders that cause a progressive decline in mental function. In 2009, it was estimated that 35.6 million people were suffering from dementia worldwide and this number is expected to be 65.7 million by 2030 and 115.4 by 2050 (1). Alzheimer’s disease (AD) is the most common cause of dementia, and accounts for 60–70% of all cases. Other common causes of dementia are dementia with Lewy bodies (DLB), vascular dementia (VaD), frontotemporal lobar degeneration (FTLD), and corticobasal degeneration (CBD). In addition to this, mixed dementias are also commonly seen (2–4).
The major risk factor for developing dementia is age, with increasing prevalence after age 65, followed by family history, environmental factors, and mutations (4). Cognitive and neuropsychiatric symptoms are the key clinical features of dementia (5).
The diagnosis of dementia is challenging and early and moderate stages of dementia are rarely detected thereby limiting the potential for early intervention. Additionally, a high number of dementia cases are left without a diagnosis (6).
It is generally accepted that there is a need for early diagnosis of dementia and many efforts have been made to develop early biomarkers with the ability to identify the pre-dementia stage of the disease before the onset of cognitive decline and brain degeneration (7, 8).
Another challenge is the differential diagnosis of dementia, as there is an overlap in the clinical features across the different dementia types (9–11). There is currently no single marker available that can differentiate between AD and other dementia types. Hence, there is a need for biomarkers that can distinguish between the dementias.
Additionally, successful development of disease-modifying drugs and prevention therapies require biomarkers that can recognize neuropathological changes in the pre-dementia stage and allow differential diagnosis. This would allow inclusion of the right patients in the clinical trials, monitoring of the treatment efficacy, and exclusion of patients that have already reached a point-of-no-return and would not have any beneficial effect of a given intervention (12, 13).
Unfortunately, the biomarker development has been hampered by the fact that tracking molecular pathological changes in the brain is a huge challenge due to the inaccessible nature of the brain. Currently imaging and CSF biomarkers provide the best method for diagnosing, staging, as well as predicting clinical progression of AD and related dementias. However their use is limited by cost, availability and by the fact that repeated brain scans and withdrawal of CSF by lumbar punctures are not advisable (14, 15). These aspects all underline the need for novel biomarkers which are easily obtainable.
The Proteopathy of Dementia
Most dementias can be designated as proteopathies characterized by aberrant processing of neuronal proteins such as fragmentations, aggregations and other post-translational modifications (PTMs) (Table 1) (3, 16).
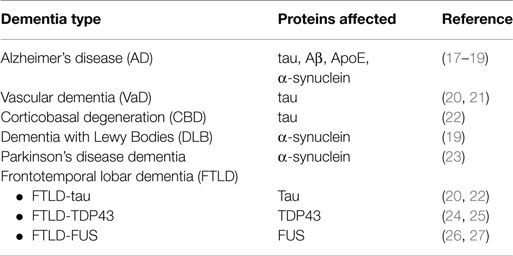
Table 1. Common types of dementia and proteins affected.
The potential of these proteins as diagnostic and prognostic biomarkers has been extensively studied at the protein level. However, these investigations have been limited by the fact that the role of each of these pathological changes throughout the development of dementia is unresolved. This is due to the intrinsic difficulty of detecting the disease before patients display symptoms, which may be 20 years before the earliest cognitive changes are detected (28). Another complicating factor in diagnosing and determining progression of dementia is the significant crossover between the proteins involved in the different types of dementia. Thus, looking solely at protein levels is too simplistic to provide enough detailed information to differentiate between different dementia types. An alternative to this is the application of PTMs as biomarkers for AD. This is not a new approach, since it has already been investigated in the development of CSF-derived AD biomarkers Aβ1–42 and phosphorylated tau (p-tau). This presents an excellent example of how understanding the molecular pathology inflicts certain protein fingerprints on key proteins, provides insight not only to central disease mechanisms, but also provides an opportunity to improve the protein’s usage in terms of diagnostic and prognostic value for a specific dementia or even a subtype of dementia.
As we have previously proposed, AD pathology and other dementias may give rise to blood circulating fragments of key neuronal proteins, thereby allowing detection of disease specific post-translationally truncated fragments in the blood (29). This would allow easier and more frequent sampling and analysis and provide earlier diagnosis and prognosis of dementia.
The present review will focus on addressing the potential of disease-specific protein fragmentation for dementia diagnosis and prognosis and how these fragments can be utilized as biomarkers to segregate between the different types of dementia, especially highlighting how the knowledge from CSF protein biomarkers can be applied to investigate blood-based biomarkers.
Status of CSF Biomarkers
The pathological alterations in the brain at the molecular level are directly reflected in the CSF, therefore this fluid has been the most investigated source for development of biomarkers for AD and related dementias. Aβ42, t-tau (total tau), p-tau, and α-synuclein are the most studied CSF biomarkers and their performance has been evaluated in several studies (30). Other biomarkers that will be described in this review are apolipoprotein E (ApoE), TAR DNA-binding protein 43 (TDP-43), fused in Sarcoma protein (FUS), and glial fibrillary acidic protein (GFAP).
Aβ42 is the main component in the extracellular amyloid plaques of AD and is a marker of amyloid precursor protein (APP) processing and plaque load. In AD, a decrease in CSF Aβ42 has been found, which is probably due to deposition in plaques (17, 31). Generation of Aβ42 is an early event in AD, hence measuring CSF Aβ42 is a very relevant strategy in prodromal AD to screen for early cases as well as monitoring disease progression. However as it is today the strategy of measuring CSF Aβ42 only provides a supplementary test to support the diagnosis once cognitive dysfunction is apparent, and it gives little information on the disease progression as this biomarker has already found a steady-state of abnormality early in the disease progression (32, 33). CSF Aβ42 is able to discriminate between AD and non-demented controls with a sensitivity of 59–96% and a specificity of 77–89% (17, 34–36). A change in Aβ42 levels has also been studied for other types of dementia and shows a slight decrease in FTLD, DLB, and VaD (32). CSF Aβ42 has been shown to predict the rate of cognitive decline in patients with very mild dementia and predict AD in subjects with mild cognitive impairment (MCI) (37, 38).
Cerebrospinal fluid t-tau is a biomarker of neuronal damage and neuronal and axonal degeneration and several studies have shown an increased level in AD patients compared with controls with a sensitivity and specificity of 70–83% and 81–92%, respectively (17, 34–36). However, CSF t-tau is not specific for AD and is also increased in other dementias such as Creutzfeldt–Jakob disease (CJD) patients and in a significant number of patients with DLB, FTLD, VaD, and CBD (20, 32).
Cerebrospinal fluid p-tau reflects aberrant phosphorylation and neurofibrillary tangle (NFT) burden. A strong increase in p-tau has been found in AD using ELISA methods that detect different phosphorylated epitopes such as p-tau(181) or p-tau(231). CSF p-tau differentiates between AD patients and controls with a sensitivity of 68–86% and a specificity of 61–73% (35, 36). A moderate increase in p-tau has also been found in CJD and DLB (17, 20). It has been reported that the use of p-tau instead of t-tau may improve the diagnostic sensitivity and differential diagnosis of AD versus DLB and FTD, respectively (34). Both t-tau and p-tau have been found to predict progression from MCI to AD (32, 39).
The combination of CSF biomarkers (t-tau/Aβ1–42 and p-tau/Aβ1–42) has been found to increase the sensitivity and specificity when compared to the single markers. The t-tau/Aβ1–42 ratio shows a potential as a preclinical biomarker since it discriminates between MCI patients that progress to AD and those that do not progress, although the CSF sampling makes it virtually useless for this purpose (36, 40, 41). Furthermore, the ratio shows promise in prediction of dementia in cognitively normal older individuals (42).
Another interesting CSF biomarker is α-synuclein. Compared to tau and Aβ1–42, little research has been done with respect to CSF levels of α-synuclein, which is the main component of Lewy bodies of DLB patients. Studies have demonstrated decreased CSF levels of α-synuclein in DLB and Parkinson’s disease (PD) when compared to controls indicating a potential diagnostic use (43, 44). In contrast to this other research groups have shown no difference in CSF levels in DLB and PD patients compared with controls and other dementias (45–47).
In both PD and DLB patients, the level of α-synuclein oligomers is increased compared to healthy patients and other types of dementias (23, 48). In PD, the ratio of oligomers of α-synuclein to total α-synuclein is also significant. There is an increase in the ratio of oligomeric/total α-synuclein when compared to other dementias (49). Recent studies have also shown significantly elevated CSF levels of α-synuclein in AD patients (50) suggesting that α-synuclein may not be specific to DLB and PD, or again indicating that mixed pathologies are common.
Although, several CSF biomarkers show a promising diagnostic and prognostic potential, there are still important drawbacks limiting their clinical utility (Table 2). An important limitation is the lack of assay standardization and global cut-off values for biomarker concentrations. The handling of CSF and use of different technological platforms and antibodies are the major reasons for significant differences in biomarker concentrations between studies (51). Fortunately, international standardization initiatives have been initiated to reduce the large variations between studies and within laboratories (52). Another limitation of CSF biomarkers is the overlap between the protein profile of different types of dementia (20). Lastly, the clinical utility of CSF biomarkers is still hampered by sample collection, which requires a lumbar puncture. Despite the fact that there is minor complications related to lumbar puncture the procedure is still regarded as invasive in the general population and repeated follow-up measurement is challenging (14, 15), and hence they are not consistently applied in clinical trials. On the other hand, the CSF proteins described here all have a pathological link to the diseases of interest, and as such are of quite some interest for the development of blood-based biomarkers.

Table 2. Advantages and drawbacks of CSF biomarkers.
