 Jacy L. Wagnon
Jacy L. Wagnon Miriam H. Meisler
Miriam H. Meisler- Department of Human Genetics, University of Michigan, Ann Arbor, MI, USA
Mutations of the voltage-gated sodium channel SCN8A have been identified in approximately 1% of nearly 1,500 children with early-infantile epileptic encephalopathies (EIEE) who have been tested by DNA sequencing. EIEE caused by mutation of SCN8A is designated EIEE13 (OMIM #614558). Affected children have seizure onset before 18 months of age as well as developmental and cognitive disabilities, movement disorders, and a high incidence of sudden death (SUDEP). EIEE13 is caused by de novo missense mutations of evolutionarily conserved residues in the Nav1.6 channel protein. One-third of the mutations are recurrent, and many occur at CpG dinucleotides. In this review, we discuss the effect of pathogenic mutations on the structure of the channel protein, the rate of recurrent mutation, and changes in channel function underlying this devastating disorder.
Introduction
Neuronal voltage-gated sodium channels contain one large, pore-forming α subunit and two smaller β subunits (1). They regulate cellular excitability by controlling the flow of sodium ions across the cell membrane. The sodium channel α subunit genes SCN1A, SCN2A, and SCN8A are broadly expressed in brain neurons, where they play a critical role in the regulation of neuronal excitability. Mutations in each of these closely related genes can result in early-infantile epileptic encephalopathy (EIEE), characterized by early-onset seizures that are refractory to treatment and accompanied by cognitive and behavioral disabilities (2, 3). Mutations of SCN1A are responsible for EIEE6 (Dravet syndrome). SCN2A mutations have been identified in EIEE11 (Ohtahara syndrome). Since 2012, more than 60 mutations of SCN8A have been identified in EIEE13 (OMIM #614558). In this article, we review the emergence of SCN8A as an important cause of EIEE.
SCN8A encodes the voltage-gated sodium channel Nav1.6, the major sodium channel at nodes of Ranvier (4) and at the axon initial segment (AIS) of excitatory and inhibitory neurons (5–7). At the AIS, Nav1.6 regulates the initiation of action potentials (8), and at nodes of Ranvier it contributes to nerve conduction velocity (9). The effects of loss of Nav1.6 have been studied in spontaneous mouse mutants. Partial or complete loss of Nav1.6 in mice causes reduced neuronal excitability as well as tremor, ataxia, and dystonia. Null mice develop hindlimb paralysis and death by 3 weeks of age (3). Heterozygous carriers of loss-of-function mutations in the mouse exhibit only mild abnormalities including disrupted sleep architecture and elevated anxiety (10, 11). On some strain backgrounds, heterozygous null mice exhibit spike–wave discharges, the hallmark of absence (non-convulsive) epilepsy, without spontaneous convulsive seizures (12).
Discovery of Patient Mutations in SCN8A
The role of SCN8A in human disease was first examined by a candidate gene approach in families segregating inherited movement disorders that resembled the defects in mutant mice. One family with a mutation of SCN8A was identified by this approach. The proband had ataxia and intellectual disability and was found to be heterozygous for a protein truncation mutation of SCN8A (13). Three additional heterozygous family members had intellectual disabilities without ataxia. It is important to note that none of the heterozygous null individuals in this family had a history of seizures, indicating that simple loss of SCN8A function is not epileptogenic. A screen of 80 patients with familial essential tremor failed to detect SCN8A variants (14). Analysis of families with inherited ataxia was also negative (Meisler, unpublished observations).
The recent introduction of exome sequencing in patients with early-onset epileptic encephalopathy resulted in identification of de novo mutation of SCN8A as an important cause of this non-familial disorder. The first de novo mutation was found in a patient with seizure onset at 6 months of age (15). More than 60 de novo mutations of SCN8A have since been identified by exome and genome sequencing and, more recently, by inclusion of SCN8A in commercial epilepsy gene sequencing panels (16–28).
Four studies utilized targeted sequencing strategies to identify pathogenic variants in large cohorts of epileptic encephalopathy patients. The proportion of EIEE cases with mutations in SCN8A was 3/500 (17), 2/264 (18), 7/683 (25), and 1/110 (26). Thus, mutations in SCN8A account for close to 1% of epileptic encephalopathy.
