 Maria Josè Sisalli
Maria Josè Sisalli Lucio Annunziato
Lucio Annunziato Antonella Scorziello
Antonella Scorziello- 1Division of Pharmacology, Department of Neuroscience, Reproductive and Odontostomatological Science, School of Medicine, Federico II University of Naples, Naples, Italy
- 2Fondazione IRCSS SDN, Naples, Italy
Ischemic preconditioning represents an important adaptation mechanism of CNS, which results in its increased tolerance to the lethal cerebral ischemia. The molecular mechanisms responsible for the induction and maintenance of ischemic tolerance in the brain are complex and not yet completely clarified. In the last 10 years, great attention has been devoted to unravel the intracellular pathways activated by preconditioning and responsible for the establishing of the tolerant phenotype. Indeed, recent papers have been published supporting the hypothesis that mitochondria might act as master regulators of preconditioning-triggered endogenous neuroprotection due to their ability to control cytosolic calcium homeostasis. More interestingly, the demonstration that functional alterations in the ability of mitochondria and endoplasmic reticulum (ER) managing calcium homeostasis during ischemia, opened a new line of research focused to the role played by mitochondria and ER cross-talk in the pathogenesis of cerebral ischemia in order to identify new molecular mechanisms involved in the ischemic tolerance. In line with these findings and considering that the expression of the three isoforms of the sodium calcium exchanger (NCX), NCX1, NCX2, and NCX3, mainly responsible for the regulation of Ca2+ homeostasis, was reduced during cerebral ischemia, it was investigated whether these proteins might play a role in neuroprotection induced by ischemic tolerance. In this review, evidence supporting the involvement of ER and mitochondria interaction within the preconditioning paradigm will be provided. In particular, the key role played by NCXs in the regulation of Ca2+-homeostasis at the different subcellular compartments will be discussed as new molecular mechanism proposed for the establishing of ischemic tolerant phenotype.
Introduction
Cerebral ischemia is a multifactorial and complex disease (1, 2). Indeed, the intracellular events activated by the loss of perfusion of the brain and responsible for neuronal damage range from impairment of intracellular homeostasis to mitochondrial dysfunction and free radical production (3–5). The complexity of these events explains the great discrepancy between the frequency of cerebral ischemic accidents and the lack of effective treatments able to inhibit or slow neuronal demise following the ischemic insult. Hence, the urgent need to identify new potential targets for the development of innovative therapeutic strategies able to defend the ischemic brain.
On these premises, in the recent years, the attention of the researchers focused on ischemic tolerance a phenomenon, also known as ischemic preconditioning (IPC), which consists of a sub-lethal anoxic insult that makes the tissue in which it occurs more resistant to a subsequent and potentially lethal ischemia (6–11). The relevance of this phenomenon is to correlate to the study of the endogenous mechanisms activated in neurons to allow cell survival after a sub-lethal ischemic stimulus. By this way, it is possible to identify new molecular targets useful to develop alternative therapeutic strategies to treat the ischemic disease. The great interest in the cerebral IPC and in the tolerance evoked by itself also comes from the similarity of this phenomenon with those clinical situations occurring in the human brain. Indeed, it is well known that transient ischemic attacks (TIAs) do not cause structural damage but appear to protect brain against a subsequent “stroke” (12, 13).
Therefore, IPC or ischemic tolerance of the brain lie in a natural adaptive process that can be mimicked by a variety of sub-lethal insults, such as transient hypoxia, spreading depression, oxidative stress, hyperthermia, or heat shock, and that increases the tissue tolerance to a subsequent, potentially lethal ischemia. This adaptive cytoprotection is a fundamental property of living cells, which allows them to survive to the exposure to potentially recurrent stressors. This phenomenon was clearly identified in the heart by Murry et al. (14) as preconditioning, or subsequently as ischemic tolerance, and in 1990 it was described also in the brain by Kitagawa et al. (15). Since then, it immediately attracted the interest of clinical and basic neuroscientists for several reasons. First, this biological process became widely recognized as a pertinent and effective experimental instrument to understand how the brain protects itself against ischemia, thereby providing an innovative approach for the discovery of novel neuroprotective strategies. Second, retrospective case-control studies showed a clinical correlate of the phenomenon discovered experimentally.
