 Celine Feillet
Celine Feillet Gijsbertus T. J. van der Horst2
Gijsbertus T. J. van der Horst2 David A. Rand
David A. Rand Franck Delaunay
Franck Delaunay- 1Circadian Systems Biology, CNRS, INSERM, Institut de Biologie Valrose, Université Nice Sophia Antipolis, Nice, France
- 2Department of Genetics, Erasmus University Medical Center, Rotterdam, Netherlands
- 3Cancer Chronotherapy Unit, University of Warwick, Coventry, UK
- 4Systems Biology Centre, University of Warwick, Coventry, UK
Uncontrolled cell proliferation is one of the key features leading to cancer. Seminal works in chronobiology have revealed that disruption of the circadian timing system in mice, either by surgical, genetic, or environmental manipulation, increased tumor development. In humans, shift work is a risk factor for cancer. Based on these observations, the link between the circadian clock and cell cycle has become intuitive. But despite identification of molecular connections between the two processes, the influence of the clock on the dynamics of the cell cycle has never been formally observed. Recently, two studies combining single live cell imaging with computational methods have shed light on robust coupling between clock and cell cycle oscillators. We recapitulate here these novel findings and integrate them with earlier results in both healthy and cancerous cells. Moreover, we propose that the cell cycle may be synchronized or slowed down through coupling with the circadian clock, which results in reduced tumor growth. More than ever, systems biology has become instrumental to understand the dynamic interaction between the circadian clock and cell cycle, which is critical in cellular coordination and for diseases such as cancer.
Clock and Cell Cycle, 2 Biological Oscillators
The Circadian Clock
The Earth’s rotation results in predictable daily variations of environmental conditions (light/dark, food, temperature). Biological clocks give an unbiased estimation of time and allow coordination of physiology in anticipation to recurring changes.
Circadian clocks are implicated in normal functioning of numerous systems (digestive, endocrine, cellular). They control 24-h rhythms, termed as “circadian rhythms” (sleep, body temperature, cortisol secretion …).
An important milestone in the field was the discovery of the mammalian circadian “conductor”: the suprachiasmatic nuclei of the hypothalamus (SCN) (1, 2). From the molecular standpoint, the SCN machinery consists of feedback loops relying on clock genes. A main loop involves CLOCK and BMAL1, which heterodimerize and activate the transcription of Period (Per1–2–3) and Cryptochrome (Cry1–2) genes. PER:CRY proteins in turn inhibit their own transcription by direct interaction with CLOCK:BMAL1. This essential loop is modulated by the REV-ERBα–β and RORα–β–γ proteins, which operate a negative and positive feedback, respectively, on Bmal1 transcription. This genetic network is also extensively regulated via post-translational processes. The molecular clockwork acts autonomously and, in the absence of resetting cues, oscillates with a period close to 24 h; it is the basis of circadian rhythmicity and defines the endogenous period of a clock (3). It is synchronized to a sharp 24-h period by environmental parameters (light/dark, feeding cycles, hormones). A rhythmic message integrating environmental informations is then generated by the SCN, and redistributed to the entire organism to synchronize physiological functions. Circadian rhythmicity and clock genes are not an exclusive property of the SCN as clock genes are rhythmically expressed in nearly all cells. Each individual cell can thus be regarded as a circadian clock (4, 5).
The Cell Cycle
Cell cycle is the process leading to cell division. It consists of two critical phases: the S phase, in which the cell replicates its DNA, and the M phase where it divides (mitosis); they are preceded by growth phases G1 and G2, respectively. Non-dividing (somatic) cells are in a quiescent state (G0). They may resume cell division, depending on environmental parameters such as growth factors, to enter G1 phase (6, 7).
Progression of the cell cycle relies on transient and sequential activation of cyclin-dependent kinases (CDKs) forming complexes with cyclins (CCN). Cell cycle successively depends on CCND/CDK4–6 (G1), CCNE/CDK2 (G1/S transition), CCNA/CDK2 (S), CCNA/CDK1 (S/G2 transition), and CCNB/CDK1 [M – (8–10)]. Activity of these enzymatic complexes finely tunes cell cycle duration, especially at critical checkpoints. Association with CDK inhibitors (CKI–P16, P27, P21) or phosphorylation by the kinase WEE1, inhibit activity of targeted CCN/CDK across the cycle. On the other hand, they are activated by phosphatases such as CDC25A–B–C (11). These activators or inhibitors are targets of proteins involved in DNA repair. Indeed, a double-strand DNA break activates the ataxia telangiectasia mutated (ATM) and check-point kinase 2 (CHK2) proteins, while a single-strand break or a replication error activate ataxia telangiectasia related (ATR) and check-point kinase 1 (CHK1) proteins (12, 13). These complexes cause a cell cycle arrest by indirect induction of CKI.