Status of Blood-Based Biomarkers
The use of blood as a source of dementia biomarkers is still under investigation. Blood is a more feasible biomarker source when compared to CSF due to its wide availability, low cost, time effectiveness, and easier sampling. Several different approaches for identification of blood biomarkers are available and these include biomarkers of the amyloid and tau pathology, biomarkers of inflammation, oxidative stress, mitochondrial dysfunction, neuronal and microvascular injury, and biomarker panels (15, 53). So far, the research has been hampered by two major challenges. The first is the complexity of blood and the large variation in samples and variation between studies. The difference in preanalytical and analytical methods is an important reason for this variation and these have been reviewed elsewhere (15). The second challenge is the fact that blood is not in direct contact with the brain. This limits the understanding of how the pathological alterations in the brain are reflected in blood analytes, as well as the absolute level of the analyte of interest in the blood. Additionally, the prevalent presence of non-specific proteins in the blood is an obstacle toward identification of disease-specific biomarkers. To overcome these limitations, the experience from the well-characterized CSF biomarkers, which in some cases are based on brain-specific pathological alterations, i.e., p-tau, may be a starting point for blood biomarker analysis. Pathological alterations in CSF proteins may be reflected in blood as a consequence of absorption of CSF into blood, by penetration due to barrier impairment in dementia or simply by diffusion (54–58). Whether a brain-derived protein can serve as a biomarker to be measured in blood will depend on the concentration, the change in concentration during disease, the molecular size and the half life in blood (57). Hence, exploring the dynamic range of brain proteins in the peripheral blood is of great interest.
The CSF biomarker tau is a brain-specific protein that can become a relevant biomarker to be measured in blood. So far, little is known about tau levels in blood and most studies have been hindered by the low abundance of the protein in blood (59). Zetterberg et al. (59) found that there was no correlation between CSF tau levels and plasma tau indicating that the clearance of tau is differently regulated (59). In healthy blood-donors tau protein concentration is in the range <10 and >100 pg/mL and the ratio between CSF:serum tau is 10:1 (57). Methods for determining tau in serum/plasma are under investigation. Few studies have reported elevated plasma tau levels in patients with AD (59, 60). The results from these studies are encouraging but highly sensitive detection methods are necessary. An ultrasensitive immunoassay for detection of plasma tau has been introduced and similar methods would be highly relevant (61).
Another CSF biomarker with potential to be a blood biomarker is Aβ. Plasma Aβ species have been examined by numerous studies but the results are contradictory. Some of these studies report high Aβ42 or Aβ40 whereas others show a decrease in AD. The overlap between patients with AD and healthy controls is also substantial. Importantly, Aβ is not brain-specific but is also expressed by other cells, and as such there is an interference of the peripheral Aβ species with the brain-derived species. Additionally, the binding of Aβ to plasma proteins and formation of Aβ oligomers may disturb the quantification by immunoassays (62, 63).
Finally, several studies have quantified plasma α-synuclein and α-synuclein oligomers in PD and DLB. However, additional studies are needed to evaluate blood α-synuclein as a valid biomarker and the high levels of α-synuclein present in red blood cells must be considered when quantifying the protein (64).
Plasma levels of ApoE, TDP-43, and GFAP have also been reported and the main results from these studies will be reviewed in the next sections.
Altogether, the inconsistent findings from plasma analyses illustrate the need for a pathology specific combination of protein and modification of this protein in order to enhance the possibility of generating a disease-specific biomarker, even more so in blood specimens than CSF.
Status of Protein Fragmentation Blood-Based Biomarkers
As mentioned identification and detection of brain-specific proteins in blood is restricted by the blood–brain-barrier, the substantial presence of non-specific proteins, and proteins from co-morbidities in the circulation. The use of post-translationally truncated protein fragments containing specific neo-epitopes as biomarkers of dementias may overcome these complexities (29, 65). Targeting protein fragments generated by disease-specific combinations of proteins and proteases opposed to detecting the intact protein could diminish the overlap between diagnostic groups. Proteolytic fragmentation of proteins is a post-translational process and several cleavage products have been identified in relation to AD and other dementias. Aβ42, Aβ40, and several other N- or C-terminally truncated Aβ peptides all represent examples of proteolytically cleaved protein fragments. Cleavage of tau, ApoE, α-synuclein, TDP-43, and GFAP has also been reported (66–70).
Although, several of the described protein fragments have been described in the literature and detected in CSF most of these have not been studied in blood. Targeting protein fragmentation by specific proteases may provide novel biomarkers for dementia and create a specific profile of each disorder based on the fragments and proteases that are involved in the pathology. Another advantage of using fragments as blood biomarkers opposed to the intact proteins may be the eased release from the central nervous system (CNS) into the periphery. The fragments may easier pass the blood-brain barrier due to their small size and be easier to detect (71–75) (Figure 1).
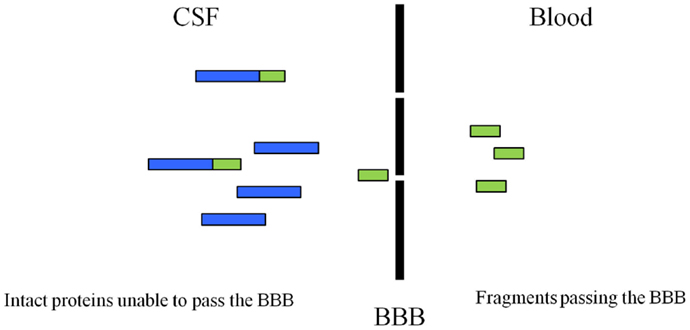
Figure 1. Illustration of how the protein fragments may be able to cross the blood–brain barrier. Protein fragments may have the advantage of crossing the barrier as these breakdown products have a smaller size when compared to the intact protein. Modified from Ref. (29).
In addition to applying disease-specific protein fragmentation to identify new biomarkers for dementia, it is important to define and validate the ability of each novel biomarker. The BIPED classification system (Burden of Disease, Investigative, Prognostic, Efficacy of Intervention and Diagnostic) is a nomenclature first used for osteoarthritis and offers categorization of biomarkers in order to improve the development and validation of biomarkers (15). The use of BIPED classification in dementia would aid in the biomarker development process from target identification to validation in clinical trials.
In the following sections, neuronal proteins involved in the proteopathy of dementias will be reviewed with emphasis on proteolytic fragmentations (Figure 2).

Figure 2. Key neuronal proteins involved in the proteopathy of different types of dementia, proteases involved in their truncation and fragments known from the literature. References can be found in the text.
Amyloid Precursor Protein
Derivatives from the full-length APP are the main components of the extracellular amyloid plaques. APPs are type 1 transmembrane proteins and exist in three isoforms in humans, APP695, APP751, and APP770. The APP695 is the main isoform in neurons and is the only isoform containing the sequence encoding Aβ (76, 77). In normal cells, APP is involved in kinase-based signaling, growth regulation, neurite outgrowth, formation of synapses and cell adhesion (33, 78). APP is cleaved by secretases and caspases at specific sites and this leads to the formation and release of several protein fragments (76, 78). The proteolytic processing of APP can follow the amyloidogenic or the non-amyloidogenic pathway. The major component of senile plaques, Aβ, is generated in the amyloidogenic pathway by sequential cleavage of APP by β-secretase and γ-secretase to generate Aβ40 and Aβ42. BACE1 (β-site APP-cleaving enzyme 1), BACE2 (β-site APP-cleaving enzyme 2), and cathepsin B have been identified as β-secretase responsible for production of Aβ. The γ-secretase activity belongs to a membrane-bound protease complex (presenilin 1, presenilin 2, nicastrin, Aph-1, and Pen-2) (76, 78). In the non-amyloidogenic processing, APP is cleaved by α-secretase which binds to and cleaves APP within the Aβ region and prevents formation of Aβ. All the identified α-secretases are from the family of disintegrin and metalloproteases (ADAMs).
The accumulation of Aβ is an early process in neurodegeneration leading to formation of oligomers, fibrils, and eventually extracellular plaques. CSF Aβ42 levels become abnormal 5–10 years or more before the diagnosis (79, 80). The concentration of CSF Aβ42 begins to increase abnormally followed by a drastic decrease. In mutation carriers (i.e., in the APP genes, presenilin 1, or presenilin 2), CSF Aβ42 levels become abnormal up to 25 years before disease onset (28). Intracellular levels of Aβ initiate synaptic dysfunction, formation of NFTs and loss of neurons. The Aβ42 is the main toxic form of Aβ, whereas Aβ40 has been shown to have neuroprotective functions (78, 81).
Aβ42 and Aβ40 have also been detected in patients with cerebral amyloid angiopathy (CAA), which can be a co-occurring disorder with AD or a separate finding. CSF levels of Aβ42 and Aβ40 are lower in patients with CAA and CAA-related inflammation (CAA-ri) than controls (82–84). Furthermore, the level of CSF anti-Aβ autoantibodies is increased in CAA-ri which shares similarities with the amyloid-related imaging abnormalities detected in AD immunization clinical trials (84). It has been suggested that the CSF anti-Aβ autoantibody concentration can be used as a biomarker during immunization clinical trials in AD (84, 85).
The Aβ peptide is subjected to further truncations by different proteases and forms peptides of various lengths. The peptides are generated by N- or C-terminal truncation of Aβ and several of these have been identified in CSF, e.g., Aβn–42 (n = 2–11), Aβ1– n (n = 13–20), Aβ1–28, Aβ1–33, Aβ1–34, and Aβ1– n (n = 37–39). These peptides have been found to be elevated in CSF of AD patients but only few are involved in plaque formation (86–89).
Recently, it was reported that some of the identified Aβ peptides in CSF are generated by an alternative APP processing pathway (90). In this pathway, APP is cleaved by α- and β-secretase without the involvement of γ-secretase. Many of the peptides derived from this pathway are elevated in CSF from AD suggesting an up-regulation of this pathway in AD as a response to the increase of the amyloidogenic pathway (90). The identified products of the alternative pathway are Aβ1-14, Aβ1-15, and Aβ1-16. Eleven other truncated peptides with C-terminal at residue 15 in the Aβ sequence and start at the N-terminal end of the β-secretase site have been identified in CSF. The peptides contain a part of the Aβ sequence but are not degradation products of Aβ because they start upstream of the β-secretase cleavage site. Several of these were found to be elevated in AD and may also be generated in the alternative processing pathway (91).
