 Annamaria Lanzillotta1
Annamaria Lanzillotta1 Vanessa Porrini1,2
Vanessa Porrini1,2 Arianna Bellucci1
Arianna Bellucci1 Marina Benarese1
Marina Benarese1 Caterina Branca1
Caterina Branca1 Edoardo Parrella1
Edoardo Parrella1 Pier Franco Spano1,2
Pier Franco Spano1,2 Marina Pizzi1,2*
Marina Pizzi1,2*- 1Department of Molecular and Translational Medicine, National Institute of Neuroscience, University of Brescia, Brescia, Italy
- 2IRCCS, San Camillo Hospital, Venice, Italy
NF-κB factors are cardinal transcriptional regulators of inflammation and apoptosis, involved in the brain programing of systemic aging and in brain damage. The composition of NF-κB active dimers and epigenetic mechanisms modulating histone acetylation, finely condition neuronal resilience to brain insults. In stroke models, the activation of NF-κB/c-Rel promotes neuroprotective effects by transcription of specific anti-apoptotic genes. Conversely, aberrant activation of NF-κB/RelA showing reduced level of total acetylation, but site-specific acetylation on lysine 310, triggers the expression of pro-apoptotic genes. Constitutive knockout of c-Rel shatters the resilience of substantia nigra (SN) dopaminergic (DA) neurons to aging and induces a parkinsonian like pathology in mice. c-rel−/− mice show increased level of aberrantly acetylated RelA in the basal ganglia, neuroinflammation, accumulation of alpha-synuclein, and iron. Moreover, they develop motor deficits responsive to l-DOPA treatment and associated with loss of DA neurons in the SN. Here, we discuss the effect of unbalanced activation of RelA and c-Rel during aging and propose novel challenges for the development of therapeutic strategies in neurodegenerative diseases.
Introduction
In the central nervous system, NF-κB transcription factor acts as a pleiotropic regulator of target genes controlling physiological function (1) as well as pathological processes associated with neurodegeneration (2, 3).
The NF-κB family of transcription factors is composed by five different members that are p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2). The RelA subunit, composing the activated p50/RelA dimer, and its post-transcriptional modifications play a pivotal role in the onset of neurodegenerative processes triggered by ischemic insults as well as glutamate or beta-amyloid toxicity (4–8). The c-Rel subunit within activated NF-κB dimers counteracts the ischemic injury acting as an innate mechanism of neuroprotection (9). The c-Rel factor is reduced in neurons exposed to oxygen–glucose deprivation (OGD), and its over-expression can limit the cell death. Moreover, the deficiency of c-Rel induces an age-related behavioral parkinsonism in mice, with degeneration of nigral dopaminergic (DA) neurons and development of a Parkinson’s disease (PD)-like neuropathology (10). Recent evidence has shown that activation of NF-κB drives the systemic and brain aging process in mice (11, 12). Notably, Tilstra and colleagues demonstrated that RelA is the most contributing subunit in degenerative changes associated with senescence in a progeroid mice model (13).
We propose that while RelA activation accompanies normal brain aging, a misbalance between RelA and c-Rel might drive pathological aging by affecting the survival of substantia nigra (SN) DA cell and turning old mice into a parkinsonian phenotype.
RelA and c-Rel: Two Opposing Regulators of Neuronal Resilience to Brain Ischemia
In the central nervous system, NF-κB factors are key players of a number of physiological processes such as neurogenesis (14), neuritogenesis (15), synaptic plasticity, learning, and memory (16–18). In recent years, a body of data has shown that NF-κB dysregulation participates to neurodegenerative mechanisms that occur in brain exposed to trauma or ischemia (19, 20), as well as in the brain of patients affected by PD (21, 22) and Alzheimer’s disease (23).
The neuronal response to external stimuli relies on a differential activation of NF-κB dimers. We found that targeting RelA or c-Rel expression by antisense oligonucleotides (5) or siRNAs (6, 9) produces opposite effects on neuron survival. While over-activated p50/RelA dimers contribute to the apoptotic program, the c-Rel containing dimers increase the resilience of injured neuronal cells (Figure 1). Neurotoxic stimuli, such as ischemia (4, 9), glutamate (5), β-amyloid (7, 24), or 1-methyl-4-phenylpyridinium (MPP+) (25, 26), induce p50/RelA dimers activation and the transcription of a panel of pro-apoptotic genes (4). Conversely, c-Rel-containing dimers are responsible for anti-apoptotic gene expression by signals promoting neuroprotection in diverse neurotoxic settings, such as S100B in models of NMDA-mediated excitotoxicity (27), agonists at mGlu5 receptors against β-amyloid- (7) and MPP+ toxicity (25) or adipocyte-derived hormone leptin in neurons exposed to OGD (28). Over-expression of c-Rel in cultured neurons promotes anti-apoptotic effects by inducing the transcription of manganese superoxide dismutase (MnSOD) and Bcl-xL (7, 29, 30). c-Rel overabundance also limits the generation of reactive oxygen species (ROS) by inducing transcription of the mitochondrial uncoupling proteins 4 (UCP4) (31), a brain-specific mitochondrial ion channel producing mild reduction of mitochondrial membrane potential and neuroprotection (32). Moreover, c-Rel can control the expression of baculoviral IAP repeat-containing protein 3 (BIRC3), named also as cIAP2, an anti-apoptotic E3 ligase, which can modulate the receptor interacting protein 1 (RIP1) activity by ubiquitination (33). Depending on its ubiquitination status, RIP1 can dictates if tumor necrosis factor (TNF)-α induces cell survival (and inflammation), or cell death pathways (34). Ubiquitinated RIP1 can recruit other kinases and finally induce NF-κB-mediated transcription of prosurvival and pro-inflammatory genes (TNF-α-dependent NF-κB activation) (35).