From a dynamical point of view, the cell cycle can be modeled either as a series of checkpoints (“domino” model) or as a biochemical oscillator where successive waves of CCN/CDK activity control its progression (14–16). In the following parts, we will use this second model as it recapitulates the rhythmic properties of the cell cycle.
Both cell cycle and molecular clocks display periodic phases of activation and repression from transcriptional to post-translational levels (17). Thus, they can be considered as biological oscillators coexisting in dividing cells.
The Circadian Gating Model
In unicellular organisms, circadian rhythms and cell division are considered as non-independent processes. In particular, the circadian system controls timing of cell division both in prokaryotic and eukaryotic species. This is the case in the cyanobacterium Synechococcus elongatus and in the flagellate alga Euglena gracilis, where the molecular clock operates as a “gating” and only allows cell division at specific circadian phases (18–20). Thus, the circadian oscillator can be viewed as an additional checkpoint for mitosis.
It was tempting to extrapolate this phenomenon to other organisms; although some research was conducted toward demonstration of circadian gating of the cell cycle in mammalian cells, results led to controversial evidence. While a study from 2004 reported that the majority of cell divisions occur in three phases of the circadian cycle (4), two more recent papers describe an absence of cell cycle regulation by the circadian clock (21, 22).
Dynamical Coupling between Clock and Cell Cycle
Faced with these contradictions, we and others have recently investigated the circadian clock-cell cycle connection by quantifying the dynamics of the two oscillators in real time, in single live mammalian cells (23, 24). Both studies used the circadian clock reporter REV-ERBα:VENUS (4). For cell cycle, Bieler et al. scored timing of division. We added a cell cycle reporter system [fluorescent ubiquitination-based cell cycle indicator, FUCCI; (25)] probing cell cycle progression. These fluorescent markers were used to quantitatively determine the properties of each oscillator in single NIH3T3 cells. Timelapse imaging combined to extensive statistical analysis and modeling exposed the dynamical properties of these biological oscillators (Figure 1A).
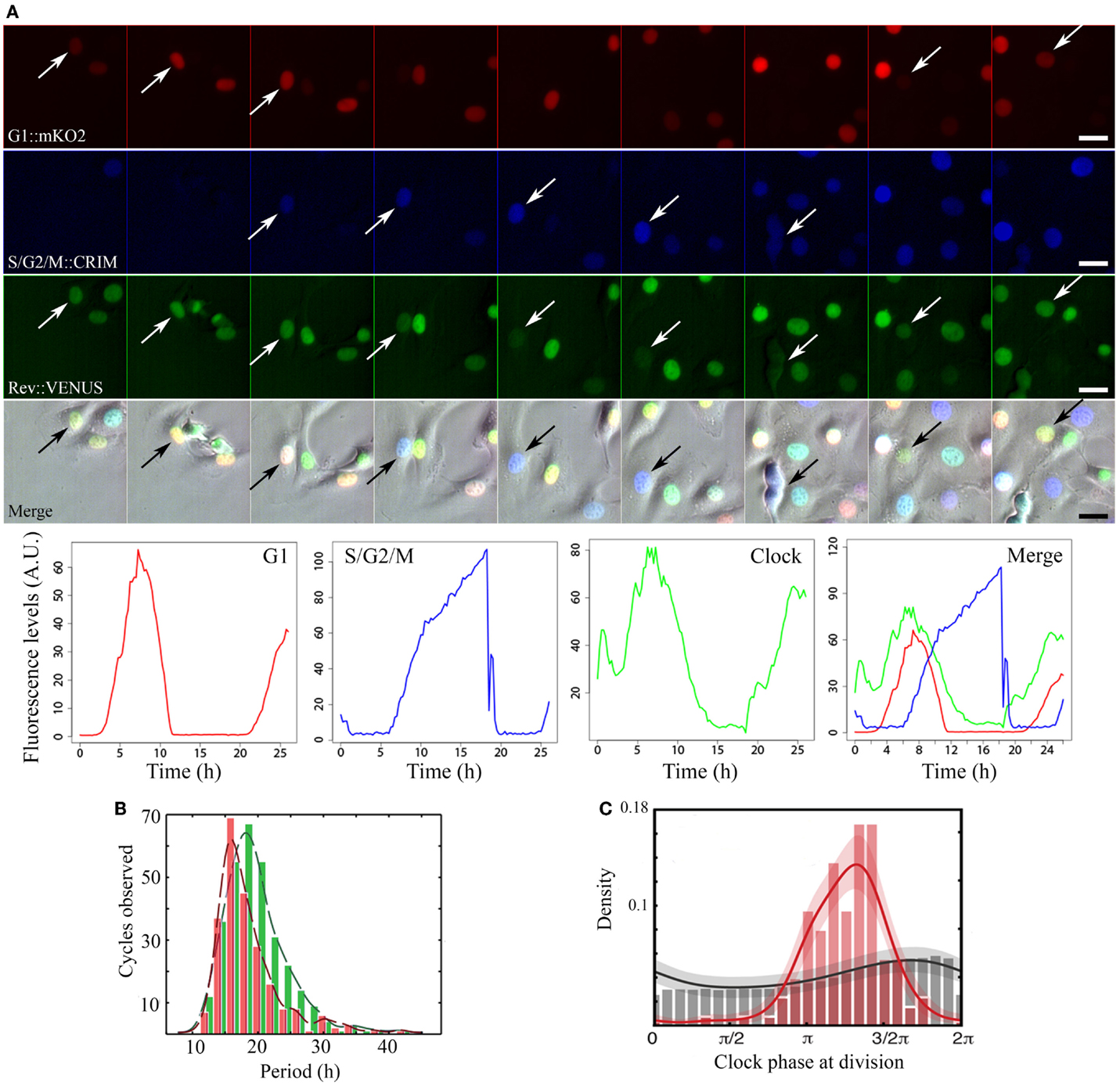