Plasma levels of Aβ42, Aβ40, and the ratio Aβ42/Aβ40 have been examined in several cross-sectional studies with AD, MCI patients, and healthy controls. The results have shown a substantial overlap between diagnostic groups and the results between studies have been contradictory (92). Aβ42 and Aβ40 have also been studied in longitudinal studies to assess their association with disease progression. Although the results are not clear between individual studies the data show that a decreased baseline level of Aβ42 predicts a greater risk of AD (92). A recent study has quantified Aβ1-17 levels in plasma and has shown significant associations with the clinical diagnosis of AD, indicating the potential of the Aβ fragments (93). The plasma levels of the remaining Aβ cleavage products have only been examined in few studies. Highly specific antibodies and robust immunoassays must be developed and used for detection of these cleavage products of different size.
Tau
Tau is the basic component of the intracellular insoluble filamentous structures, also referred to as NFTs. The tau protein belongs to the family of microtubule-associated proteins and binds to, stabilizes, and promotes the assembly of microtubules. Tau is also involved in signaling pathways and cytoskeletal organization (94).
Tau is mainly expressed in the central and peripheral nervous system and most abundant in axons. There are six isoforms in the adult human brain, which vary in size and have either three or four microtubules-binding domains. The six forms each show functional differences (95, 96). The ratio between tau containing three and four domains is 1:1 in normal human brain but this ratio is altered in the different tauopathies. Additionally, different isoforms of tau are involved in the different tauopathies and affect distinct brain regions, hence it has been suggested that the isoform profiles can be used to classify the different tauopathies (97, 98). Besides AD, the tauopathies include FTLD, progressive supranuclear palsy (PSP), CBD, and prion diseases (20, 98).
In AD, the concentration of CSF t-tau and p-tau become abnormal after Aβ42 and their levels increase progressively up to the time of diagnosis. Thus, tau levels are higher in MCI patients with an early conversion compared with late converters (79, 80). Increased CSF levels of tau are increased 15 years before symptoms in mutation carriers (28).
The conversion of soluble tau protein to insoluble inclusions is a central event in AD and other tauopathies. Formation of inclusions is mediated by protein aggregation and misfolding. The aggregates have been shown to be self-propagating and spread from one neuron to another (99). Tau aggregation and misfolding are induced by abnormal phosphorylation and proteolytic cleavage. Hyperphosphorylated tau is the main component of NFTs and several kinases and phosphatases have been associated with this. A level of phosphorylation occurs at normal state but in disease state, an abnormal level of phosphorylation is seen and results in a low-binding affinity to tubulin promoting disassembly of microtubules (94, 96).
Although the presence of t-tau and p-tau in CSF has been investigated in several studies, the nature of the protein in CSF is not fully known. A number of studies have suggested the presence of different tau and p-tau fragments in CSF (94, 95) and a recent study has reported that CSF tau and p-tau occur as various N-terminal and mid-domain fragments (67). The level of specific fragments were significantly elevated in AD patients when compared to controls and showed a diagnostic potential but the fragments still remain to be measured in other dementias (67).
Plasma levels of t-tau and tau fragments have only been assessed in few studies. It has been demonstrated that plasma t-tau levels are elevated in AD patients but with an overlap with control subjects (59). Hence, the diagnostic utility of plasma t-tau is not clear. Recently, the presence of protease generated fragments of tau has been shown in serum (75, 100, 101). The fragments have been shown to correlate with symptoms in AD patients and predict the disease progression in early AD (100, 101), indicating the pathological relevance of fragmentations.
It is a possibility that the assays for t-tau may also detect certain fragments of tau and as multiple systems are in use for detecting t-tau, this is most likely different from assay to assay depending on the antibodies used. Unless an assay is constructed as a sandwich ELISA with antibodies detecting the N- and C-terminal sequences, there is this possibility.
Furthermore, it must be noticed that the relative concentration of the protein determined in the clinical studies is a result of the specific calibrators used in the different assays.
In dementia, tau is cleaved by caspases and calpains, but other proteases have also been detected including thrombin, cathepsins, and puromycin-sensitive aminopeptidase (102). It has been found that certain proteolytic fragments of tau are specific for the different tauopathies suggesting that different proteases may be specific to individual tauopathies (102). Several tau fragments have been reported and the most studied are caspase-generated tau fragments cleaved at D13, E391, and D421 as well as a calpain-cleaved fragment of 17 kDa which are associated with AD (66, 103). The majority of the reported fragments have only been analyzed in vitro, in AD-affected brains or transgenic animals (94).
Apolipoprotein E
The ε4 allele of ApoE is known to be associated with the risk of developing AD. ApoE is a major transport protein of cholesterols and other lipids in plasma and in the brain. It is most abundant in the brain and the liver (104). In the CNS, ApoE is mainly synthesized in astrocytes but is also present in lower concentration in some neurons, activated microglia, oligodendrocytes, and ependymal layer cells. In neurons, the synthesis of ApoE is induced under neuronal stress and damage and has been detected in cortical and hippocampal neurons (105). In the normal brain, ApoE is associated with the maintenance and repair of neurons and involved in the cholesterol homeostasis (106). ApoE is a polymorphic protein with the main isoforms being ε2, ε3, and ε4. The three isoforms differ by single amino acid substitutions at positions 112 and 158 (104, 107). The ApoE ε4 allele is a risk factor for late-onset familial and sporadic AD (18, 108). Around 10–15% of the general population has the ε4 allele, whereas the prevalence is 40–65% in AD patients. The majority of the general population is homozygous for the ApoE ε3 allele. The third common isoforms ε2 is present in 5–10% of the population. The ApoE ε2 allele has protective effects on the cognition and has been associated with reduced AD-related disease burden (109, 110).
Homozygosity for ApoE ε4 leads to a 50–90% risk of developing AD by the age 85, whereas individuals with one copy have a risk of 45%. For individuals with no ApoE ε4 alleles the risk is about 20% (18, 111). ApoE has been found to be co-localized with amyloid plaques and NFTs (105). Several mechanisms have been proposed for the role of ApoE ε4 in the pathology of AD including regulation of the deposition and clearance of Aβ and amyloid plaques, regulation of phosphorylation and assembly of tau into NFTs, dysfunction of the neuronal signaling pathways, induction of Aβ-regulated lysosomal leakage, increased atherosclerosis and vascular inflammation in AD, and apoptosis in neurons (105, 112). However, its exact role in the AD pathology still remains unclear (105). Besides AD, the ε4 allele has also been associated with CAA, hemorrhages, tauopathies, DLB, PD, and multiple sclerosis (113–116).
The CSF, ApoE levels have been determined by several studies and some have found decreased levels in CSF of AD patients whereas other studies have shown an increase (117). Increased CSF levels of ApoE were also detected in DLB and PD patients (118).
Plasma ApoE levels have also been reported but as seen with the CSF measurements the results have been inconsistent. A study by Taddei et al. (119) reported increased plasma ApoE levels in AD patients compared to controls. In contrast to this, the Australian Imaging, Biomarkers and Lifestyle (AIBL) study showed decreased plasma levels of ApoE and ApoE ε4 in AD patients and showed a correlation with the disease level (120). Two other studies based on the Rotterdam study and apoEurope Study, respectively, also observed decreased ApoE levels in AD patients compared to controls (121, 122). However, this difference was not significant in the Rotterdam study when adjusted for ApoE genotype, age, and gender (121). Finally, a recent study has shown that low plasma ApoE levels are associated with the risk of developing AD independent of the ApoE genotype, indicating the potential of this biomarker as a preclinical marker (123).
Aberrant proteolytic cleavage of ApoE plays an important role in the AD pathology associated with ApoE. ApoE is subjected to intracellular proteolytic cleavage and generates neurotoxic fragments. The fragments have been detected in cultured neurons and AD brains and have been shown to induce tau phosphorylation and formation of NFT-like aggregates in CNS neurons with p-tau and phosphorylated neurofilaments (124, 125). In addition, the fragments impair the function of mitochondria in neurons and promote neurodegeneration. The level of ApoE fragments is elevated in AD brains compared to cognitively normal controls (68). Importantly, ApoE ε4 is more susceptible to fragmentation than ApoE ε3 (124, 126). Among the fragments, a 22 kDa N-terminally peptide has been detected in brain tissue and CSF. Interestingly, the ApoE ε4-derived 22 kDa fragment has been found to be more neurotoxic than the corresponding ApoE ε3-derived fragment (68). Several C-terminally truncated ApoE fragments of different lengths have also been detected in AD brains. One of these is the apoE4 (Δ272–299) fragment which interacts with p-tau and phosphorylated neurofilament to form inclusions (124). A neuro-specific chymotrypsin like protease has been suggested to be involved in the formation of these fragments but further studies are needed (127).
So far, there are no studies on plasma levels of ApoE fragments and their correlation with AD or other dementias.
α-Synuclein
α-synuclein is a small protein located in both the CNS and the peripheral nervous system. It can be found specifically bound to the membrane of pre-synaptic vesicles and very little α-synuclein is distributed throughout the rest of the nerve (128). α-synuclein is also expressed in other tissues including red blood cells (64), kidney, lung, heart, and liver (129). The specific function of α-synuclein is unknown but it is implicated in a number of dementias including AD, DLB, and PD. α-synuclein aggregates to form a component of Lewy bodies that can be found in the cytoplasm of neurons. These aggregates are observed in the dementias mentioned above except for AD and are believed to be the key step in progression of neurdegeneration in synucleionopathies. There is, however, evidence that suggests α-synuclein plays a role in the aggregation of tau, which is observed in AD (130, 131). Furthermore, increased levels of soluble α-synuclein have been found in AD brains in patients in absence of LBD pathology and the levels showed a correlation with cognitive decline (132).
Cerebrospinal fluid levels of α-synuclein and its oligomers have been assessed in several types of dementia. The differential performance of α-synuclein has been inconsistent in different clinical studies. A number of studies have shown that CSF α-synuclein levels are lower in DLB and PD patients than those with AD and other dementias (43, 44, 133), whereas others have concluded that CSF α-synuclein does not discriminate between dementias (46). The levels of CSF α-synuclein oligomers are increased in DLB and PD compared with controls and AD patients (48).