Shared Clinical Features of EIEE13 Patients
Most patients with EIEE13 have seizure onset between birth and 12 months of age, with a median age of 4 months. Several patients exhibited seizures within a week or two of birth. Generalized tonic and tonic–clonic seizures are present in most patients. Atonic, myoclonic, and focal and absence seizures are not uncommon. Febrile seizures are rare in EIEE13, providing a useful distinction between EIEE13 and Dravet syndrome. Prior to seizure onset, development is normal in approximately half of patients. After seizure onset, developmental regression often results in mild to severe intellectual disability. A distinguishing feature of EIEE13 is the movement disorders, which range from mild ataxia to choreoathetosis and, in several patients, quadriplegia. Hypotonia and hypertonia are frequent, sometimes in the same patient. SUDEP has been reported in 5/40 patients.
EEG at or near the time of seizure onset is normal in approximately 50% of cases. In the following months, most individuals develop EEG abnormalities including focal or multi-focal sharp waves or spikes, moderate to severe background slowing, and spike–wave complexes. Hypsarrhythmia is rare.
MRI scans usually appear normal at onset of EIEE13. After seizure onset, the MRI remains normal in approximately 50% of patients. The most common MRI abnormality is mild diffuse atrophy or mild to moderate cerebral atrophy. Cerebellar atrophy and thinning of the corpus callosum has also been reported. The extent of cerebral and cerebellar atrophy appears to be correlated with severity of the movement disorder. Under-developed cortex and defective myelination has been seen in a few cases. Microcephaly is rare.
Drug Responses in Patients with EIEE13
Seizures in EIEE13 patients are typically refractory to treatment, but up to 1/3 of patients have enjoyed seizure-free periods. Approximately, 40% of patients for whom we have detailed clinical histories have responded favorably to anticonvulsants that directly modulate sodium channel activity (“sodium channel blockers”) including valproic acid, phenytoin, carbamazepine, oxcarbazepine, lamotrigine, and topiramate (Table 1). Monotherapy with sodium channel blockers has been effective for some patients. Sodium channel blockers have been effective in patients with either gain-of-function or apparent loss-of-function mutations. Carbamazepine and its derivative oxcarbazepine have been useful in the largest number of patients. Patients carrying the identical SCN8A mutation can differ in their drug responses, indicating a significant role for modifier genes and/or stochastic developmental processes (Tables 1 and 2).
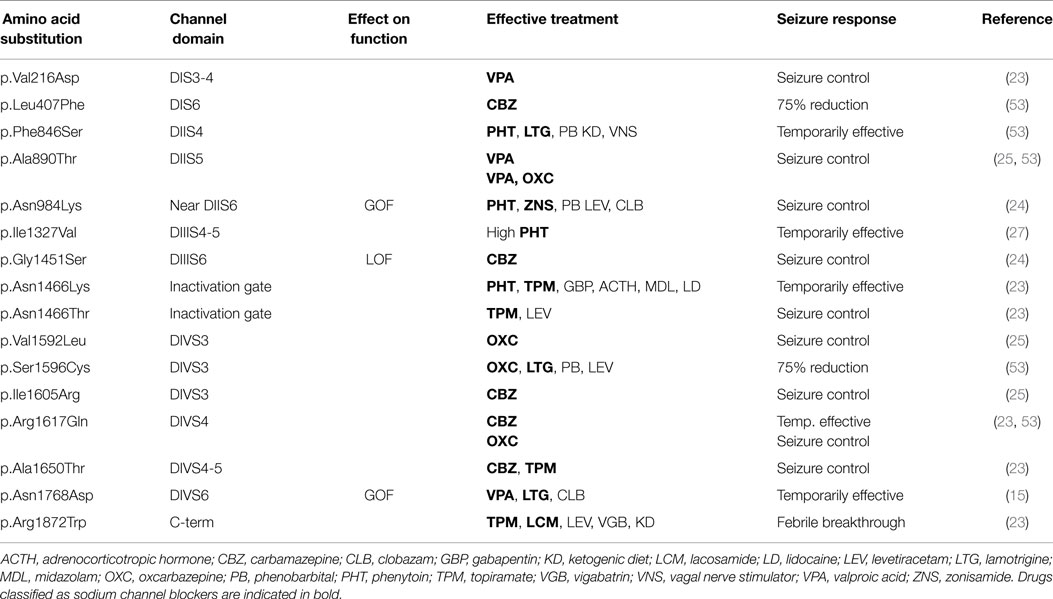
Table 1. Reported drug response in epileptic encephalopathy due to SCN8A mutations.