Figure 1. Image acquisition, fluorescence quantification, period, and phase dynamics in non-synchronized NIH3T3-REVERBα:VENUS_FUCCI cells. (A) Time series of a representative single cycle for the various fluorescent reporters and respective quantified traces: from top to bottom: Cell Cycle_G1 (red) = CDT1:mKO2, Cell Cycle_S/G2/M (blue) = GEMININ:E2CRIMSON, Circadian Clock (green) = REVERBα:VENUS, Merge = fluorescent channels combined with the corresponding brightfield image. Arrows point to tracked cell nuclei. Images are 2.5 h apart. Traces at the bottom have been plotted from measured intensities extracted from tracking with the LineageTracker plugin for ImageJ. (B) Histograms showing distribution of periods for both the clock (green) and cell cycle (red) in the whole population. In non-synchronized cells, mean clock period (19.4 ± 0.5 h) is not significantly different from mean cell cycle period: (18.6 ± 0.6 h). (C) Phase histograms for the same cells. Gray histogram and trace show random background densities. Colored histogram and trace show the observed phase of the clock at division. We observe a preferred clock phase for performing cell division (phase locking).
In cells where neither clock nor cell cycle was synchronized by external cues, they appear robustly coupled, with a 1:1 ratio between their respective periods, over a wide range of durations (18–27 h; Figures 1B,C). A clear shortening of the circadian period occurred in dividing cells compared to non-dividing cells thus revealing an influence of cell cycle on the clock. Mathematical analysis and stochastic modeling unambiguously showed that coupling rather than gating governs cell cycle and circadian interaction in NIH3T3 cells. It also revealed that the clock reporter reproducibly peaked about 5 h after cell division. This feature is termed as phase locking (Figure 1C). Changing cell cycle duration impacted on circadian cycles, but 1:1 locking was resilient to such changes (23, 24). Additionally, inhibition of the cell cycle at the G1/S or G2/M transitions lengthened circadian intervals and delayed division phase. Bieler et al. looked at the reverse interaction by changing circadian period. This did not affect cell cycle length but advanced division with respect to circadian phase. The authors thus proposed a unidirectional coupling from the cell cycle to the circadian clock (23).
The above experiments were performed in absence of external cues. However, in vivo cellular clocks are subjected to synchronizing messages (e.g., corticosteroids, temperature). We therefore investigated coupling after a 2 h treatment with dexamethasone, which resets the circadian clock. We observed two distinct dynamical behaviors coexisting within the cell population. Whereas one sub-population kept a 1:1 phase locking, the ratio of cell cycle and clock periods was fixed to 3:2 in the other one (i.e., 3 cell cycles for 2 clock cycles). Moreover, when projecting the timing of mitosis across the whole experiment, we observed a clear clustering of cell division suggesting that the cell cycle was synchronized by physiological cues via the circadian clock. We thus inferred a bidirectional coupling between the two oscillators, supported by mathematical modeling (24).