The plasma levels of α-synuclein and its oligomers have been quantified in DLB and PD patients by several studies. Increased plasma levels of α-synuclein and oligomers were seen in patients with PD when compared to controls (134–137). However, contradictory results were observed in other investigations (138, 139). Similarly, the level of plasma α-synuclein oligomers was higher in DLB patients than controls whereas the α-synuclein levels were lower in DLB than AD patients and controls (134, 139).
A lot of focus has been on aggregation of the intact α-synuclein, however more recently studies suggest that fragmentation of α-synuclein is significant in the pathology of synucleinopathies. Fragments of α-synuclein have been identified in brains of PD and DLB patients (69, 141). One protease of interest is calpain, which has been observed to create cleavage products that can induce aggregation of α-synuclein in vitro. Calpain cleaves α-synuclein in the N- and C-terminal regions (140). MMPs also play a role in α-synuclein aggregation and therefore Lewy Body formation. Partial cleavage with either MMP-1 or MMP-3 increases aggregation of the protein (141) and both proteases are elevated in PD brains (142, 143). Neurosin is another protease of interest, especially as it is found within amyloid plaques in AD (144). Neurosin has also been identified in CSF and has been found to be lower in patients with synucleinopathies compared to those with AD and healthy patients (145). Finally, cathepsins are known to be involved in the proteolysis of α-synuclein (146). The presence of α-synuclein fragments in CSF and plasma remains to be investigated.
TAR DNA-Binding Protein 43 and Fused in Sarcoma Protein
TAR DNA-binding protein 43 is a nuclear protein that functions in regulation of transcription and exon splicing (24, 147). TDP-43 is known as the key protein in the pathogenesis of FTLD with ubiquitin-positive, tau-negative inclusions. FTLD is the second most common type of dementia after AD with an onset before 65 years of age (148) and differentiation between AD and FTLD can be challenging as they share several clinical features (149).
In FTLD, TDP-43 is post-translationally modified by aberrant ubiquitination, hyperphosphorylation, and proteolytic cleavage at the N-terminus (24, 25). In addition, TDP-43 is translocated from the nucleus and generates cytoplasmic insoluble inclusions containing ubiquitinated and aberrantly phosphorylated TDP-43 (24).
TAR DNA-binding protein 43 neuronal and glial inclusions have been detected in AD and several types of PD (150). TDP-43 inclusions are found in 25–30% of all sporadic AD patients and 14% of familial AD patients. The presence of TDP-43 in AD brains has been shown to give greater brain atrophy and more deficits when compared to AD patients without TDP-43 inclusions (151). In addition, caspase 3-cleaved TDP-43 has been detected in AD brains and it is proposed to be associated with neurodegeneration (70). This suggests that TDP-43 in combination with specific AD biomarkers can be used to identify patients with the risk to develop severe clinical deficits.
TAR DNA-binding protein 43 levels are detectable in CSF and were found to be elevated in FTLD patients when compared to controls (152, 153). TDP-43 has also been detected in plasma and the levels were increased in FTLD and a subset of AD patients (154, 155).
Fragmentation of TDP-43 has been observed. The N-terminal cleavage of TDP-43 generates C-terminal fragments, but the cleavage sites and their function in the pathology of FTLD are not fully known. In an in vitro study, two caspase-generated C-terminal fragments of 25 and 35 kDa were identified (156). The 25 kDa fragment of TDP-43 was found to induce the formation of intra-cellular toxic, insoluble and ubiquitin- and phospho-positive aggregations. Hence, protease cleavage initiates the translocation of TDP-43 from the nucleus to cytoplasm and induces formation of toxic insoluble inclusions (25). Caspase 3, 7, 6, and 8 have all been associated with TDP-43 cleavage (156).
The TDP-43 fragments have not been investigated in CSF or plasma.
TAR DNA-binding protein 43 and its fragments are potential biomarkers for tau-negative FTLD and can be used in the differential diagnosis of dementia and aid in the separation between tau-negative FTLD and tauopathies.
Another protein with implication for the differential diagnosis of dementia is the RNA-binding protein fused in sarcoma. The FUS protein is the pathological protein in 10–20% of sporadic FTLD patients (FTLD-FUS), which are negative for TDP-43 (26, 27, 157). The FUS protein binds to DNA and RNA and is associated with several cellular processes such as cell proliferation, DNA repair, transcription regulation, RNA splicing and transport of RNA (158–162). FUS is ubiquitously expressed in the nucleus and cytoplasm in most cell types and in neurons and glial cells it is primarily expressed in the nucleus (163). In FTLD, the FUS protein is mostly present in the cytoplasm whereas the FUS levels in the nucleus are decreased indicating a delocalization of the protein. The delocalization and accumulation of FUS lead to formation of cytoplasmic inclusions that are the characteristics of FTLD-FUS (26, 150). In addition, a mouse model has shown that overexpression of the FUS protein results in neurodegeneration (164).
To the best our knowledge neither the levels of FUS in CSF and plasma nor its fragmentation have been reported.
Glial Fibrillary Acidic Protein
Glial Fibrillary Acidic Protein is a type III intermediate filament (IF) protein constituting a part of the cytoskeleton in specific cell types. Besides the pivotal role of GFAP in the structural properties of these cells, it is involved in several fundamental cellular activities including motility (165), autophagy (166), synapse formation (167), and myelination (168).
Although it was originally considered an astrocyte-specific marker (169), GFAP has subsequently been demonstrated in glial and non-glial cells of the periphery (170–173). GFAP has been observed in virtually all areas of the brain but is mainly expressed in hippocampal regions (174–176) as well as the subventricular zone and olfactory system of both non-demented elders and patients with dementia (174–177). Multiple splice variants exist and in human hippocampal AD tissue many of these isoforms show differential transcript levels (176).
Differential transcript levels of GFAP isoforms may affect cellular function and/or morphology (165) as analysis of in vitro transfection suggests that GFAP isoforms differ in their ability to form functioning IFs (174, 176, 178, 179). In general, little is known about the role of GFAP in AD and other dementias. GFAP is known to interact with proteins involved in cleavage of APP (180, 181) as well as proteins modulating chaperone mediated autophagy (CMA) (166). GFAP may both inhibit and promote CMA and the phosphorylation state of GFAP is suggested to influence this balance (166). Incomplete CMA of tau is suggested to promote tau aggregation (182) which is a hallmark of several tauopathies including AD (103).
Studies have shown a correlation between increased expression levels of GFAP within brain regions involved in memory and the neuropathological changes of AD such as Aβ deposits and NFTs (183–187). Also, disease duration and progression of AD has been shown to correlate strongly with up-regulation of GFAP in the temporal lobe of AD patients (176, 184, 188).
In CSF, levels of GFAP have been observed to be increased in AD patients compared to controls (189–192). Furthermore, GFAP levels were found to increase with AD severity (189). In most studies, increased GFAP levels were independent of age, however, Rosengren et al. (190), observed a correlation between these two parameters (190).
Cerebrospinal fluid GFAP levels are also increased in patients with other neurological disorders and brain injuries such as CJD (191, 192), stroke (193, 194), and traumatic brain injury (195, 196). Regardless of this general increase in GFAP levels observed in these disorders and injuries, GFAP may be applied in context with other biomarkers for differential diagnosis, e.g., GFAP, together with the glial-specific S100 calcium binding protein B (S100β) may hold the potential to distinguish between CJD and AD (191).
In a recent study, GFAP was measured in plasma. Patients covering a broad spectrum of neurological diseases, including several forms of dementia, were included. Plasma levels of GFAP were found to be independent of age and evenly distributed between genders. No disease category displayed consistently increased levels of GFAP (197).
In vitro, GFAP is cleaved by caspase 6 at VELD225. The result is a C-terminal fragment of GFAP unable to assemble into filaments and an N-terminal fragment of GFAP perturbing in vitro filament assembling and promoting inter-filament aggregation (198). Caspase 3 is suggested to cleave GFAP at DLTD266. Cleaved GFAP has been shown to co-localize with caspase 3 in apoptotic astrocytes around blood vessels as well as plaque-rich regions of specific areas in the human AD brain (199). Furthermore, studies have shown calpain I-mediated cleavage products of GFAP in human brain as well as in CSF following traumatic brain injury (200, 201). Taken together, these data suggest that GFAP is a target of calpain I, caspase 3, and caspase 6 and that astrocyte injury and damage in the AD brain may involve cleavage of GFAP.
Conclusive Remarks
In the last decades several biomarker candidates have been developed and evaluated for AD and related dementias. Given the multiplicity of proteins involved in AD and related dementias as well as the overlap in pathological features between the different dementias it has to be acknowledged that so far no single biomarker permits an accurate and differential diagnosis. The diagnostic performance of the identified biomarkers could be improved by focusing on the pathological fragmentation of these proteins.
Although further studies are needed to evaluate the performance of protein fragmentation biomarkers, we believe that these biomarkers either alone or in combination with other biomarkers have a clinical potential.
Conflict of Interest Statement
All authors are employed by Nordic Bioscience Biomarkers and Research. Kim Henriksen and Morten Asser Karsdal hold patents on biomarkers of neurodegeneration. Morten Asser Karsdal holds stock in Nordic Bioscience.