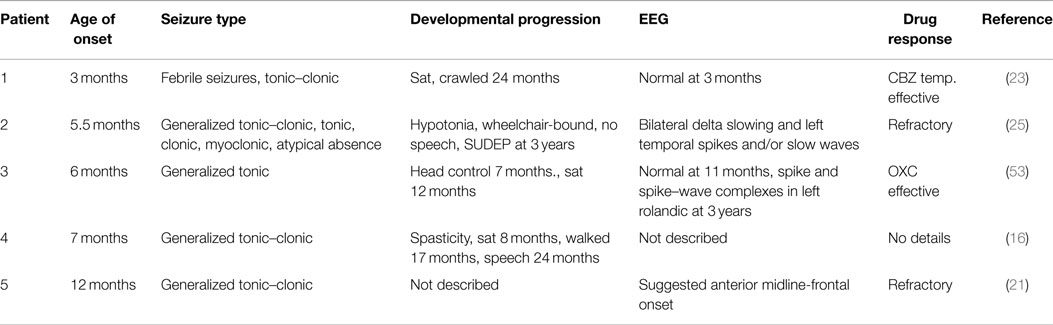
Table 2. Phenotypic variability in patients with the p.Arg1617Gln mutation in SCN8A.
The limited clinical evidence suggests that sodium channel blockers may be the best first line treatments for patients with SCN8A mutations, rather than modulators of GABA signaling or other synaptic function. By contrast, sodium channel blockers usually exacerbate seizures in patients with Dravet syndrome, which is caused by a deficiency of SCN1A activity. EIEE13, which is due to overactivity of SCN8A, has not been reported to be exacerbated by sodium channel blockers. Seizure exacerbation in EIEE13 patients has been observed with administration of levetiracetam. The ketogenic diet is reported to be effective for as many as 1/3 of patients with Dravet Syndrome. The response of patients with EIEE13 is not yet known. Early diagnostic DNA sequencing to distinguish between EIEE13 and Dravet Syndrome is thus very important for the selection of treatment.
Recurrent Mutations of SCN8A
The known mutations of SCN8A in EIEE13 are missense mutations that arose de novo in the patient or, in two cases, were inherited from a mosaic parent. All of the missense mutations result in substitution of an evolutionarily conserved amino acid residue. The locations of these mutations are shown in Figure 1. One-third of the mutations have reoccurred in two or more unrelated individuals (Figure 1, open circles). The molecular alterations in the recurrent mutations are indicated in Table 3. There have been five occurrences of the missense mutation p.Arg1617Gln and nine mutations of arginine residue 1872, four with substitution of glutamine, three with substitution of tryptophan, and two with substitution of leucine.
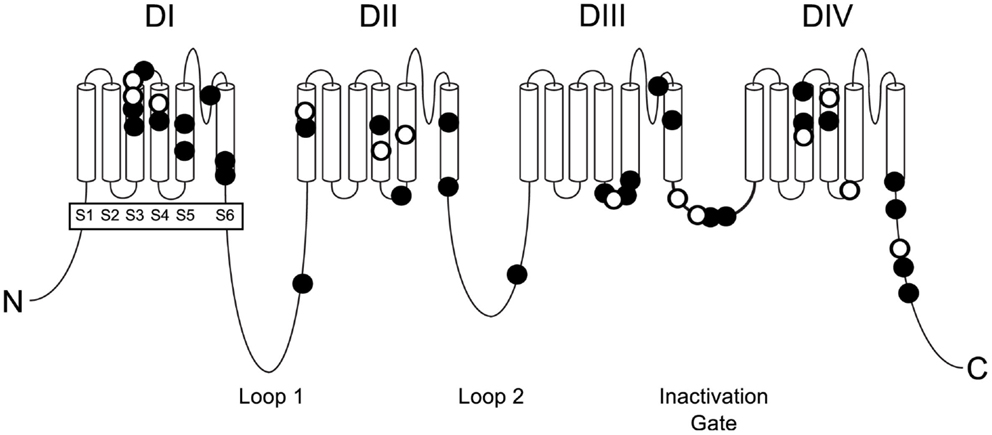
Figure 1. Positions of missense mutations of SCN8A in epileptic encephalopathy. The four homologous domains of Nav1.6 (DI to DIV) each contain six transmembrane segments (S1 to S6). The large inter-domain cytoplasmic loops 1 and 2 are evolutionarily less well-conserved than the transmembrane and linker domains. Loop 3 functions as the inactivation gate and is very highly conserved. Closed symbols, EIEE13 mutations in a single patient; open symbols, recurrent mutations.