Although the team of Naef concluded to a unidirectional coupling whereas we concluded to a bidirectional coupling, both studies led us to reject the concept of “gating” of the cell cycle by the clock in mammalian cells, usually put forward without theoretical support (23, 24). In the end, it seems that the cell cycle is capable of impacting on the circadian clock and vice versa, the dominant influence being dependent on the environment of the cell.
Molecular Coupling between Clock and Cell Cycle
Molecular mechanisms underlying progression of the clock and cell cycle have been extensively studied. Yet, little is known about candidates that could underlie coupling between the two oscillators.
Influence from the Circadian Clock on the Cell Cycle
The molecular clock impacts on the cell cycle, by transcriptional control or direct protein–protein interactions. For instance, in G1, the CKI P21 is transcriptionally regulated by REV-ERBα and RORα/γ (26). At G1/S transition, NONO regulates the p16-Ink4A check-point gene in a PER-dependent fashion (27). Transcription of the WEE1 kinase (G2/M transition) is tightly controlled by the CLOCK:BMAL1 dimer (28).
At the post-translational level, CRY modulates CHK1/ATR (G1/S transition checkpoint) by interacting with TIMELESS (TIM) in a time-of-day-dependent manner. PER and TIM also regulate the G2/M transition via interactions with CHK2-ATM (12, 13, 29, 30).
Other clock-controlled cell cycle regulators include known oncogenes (c-Myc, Mdm2, and β-catenin), cyclins (CCND1, B, and A), and tumor suppressor p53. Many key cell cycle regulators, such as Cdk4, Itga6, Wnt3, LHx2, Tcf4, Sox 9, and Smad7 are also directly clock-regulated (31). This multitude of interactions gives a reasonable explanation why loss of coupling between clock and cell cycle may play a key role in carcinogenesis and abnormal growth in vivo (see below).
Influence of the Cell Cycle on the Circadian Clock
Apart from transcription silencing occurring at mitosis (32), which is bound to influence the circadian feedback loops, molecular evidence of cell cycle-dependent control of the circadian clock is sparse. Nevertheless, DNA damage can phase advance cellular and behavioral circadian rhythms in a dose- and time-dependent manner. The underlying mechanism involves ATM-mediated damage signaling, possibly through interaction with the PER and TIM proteins (33).
More recently, an impact of the tumor suppressors P53 and promyelocytic leukemia (PML) proteins on circadian function was proposed: Per2 transcription is repressed by P53, which prevents binding of CLOCK:BMAL1. At the post translational level, PML physically interacts with PER2, and promotes its nuclear localization. These molecular connections are translated into altered circadian behavior (34, 35). As a whole, these results point to a global influence of cell cycle regulation on circadian function.
Among these molecular interactions, only p53 has been found to impact on clock and cell cycle in a bidirectional way. Thus, it may participate in coupling between the two oscillators, which remains to be confirmed.
Cellular Consequences of Coupling in Healthy Cells
A major impact of clock and cell cycle coupling on cell physiology resides in timed mitoses. For instance, about 1/6th of human epidermal cells divide daily, mainly at night (36), which is determined by intrinsic Bmal1 expression (37). Rhythmicity in cell cycle parameters was also found in the hematopoietic and immune systems (spleen, thymus, bone marrow), gastro-intestinal tract (colon, liver), skin, and cornea of rodents and/or humans (38). DNA synthesis and mitosis rhythmicity seem impervious to ablation of adrenals, medulla, pituitary, or even SCN, although sometimes displaying altered characteristics (38–40). Hence, if systemic circadian cues are required to coordinate cell divisions in the whole organism, local clock/cell cycle coupling likely governs rhythmic mitosis at the cellular and tissue levels.
In line with this hypothesis, different populations of epidermal stem cells were found to express clock genes in opposite phases, which results in a differential propensity for activation. Specific disruption of the circadian clock in these cells led to premature epidermal aging, which confirms that local coupling is necessary to ensure tissue integrity (41).
Coupling even manifests itself in a post-mitotic tissue such as the adult brain, where quiescent neural progenitors in the hippocampus show rhythmic proliferation. Per2 and Bmal1 play a critical role in this rhythmic neurogenesis, impacting on cognitive function. Mathematical modeling pointed to clock-driven p21 expression as a trigger for cell cycle progression through regulation of the CCND/CDK4–6 complexes (42).