Molecular Mechanisms Involved in Ischemic Preconditioning
Among the molecular mechanisms underlining ischemic tolerance, the substantial changes in gene expression play a crucial role, suggesting that preconditioning is able to stimulate a genomic reprograming of cells which in turn is responsible for the cytoprotection and cellular survival described in those tissues interested by the ischemic event (16, 17). The effects in the cell genome, occurring after IPC, represent the signature of the complex interplay of multiple signaling pathways. It is interesting to underline that they are highly specialized pathways in different cell types of the brain and concur to the cellular and systemic response activated in the tissue to combat the noxious stimulus. Indeed, it has been reported that hundreds of genes undergo upregulation or downregulation in response to IPC stimuli (18–20), and that these changes in gene expression differ between the harmful ischemic insult and the sub-lethal IPC. In particular, preconditioning seems to attenuate the response to ischemia (19), and the tolerance established modifies the expression of genes involved in the suppression of metabolic pathways, in the control of immune responses, in the modulation of ion-channel activity, and in the changes of blood coagulation parameters (19).
As recently reported, three phases, temporally consecutive of the preconditioning phenomenon have been identified as induction, transduction, and tolerance.
Induction and Transduction Mechanisms of Tolerance
The induction of tolerance requires that the preconditioning stimulus has to be recognized by molecular sensors as a sign of a more severe phenomenon that will occur. This implicated the identification of numerous types of sensors, including neurotransmitters, neuromodulators, cytokines, and toll-like receptors (21) as well as ion channels and redox-sensitive enzymes (22–24) which in turn will activate subsequent transduction pathways responsible for initiating the adaptive response. The activation of the transduction phase also implies the involvement of different transducers depending, at least in part, on the nature of the preconditioning stimulus. They include members of mitogen-activated protein kinases (MAPKs) and their phosphorylated Ras, Raf, MEK, and ERK subfamilies (7, 22, 24, 25); mitochondrial ATP-sensitive K+ (KATP) channels (26, 27) Akt (28–31); and the protein kinase C-ε isoform (32). The possibility that the nitric oxide (NO)-based adaptive response to hypoxia in Drosophila (33) is evolutionarily conserved suggests that this multifunctional modulator might be a logical choice as an autocrine and paracrine mediator of preconditioning stress. Indeed, pharmacological and genetic evidence supporting the involvement of NO in the transduction process is continuing to mount. As matter of fact, it has been reported that NO can exert a neuroprotective role during preconditioning (7, 34), and we have recently proposed that NO-induced neuroprotection is associated to preservation of mitochondrial function (24). On the other hand, preconditioning positively affects the integrity of mitochondrial oxidative phosphorylation after cerebral ischemia, an effect restricted to the delayed window of preconditioning (35). Moreover, preconditioning prevents mitochondrial swelling, preserves membrane integrity fluidity, and protects mitochondrial energy metabolism during cerebral ischemia avoiding ATP consumption (36).
Given the redox sensitivity of many of those above-mentioned kinases and transcription factors, reactive oxygen species might also be contemplated within transducers (37–39). Furthermore, adenosine, another prototypical paracrine mediator and “retaliatory metabolite,” whose production is linked to ATP degradation, might be considered a transducer responsible for tolerance induction, in some in vitro and in vivo models (26, 27, 40, 41).
It is worth of note that although in general, mitochondria have been considered important mediators of endogenous neuroprotection, the mechanisms by which they might “integrate” cytoprotective signaling of preconditioning remain to be fully elucidated.
Role of Mitochondria in Ischemic Tolerance
It is well known that mitochondria are also able to sense and shape cytosolic Ca2+ signals by taking up and subsequently releasing Ca2+ ions during physiological and pathological Ca2+ elevations (42). Mitochondrial function is strictly dependent on maintaining of mitochondrial calcium homeostasis. In fact, thanks to the activity of Ca2+-sensitive mitochondrial dehydrogenases these organelles can regulate oxidative phosphorylation and ATP synthesis during conditions of high cellular demand (43). Therefore, to explore the relationship between mitochondrial oxidative capacity and calcium buffering activity during IPC might represent an attractive strategy mediating neuroprotection in ischemic conditions. In particular, considering that the influence of Ca2+ in the regulation of mitochondrial function is highly dependent on the spatiotemporal distribution of [Ca2+]i (44, 45), the assumption that during IPC an interaction between mitochondria and ER might affect mitochondrial metabolic properties, and in turn neuronal survival has been also investigated (31). This hypothesis is supported by the concept that microdomains of high [Ca2+]i have been identified near Ca2+ channels on the plasma membrane and ER (46).