References
2. Karantzoulis S, Galvin JE. Distinguishing Alzheimer’s disease from other major forms of dementia. Expert Rev Neurother (2011) 11:1579–91. doi:10.1586/ern.11.155
4. Borroni B, Di Luca M, Padovani A. Predicting Alzheimer dementia in mild cognitive impairment patients. Are biomarkers useful? Eur J Pharmacol (2006) 545:73–80. doi:10.1016/j.ejphar.2006.06.023
5. Assal F, Cummings JL. Neuropsychiatric symptoms in the dementias. Curr Opin Neurol (2002) 15:445–50. doi:10.1097/00019052-200208000-00007
6. Mistur R, Mosconi L, De Santi S, Guzman M, Li Y, Tsui W, et al. Current challenges for the early detection of Alzheimer’s disease: brain imaging and CSF studies. J Clin Neurol (2009) 5:153–66. doi:10.3988/jcn.2009.5.4.153
7. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7(3):263–9. doi:10.1016/j.jalz.2011.03.005
8. Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, et al. “Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration,”. Acta Neuropathol (2007) 114:5–22. doi:10.1007/s00401-007-0237-2
9. Hohl U, Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. Diagnostic accuracy of dementia with Lewy bodies. Arch Neurol (2000) 57:347–51. doi:10.1001/archneur.57.3.347
10. Woodward M, Jacova C, Black SE, Kertesz A, Mackenzie IR, Feldman H, et al. “Differentiating the frontal variant of Alzheimer’s disease,”. Int J Geriatr Psychiatry (2010) 25:732–8. doi:10.1002/gps.2415
11. Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord (1999) 13(Suppl 3):S115–23. doi:10.1097/00002093-199912003-00017
12. Cummings JL. Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement (2011) 7(3):e13–44. doi:10.1016/j.jalz.2010.06.004
13. Blennow K. Biomarkers in Alzheimer’s disease drug development. Nat Med (2010) 16:1218–22. doi:10.1038/nm.2221
14. Schneider P, Hampel H, Buerger K. Biological marker candidates of Alzheimer’s disease in blood, plasma, and serum. CNS Neurosci Ther (2009) 15:358–74. doi:10.1111/j.1755-5949.2009.00104.x
15. Henriksen K, O’Bryant SE, Hampel H, Trojanowski JQ, Montine TJ, Jeromin A, et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement (2014) 10:115–31. doi:10.1016/j.jalz.2013.01.013
16. Ingelsson M, Hyman BT. Disordered proteins in dementia. Ann Med (2002) 34:259–71. doi:10.1080/078538902320322529
17. Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx (2004) 1:213–25. doi:10.1602/neurorx.1.2.213
18. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (1993) 261:921–3. doi:10.1126/science.8346443
19. Mukaetova-Ladinska EB, Milne J, Andras A, Abdel-All Z, Cerejeira J, Greally E, et al. Alpha- and gamma-synuclein proteins are present in cerebrospinal fluid and are increased in aged subjects with neurodegenerative and vascular changes. Dement Geriatr Cogn Disord (2008) 26:32–42. doi:10.1159/000141039
20. Schoonenboom NSM, Reesink FE, Verwey NA, Kester MI, Teunissen CE, Van De Ven PM, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology (2012) 78(1):47–54. doi:10.1212/WNL.0b013e31823ed0f0
21. Kaerst L, Kuhlmann A, Wedekind D, Stoeck K, Lange P, Zerr I. Cerebrospinal fluid biomarkers in Alzheimer’s disease, vascular dementia and ischemic stroke patients: a critical analysis. J Neurol (2013) 260:2722–7. doi:10.1007/s00415-013-7047-3
22. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology (2006) 66:41–8. doi:10.1212/01.wnl.0000191307.69661.c3
23. Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T, et al. Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology (2010) 75:1766–72. doi:10.1212/WNL.0b013e3181fd613b
24. Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem (2009) 284:8516–24. doi:10.1074/jbc.M809462200
25. Zhang Y-J, Xu Y-F, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A (2009) 106:7607–12. doi:10.1073/pnas.0900688106
26. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, MacKenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain (2009) 132:2922–31. doi:10.1093/brain/awp214
27. Roeber S, Mackenzie IRA, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol (2008) 116:147–57. doi:10.1007/s00401-008-0395-x
28. Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med (2012) 367:795–804. doi:10.1056/NEJMoa1202753
29. Wang Y, Sørensen MG, Zheng Q, Zhang C, Karsdal MA, Henriksen K. Will posttranslational modifications of brain proteins provide novel serological markers for dementias? Int J Alzheimers Dis (2012) 2012:209409. doi:10.1155/2012/209409
30. Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol (2003) 2:605–13. doi:10.1016/S1474-4422(03)00530-1
31. Mattsson N, Blennow K, Zetterberg H. CSF biomarkers: pinpointing Alzheimer pathogenesis. Ann N Y Acad Sci (2009) 1180:28–35. doi:10.1111/j.1749-6632.2009.04944.x
32. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol (2006) 5:228–34. doi:10.1016/S1474-4422(06)70355-6
33. Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem (2008) 283:29615–9. doi:10.1074/jbc.R800019200
34. Hampel H, Teipel SJ. Total and phosphorylated tau proteins: evaluation as core biomarker candidates in frontotemporal dementia. Dement Geriatr Cogn Disord (2004) 17(4):350–4. doi:10.1159/000077170
35. Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol (2009) 65:403–13. doi:10.1002/ana.21610
36. Duits FH, Teunissen CE, Bouwman FH, Visser PJ, Mattsson N, Zetterberg H, et al. The cerebrospinal fluid ‘Alzheimer profile’: easily said, but what does it mean? Alzheimers Dement (2014) 10(6):713–23.e2. doi:10.1016/j.jalz.2013.12.023
37. Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang J, Otto M, et al. Value of CSF beta-amyloid1-42 and tau as predictors of Alzheimer’s disease in patients with mild cognitive impairment. Mol Psychiatry (2004) 9:705–10. doi:10.1038/sj.mp.4001473
38. Snider BJ, Fagan AM, Roe C, Shah AR, Grant EA, Xiong C, et al. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch Neurol (2009) 66:638–45. doi:10.1001/archneurol.2009.55
39. Andreasen N, Vanmechelen E, Vanderstichele H, Davidsson P, Blennow K. Cerebrospinal fluid levels of total-tau, phospho-tau and A-beta 42 predicts development of Alzheimer’s disease in patients with mild cognitive impairment. Acta Neurol Scand Suppl (2003) 179:47–51. doi:10.1034/j.1600-0404.107.s179.9.x
40. Hulstaert F, Blennow K, Ivanoiu A, Schoonderwaldt HC, Riemenschneider M, De Deyn PP, et al. Improved discrimination of AD patients using beta-amyloid(1-42) and tau levels in CSF. Neurology (1999) 52:1555–62. doi:10.1212/WNL.52.8.1555
41. Maddalena A, Papassotiropoulos A, üller-Tillmanns BM, Jung HH, Hegi T, Nitsch RM, et al. Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to beta-amyloid peptide42. Arch Neurol (2003) 60:1202–6. doi:10.1001/archneur.60.9.1202
42. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol (2007) 64:343–9. doi:10.1001/archneur.64.3.noc60123
43. Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, et al. Decreased-alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun (2006) 349:162–6. doi:10.1016/j.bbrc.2006.08.024
44. Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, et al. Direct quantification of CSF α-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol (2008) 213:315–25. doi:10.1016/j.expneurol.2008.06.004
45. Aerts MB, Esselink RAJ, Abdo WF, Bloem BR, Verbeek MM. CSF-alpha-synuclein does not differentiate between parkinsonian disorders. Neurobiol Aging (2012) 33:430.e1–3. doi:10.1016/j.neurobiolaging.2010.12.001
46. Spies PE, Melis RJF, Sjögren MJC, Rikkert MGMO, Verbeek MM. Cerebrospinal fluid α-synuclein does not discriminate between dementia disorders. J Alzheimers Dis (2009) 16:363–9. doi:10.3233/JAD-2009-0955
47. Reesink FE, Lemstra AW, Van Dijk KD, Berendse HW, Van De Berg WDJ, Klein M, et al. CSF-alpha-synuclein does not discriminate dementia with Lewy bodies from Alzheimer’s disease. J Alzheimers Dis (2010) 22:87–95. doi:10.3233/JAD-2010-100186
48. Hansson O, Hall S, Öhrfelt A, Zetterberg H, Blennow K, Minthon L, et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res Ther (2014) 6:25. doi:10.1186/alzrt255
49. Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord (2014) 29:1019–27. doi:10.1002/mds.25772
50. Slaets S, Vanmechelen E, Le Bastard N, Decraemer H, Vandijck M, Martin JJ, et al. Increased CSF-alpha-synuclein levels in Alzheimer’s disease: correlation with tau levels. Alzheimers Dement (2014) 10(5 Suppl):S290–8. doi:10.1016/j.jalz.2013.10.004
51. Rosén C, Hansson O, Blennow K, Zetterberg H. Fluid biomarkers in Alzheimer’s disease - current concepts. Mol Neurodegener (2013) 8:20. doi:10.1186/1750-1326-8-20
52. Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, et al. The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement (2011) 7:386–95.e6. doi:10.1016/j.jalz.2011.05.2243
53. Snyder HM, Carrillo MC, Grodstein F, Henriksen K, Jeromin A, Lovestone S, et al. Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement (2014) 10:109–14. doi:10.1016/j.jalz.2013.10.007
54. Marques F, Sousa JC, Sousa N, Palha JA. Blood – brain-barriers in aging and in Alzheimer’s disease. Mol Neurodegener (2013) 8(1):38. doi:10.1186/1750-1326-8-38
55. Sharma HS, Castellani RJ, Smith MA, Sharma A. The blood-brain barrier in Alzheimer’s disease. Novel therapeutic targets and nanodrug delivery. Int Rev Neurobiol (2012) 102:47–90. doi:10.1016/B978-0-12-386986-9.00003-X
56. Desai BS, Monahan AJ, Carvey PM, Hendey B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. Cell Transplant (2007) 16:285–99. doi:10.3727/000000007783464731