In the adult liver, only 1% of hepatocytes show rhythmic mitoses under normal conditions. After partial hepatectomy, however, most of the remaining hepatocytes enter cell cycle synchronously, in a clock-dependent manner, which will restore liver mass within few days. In arrhythmic Cry deficient mice, liver regeneration still occurs, although full recovery of liver mass is delayed (28). A more common situation requiring controlled cell division at an adult age is wound repair: after skin incision, wound healing defects were found in mice lacking NONO, a possible link between clock and cell cycle. (27).
Thus, crosstalk between the clock and cell cycle oscillators is required for timed and optimal organization of cell proliferation both in healthy tissue and in times of challenge such as regeneration.
Importance of Coupling in Times of Challenge: From Healthy Cells to Pathology
A major interest of the interplay between the clock and cell cycle is that a dysfunction of either system can lead to diseases such as cancer. Among the hallmarks of cancer, genome instability and mutations in cell cycle genes are a recurring enabling factor (43). Indeed, mutation in Cyclin, Cdk, or Cki genes was found in 90% of human cancers (44). However, evidence is increasing that cancer cells also display a deregulation of the circadian clockwork, which may promote abnormal proliferation (31).
Circadian Disruption and Cell Cycle Deregulation
Although mutations in clock genes are frequent in human cancer cells, it is unclear whether it may be a primary cause of cancer development. Per2 mutant mice display tumor-prone phenotypes and deregulation of various cyclins, proto-oncogenes, and tumor suppressors (45). On the other hand, Cry1/Cry2 KO mice do not show increased cancer development after γ-irradiation (46). Thus, deregulation of the core clock cannot fully account for the observed phenotype in Per2 mutant mice. Additionally, mutation of Bmal1 or Clock does not lead to enhanced cancer development (47, 48). These results are subject to debate, as a more recent study showed that Per1−/− Per2−/−, Cry1−/− Cry2−/−, and Bmal1−/+ mice have increased spontaneous and radiation-induced tumor development (49). So it remains unclear whether a mutation in clock genes is by itself sufficient to trigger cancer development. These observed cancer-prone phenotypes might be due to a non-circadian function of these genes.
If clock gene mutations do not induce cancer per se, systemic environmental disruption of the circadian function may impact on cancer development and cell proliferation. Constant light exposure or ablation of the pineal gland results in perturbation of endocrine rhythms and increase carcinogenesis in liver and mammary glands of rodents (50, 51). In line with these results, circadian disruption following SCN lesions or chronic jetlag increases proliferation of implanted Glasgow osteosarcoma or pancreatic adenocarcinoma (52, 53). Chronic jetlag also promotes tumor development in the liver of mice exposed to the hepatic carcinogen diethylnitrosamine (54).
The latter finding is of particular interest when considering cancer development in humans. As already mentioned, circadian alterations at the molecular level have been found in numerous human cancers. At the epidemiological level, studies involving shift workers have associated circadian disruption with an increased risk of cancer development (31). Since 2007, the International Agency for Research on Cancer even listed “shiftwork that involves circadian disruption” as a “probable carcinogen.”
Cell Cycle Deregulation and Cancer Lead to Circadian Disruption
Many human cancer cell lines (including breast, prostate, colon, liver, and lung), which by definition show abnormal cell cycle progression and proliferation, also display abnormal circadian gene expression (31). In the context of bidirectional interaction between the clock and cell cycle oscillators, it is tricky to decipher which alteration is the cause or consequence. If mutations in cell cycle genes are the primary element, it will in turn impact on the circadian clock. Another possibility is that cell cycle and circadian functions will be synergistically disrupted as a consequence of their coupling.
At the physiological level, recent studies have explored circadian rhythmicity in metastatic colorectal or breast cancer patients (rest activity and/or salivary cortisol rhythms). All studies concluded to large disparities in behavioral and endocrine rhythmicity in these patients, ranging from robust to absent rhythms. Interestingly, the outcome of the disease in patients with damped or abnormal circadian rhythmicity is generally unfavorable, independently of other prognostic factors (38).
Circadian Lifestyle and Slowing Down Tumor Progression: Perspectives of Coupling
Temporal restricted feeding (RF), also known as meal timing, powerfully entrains peripheral clocks (55). It is also capable of restoring circadian rhythms in peripheral tissues of otherwise arrhythmic mice (56). Tumor cells often show abnormal circadian rhythms in LD and ad libitum feeding conditions, but they are not blind to cyclic metabolic cues. Two studies tested the impact of RF on growth rate of implanted Glasgow osteosarcoma or P03 pancreatic adenocarcinoma in mice (57, 58). At the physiological level, this treatment increased the amplitude of body temperature rhythms, known to be a powerful circadian synchronizer (59, 60). Interestingly, tumor size was significantly reduced and survival was prolonged in mice submitted to RF. Thus, reinforcement of host’s circadian rhythms may lead to improved host-mediated tumor control or alteration of the tumor circadian clock, which in the end slows down tumor progression.