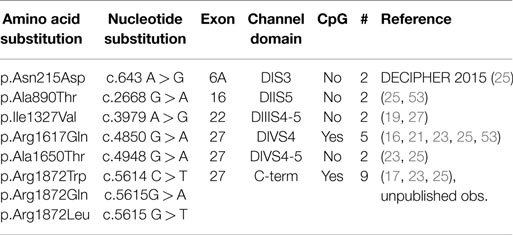
Table 3. Recurrent de novo mutations of SCN8A in epileptic encephalopathy.
One-third of the recurrent mutations are located at CpG dinucleotide residues, e.g., in arginine codons 1872 (CGG) and 1617 (CGA). CpG dinucleotides are mutation hotspots that can undergo enzymatic methylation of cytosine followed by spontaneous deamination of the methylated C to form thymine (Figure 2). Both of these arginine codons contain CpG sequences on coding and non-coding strands, and CpG deamination at these sites can account for most of the observed amino acid substitutions (Figure 2).
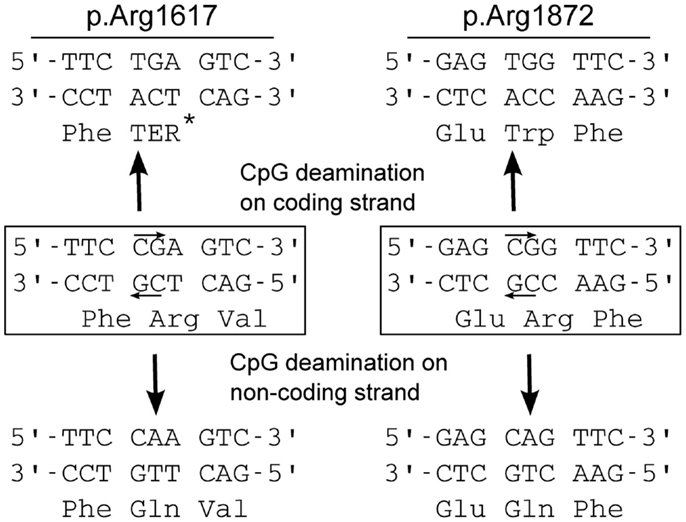
Figure 2. Mechanism for recurrent mutation of SCN8A at CpG dinucleotides. Arginine 1617 is encoded by a CGA codon, and arginine 1872 is encoded by a CGG codon. Both contain CpG dinucleotides on coding and non-coding strands (small arrows). Cytosine methylation followed by deamination on either strand leads to mutation: at arginine 1617, replacement by the premature termination codon (TGA, Ter) or glutamine (Gln); at arginine 1872, replacement by tryptophan (Trp) or glutamine (Gln). The premature termination mutation p.Arg1617Ter has not been seen in EIEE13 patients, and would probably not cause seizures (see text).
Clinical information is available for five patients with the p.Arg1617Gln mutation. Remarkably, these patients differ in many respects, including age of seizure onset, seizure type, severity of co-morbidities, and drug response (Table 2). These observations emphasize the important role of genetic background, including variation in other ion channel genes, on the clinical outcome for any individual patient.
Recurrent mutations of SCN1A have also been observed in Dravet syndrome. Recurrent mutations account for 25% of pathogenic mutations, and 1/3 of recurrent mutations are located at CpG dinucleotides (29). Recurrent deletions of short direct repeats that are susceptible to slipped-strand mispairing account for generation of some null alleles of SCN1A.
Sodium channels are among the most highly conserved proteins in the mammalian genome. SCN8A ranks among the 2% of human proteins that are least tolerant of variation [see dataset S2 in Ref. (30)]. The pathogenic missense mutations of SCN8A identify a subset of residues whose substitution results in elevated channel activity.