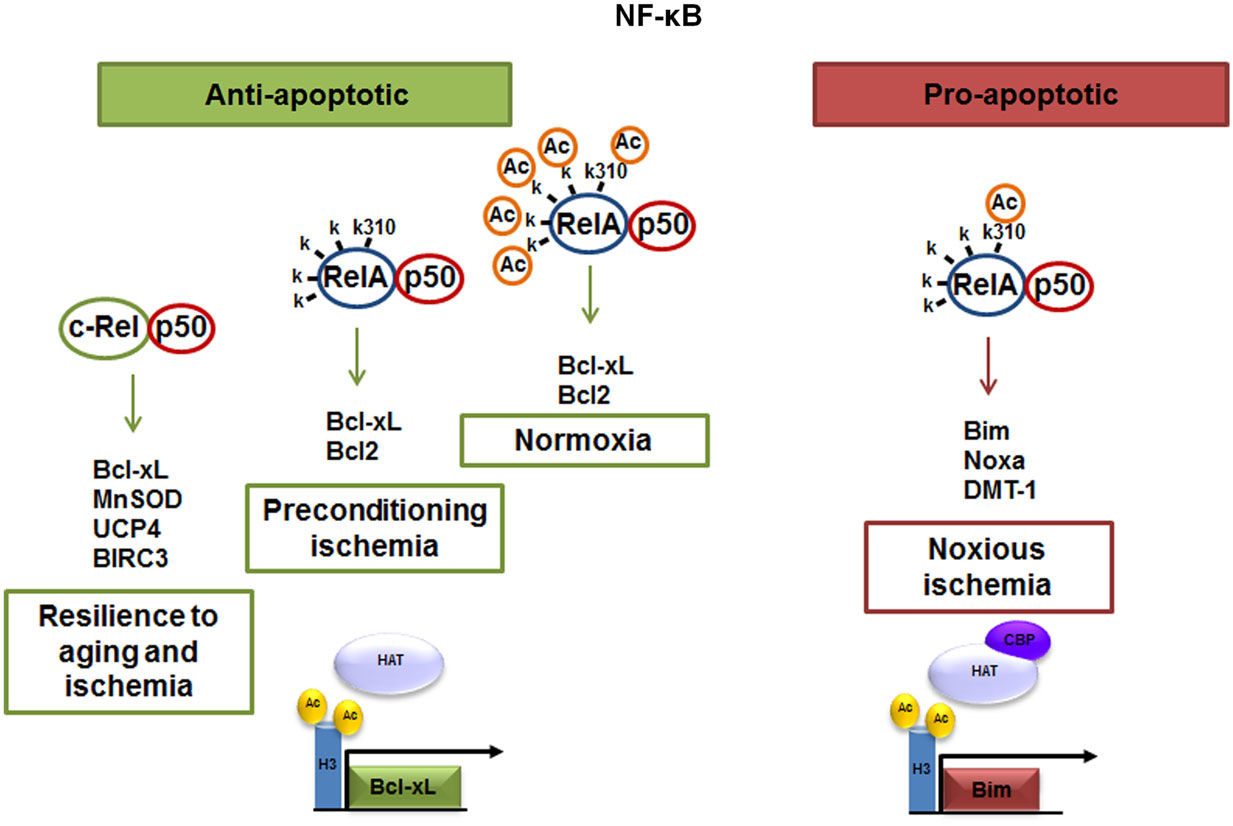
Figure 1. The p50/RelA and p50/c-Rel dimers regulate neuronal survival. Anti-apoptotic effects of NF-κB can be mediated by c-Rel containing dimers, which enhance neuronal resilience to oxidative stress by inducing Bcl-xL, MnSOD, UCP4, and BIRC3 expression. NF-κB anti-apoptotic effects can also be mediated by p50/RelA during preconditioning ischemia through the transcription of Bcl-xL. The pro-apoptotic effect elicited by NF-κB p50/RelA dimer in noxious ischemia is dependent on changes in the RelA acetylation state. A lower grade of RelA general acetylation, respect to control condition, but a site-specific acetylation on Lys 310 residue addresses the NF-κB binding toward Bim, Noxa, and DMT1 promoter.
The elucidation of the dual effects of NF-κB activation on neuron survival was more evident in studies of severe brain ischemia. The activation of p50/RelA rapidly occurs in neurons and glial cells and has been implicated in pathogenesis of post-ischemic injury (36– 38). In brain ischemic tissue of mice subjected to permanent middle cerebral artery occlusion (MCAO) and in primary cortical neurons exposed to OGD, NF-κB followed a similar pattern of activation (8, 39) characterized by increased nuclear translocation of p50/RelA dimer (4, 36) and decreased translocation of c-Rel-containing dimers (9). In these conditions, NF-κB activity was associated with an unbalance expression of pro-apoptotic RelA target genes, i.e., an increased expression of the pro-apoptotic members of Bcl-2 family genes (4) and reduced level of the anti-apoptotic member Bcl-xL (9, 40). During brain ischemia, RelA induced the expression of the 1B isoform of the divalent metal transporter-1 (1B/DMT1), the membrane carrier responsible for iron accumulation and brain damage after injury (41). The RelA-induced 1B/DMT1 expression acted as an upstream mechanism responsible for iron accumulation and contributing to neuronal cell death. While the over-expression of RelA increased cell death, the over-expression of c-Rel prevented neuronal loss in cortical neurons exposed to OGD, by increasing the transcription of Bcl-xL gene (9, 39). Knocking-down c-Rel expression exacerbated neuronal susceptibility to OGD-mediated damage. Under brain ischemia, mice deficient for the c-Rel factor appeared insensitive to neuroprotective activity of leptin, a c-Rel inducer capable to limit cortical damage in wild-type mice (24, 28). These data strongly suggested that inhibition of c-Rel-containing dimers and activation of p50/RelA are key events in the pathogenesis of post-ischemic brain injury. In spite of these premises, p50/RelA activation per se appeared to be insufficient to drive pro-apoptotic transcription during brain ischemia. A similar pattern of p50/RelA nuclear translocation was found in mice exposed to a brief preconditioning ischemia (8) generating brain tolerance to a subsequent lethal ischemic injury (42). In neuronal cells, likewise in tumor cells, gene targeting by p50/RelA is finely regulated by post-transcriptional modification of RelA subunit, such as phosphorylation and acetylation (43). These modifications shape the strength and specificity of the NF-κB–DNA binding and final transcriptional responses. On this line, we investigated whether the activation of the p50/RelA dimer, in preconditioning or in lethal ischemia, differs in the RelA acetylation state (44).