Looking at this data with the newly discovered bidirectional coupling between clock and cell cycle, we would like to put forward the possibility that the cell cycle may be synchronized/slowed down through coupling with the circadian clock. Hence, it would be possible to use a known circadian synchronizer such as timed feeding or high-amplitude temperature cycles to re-entrain or reinforce the circadian clock in tumor cells. These cells tend to proliferate at a high rate, with periods shorter than 24 h. Entraining the circadian clock in these cells could slow down cell cycle, through coupling between the two oscillators. This would, in the end, delay tumor progression.
Conclusion and Perspectives
In mammals, the circadian and cell cycle oscillators were long considered as two completely independent entities, or were at best regarded as a circadian clock gating cell cycle progression. These views are seriously challenged by the recently established bidirectionality of clock and cell cycle interactions. At the moment, only P53 has been identified as capable of impinging on both clock and cell cycle, but its role in coupling needs to be confirmed. Given the complexity of these oscillatory circuits, additional candidates likely exist, but remain to be found.
In terms of physiology, the consequences of coupling in healthy cells are obvious. But coupling becomes critical when considering a pathology such as cancer, where circadian and cell cycle phenomena are intertwined. The benefits of chronotherapeutics for cancer treatment is well established (38). They are a consequence of circadian fluctuations in toxicity and efficacy of cancer drugs (and drug metabolism in general). Considering that reinforcing the circadian clock through a change in lifestyle (e.g., meal timing) is capable of slowing down tumor progression, probably through coupling with the cell cycle, we may be able to propose individualized life schedules that will potentiate the effect of chronotherapy, to reduce tumor progression without additional pharmacological intervention. Not to say that reinforcing circadian timing will also alleviate dampening of circadian rhythmicity, which is often a complaint from cancer patients. This long-term perspective may have a high impact on human health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in our laboratories is supported by University Nice Sophia Antipolis, CNRS, INSERM, and University of Warwick, and grants from the ANR (Grants No. ANR-2009-SYSB-002-02, ANR-14-CE09-0011-01 and the “Investments for the Future” LABEX SIGNALIFE program ANR-11-LABX-0028-01), the EPSRC (Grant No. GR/S29256/01) and the BBSRC (Grants No. BB/I004521/1 and BB/F005814/1).
References
1. Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res (1972) 42:201–6. doi: 10.1016/0006-8993(72)90054-6
2. Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci U S A (1972) 69:1583–6. doi:10.1073/pnas.69.6.1583
3. Buhr ED, Takahashi JS. Molecular components of the mammalian circadian clock. Handb Exp Pharmacol (2013):3–27. doi:10.1007/978-3-642-25950-0_1
4. Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell (2004) 119:693–705. doi:10.1016/j.cell.2004.11.015
5. Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol (2004) 14:2289–95. doi:10.1016/j.cub.2004.11.057
6. Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A (1974) 71:1286–90. doi:10.1073/pnas.71.4.1286
7. Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle (2002) 1:103–10. doi:10.4161/cc.1.2.108
8. Schafer KA. The cell cycle: a review. Vet Pathol (1998) 35:461–78. doi:10.1177/030098589803500601
9. Nurse P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem (2002) 3:596–603. doi:10.1002/1439-7633(20020703)3:7<596::AID-CBIC596>3.0.CO;2-U
10. Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci (2005) 30:630–41. doi:10.1016/j.tibs.2005.09.005
12. Yang X, Wood PA, Hrushesky WJ. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J Biol Chem (2010) 285:3030–4. doi:10.1074/jbc.M109.050237
13. Kang TH, Leem SH. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res (2014) 42:4427–34. doi:10.1093/nar/gku094
14. Goldbeter A. A minimal cascade model for the mitotic oscillator involving cyclin and cdc2 kinase. Proc Natl Acad Sci U S A (1991) 88:9107–11. doi:10.1073/pnas.88.20.9107
15. Tyson JJ. Modeling the cell division cycle: cdc2 and cyclin interactions. Proc Natl Acad Sci U S A (1991) 88:7328–32. doi:10.1073/pnas.88.16.7328