57. Reiber H. Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci (2003) 21:79–96.
58. Duong T, Pommier EC, Scheibel AB. Immunodetection of the amyloid P component in Alzheimer’s disease. Acta Neuropathol (1989) 78:429–37. doi:10.1007/BF00688180
59. Zetterberg H, Wilson D, Andreasson U, Minthon L, Blennow K, Randall J, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther (2013) 5:9. doi:10.1186/alzrt163
60. Chiu MJ, Chen YF, Chen TF, Yang SY, Yang FPG, Tseng TW, et al. Plasma tau as a window to the brain-negative associations with brain ume and memory function in mild cognitive impairment and early Alzheimer’s disease. Hum Brain Mapp (2014) 35:3132–42. doi:10.1002/hbm.22390
61. Randall J, Mörtberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation (2013) 84(3):351–6. doi:10.1016/j.resuscitation.2012.07.027
62. Hansson O, Stomrud E, Vanmechelen E, Östling S, Gustafson DR, Zetterberg H, et al. Evaluation of plasma Aβ as predictor of Alzheimer’s disease in older individuals without dementia: a population-based study. J Alzheimers Dis (2012) 28:231–8. doi:10.3233/JAD-2011-111418
63. Zetterberg H, Blennow K. Plasma Abeta in Alzheimer’s disease-up or down? Lancet Neurol (2006) 5:638–9. doi:10.1016/S1474-4422(06)70503-8
64. Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis (2008) 5:55–9. doi:10.1159/000112832
65. Karsdal MA, Henriksen K, Leeming DJ, Woodworth T, Vassiliadis E, Bay-Jensen AC. Novel combinations of post-translational modification (PTM) neo-epitopes provide tissue-specific biochemical markers-are they the cause or the consequence of the disease? Clin Biochem (2010) 43(10–11):793–804. doi:10.1016/j.clinbiochem.2010.03.015
66. Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int (2011) 58:458–71. doi:10.1016/j.neuint.2010.12.023
67. Meredith JE, Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ, et al. Characterization of novel CSF tau and ptau biomarkers for Alzheimer’s disease. PLoS One (2013) 8:e76523. doi:10.1371/journal.pone.0076523
68. Marques MA, Tolar M, Harmony JA, Crutcher KA. A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. Neuroreport (1996) 7:2529–32. doi:10.1097/00001756-199611040-00025
69. Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem (2005) 280:22670–8. doi:10.1074/jbc.M501508200
70. Rohn TT. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain Res (2008) 1228:189–98. doi:10.1016/j.brainres.2008.06.094
71. Shayo M, McLay RN, Kastin AJ, Banks WA. The putative blood-brain barrier transporter for the beta-amyloid binding protein apolipoprotein j is saturated at physiological concentrations. Life Sci (1997) 60:L115–8. doi:10.1016/S0024-3205(96)00685-6
72. Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, et al. Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vascul Pharmacol (2002) 38:303–13. doi:10.1016/S1537-1891(02)00198-2
73. Kastin AJ, Pan W, Maness LM, Banks WA. Peptides crossing the blood-brain barrier: some unusual observations. Brain Res (1999) 848:96–100. doi:10.1016/S0006-8993(99)01961-7
74. Pan W, Kastin AJ. Polypeptide delivery across the blood-brain barrier. Curr Drug Targets CNS Neurol Disord (2004) 3:131–6. doi:10.2174/1568007043482525
75. Shahim P, Linemann T, Inekci D, Karsdal MA, Blennow K, Tegner Y, et al. Serum tau fragments predict return to play in concussed professional ice hockey players. J Neurotrauma (2015). doi:10.1089/neu.2014.3741
76. Brunholz S, Sisodia S, Lorenzo A, Deyts C, Kins S, Morfini G. Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp Brain Res (2012) 217:353–64. doi:10.1007/s00221-011-2870-1
77. De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev (2010) 90:465–94. doi:10.1152/physrev.00023.2009
78. Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain (2011) 4:3. doi:10.1186/1756-6606-4-3
79. Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of Tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry (2012) 69:98–106. doi:10.1001/archgenpsychiatry.2011.155
80. Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol (2013) 12(2):207–16. doi:10.1016/S1474-4422(12)70291-0
81. Zou K, Kim D, Kakio A, Byun K, Gong J-S, Kim J, et al. Amyloid beta-protein (Abeta)1-40 protects neurons from damage induced by Abeta1-42 in culture and in rat brain. J Neurochem (2003) 87:609–19. doi:10.1046/j.1471-4159.2003.02018.x
82. Verbeek MM, Kremer BPH, Rikkert MO, Van Domburg PHMF, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann Neurol (2009) 66(2):245–9. doi:10.1002/ana.21694
83. Bogner S, Bernreuther C, Matschke J, Barrera-Ocampo A, Sepulveda-Falla D, Leypoldt F, et al. Immune activation in amyloid-β-related angiitis correlates with decreased parenchymal amyloid-β plaque load. Neurodegener Dis (2013) 13(1):38–44. doi:10.1159/000352020
84. Piazza F, Greenberg SM, Savoiardo M, Gardinetti M, Chiapparini L, Raicher I, et al. Anti-amyloid β autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol (2013) 73(4):449–58. doi:10.1002/ana.23857
85. Boncoraglioa GB, Piazza F, Savoiardod M, Farinad L, DiFrancescobe JC, Prionif S, et al. Prodromal Alzheimer’s disease presenting as cerebral amyloid angiopathy-related inflammation with spontaneous amyloid-related imaging abnormalities and high cerebrospinal fluid anti-beta autoantibodies. J Alzheimers Dis (2015) 45:363–7. doi:10.3233/JAD-142376
86. Bibl M, Gallus M, Welge V, Esselmann H, Wiltfang J. Aminoterminally truncated and oxidized amyloid-β peptides in the cerebrospinal fluid of Alzheimer’s disease patients. J Alzheimers Dis (2012) 29(4):809–16. doi:10.3233/JAD-2012-111796
87. Larner AJ. Hypothesis: amyloid beta-peptides truncated at the N-terminus contribute to the pathogenesis of Alzheimer ‘s disease. Neurobiol Aging (1999) 20:65–9.
88. Portelius E, Tran AJ, Andreasson U, Persson R, Brinkmann G, Zetterberg H, et al. Characterization of amyloid-beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J Proteome Res (2007) 6:4433–9. doi:10.1021/pr0703627
89. Portelius E, Bogdanovic N, Gustavsson MK, Kmann I, Brinkmalm G, Zetterberg H, et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol (2010) 120:185–93. doi:10.1007/s00401-010-0690-1
90. Portelius E, Mattsson N, Andreasson U, Blennow K, Zetterberg H. Novel Aβ isoforms in Alzheimer’s disease – their role in diagnosis and treatment. Curr Pharm Des (2011) 17:2594–602. doi:10.2174/138161211797416039
91. Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, et al. A novel pathway for amyloid precursor protein processing. Neurobiol Aging (2011) 32:1090–8. doi:10.1016/j.neurobiolaging.2009.06.002
92. Thambisetty M, Lovestone S. Blood-based biomarkers of Alzheimer’s disease: challenging but feasible. Biomark Med (2010) 4:65–79. doi:10.2217/bmm.09.84
93. Perez-Grijalba SM, Pesini V, Allue P, Sarasa JA, Montanes L, Lacosta M, et al. A-beta (1-17) is a major amyloid-beta fragment isoform in cerebrospinal fluid and blood with possible diagnostic value in Alzheimer’s disease. J Alzheimers Dis (2015) 43(1):47–56. doi:10.3233/JAD-140156
94. Iqbal K, Del A, Alonso C, Chen S, Chohan MO, El-Akkad E, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta (2005) 1739:198–210. doi:10.1016/j.bbadis.2004.09.008
95. Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev (2000) 33:95–130. doi:10.1016/S0165-0173(00)00019-9
96. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol (2013) 12:609–22. doi:10.1016/S1474-4422(13)70090-5