Location and Functional Consequences of SCN8A Mutations
Sodium channel α subunits contain four homologous domains (DI-DIV), each with six highly conserved transmembrane segments designated S1–S6 (Figure 1). Less-conserved cytoplasmic loops separate domains DI and DII, and domains DII and DIII. Domains III and IV are separated by a short, highly conserved inactivation gate (Figure 1). The proximal 2/3 of the C-terminus (residues 1767–1912) harbors several binding sites for interacting proteins, including sodium channel beta subunit β1 (31), intracellular FGFs (32–34), and calmodulin (35). The final 1/3 of the C-terminus, downstream of the calmodulin-binding motif, is disordered and not well-conserved. Most of the mutated residues in EIEE13 are located in the highly conserved portions of the protein: the domains containing transmembrane segments, inactivation gate, and proximal 2/3 of the C-terminus of Nav1.6 (Figure 3). In contrast, the non-pathogenic variants reported in the ExAC database of more than 60,000 exomes [Exome Aggregation Consortium, Cambridge, MA, USA (URL: http//exac.broadinstitute.org), accessed 04/2015] are concentrated in the non-conserved cytoplasmic loops of the channel (Figure 3).
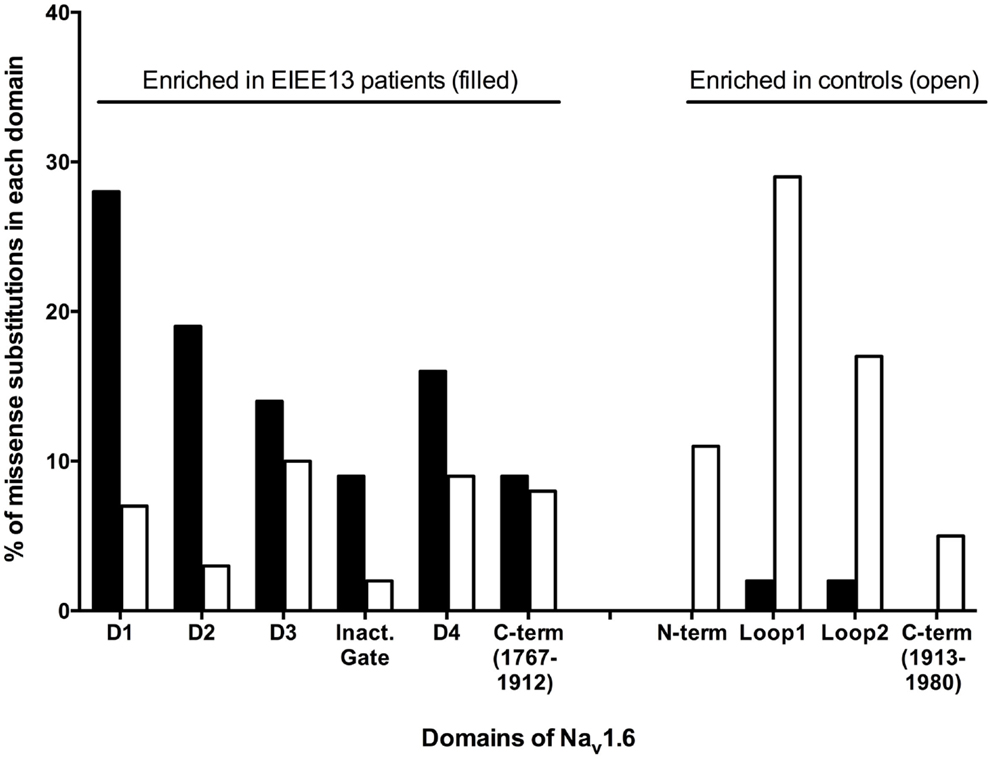
Figure 3. Differential distribution of pathogenic and non-pathogenic variants in the protein domains of SCN8A. Mutations of patients with EIEE13 are compared with variation in the ExAC database (exac.broadinstitute.org, accessed 04/2015), which excludes severe pediatric disease. Each amino acid substitution was counted once. Data were available for 43 mutations in EIEE13 patients and 228 variants from the ExAC database. Filled bars, EIEE13 patients; open bars, ExAC controls; Inact. gate, inactivation gate.