16. Novak B, Tyson JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci (1993) 106(Pt 4):1153–68.
17. Hunt T, Sassone-Corsi P. Riding tandem: circadian clocks and the cell cycle. Cell (2007) 129:461–4. doi:10.1016/j.cell.2007.04.015
18. Mori T, Johnson CH. Circadian control of cell division in unicellular organisms. Prog Cell Cycle Res (2000) 4:185–92. doi:10.1007/978-1-4615-4253-7_16
19. Bolige A, Hagiwara SY, Zhang Y, Goto K. Circadian G2 arrest as related to circadian gating of cell population growth in Euglena. Plant Cell Physiol (2005) 46:931–6. doi:10.1093/pcp/pci100
20. Dong G, Kim YI, Golden SS. Simplicity and complexity in the cyanobacterial circadian clock mechanism. Curr Opin Genet Dev (2010) 20:619–25. doi:10.1016/j.gde.2010.09.002
21. Pendergast JS, Yeom M, Reyes BA, Ohmiya Y, Yamazaki S. Disconnected circadian and cell cycles in a tumor-driven cell line. Commun Integr Biol (2010) 3:536–9. doi:10.4161/cib.3.6.12841
22. Yeom M, Pendergast JS, Ohmiya Y, Yamazaki S. Circadian-independent cell mitosis in immortalized fibroblasts. Proc Natl Acad Sci U S A (2010) 107:9665–70. doi:10.1073/pnas.0914078107
23. Bieler J, Cannavo R, Gustafson K, Gobet C, Gatfield D, Naef F. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol Syst Biol (2014) 10:739. doi:10.15252/msb.20145218
24. Feillet C, Krusche P, Tamanini F, Janssens RC, Downey MJ, Martin P, et al. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc Natl Acad Sci U S A (2014) 111:9828–33. doi:10.1073/pnas.1320474111
25. Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell (2008) 132:487–98. doi:10.1016/j.cell.2007.12.033
26. Gréchez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem (2008) 283:4535–42. doi:10.1074/jbc.M705576200
27. Kowalska E, Ripperger JA, Hoegger DC, Bruegger P, Buch T, Birchler T, et al. NONO couples the circadian clock to the cell cycle. Proc Natl Acad Sci U S A (2013) 110:1592–9. doi:10.1073/pnas.1213317110
28. Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science (2003) 302:255–9. doi:10.1126/science.1086271
29. Unsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol (2005) 25:3109–16. doi:10.1128/MCB.25.8.3109-3116.2005
30. Kondratov RV, Antoch MP. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol (2007) 17:311–7. doi:10.1016/j.tcb.2007.07.001
31. Fu L, Kettner NM. The circadian clock in cancer development and therapy. Prog Mol Biol Transl Sci (2013) 119:221–82. doi:10.1016/B978-0-12-396971-2.00009-9
32. Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends Biochem Sci (1997) 22:197–202. doi:10.1016/S0968-0004(97)01045-1
33. Oklejewicz M, Destici E, Tamanini F, Hut RA, Janssens R, Van Der Horst GT. Phase resetting of the mammalian circadian clock by DNA damage. Curr Biol (2008) 18:286–91. doi:10.1016/j.cub.2008.01.047
34. Miki T, Xu Z, Chen-Goodspeed M, Liu M, Van Oort-Jansen A, Rea MA, et al. PML regulates PER2 nucrlear localization and circadian function. EMBO J (2012) 31:1427–39. doi:10.1038/emboj.2012.1
35. Miki T, Matsumoto T, Zhao Z, Lee CC. p53 regulates Period2 expression and the circadian clock. Nat Commun (2013) 4:2444. doi:10.1038/ncomms3444
36. Scheving LE. Mitotic activity in the human epidermis. Anat Rec (1959) 135:7–19. doi:10.1002/ar.1091350103
37. Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, et al. Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc Natl Acad Sci U S A (2012) 109:11758–63. doi:10.1073/pnas.1209592109
38. Lévi F, Filipski E, Iurisci I, Li XM, Innominato P. Cross-talks between circadian timing system and cell division cycle determine cancer biology and therapeutics. Cold Spring Harb Symp Quant Biol (2007) 72:465–75. doi:10.1101/sqb.2007.72.030
39. Scheving LE, Tsai TH, Powell EW, Pasley JN, Halberg F, Dunn J. Bilateral lesions of suprachiasmatic nuclei affect circadian rhythms in [3H]-thymidine incorporation into deoxyribonucleic acid in mouse intestinal tract, mitotic index of corneal epithelium, and serum corticosterone. Anat Rec (1983) 205:239–49. doi:10.1002/ar.1092050302