RelA Acetylation is a Dynamic Process Which Tunes p50/RelA-Mediated Pro-Apoptotic Transcription in Brain Ischemia and is Modulated by Epigenetic Drugs
Acetylation is the key post-translational modification of histones that controls the accessibility of chromatin to the transcriptional machinery and plays an essential role in gene activation (45). Lysine acetylation is reversible and controlled by the opposing activities of histone acetyltransferase (HAT) and histone deacetylase (HDAC).
Besides histones, diverse non-histone proteins, including transcription factors NF-κB, are modified by HAT co-activators and HDACs (46). Acetylation of RelA on specific lysine residues (K122, 123, 218, 221, and 310) is a dynamic process that differently affects the RelA interaction with IκBα, the DNA-binding ability and the transcriptional activity of the protein (43, 47).
Members of class I HDACs, particularly HDAC1, HDAC2, and HDAC3, inhibited by vorinostat and entinostat (MS-275) are the most responsible for the general deacetylation of RelA (43, 48). Conversely, sirtuin 1 (SIRT1), an atypical class III HDAC that requires nicotinamide adenosine dinucleotide (NAD+) rather than zinc as a co-factor (49), activated by resveratrol, selectively deacetylates RelA at lysine 310 (K310) residue (8, 50).
Our studies have shown that mechanisms affecting the acetylation state of RelA might discriminate between protective and neurotoxic activation of NF-κB during ischemia (8). Protective ischemic preconditioning and harmful ischemia induced similar levels of p50/RelA activation, but only the ischemic injury induced an atypical RelA acetylation. The RelA that translocated to the nucleus in primary cortical neurons exposed to preconditioning OGD, or in cortices of mice subjected to preconditioning MCAO, showed a general deacetylation that paralleled the deacetylation on the K310 residue. Conversely, RelA activated in neurons exposed to lethal OGD or in cortices of mice subjected to noxious ischemia displayed a general deacetylation, but site-specific acetylation on the K310 residue. This suggested that a mismatch between general and “site-specific” (K310) acetylation of RelA – where total acetylation decreases and K310 acetylation increases – could be responsible for pro-apoptotic transcription in ischemic conditions (8). The relevance of K310 acetylation to RelA-mediated effects during ischemia was demonstrated by mutagenesis analysis. The substitution of lysine with arginine at the RelA 310 residue impaired the acetylation at this site. In cells expressing the mutated RelA subunit, the OGD-mediated DMT1 transcription and the cell damage were totally prevented (8, 41). By undergoing such aberrant acetylation, RelA detached from the anti-apoptotic Bcl-xL promoter to bind the pro-apoptotic Bim promoter (44). In addition to changing the acetylation state of RelA, lethal ischemia produced a significant reduction of H3 histone acetylation (44), in line with previous evidence (51).
Prompted by these findings and in order to correct altered acetylation of RelA and histones after brain ischemia, we studied the association of the specific class I HDAC inhibitor MS-275 (52), and resveratrol (53). MS-275 is a synthetic benzamide derivative that currently is under clinical evaluation for cancer therapy (54). MS-275 has been shown to inhibit HDAC 1-3 with excellent pharmacokinetic properties (52).
Resveratrol is a polyphenol with multiple activities, including anti-oxidant, anti-tumorigenic, and neuroprotective activity (53, 55). In various models of brain ischemia, resveratrol delayed axonal degeneration after injury and mitigated the formation of free radical species as well as mitochondria-mediated apoptosis (56–59).
Widely known mechanisms of resveratrol action include the activation of the longevity factors SIRT1 (60) and AMP-activated kinase (AMPK), a serine–threonine kinase that acts as a key metabolic and stress sensor/effector (61). We found that treatments with either MS-275 or resveratrol in the post-ischemic period of mice subjected to MCAO decreased the infarct volume and displayed a significant neuroprotective activity in cortical neurons exposed to OGD (44). What’s more, we showed that the combination of MS-275 and resveratrol at sub-threshold doses elicited a synergistic effect leading to maximal neuroprotection in both the animal and the cellular models of brain ischemia. MS-275 at the highest concentration tested, 1 μM, increased acetylation of H3 histones on K9/18 residues in neurons exposed to OGD. Resveratrol, unable to modify per se the H3 acetylation, produced a synergistic acetylation of H3 K9/18 when used in combination with MS-275.