Five mutations of SCN8A have been tested in functional assays in transfected cells (Table 4). Three of these have dramatic gain-of-function effects. p.Asn1768Asp causes impaired channel inactivation and elevated persistent sodium current (15). As predicted by these experiments, transfection of primary hippocampal neurons with the mutant Nav1.6 cDNA increases neuronal excitability and promotes spontaneous firing (15). Similar missense mutations causing increased persistent current have also been identified in three other neuronal sodium channels. SCN1A mutations with increased persistent current were observed in patients with generalized epilepsy with febrile seizures plus (GEFS+) and Dravet Syndrome (36–38). SCN2A mutations with delayed inactivation and increased persistent current were described in both human and mouse (39–43). An SCN3A mutation with increased persistent current was identified in a child with pediatric partial epilepsy (44, 45). Thus, increased persistent current is a common pathological mechanism in epilepsy due to sodium channel mutations.
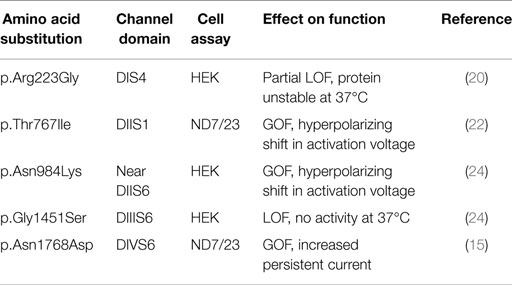
Table 4. Functional effects of five SCN8A mutations in patients with epileptic encephalopathy.
Two additional SCN8A mutations, p.Thr767Ile and p.Asn 984Lys, cause a hyperpolarized shift in the voltage-dependence of activation, which is predicted to result in premature neuronal firing (22, 24). Hyperpolarized shifts in activation have also been observed in missense mutations of SCN1A and SCN2A that cause early-onset epilepsy (46–48). Elevated activity of Nav1.6 thus appears to be the cause of seizures in these two cases.
The two mutations of SCN8A with apparent loss-of-function in transfected cell assays are puzzling. Simple loss-of-function mutations caused by protein truncation do not cause seizures in human or mouse (3, 13). It seems most likely that these two patient mutations actually exhibit gain-of-function in vivo. p.Gly1451Ser encodes a stable protein that lacks channel activity in transfected cells (24). p.Arg223Gly, results in protein instability at 37°C, greatly reducing current density but at 30°C, partial function is restored (20). At 30°C, activation and inactivation kinetics do not differ from wildtype but the mutant channel generates elevated ramp current in response to slow ramp depolarizations (20). Increased ramp current is a gain-of-function property that has been observed in a mutation of Nav1.3 in pediatric partial epilepsy (44, 45). Because the p.Arg223Gly mutation affects the second arginine residue in the DIS4 voltage sensor, it may also cause increased gating pore current, as observed for pathogenic mutations of Nav1.4 in periodic paralysis and Nav1.5 in dilated cardiomyopathy (49, 50). Thus, these two apparently inactive channel proteins may exhibit gain-of-function features in vivo. Thorough analysis of additional patient mutations will be required for definition of the relationship between genotype and phenotype of missense substitutions in SCN8A.
Modeling SCN8A Epileptic Encephalopathy in the Mouse
The earliest models of Scn8a dysfunction in the mouse had partial or complete loss of channel activity, and did not exhibit seizures (3). To generate a mouse model of EIEE13, we introduced the patient mutation p.Asn1768Asp into the mouse genome (51). Heterozygous Scn8aN1768D/+ mice recapitulate several features of EIEE13, including spontaneous convulsive seizures, ataxia, and sudden death (52). In heterozygous mutant mice, seizure onset occurs at 2–4 months of age. The mice may have several generalized tonic–clonic seizures per day, and progress to sudden death within 1 month of seizure onset.
Like many patients, the heterozygous Scn8aN1768D/+ mice have normal EEG prior to seizure onset (52). After seizure onset, they exhibit sporadic, semiperiodic biphasic slow wave or sharp wave–slow wave complexes. In addition, there are multiple daily interictal epileptiform discharges with diffuse polyspikes or single spike–wave discharges, similar to those in EIEE13. These discharges are frequently accompanied by myoclonic jerks or spasms. Unlike the patients, the mice do not demonstrate impaired learning or behavior, brain atrophy, or structural abnormalities of the brain. This novel Scn8a mouse model will be useful for investigation of pathogenesis and mechanisms of SUDEP. It will also be useful for evaluation of new therapies.