40. Filipski E, King VM, Etienne MC, Li X, Claustrat B, Granda TG, et al. Persistent twenty-four hour changes in liver and bone marrow despite suprachiasmatic nuclei ablation in mice. Am J Physiol Regul Integr Comp Physiol (2004) 287:R844–51. doi:10.1152/ajpregu.00085.2004
41. Janich P, Pascual G, Merlos-Suárez A, Batlle E, Ripperger J, Albrecht U, et al. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature (2011) 480:209–14. doi:10.1038/nature10649
42. Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kærn M, Cheng HY. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep (2013) 5:961–73. doi:10.1016/j.celrep.2013.10.037
43. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
44. Bonelli P, Tuccillo FM, Borrelli A, Schiattarella A, Buonaguro FM. CDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivity. Biomed Res Int (2014) 2014:361020. doi:10.1155/2014/361020
45. Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell (2002) 111:41–50. doi:10.1016/S0092-8674(02)01223-0
46. Gauger MA, Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res (2005) 65:6828–34. doi:10.1158/0008-5472.CAN-05-1119
47. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev (2006) 20:1868–73. doi:10.1101/gad.1432206
48. Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, et al. Disruption of the circadian clock due to the clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle (2008) 7:1197–204. doi:10.4161/cc.7.9.5886
49. Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One (2010) 5:e10995. doi:10.1371/journal.pone.0010995
50. Shah PN, Mhatre MC, Kothari LS. Effect of melatonin on mammary carcinogenesis in intact and pinealectomized rats in varying photoperiods. Cancer Res (1984) 44:3403–7.
51. Van Den Heiligenberg S, Deprés-Brummer P, Barbason H, Claustrat B, Reynes M, Lévi F. The tumor promoting effect of constant light exposure on diethylnitrosamine-induced hepatocarcinogenesis in rats. Life Sci (1999) 64:2523–34. doi:10.1016/S0024-3205(99)00210-6
52. Filipski E, King VM, Li X, Granda TG, Mormont MC, Liu X, et al. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst (2002) 94:690–7. doi:10.1093/jnci/94.9.690
53. Filipski E, Delaunay F, King VM, Wu MW, Claustrat B, Gréchez-Cassiau A, et al. Effects of chronic jet lag on tumor progression in mice. Cancer Res (2004) 64:7879–85. doi:10.1158/0008-5472.CAN-04-0674
54. Filipski E, Subramanian P, Carrière J, Guettier C, Barbason H, Lévi F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat Res (2009) 680:95–105. doi:10.1016/j.mrgentox.2009.10.002
55. Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev (2000) 14:2950–61. doi:10.1101/gad.183500
56. Vollmers C, Gill S, Ditacchio L, Pulivarthy SR, Le HD, Panda S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci U S A (2009) 106:21453–8. doi:10.1073/pnas.0909591106
57. Wu MW, Li XM, Xian LJ, Lévi F. Effects of meal timing on tumor progression in mice. Life Sci (2004) 75:1181–93. doi:10.1016/j.lfs.2004.02.014
58. Li XM, Delaunay F, Dulong S, Claustrat B, Zampera S, Fujii Y, et al. Cancer inhibition through circadian reprogramming of tumor transcriptome with meal timing. Cancer Res (2010) 70:3351–60. doi:10.1158/0008-5472.CAN-09-4235
59. Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science (2010) 330:379–85. doi:10.1126/science.1195262
Keywords: circadian desynchrony, cell cycle, coupling, cancer, mathematical modeling
Citation: Feillet C, van der Horst GTJ, Levi F, Rand DA and Delaunay F (2015) Coupling between the circadian clock and cell cycle oscillators: implication for healthy cells and malignant growth. Front. Neurol. 6:96. doi: 10.3389/fneur.2015.00096
Received: 20 February 2015; Paper pending published: 25 March 2015;
Accepted: 20 April 2015; Published: 11 May 2015
Edited by:
Urs Albrecht, University of Fribourg, SwitzerlandReviewed by:
Steven Brown, University of Zürich, SwitzerlandGeorg A. Bjarnason, Sunnybrook Odette Cancer Centre, Canada
Copyright: © 2015 Feillet, van der Horst, Levi, Rand and Delaunay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franck Delaunay, Circadian Systems Biology, CNRS, INSERM, Institut de Biologie Valrose, Université Nice Sophia Antipolis, 28 Avenue de Valrose, Nice Cedex 2 06108, France,ZnJhbmNrLmRlbGF1bmF5QHVuaWNlLmZy