Notably, the synergistic effect produced by co-administration of low doses of MS-275 (0, 1 μM) and resveratrol (3 μM) was sustained by AMPK activation by resveratrol. This could be ascribed to the fact that AMPK can activate many catabolic pathways to produce ATP and acetyl-CoA (62), the fundamental co-factor for HAT activity. AMPK has also been found to indirectly support the resveratrol-dependent SIRT1 induction by inducing NAD+ generation (61). As a consequence of AMPK-mediated enhancement of HAT and SIRT1 activity, the combination of MS-275 and resveratrol reversed the mismatch of RelA acetylation state in neurons exposed to OGD by, respectively, enhancing the RelA general acetylation and by reducing the acetylation at the K310 residue. The neuroprotective effect and transcription of anti-apoptotic genes observed following the treatment with the drug combination appeared closely related to the restored optimal RelA acetylation state (8, 63). The protective and transcriptional effects produced by resveratrol and MS-275 in cortical neurons were entirely reproduced in the mouse MCAO model. The combination of sub-threshold doses of the drugs, administered during the reperfusion period, elicited a synergistic effect that limited the cerebral infarct volume and the subsequent neurological deficits. MS-275 and resveratrol in combination showed a long-lasting efficacy as the beneficial effects were still evident 72 h after the injury. Moreover, they displayed a wide therapeutic window as their efficacy was evident when administered within 7 h after the ischemic onset. The treatment induced a transcriptional switch from pro- to anti-apoptotic genes. The RelA binding shifted from the Bim to the Bcl-xL promoter and the acetylation of associated histones changed accordingly. H3 acetylation decreased at the Bim and increased at the Bcl-xL gene.
Recently, we evaluated the acetylation of histone residues at the brain-derived neurotrophic factor (BDNF) IV promoter in primary mouse cortical neurons exposed to OGD and treated with the synergistic combination of MS-275 and resveratrol (64).
We focused on promoter IV, which is known to be important for synaptic plasticity, both during neuronal development and in the adult brain (65). In the cortex, the promoter IV-dependent BDNF transcription accounts for the majority of the neuronal activity-induced BDNF expression (66, 67). Several studies have proposed BDNF as possible mediators of the beneficial effects of HDAC inhibitors in nervous system disorders (68–70). A ChIP analysis in cortical neurons showed that histones at the BDNF promoter IV were deacetylated after OGD exposure. Treatment in the post-OGD period with the combination of MS-275 and resveratrol significantly increased acetylation at H3K9/18 and H4K12 histones (64). These histone modifications may act cooperatively and possibly in parallel to other histone modifications to increase BDNF expression.
It can be proposed that neuroprotection elicited by MS-275 and resveratrol treatment is also closely related to modulation of BDNF expression and may improve neurologic function by enhancing neuronal plasticity.
All together, these data provide evidence that a pharmacological intervention targeting the epigenetic machinery can represents a promising strategy to limit post-ischemic injury with an extended therapeutic window.
c-Rel Deficiency Causes a Progressive Late-Onset Parkinsonism in Mice
Following the evidence that RelA and c-Rel play opposing effects on neuron survival, and prevalence of p50/RelA activation versus p50/c-Rel triggers apoptotic cell death in brain ischemia (9), we tested whether a constitutive defect in c-Rel protein might affect the brain aging. Behavioral and pathological analyses of c-Rel knockout mice were performed at 2, 12, and 18 months of age and c-rel−/− mice showed to develop a PD-like syndrome and pathology with aging (10). Besides c-Rel subunit, other NF-κB subunits have been previously investigated for possible correlation with the onset of a parkinsonian pathology. Increased levels of RelA have been detected in the brains of MPTP-intoxicated mice (21) as well as in the brain of subjects affected by PD (21, 22, 71). This increase was evident both in neuronal and glial cells of the SN, suggesting a role of RelA activation in neuronal cell loss and neuroinflammatory response associated with PD progression. The role of the other NF-κB subunits in PD remains unclear. The p50−/− mice treated with MPTP did not behave differently from wild-type mice (72), suggesting no, or only minor, role for p50 in the regulation of SN neuron resilience.
In 18-month-old c-rel−/− mice, the analysis of tyrosine hydroxylase (TH)-positive cells of the SN pars compacta (SNc) revealed a loss of DA neurons that paralleled the total loss of Nissl-stained cells. No significant change in the estimated number of DA cells was evident in 2- or 12-month-old c-rel−/− mice compared to age-matched controls. Notably, the loss of SNc DA neurons was associated with a decrease of TH-positive fibers and reduction of dopamine transporter (DAT) and dopamine content in the striatum. The 18-month-old c-rel−/− mice displayed no significant degeneration in the other brain areas examined, the nucleus basalis magnocellularis and the medial septal area, or in the ventral tegmental area that is generally spared in PD. Additional examination of SNc in aged c-rel−/− mice revealed a marked immunoreactivity for α-synuclein, the main protein constituent of Lewy bodies and Lewy neuritis and the key pathological feature of PD (73). Of note, fibrillary aggregated α-synuclein, detected by Thioflavin-S labeling, was present in the spared DA neurons of the SNc. Accumulation of insoluble α-synuclein in the mesencephalon was confirmed by the presence of a monomeric α-synuclein in the urea/SDS extracts used to solubilize the insoluble fraction. Neuroinflammation characterized by chronic microglial reactivity, RelA activation, and iron accumulation are also important features of the PD neuropathology (74, 75). As observed in PD brain (76), SNc and striatum of aged c-rel−/− mice showed marked signs of microglia activation with increased number, swollen cell bodies, and thick processes of CD11b-positive cells. Both SNc and striatum of aged c-rel−/− mice displayed elevated levels of iron and increase of the iron transporter DMT1 that, as mentioned above, is a transcriptional target of aberrantly acetylated RelA. Preliminary investigation on NF-κB in striatal extracts of 18-month-old c-rel−/− mice confirmed, indeed, the presence of aberrantly acetylated RelA as reported in Figure 2. The immunoprecipitated RelA showed reduced level of general acetylation associated with increased site-specific acetylation at the K310 residue. These changes occurred without any significant variation in the cellular amount of RelA. This finding strongly suggests that a misbalance between c-Rel and RelA can evolve during aging in c-rel−/− mice to produce changes in RelA acetylation, microglia activation, and neuronal apoptosis.