Conclusion
Large-scale sequencing of patient exomes has revealed the role of SCN8A in human epilepsy. Mutation of SCN8A accounts for approximately 1% of EIEE. The predominance of de novo mutations in children with EIEE13 reflects the severity of the disorder, which prevents transmission to the next generation. One-third of the mutations in EIEE13 are recurrent, and most occur in highly conserved regions of the channel. Further analysis of the functional effects of additional SCN8A mutations will be required to generate a comprehensive view of structure/function relationships. Functional differences among pathogenic mutations of SCN8A will have implications for selection of treatment for individual patients and for development of new therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work on human and mouse SCN8A is supported by NIH grant R01 NS34509 to MHM. JLW acknowledges support from a Dravet Syndrome Foundation Postdoctoral Fellowship.
References
1. Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol (2012) 590:2577–89. doi: 10.1113/jphysiol.2011.224204
2. Meisler MH, O’brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol (2010) 588:1841–8. doi:10.1113/jphysiol.2010.188482
3. O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet (2013) 4:213. doi:10.3389/fgene.2013.00213
4. Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A (2000) 97:5616–20. doi:10.1073/pnas.090034797
5. Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci (2008) 28:14329–40. doi:10.1523/JNEUROSCI.4833-08.2008
6. Li T, Tian C, Scalmani P, Frassoni C, Mantegazza M, Wang Y, et al. Action potential initiation in neocortical inhibitory interneurons. PLoS Biol (2014) 12:e1001944. doi:10.1371/journal.pbio.1001944
7. Tian C, Wang K, Ke W, Guo H, Shu Y. Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front Cell Neurosci (2014) 8:297. doi:10.3389/fncel.2014.00297
8. Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci (2009) 12:996–1002. doi:10.1038/nn.2359
9. Kearney JA, Buchner DA, De Haan G, Adamska M, Levin SI, Furay AR, et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Hum Mol Genet (2002) 11:2765–75. doi:10.1093/hmg/11.22.2765
10. McKinney BC, Chow CY, Meisler MH, Murphy GG. Exaggerated emotional behavior in mice heterozygous null for the sodium channel Scn8a (Nav1.6). Genes Brain Behav (2008) 7:629–38. doi:10.1111/j.1601-183X.2008.00399.x
11. Papale LA, Paul KN, Sawyer NT, Manns JR, Tufik S, Escayg A. Dysfunction of the Scn8a voltage-gated sodium channel alters sleep architecture, reduces diurnal corticosterone levels, and enhances spatial memory. J Biol Chem (2010) 285:16553–61. doi:10.1074/jbc.M109.090084
12. Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, et al. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum Mol Genet (2009) 18:1633–41. doi:10.1093/hmg/ddp081
13. Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet (2006) 43:527–30. doi:10.1136/jmg.2005.035667
14. Sharkey LM, Jones JM, Hedera P, Meisler MH. Evaluation of SCN8A as a candidate gene for autosomal dominant essential tremor. Parkinsonism Relat Disord (2009) 15:321–3. doi:10.1016/j.parkreldis.2008.06.010
15. Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet (2012) 90:502–10. doi:10.1016/j.ajhg.2012.01.006
16. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet (2012) 380:1674–82. doi:10.1016/S0140-6736(12)61480-9
17. Carvill GL, Heavin SB, Yendle SC, Mcmahon JM, O’Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet (2013) 45:825–30. doi:10.1038/ng.2646
18. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature (2013) 501:217–21. doi:10.1038/nature12439
19. Vaher U, Noukas M, Nikopensius T, Kals M, Annilo T, Nelis M, et al. De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol (2013) 29:202–6. doi:10.1177/0883073813511300
20. de Kovel CG, Meisler MH, Brilstra EH, Van Berkestijn FM, Van ‘T Slot R, Van Lieshout S, et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res (2014) 108:1511–8. doi:10.1016/j.eplepsyres.2014.08.020
21. Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Warman Chardon J, et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet (2014). doi:10.1111/cge.12464
22. Estacion M, O’Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis (2014) 69:117–23. doi:10.1016/j.nbd.2014.05.017
23. Ohba C, Kato M, Takahashi S, Lerman-Sagie T, Lev D, Terashima H, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia (2014) 55:994–1000. doi:10.1111/epi.12668
24. Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans MC, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet (2015) 52:330–7. doi:10.1136/jmedgenet-2014-102813
25. Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology (2015) 84:480–9. doi:10.1212/WNL.0000000000001211
26. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia (2015). doi:10.1111/epi.12954
27. Singh R, Jayapal S, Goyal S, Jungbluth H, Lascelles K. Early-onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure (2015) 26:69–71. doi:10.1016/j.seizure.2015.01.017
28. The Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature (2015) 519:223–8. doi:10.1038/nature14135
29. Kearney JA, Wiste AK, Stephani U, Trudeau MM, Siegel A, Ramachandrannair R, et al. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr Neurol (2006) 34:116–20. doi:10.1016/j.pediatrneurol.2005.07.009
30. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet (2013) 9:e1003709. doi:10.1371/journal.pgen.1003709
31. Spampanato J, Kearney JA, De Haan G, Mcewen DP, Escayg A, Aradi I, et al. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci (2004) 24:10022–34. doi:10.1523/JNEUROSCI.2034-04.2004
32. Liu CJ, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem (2003) 278:1029–36. doi:10.1074/jbc.M207074200
33. Wittmack EK, Rush AM, Craner MJ, Goldfarb M, Waxman SG, Dib-Hajj SD. Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J Neurosci (2004) 24:6765–75. doi:10.1523/JNEUROSCI.1628-04.2004
34. Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, et al. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci (2009) 42:90–101. doi:10.1016/j.mcn.2009.05.007
35. Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett (2002) 513:107–13. doi:10.1016/S0014-5793(01)03239-2
36. Escayg A, Macdonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet (2000) 24:343–5. doi:10.1038/74159
37. Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL Jr. Molecular basis of an inherited epilepsy. Neuron (2002) 34:877–84. doi:10.1016/S0896-6273(02)00714-6
38. Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL Jr. Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci U S A (2004) 101:11147–52. doi:10.1073/pnas.0402482101
39. Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience (2001) 102:307–17. doi:10.1016/S0306-4522(00)00479-6
40. Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A (2001) 98:6384–9. doi:10.1073/pnas.111065098
41. Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology (2010) 75:1454–8. doi:10.1212/WNL.0b013e3181f8812e
42. Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain (2010) 133:1403–14. doi:10.1093/brain/awq057
43. Lauxmann S, Boutry-Kryza N, Rivier C, Mueller S, Hedrich UB, Maljevic S, et al. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na(+) current. Epilepsia (2013) 54:e117–21. doi:10.1111/epi.12241
44. Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett (2008) 433:65–70. doi:10.1016/j.neulet.2007.12.064
45. Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol (2010) 224:362–8. doi:10.1016/j.expneurol.2010.04.012
46. Spampanato J, Escayg A, Meisler MH, Goldin AL. Generalized epilepsy with febrile seizures plus type 2 mutation W1204R alters voltage-dependent gating of Na(v)1.1 sodium channels. Neuroscience (2003) 116:37–48. doi:10.1016/S0306-4522(02)00698-X
47. Rhodes TH, Vanoye CG, Ohmori I, Ogiwara I, Yamakawa K, George AL Jr. Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures. J Physiol (2005) 569:433–45. doi:10.1113/jphysiol.2005.094326
48. Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology (2009) 73:1046–53. doi:10.1212/WNL.0b013e3181b9cebc
49. Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature (2007) 446:76–8. doi:10.1038/nature05598
50. Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, et al. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One (2012) 7:e38331. doi:10.1371/journal.pone.0038331
51. Jones JM, Meisler MH. Modeling human epilepsy by TALEN targeting of mouse sodium channel Scn8a. Genesis (2014) 52:141–8. doi:10.1002/dvg.22731
52. Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet (2015) 24:506–15. doi:10.1093/hmg/ddu470
Keywords: sodium channel, epilepsy, seizures, SUDEP, CpG, Dravet syndrome, mutation
Citation: Wagnon JL and Meisler MH (2015) Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front. Neurol. 6:104. doi: 10.3389/fneur.2015.00104
Received: 01 April 2015; Accepted: 27 April 2015;
Published: 15 May 2015
Edited by:
Jean François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Saïd Bendahhou, Centre National de la Recherche Scientifique, FranceSaikat Chakraborty, Stony Brook Medicine, USA
Copyright: © 2015 Wagnon and Meisler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam H. Meisler, Department of Human Genetics, University of Michigan, 4909 Buhl Box 5618, Ann Arbor, MI 48109-5618, USA,bWVpc2xlcm1AdW1pY2guZWR1