97. Espinoza M, de Silva R, Dickson DW, Davies P. Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis (2008) 14:1–16.
98. Buée L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol (1999) 9:681–93. doi:10.1111/j.1750-3639.1999.tb00550.x
99. Mohamed NV, Herrou T, Plouffe V, Piperno N, Leclerc N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur J Neurosci (2013) 37:1939–48. doi:10.1111/ejn.12229
100. Henriksen K, Byrjalsen I, Christiansen C, Karsdal MA. Relationship between serum levels of tau fragments and clinical progression of Alzheimer ‘ s disease,”. J Alzheimers Dis (2015) 43(4):1331–41. doi:10.3233/JAD-140984
101. Henriksen K, Wang Y, Sørensen MG, Barascuk N, Suhy J, Pedersen JT, et al. An enzyme-generated fragment of tau measured in serum shows an inverse correlation to cognitive function. PLoS One (2013) 8(5):e64990. doi:10.1371/journal.pone.0064990
102. Hanger DP, Wray S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem Soc Trans (2010) 38(4):1016–20. doi:10.1042/BST0381016
103. Wang Y, Garg S, Mandelkow E-M, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans (2010) 38:955–61. doi:10.1042/BST0380955
104. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science (1988) 240:622–30. doi:10.1126/science.3283935
105. Huang Y, Weisgraber K, Mucke L, Mahley R. Apolipoprotein E. J Mol Neurosci (2004) 23:189–204. doi:10.1385/JMN:23:3:189
106. Leduc V, Jasmin-Bélanger S, Poirier J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol Med (2010) 16:469–77. doi:10.1016/j.molmed.2010.07.008
107. Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci (2006) 31:445–54. doi:10.1016/j.tibs.2006.06.008
108. Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci (2008) 9:768–78. doi:10.1038/nrn2494
109. Suri S, Heise V, Trachtenberg AJ, Mackay CE. The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE e2. Neurosci Biobehav Rev (2013) 37(10):2878–86. doi:10.1016/j.neubiorev.2013.10.010
110. Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEε2 is associated with milder clinical and pathological Alzheimer’s disease. Ann Neurol (2015). doi:10.1002/ana.24369
111. Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang SJ, et al. Astroglial regulation of apolipoprotein E expression in neuronal cells – implications for Alzheimer’s disease. J Biol Chem (2004) 279:3862–8. doi:10.1074/jbc.M309475200
112. Tibolla G, Norata GD, Meda C, Arnaboldi L, Uboldi P, Piazza F, et al. Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis (2010) 210(1):78–87. doi:10.1016/j.atherosclerosis.2009.10.040
113. Greenberg SM, Briggs ME, Hyman BT, Kokoris GJ, Takis C, Kanter DS, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke (1996) 27:1333–7. doi:10.1161/01.STR.27.8.1333
114. Pinholt M, Frederiksen JL, Christiansen M. The association between apolipoprotein E and multiple sclerosis. Eur J Neurol (2006) 13:573–80. doi:10.1111/j.1468-1331.2006.01360.x
115. Josephs KA, Tsuboi Y, Cookson N, Watt H, Dickson DW. Apolipoprotein E epsilon 4 is a determinant for Alzheimer-type pathologic features in tauopathies, synucleinopathies, and frontotemporal degeneration. Arch Neurol (2004) 61:1579–84. doi:10.1001/archneur.61.10.1579
116. Martinez M, Brice A, Vaughan JR, Zimprich A, Breteler MMB, Meco G, et al. Apolipoprotein E4 is probably responsible for the chromosome 19 linkage peak for Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet (2005) 136B:72–4. doi:10.1002/ajmg.b.30196
117. Hesse C, Larsson H, Fredman P, Minthon L, Andreasen N, Davidsson P, et al. Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res (2000) 25:511–7. doi:10.1023/A:1007516210548
118. Vijayaraghavan S, Maetzler W, Reimold M, Lithner CU, Liepelt-Scarfone I, Berg D, et al. High apolipoprotein E in cerebrospinal fluid of patients with Lewy body disorders is associated with dementia. Alzheimers Dement (2014) 10:530–40. doi:10.1016/j.jalz.2013.03.010
119. Taddei K, Clarnette R, Gandy SE, Martins RN. Increased plasma apolipoprotein E (apoE) levels in Alzheimer’s disease. Neurosci Lett (1997) 223:29–32. doi:10.1016/S0304-3940(97)13394-8
120. Gupta VB, Laws SM, Villemagne VL, Ames D, Bush AI, Ellis KA, et al. Plasma apolipoprotein e and Alzheimer disease risk: the AIBL study of aging. Neurology (2011) 76:1091–8. doi:10.1212/WNL.0b013e318211c352
121. Slooter AJC, De Knijff P, Hofman A, Cruts M, Breteler MMB, Van Broeckhoven C, et al. Serum apolipoprotein e level is not increased in Alzheimer’s disease: the Rotterdam study. Neurosci Lett (1998) 248:21–4. doi:10.1016/S0304-3940(98)00339-5
122. Siest G, Bertrand P, Qin B, Herbeth B, Serot JM, Masana L, et al. Apolipoprotein E polymorphism and serum concentration in Alzheimer’s disease in nine European centres: the ApoEurope study. ApoEurope group. Clin Chem Lab Med (2000) 38:721–30. doi:10.1515/CCLM.2000.102
123. Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol (2015) 77:301–11. doi:10.1002/ana.24326
124. Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A (2001) 98:8838–43. doi:10.1073/pnas.151254698
125. Wellnitz S, Friedlein A, Bonanni C, Anquez V, Goepfert F, Loetscher H, et al. A 13 kDa carboxy-terminal fragment of ApoE stabilizes Abeta hexamers. J Neurochem (2005) 94:1351–60. doi:10.1111/j.1471-4159.2005.03295.x
126. Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, et al. “Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice.,”. J Neurosci (2004) 24:2527–34. doi:10.1523/JNEUROSCI.4315-03.2004
127. Tamboli IY, Heo D, Rebeck GW. Extracellular proteolysis of apolipoprotein e (apoE) by secreted serine neuronal protease. PLoS One (2014) 9:e93120. doi:10.1371/journal.pone.0093120
128. Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron (1995) 14:467–75. doi:10.1016/0896-6273(95)90302-X
129. Ltic S, Perovic M, Mladenovic A, Raicevic N, Ruzdijic S, Rakic L, et al. Alpha-synuclein is expressed in different tissues during human fetal development. J Mol Neurosci (2004) 22:199–204. doi:10.1385/JMN:22:3:199
130. Jensen PH, Hager H, Nielsen MS, Højrup P, Gliemann J, Jakes R. α-synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem (1999) 274:25481–9. doi:10.1074/jbc.274.36.25481
131. Iqbal K, Flory M, Khatoon S, Soininen H, Pirttila T, Lehtovirta M, et al. Subgroups of Alzheimer’s disease based on cerebrospinal fluid molecular markers. Ann Neurol (2005) 58:748–57. doi:10.1002/ana.20639
132. Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci (2012) 32:10253–66. doi:10.1523/JNEUROSCI.0581-12.2012
133. Kasuga K, Tokutake T, Ishikawa A, Uchiyama T, Tokuda T, Onodera O, et al. Differential levels of alpha-synuclein, beta-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer’s disease. J Neurol Neurosurg Psychiatry (2010) 81:608–10. doi:10.1136/jnnp.2009.197483
134. El-Agnaf OMA, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J (2006) 20:419–25. doi:10.1096/fj.03-1449com
135. Lee PH, Lee G, Park HJ, Bang OY, Joo IS, Huh K. The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm (2006) 113:1435–9. doi:10.1007/s00702-005-0427-9
136. Duran R, Barrero FJ, Morales B, Luna JD, Ramirez M, Vives F. Plasma alpha-synuclein in patients with Parkinson’s disease with and without treatment. Mov Disord (2010) 25:489–93. doi:10.1002/mds.22928
137. Foulds PG, Mitchell JD, Parker A, Turner R, Green G, Diggle P, et al. Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J (2011) 25(12):4127–37. doi:10.1096/fj.10-179192
138. Park MJ, Cheon SM, Bae HR, Kim SH, Kim JW. Elevated levels of α-synuclein oligomer in the cerebrospinal fluid of drug-naïve patients with Parkinson’s disease. J Clin Neurol (2011) 7:215–22. doi:10.3988/jcn.2011.7.4.215
139. Laske C, Fallgatter AJ, Stransky E, Hagen K, Berg D, Maetzler W. Decreased alpha-synuclein serum levels in patients with Lewy body dementia compared to Alzheimer’s disease patients and control subjects. Dement Geriatr Cogn Disord (2011) 31:413–6. doi:10.1159/000329763
140. Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol (2007) 170:1725–38. doi:10.2353/ajpath.2007.061232
141. Levin J, Giese A, Boetzel K, Israel L. T. Högen, G. Nübling, H. Kretzschmar, and S. Lorenzl, “Increased α-synuclein aggregation following limited cleavage by certain matrix metalloproteinases,”. Exp Neurol (2009) 215:201–8. doi:10.1016/j.expneurol.2008.10.010
142. Lorenzl S, Albers DS, Narr S, Chirichigno J, Beal MF. Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson’s disease. Exp Neurol (2002) 178:13–20. doi:10.1006/exnr.2002.8019
143. Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ, et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J (2007) 21:179–87. doi:10.1096/fj.06-5865com
144. Ogawa K, Yamada T, Tsujioka Y, Taguchi J, Takahashi M, Tsuboi Y, et al. Localization of a novel type trypsin-like serine protease, neurosin, in brain tissues of Alzheimer’s disease and Parkinson’s disease. Psychiatry Clin Neurosci (2000) 54:419–26. doi:10.1046/j.1440-1819.2000.00731.x
145. Wennström M, Surova Y, Hall S, Nilsson C, Minthon L, Boström F, et al. Low CSF levels of both α-synuclein and the α-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS One (2013) 8:e53250. doi:10.1371/journal.pone.0053250
146. Cookson MR. α-Synuclein and neuronal cell death. Mol Neurodegener (2009) 4:9. doi:10.1186/1750-1326-4-9
147. Hu WT, Grossman M. TDP-43 and frontotemporal dementia. Curr Neurol Neurosci Rep (2009) 9:353–8. doi:10.1007/s11910-009-0052-3
148. Snowden JS. Frontotemporal dementia. Br J Psychiatry (2002) 180:140–3. doi:10.1192/bjp.180.2.140
149. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain (2011) 134:2456–77. doi:10.1093/brain/awr179
150. Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet (2010) 19:R46–64. doi:10.1093/hmg/ddq137
151. Wilson AC, Dugger BN, Dickson DW, Wang D-S. TDP-43 in aging and Alzheimer’s disease - a review. Int J Clin Exp Pathol (2011) 4:147–55.
152. Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CAF, Lehnert S, et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol (2008) 65:1481–7. doi:10.1001/archneur.65.11.1481
153. Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol (2009) 117:55–62. doi:10.1007/s00401-008-0456-1
154. Foulds PG, Davidson Y, Mishra M, Hobson DJ, Humphreys KM, Taylor M, et al. Plasma phosphorylated-TDP-43 protein levels correlate with brain pathology in frontotemporal lobar degeneration. Acta Neuropathol (2009) 118:647–58. doi:10.1007/s00401-009-0594-0
155. Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown SM, Neary D, et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol (2008) 116:141–6. doi:10.1007/s00401-008-0389-8
156. De Marco G, Lomartire A, Mandili G, Lupino E, Buccinnà B, Ramondetti C, et al. Reduced cellular Ca2+ availability enhances TDP-43 cleavage by apoptotic caspases. Biochim Biophys Acta (2014) 1843:725–34. doi:10.1016/j.bbamcr.2014.01.010
157. Mackenzie IRA, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain (2008) 131:1282–93. doi:10.1093/brain/awn061
158. Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci (1997) 110(Pt 1):1741–50.