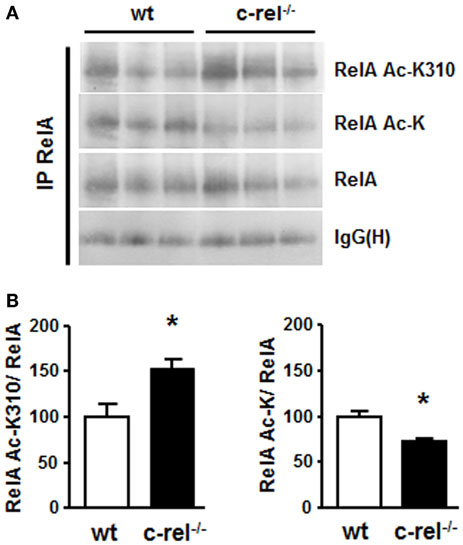
Figure 2. RelA acetylation in the striatum of c-rel−/− and wt mice. (A) Representative picture of the immunoprecipitation analysis of RelA acetylation in total proteins of caudatus putamen. RelA acetylation at K310 residue increased, while total RelA acetylation was reduced in striatal total extracts of 18-month-old c-rel−/− mice. No significant change was detected in the total RelA content. (B) Values from densitometry analysis of immunoblots are expressed as a percentage of the wt value. The signal given by IgG(H) is used as a control for the quality of the immunoprecipitation. Bars depict the mean ± SEM (n = 4 animals per group), *p < 0.01 versus wt value.
Because inflammatory and neurotoxic activation of microglia have been also reported to be triggered by acetyl-RelA (K310) (77), it is feasible that microglia activation participate to the neurodegenerative process in c-rel−/− mice. The extensive analysis of the neuroinflammatory profile of c-rel−/− mice along with disease progression will reveal the exact entity of this inflammatory process and the specific participation of innate and adaptive immunity.
The neurochemical changes observed in aged c-rel−/− mice were also accompanied by the onset of motor deficits. A significant impairment in spontaneous motor activity was evident in c-rel−/− mice at 18 months, but not in younger mice as previously shown (17, 18). Indeed, either monitored for 1 h or six consecutive days to avoid stress-related bias, 18-month-old c-rel−/− mice displayed a lower locomotor activity. Furthermore, the gaiting analysis supported the presence of a locomotor dysfunction related to bradykinesia and rigidity. Noteworthy, the treatment with l-DOPA plus benserazide, a cocktail that is considered the gold standard for PD therapy, totally reversed the locomotor deficits and normalized most of the gaiting parameters.
Despite these findings, how the constitutive c-Rel deficiency can specifically affect DA neurons of SNc is still an open question (10). The selective vulnerability of SNc neurons in PD has been attributed to the peculiar “energy-demanding” physiology of these cells (78), which display enormous axonal field and impressive number of synapses for each axon (79). Moreover, during their pacemaking activity, SNc DA neurons, but not the ventral tegmental area neurons, generate autonomous action potentials by unusual engaging of L-type Ca2+ channels, which require subsequent activation of ATP-dependent Ca2+ pumps to maintain Ca2+ homeostasis (80). The energy production by mitochondria and endoplasmic reticulum in SNc DA neurons associates with the generation of large amounts of ROS that are constantly neutralized by anti-oxidant systems, including SODs catalases, glutathione peroxidase, and UCP4 and UCP5 (81). It can be inferred that in the absence of c-Rel a reduced expression of UCP4 (31) and MnSOD (7, 29) might enhance ROS accumulation during aging in SNc neurons (82), and synergize with reduced expression of anti-apoptotic Bcl-xL (9, 30) to affect neuronal resilience. Also, it is conceivable that mitochondria impairment associated with c-Rel deficiency may switch the acetylation state of RelA during aging to elevate Bim, DMT1, and iron (75) as well as the intracellular levels of α-synuclein (83). In turn, these events lead to α-synuclein aggregation (83), microglia activation, and neuronal damage (84). All together, these findings point to a role of c-Rel in the regulation of SNc susceptibility to aging.
Finally, latest results (unpublished results) indicate that at a premotor phase (7–12 months) when no loss of SNc DA neurons is evident yet, c-rel−/− mice display olfactory deficits, gut dysfunctions, and reduced DAT immunoreactivity in the striatum. This disease progression mimics the pathological and clinical progression observed in PD patients that at premotor stage of the disease show constipation, hyposmia (85), and reduced DAT imaging by PET or SPECT scan. These findings further strengthen the notion that c-rel−/− mice represent an innovative disease model suitable both for studies aimed at dissecting the mechanisms of PD onset and to test novel therapeutic approaches for intervention at the premotor stages of the disorder.
Conclusion
Although NF-κB factors are transcriptional regulators of inflammation and apoptosis, their relevance in aging-related neurodegeneration is still underestimated.
Activation of RelA has been proposed to lead the systemic aging process in mice (86, 87), being negligible in the hypothalamus of young mice and progressively increasing, earlier in microglia and later in neuronal cells, as the mice become older (12). The genetic depletion of one allele of RelA attenuated the behavioral signs of neurodegeneration and extended the healthspan in a progeroid mouse model (13).