159. Perrotti D, Bonatti S, Trotta R, Martinez R, Skorski T, Salomoni P, et al. TLS/FUS, a pro-oncogene inved in multiple chromosomal translocations, is a novel regulator of BCR/ABL-mediated leukemogenesis. EMBO J (1998) 17:4442–55. doi:10.1093/emboj/17.15.4442
160. Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS. Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene (1999) 18:4515–21. doi:10.1038/sj.onc.1203048
161. Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem (1999) 274:34337–42. doi:10.1074/jbc.274.48.34337
162. Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins inved in RNA splicing. J Biol Chem (1998) 273:27761–4. doi:10.1074/jbc.273.43.27761
163. Andersson MK, Ståhlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, et al. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and invement in cell spreading and stress response. BMC Cell Biol (2008) 9:37. doi:10.1186/1471-2121-9-37
164. Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol (2013) 125:273–88. doi:10.1007/s00401-012-1043-z
165. Yoshida T, Tomozawa Y, Arisato T, Okamoto Y, Hirano H, Nakagawa M. The functional alteration of mutant GFAP depends on the location of the domain: morphological and functional studies using astrocytoma-derived cells. J Hum Genet (2007) 52:362–9. doi:10.1007/s10038-007-0124-7
166. Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo1 AM. Identification of regulators of chaperone-mediated autophagy Mol Cell (2010) 39:535–47. doi:10.1016/j.molcel.2010.08.004
167. Emirandetti A, Graciele ZR, Sabha M Jr., de Oliveira AL. Astrocyte reactivity influences the number of presynaptic terminals apposed to spinal motoneurons after axotomy. Brain Res (2006) 1095:35–42. doi:10.1016/j.brainres.2006.04.021
168. Giménez YRM, Langa F, Menet V, Privat A. Comparative anatomy of the cerebellar cortex in mice lacking vimentin, GFAP, and both vimentin and GFAP. Glia (2000) 31:69–83. doi:10.1002/(sici)1098-1136(200007)31:1<69::aid-glia70>3.0.co;2-w
169. Eliasson C, Sahlgren C, Berthold CH, Stakeberg J, Celis JE, Betsholtz C, et al. Intermediate filament protein partnership in astrocytes. J Biol Chem (1999) 274:23996–4006. doi:10.1074/jbc.274.34.23996
170. Kato H, Yamamoto T, Yamamoto H, Ohi R, So N, Iwasaki Y. Immunocytochemical characterization of supporting cells in the enteric nervous system in Hirschsprung’s disease. J Pediatr Surg (1990) 25:514–9. doi:10.1016/0022-3468(90)90563-O
171. Riol H, Tardy M, Rolland B, Levesque G, Murthy MR. Detection of the peripheral nervous system (PNS)-type glial fibrillary acidic protein (GFAP) and its mRNA in human lymphocytes. J Neurosci Res (1997) 48:53–62. doi:10.1002/(SICI)1097-4547(19970401)48:1%3C53::AID-JNR5%3E3.0.CO;2-D
172. Hainfellner JA, Voigtlander T, Strobel T, Mazal PR, Maddalena AS, Aguzzi A, et al. Fibroblasts can express glial fibrillary acidic protein (GFAP) in vivo. J Neuropathol Exp Neurol (2001) 60:449–61.
173. Carotti S, Morini S, Corradini SG, Burza MA, Molinaro A, Carpino G, et al. Glial fibrillary acidic protein as an early marker of hepatic stellate cell activation in chronic and posttransplant recurrent hepatitis C. Liver Transpl (2008) 14:806–14. doi:10.1002/lt.21436
174. Roelofs RF, Fischer DF, Houtman SH, Sluijs JA, Van Haren W, van Leeuwen FW, et al. Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia (2005) 52:289e300. doi:10.1002/glia.20243
175. Middeldorp J, van den Berge SA, Aronica E, Speijer D, Hol EM. Specific human astrocyte subtype revealed by affinity purified GFAP+1 antibody. Unpurified serum cross-reacts with neurofilament-L in Alzheimer PLoS One (2009) 4:e7663. doi:10.1371/journal.pone.0007663
176. Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi EJ, Moeton M, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol Aging (2014) 35:492–510. doi:10.1016/j.neurobiolaging.2013.09.035
177. van den Berge SA, Middeldorp J, Eleana ZC, Curtis MA, Leonard BW, Mastroeni D, et al. Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP-delta. Aging Cell (2010) 9:313–26. doi:10.1111/j.1474-9726.2010.00556.x
178. Perng MD, Wen SF, Gibbon T, Middeldorp J, Sluijs J, Hol EM, et al. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association. Mol Biol Cell (2008) 19:4521–33. doi:10.1091/mbc.E08-03-0284
179. Blechingberg J, Holm IE, Nielsen KB, Jensen TH, Jorgensen AL, Nielsen AL. Identification and characterization of GFAPkappa, a novel glial fibrillary acidic protein isoform. Glia (2007) 55:497–507. doi:10.1002/glia.20475
180. Nielsen AL, Holm IE, Johansen M, Bonven B, Jorgensen P, Jorgensen ALA. new splice variant of glial fibrillary acidic protein. GFAP epsilon, interacts with the presenilin proteins J Biol Chem (2002) 277:29983–91. doi:10.1074/jbc.M112121200
181. Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nature Cell Biol (2000) 2:463–5. doi:10.1038/35017108
182. Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet (2009) 18(21):4153–70. doi:10.1093/hmg/ddp367
183. Wharton SB, O’Callaghan JP, Savva GM, Nicoll JA, Matthews F, Simpson JE, et al. Population variation in Glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord (2009) 27:465–73. doi:10.1159/000217729
184. Muramori F, Kobayashi K, Nakamura IA. quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer’s disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychiatry Clin Neurosci (1998) 52:593–9. doi:10.1111/j.1440-1819.1998.tb02706.x
185. Porchet R, Probst A, Bouras C, Draberova E, Draber P, Riederer BM. Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics (2003) 3:1476–85. doi:10.1002/pmic.200300456
186. Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, et al. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging (2010) 31:578–90. doi:10.1016/j.neurobiolaging.2008.05.015
187. Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem (1997) 68:430–3. doi:10.1046/j.1471-4159.1997.68010430.x
188. Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology (2004) 62:925–31. doi:10.1212/01.WNL.0000115115.98960.37
189. Fukuyama R, Izumoto T, Fushiki S. The cerebrospinal fluid level of glial fibrillary acidic protein is increased in cerebrospinal fluid from Alzheimer’s disease patients and correlates with severity of dementia. Eur Neurol (2001) 46(1):35–8. doi:10.1159/000050753
190. Rosengren LE, Wikkelsø C, Hagberg L. A sensitive ELISA for glial fibrillary acidic protein: application in CSF of adults.,”. J Neurosci Methods (1994) 51(2):197–204. doi:10.1016/0165-0270(94)90011-6
191. Jesse S, Steinacker P, Cepek L, Arnim CV, Tumani H, Lehnert S, et al. Glial fibrillary acidic protein and protein S-100B: different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. J Alzheimers Dis (2009) 17(3):541–51. doi:10.3233/JAD-2009-1075
192. Van Eijk JJ, Van Everbroeck B, Abdo WF, Kremer BPH, Verbeek MM. CSF neurofilament proteins levels are elevated in sporadic Creutzfeldt-Jakob disease. J Alzheimers Dis (2010) 21(2):569–76. doi:10.3233/JAD-2010-090649
193. Brouns R, De Vil B, Cras P, De Surgeloose D, Mariën P, De Deyn PP. Neurobiochemical markers of brain damage in cerebrospinal fluid of acute ischemic stroke patients. Clin Chem (2010) 56(3):451–8. doi:10.1373/clinchem.2009.134122
194. Aurell A, Rosengren LE, Karlsson B, Olsson JE, Zbornikova V, Haglid KG. Determination of S-100 and glial fibrillary acidic protein concentrations in cerebrospinal fluid after brain infarction. Stroke (1991) 22(10):1254–8. doi:10.1161/01.STR.22.10.1254
195. Neselius S, Brisby H, Theodorsson A, Blennow K, Zetterberg H, Marcusson J. Csf-biomarkers in olympic boxing: diagnosis and effects of repetitive head trauma. PLoS One (2012) 7(4):e33606. doi:10.1371/journal.pone.0033606
196. Fraser DD, Close TE, Rose KL, Ward R, Mehl M, Farrell C, et al. Severe traumatic brain injury in children elevates glial fibrillary acidic protein in cerebrospinal fluid and serum. Pediatr Crit Care Med (2011) 12(3):319–24. doi:10.1097/PCC.0b013e3181e8b32d
197. Mayer CA, Brunkhorst R, Niessner M, Pfeilschifter W, Steinmetz H, Foerch C. Blood levels of glial fibrillary acidic protein (GFAP) in patients with neurological diseases. PLoS One (2013) 8(4):e62101. doi:10.1371/journal.pone.0062101
198. Chen MH, Hagemann TL, Quinlan RA, Messing A, Perng MD. Caspase cleavage of GFAP produces an assembly-compromised proteolytic fragment that promotes filament aggregation. ASN Neuro (2013) 5(5):e00125. doi:10.1042/AN20130032
199. Mouser PE, Head E, Ha KH, Rohn TT. Caspase-mediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer’s disease brain. Am J Pathol (2006) 168:936–46. doi:10.2353/ajpath.2006.050798
200. Lee YB, Du S, Rhim H, Lee EB, Markelonis GJ, Oh TH. Rapid increase in immunoreactivity to GFAP in astrocytes in vitro induced by acidic pH is mediated by calcium influx and calpain I. Brain Res (2000) 864(2):220–9. doi:10.1016/S0006-8993(00)02180-6
Keywords: dementia, Alzheimer’s disease, biomarkers, blood, post-translational modifications
Citation: Inekci D, Jonesco DS, Kennard S, Karsdal MA and Henriksen K (2015) The potential of pathological protein fragmentation in blood-based biomarker development for dementia – with emphasis on Alzheimer’s disease. Front. Neurol. 6:90. doi: 10.3389/fneur.2015.00090
Received: 02 February 2015; Accepted: 10 April 2015;
Published: 11 May 2015
Edited by:
Charlotte Elisabeth Teunissen, VU University Medical Center Amsterdam, NetherlandsReviewed by:
Alison Louise Baird, University of Oxford, UKFabrizio Piazza, University of Milano Bicocca, Italy
Andreas Jeromin, Quanterix, USA
Copyright: © 2015 Inekci, Jonesco, Kennard, Karsdal and Henriksen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim Henriksen, Nordic Bioscience A/S, Herlev Hovedgade 207, Herlev DK 2730, Denmark,a2hAbm9yZGljYmlvc2NpZW5jZS5jb20=