These stimulating results could be reread in the light of recent evidence showing that protective versus harmful p50/RelA activation strongly depends on the acetylation state of RelA. Activation of RelA subunit displaying reduced grade of general acetylation, but site-specific acetylation of K310, triggers apoptotic gene expression in brain ischemia. In the absence of such a mismatch, the activation of RelA is neuroprotective, as observed in preconditioning ischemia.
Whether a dysregulation of the RelA acetylation state is also involved in normal aging, or just in pathological aging, remains to be established. What seems promising is that aberrant RelA acetylation, more than RelA nuclear translocation, can be a suitable target. It is corrected by the synergistic association of HDAC inhibitors and resveratrol to produce neuroprotection.
While evidence suggests that RelA activation marks physiological elderly in mice, we show that the deficiency of c-Rel leads to a parkinsonian phenotype with aging (10). The degeneration of DA neurons in the SNc, the microglia activation and the α-synuclein pathology are associated with development of an l-DOPA-responsive parkinsonism. Intriguingly, in the basal ganglia of aged c-Rel deficient mice, but not in aged matched controls, the acetylation state of RelA was reminding the one observed in lethal ischemia.
This body of evidence supports the premise that the balance between c-Rel- and RelA-mediated transcription may be at the crossroad between normal and pathological aging of the mammal brain. A defect of c-Rel activity, associated with higher RelA activation, reduces SNc resilience to aging hereby leading to a late-onset form of parkinsonism. Validation of the impact of c-Rel activity in PD onset and progression is now the challenge of ongoing studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Guest Associate Editor Giuseppe Pignataro declares that, despite having collaborated previously with the authors Annamaria Lanzillotta, Caterina Branca, Pier Franco Spano and Marina Pizzi, the review process was handled objectively and no conflict of interest exists.
Acknowledgments
This work was supported by Ex 60% 2008–2011 from the University of Brescia; NEDD Project (CUP H81J09002660007), Regione Lombardia, Italy; Ricerca Finalizzata Ministero della Salute Italy RF-2010-2315142; Marie Curie Actions – ITN nEUROinflammation, FP7 Project No. 607962 Fondazione CARIPLO 2014-0769, Italy.
Abbreviations
1B/DMT1, 1B isoform of divalent metal transporter-1; AMPK, AMP-activated kinase; BDNF, brain-derived neurotrophic factor; BIRC3, baculoviral IAP repeat-containing protein 3; DA, dopaminergic; HAT, histone acetyltransferase; HDAC, histone deacetylase; K310, lysine 310; MCAO, middle cerebral artery occlusion; MnSOD, superoxide dismutase; MPP+, 1-methyl-4-phenylpyridinium; NAD+, nicotinamide adenosine dinucleotide; OGD, oxygen–glucose deprivation; PD, Parkinson’s disease; RIP1, receptor interacting protein; ROS, reactive oxygen species; SIRT1, sirtuin 1; SN, substantia nigra; SNc, SN pars compacta; TH, tyrosine hydroxylase; TNF, tumor necrosis factor; UCP4, mitochondrial uncoupling protein 4.
References
1. Crampton SJ, O’Keeffe GW. NF-κB: emerging roles in hippocampal development and function. Int J Biochem Cell Biol (2013) 45(8):1821–4. doi: 10.1016/j.biocel.2013.05.037
2. Pizzi M, Spano P. Distinct roles of diverse nuclear factor-kappaB complexes in neuropathological mechanisms. Eur J Pharmacol (2006) 545(1):22–8. doi:10.1016/j.ejphar.2006.06.027
3. Camandola S, Mattson MP. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets (2007) 11(2):123–32. doi:10.1517/14728222.11.2.123
4. Inta I, Paxian S, Maegele I, Zhang W, Pizzi M, Spano P, et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J Neurosci (2006) 26(50):12896–903. doi:10.1523/JNEUROSCI.3670-06.2006
5. Pizzi M, Boroni F, Bianchetti A, Moraitis C, Sarnico I, Benarese M, et al. Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. Eur J Neurosci (2002) 16(12):2342–50. doi:10.1046/j.1460-9568.2002.02403.x
6. Pizzi M, Sarnico I, Boroni F, Benetti A, Benarese M, Spano PF. Inhibition of IkappaBalpha phosphorylation prevents glutamate-induced NF-kappaB activation and neuronal cell death. Acta Neurochir Suppl (2005) 93:59–63. doi:10.1007/3-211-27577-0_8
7. Pizzi M, Sarnico I, Boroni F, Benarese M, Steimberg N, Mazzoleni G, et al. NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death Differ (2005) 12(7):761–72. doi:10.1038/sj.cdd.4401598
8. Lanzillotta A, Sarnico I, Ingrassia R, Boroni F, Branca C, Benarese M, et al. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis (2010) 1:e96. doi:10.1038/cddis.2010.76
9. Sarnico I, Lanzillotta A, Boroni F, Benarese M, Alghisi M, Schwaninger M, et al. NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem (2009) 108(2):475–85. doi:10.1111/j.1471-4159.2008.05783.x
10. Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, et al. Late-onset parkinsonism in NFκB/c-Rel-deficient mice. Brain (2012) 135(Pt 9):2750–65. doi:10.1093/brain/aws193
11. Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev (2007) 21(24):3244–57. doi:10.1101/gad.1588507
12. Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature (2013) 497(7448):211–6. doi:10.1038/nature12143
13. Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest (2012) 122(7):2601–12. doi:10.1172/JCI45785
14. Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A (2010) 107(6):2669–74. doi:10.1073/pnas.0910658107
15. Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol (2007) 9(9):1081–8. doi:10.1038/ncb1629
16. Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, et al. A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J Neurosci (2004) 24(16):3933–43. doi:10.1523/JNEUROSCI.5646-03.2004
17. Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD. c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem (2008) 15(7):539–49. doi:10.1101/lm.866408
18. O’Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, et al. Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci (2006) 26(18):4870–9. doi:10.1523/JNEUROSCI.4527-05.2006
19. Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci (1998) 18(9):3251–60.
20. Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med (1999) 5(5):554–9. doi:10.1038/6458
21. Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, et al. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A (2007) 104(47):18754–9. doi:10.1073/pnas.0704908104
22. Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, et al. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A (1997) 94(14):7531–6. doi:10.1073/pnas.94.14.7531
23. Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A (1997) 94(6):2642–7. doi:10.1073/pnas.94.6.2642
24. Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, et al. NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur J Neurosci (2006) 23(7):1711–20. doi:10.1111/j.1460-9568.2006.04722.x
25. Sarnico I, Boroni F, Benarese M, Sigala S, Lanzillotta A, Battistin L, et al. Activation of NF-kappaB p65/c-Rel dimer is associated with neuroprotection elicited by mGlu5 receptor agonists against MPP(+) toxicity in SK-N-SH cells. J Neural Transm (2008) 115(5):669–76. doi:10.1007/s00702-007-0007-2
26. Yang HJ, Wang L, Xia YY, Chang PN, Feng ZW. NF-kappaB mediates MPP+-induced apoptotic cell death in neuroblastoma cells SH-EP1 through JNK and c-Jun/AP-1. Neurochem Int (2010) 56(1):128–34. doi:10.1016/j.neuint.2009.09.010
27. Kögel D, Peters M, König HG, Hashemi SM, Bui NT, Arolt V, et al. S100B potently activates p65/c-Rel transcriptional complexes in hippocampal neurons: clinical implications for the role of S100B in excitotoxic brain injury. Neuroscience (2004) 127(4):913–20. doi:10.1016/j.neuroscience.2004.06.013
28. Valerio A, Dossena M, Bertolotti P, Boroni F, Sarnico I, Faraco G, et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke (2009) 40(2):610–7. doi:10.1161/STROKEAHA.108.528588
29. Bernard D, Quatannens B, Begue A, Vandenbunder B, Abbadie C. Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res (2001) 61(6):2656–64.
30. Chen C, Edelstein LC, Gélinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol (2000) 20(8):2687–95. doi:10.1128/MCB.20.8.2687-2695.2000
31. Ho JW, Ho PW, Liu HF, So DH, Chan KH, Tse ZH, et al. UCP4 is a target effector of the NF-κB c-Rel prosurvival pathway against oxidative stress. Free Radic Biol Med (2012) 53(2):383–94. doi:10.1016/j.freeradbiomed.2012.05.002
32. Echtay KS. Mitochondrial uncoupling proteins – what is their physiological role? Free Radic Biol Med (2007) 43(10):1351–71. doi:10.1016/j.freeradbiomed.2007.08.011
33. Calandria JM, Asatryan A, Balaszczuk V, Knott EJ, Jun BK, Mukherjee PK, et al. NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival. Cell Death Differ (2015). doi:10.1038/cdd.2014.233
34. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell (2008) 30(6):689–700. doi:10.1016/j.molcel.2008.05.014
35. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A (2008) 105(33):11778–83. doi:10.1073/pnas.0711122105
36. Crack PJ, Taylor JM, Ali U, Mansell A, Hertzog PJ. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke (2006) 37(6):1533–8. doi:10.1161/01.STR.0000221708.17159.64
37. Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nat Med (2005) 11(12):1322–9. doi:10.1038/nm1323
38. Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke (2004) 35(4):987–91. doi:10.1161/01.STR.0000120732.45951.26
39. Pizzi M, Sarnico I, Lanzillotta A, Battistin L, Spano P. Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J (2009) 276(1):27–35. doi:10.1111/j.1742-4658.2008.06767.x
40. Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, et al. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci (2002) 22(13):5423–31.
41. Ingrassia R, Lanzillotta A, Sarnico I, Benarese M, Blasi F, Borgese L, et al. 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-κB/RelA acetylation at Lys310. PLoS One (2012) 7(5):e38019. doi:10.1371/journal.pone.0038019
42. Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci (2001) 21(13):4668–77.
43. Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol (2004) 5(5):392–401. doi:10.1038/nrm1368
44. Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, et al. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis (2013) 49:177–89. doi:10.1016/j.nbd.2012.08.018
45. Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry (2009) 65(3):191–7. doi:10.1016/j.biopsych.2008.09.002
46. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi:10.1038/nrg2485
47. Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem (2003) 278(4):2758–66. doi:10.1074/jbc.M209572200
48. Ashburner BP, Westerheide SD, Baldwin AS. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol (2001) 21(20):7065–77. doi:10.1128/MCB.21.20.7065-7077.2001
49. Buck SW, Gallo CM, Smith JS. Diversity in the Sir2 family of protein deacetylases. J Leukoc Biol (2004) 75(6):939–50. doi:10.1189/jlb.0903424
50. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J (2004) 23(12):2369–80. doi:10.1038/sj.emboj.7600244
51. Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol (2006) 70(6):1876–84. doi:10.1124/mol.106.027912
52. Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A (2006) 103(5):1587–92. doi:10.1073/pnas.0510341103
53. Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov (2006) 5(6):493–506. doi:10.1038/nrd2060
54. Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol (2010) 3:5. doi:10.1186/1756-8722-3-5
55. Yu W, Fu YC, Wang W. Cellular and molecular effects of resveratrol in health and disease. J Cell Biochem (2012) 113(3):752–9. doi:10.1002/jcb.23431
56. Agrawal M, Kumar V, Kashyap MP, Khanna VK, Randhawa GS, Pant AB. Ischemic insult induced apoptotic changes in PC12 cells: protection by trans resveratrol. Eur J Pharmacol (2011) 666(1–3):5–11. doi:10.1016/j.ejphar.2011.05.015
57. Li H, Yan Z, Zhu J, Yang J, He J. Neuroprotective effects of resveratrol on ischemic injury mediated by improving brain energy metabolism and alleviating oxidative stress in rats. Neuropharmacology (2011) 60(2–3):252–8. doi:10.1016/j.neuropharm.2010.09.005
58. Morris KC, Lin HW, Thompson JW, Perez-Pinzon MA. Pathways for ischemic cytoprotection: role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J Cereb Blood Flow Metab (2011) 31(4):1003–19. doi:10.1038/jcbfm.2010.229
59. Ren J, Fan C, Chen N, Huang J, Yang Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem Res (2011) 36(12):2352–62. doi:10.1007/s11064-011-0561-8
60. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature (2003) 425(6954):191–6. doi:10.1038/nature01960
61. Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab (2010) 298(4):E751–60. doi:10.1152/ajpendo.00745.2009
62. Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem (1999) 72(4):1707–16. doi:10.1046/j.1471-4159.1999.721707.x
63. Raval AP, Dave KR, Pérez-Pinzón MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab (2006) 26(9):1141–7. doi:10.1038/sj.jcbfm.9600262
64. Lanzillotta A, Porrini V, Bellucci A, Benarese M, Branca C, Parrella E, et al. NF-κB unbalance and dysfunction in acute and age-related neurodegenerative disease. EJND (2014). (in press).
65. Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron (2008) 60(4):610–24. doi:10.1016/j.neuron.2008.09.024
66. Tao J, Ji F, Liu B, Wang F, Dong F, Zhu Y. Improvement of deficits by transplantation of lentiviral vector-modified human amniotic mesenchymal cells after cerebral ischemia in rats. Brain Res (2012) 1448:1–10. doi:10.1016/j.brainres.2012.01.069
67. Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron (1993) 10(3):475–89. doi:10.1016/0896-6273(93)90335-O
68. Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry (2006) 11(12):1116–25. doi:10.1038/sj.mp.4001893
69. Chiu CT, Liu G, Leeds P, Chuang DM. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology (2011) 36(12):2406–21. doi:10.1038/npp.2011.128
70. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry (2009) 14(1):51–9. doi:10.1038/sj.mp.4002099
71. Pranski E, Van Sanford CD, Dalal N, Orr AL, Karmali D, Cooper DS, et al. NF-κB activity is inversely correlated to RNF11 expression in Parkinson’s disease. Neurosci Lett (2013) 547:16–20. doi:10.1016/j.neulet.2013.04.056
72. Teismann P, Schwaninger M, Weih F, Ferger B. Nuclear factor-kappaB activation is not involved in a MPTP model of Parkinson’s disease. Neuroreport (2001) 12(5):1049–53. doi:10.1097/00001756-200104170-00037
73. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A (1998) 95(11):6469–73. doi:10.1073/pnas.95.11.6469
74. Kannarkat GT, Boss JM, Tansey MG. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis (2013) 3(4):493–514. doi:10.3233/JPD-130250
75. Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A (2008) 105(47):18578–83. doi:10.1073/pnas.0804373105
76. Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol (2009) 8(4):382–97. doi:10.1016/S1474-4422(09)70062-6
77. Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem (2005) 280(48):40364–74. doi:10.1074/jbc.M509329200
78. Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience (2011) 198:221–31. doi:10.1016/j.neuroscience.2011.08.045
79. Arbuthnott GW, Wickens J. Space, time and dopamine. Trends Neurosci (2007) 30(2):62–9. doi:10.1016/j.tins.2006.12.003
80. Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol (2000) 83(5):3084–100.
81. Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem (2006) 97(6):1634–58. doi:10.1111/j.1471-4159.2006.03907.x
82. Cardozo-Pelaez F, Song S, Parthasarathy A, Hazzi C, Naidu K, Sanchez-Ramos J. Oxidative DNA damage in the aging mouse brain. Mov Disord (1999) 14(6):972–80. doi:10.1002/1531-8257(199911)14:6<972::AID-MDS1010>3.0.CO;2-0
83. Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem (2007) 103(1):17–37. doi:10.1111/j.1471-4159.2007.04764.x
84. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J (2005) 19(6):533–42. doi:10.1096/fj.04-2751com
85. Simuni T, Sethi K. Nonmotor manifestations of Parkinson’s disease. Ann Neurol (2008) 64(Suppl 2):S65–80. doi:10.1002/ana.21472
86. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell (2009) 136(1):62–74. doi:10.1016/j.cell.2008.10.052
Keywords: NF-κB, epigenetic drugs, BDNF, c-Rel deficient mice, RelA (K310)
Citation: Lanzillotta A, Porrini V, Bellucci A, Benarese M, Branca C, Parrella E, Spano PF and Pizzi M (2015) NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front. Neurol. 6:98. doi: 10.3389/fneur.2015.00098
Received: 30 December 2014; Accepted: 21 April 2015;
Published: 20 May 2015
Edited by:
Giuseppe Pignataro, Federico II University of Naples, ItalyReviewed by:
Mariagrazia Grilli, University of Piemonte Orientale, ItalyMarkus Schwaninger, University of Lübeck, Germany
Copyright: © 2015 Lanzillotta, Porrini, Bellucci, Benarese, Branca, Parrella, Spano and Pizzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Pizzi, Department of Molecular and Translational Medicine, Section of Pharmacology, School of Medicine, University of Brescia, V. le Europa, 11, Brescia 25123, Italy,bWFyaW5hLnBpenppQHVuaWJzLml0