Cellular Ionic Homeostasis and Energy Metabolism during Ischemic Preconditioning
It has been widely described that IPC activates intracellular biological responses prior to a potential lethal insult (17, 47–49), and that these events make the tissue in which they occur more resistant to the subsequent severe ischemia (17, 47, 50–52). In this regard, it is possible to speculate that an increase of energy metabolism or a latency in anoxic depolarization following the onset of ischemic insult might represent one of the mechanisms by which tissues strengthen their tolerance when exposed to a sub-lethal insult. Indeed, it has been reported that a reduction in energy demand and in the activity of ion channels represents determinant factors for ischemic tolerance establishment (19, 53, 54). To this aim, in vitro experiments performed in cortical neurons demonstrated that the exposure to brief non-injurious oxygen and glucose deprivation (OGD) causes an impairment in voltage-gated potassium channels (55). Similarly, the impairment of Na+/K+-ATPase activity occurring in rat hippocampal and cortical neurons exposed to global forebrain ischemia is counteracted by IPC (56). Interestingly, in the last years, it has been shown that some integral plasma membrane proteins, involved in the control of Ca2+ and Na+ ion influx or efflux, the sodium calcium exchangers (NCXs) might function as crucial players in the pathogenesis of brain ischemic damage (57). Therefore, these proteins, by regulating Na+ and Ca2+ homeostasis, may represent more suitable molecular targets for therapeutic intervention in ischemic stroke. As matter of fact, in vivo experiments performed in gerbils demonstrated that during IPC, an increase in Ca2+-ATPase activity and an enhancement in mitochondrial calcium sequestration occur in CA1 hippocampal neurons (47). In line with this findings, intracellular calcium measurements in hippocampal neurons of preconditioned gerbils showed that the increase in [Ca2+]i occurring after anoxic and aglycemic episodes was markedly reduced in the ischemic tolerant animals (58). However, the molecular mechanisms responsible for this effect are still matter of debate and not completely clarified. As recently proposed by Kato et al. (59), a possible explanation could be related to the increased expression of the Ca2+-ATPase isoform 1 [plasma membrane calcium ATPase 1 (PMCA-1)]. Moreover, considering that mitochondria play a crucial role in the regulation of intracellular Ca2+ homeostasis due to their high capacity to sequestrate Ca2+ both in physiological and in pathological conditions, they have been included among the possible intracellular mechanisms triggered by preconditioning stimuli. Indeed, although it is widely reported that the excessive amount of Ca2+, as it occurs during ischemia, causes an impairment of mitochondrial function with consequent massive uncoupling of oxidative phosphorylation, mitochondrial depolarization and mitochondrial permeability transitional pore (MPTP) opening, reversal of the action of ATP synthase and cell death, and studies performed both in the heart and in the brain demonstrated that the inhibition of MPTP opening and its signaling cascade represent crucial events responsible for cytoprotection observed in IPC (53, 60). What are the molecular mechanisms underlining these effects is still object of investigation. Among them, nitrite and protein kinases have been proposed as possible MPTP regulators (61–64) Table 1.
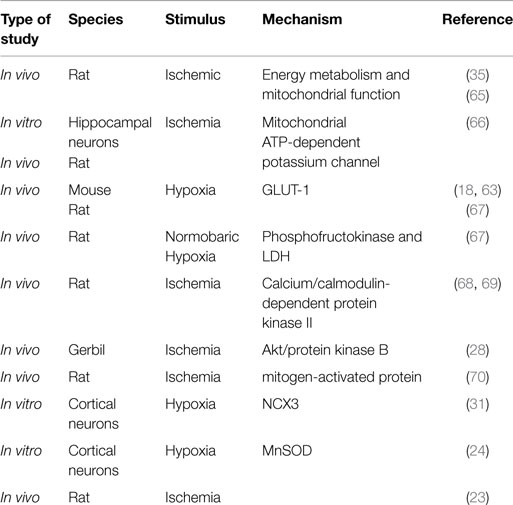
Table 1. Mitochondrial effectors of preconditioning-induced neuroprotection.
More interestingly, the hypothesis that a modulation of the expression and activity of the NCXs might have a role in the regulation of calcium and sodium homeostasis during ischemic tolerance has been recently suggested (31, 50, 71). This might be relating to the ability of Na+/Ca2+ exchanger to work in concert with selective ion channels and ATP-dependent pumps, in maintaining the physiological cytosolic concentrations of these two ions (72).
NCXs Functional Properties in Ischemic Brain
In the brain, unlike other tissues, NCX is present in three different gene products, named NCX1, NCX2, and NCX3, with a distinct distribution pattern in different brain regions (73). Under physiological conditions, NCX works primary extruding Ca2+ in response to a depolarization or to an increase in intracellular Ca2+ concentrations coupled to receptor stimulation (74). However, during hypoxic conditions, when the dysfunction of the two plasma membrane ATP-dependent pumps Na+/K+ ATPase and Ca2+ ATPase occurs, NCX assumes a relevant role in controlling the intracellular homeostasis of these two cations, since it is able to operate promoting Na+ ions extrusion and Ca2+ influx (74, 75). Although this reverse mode of operation in the early phase of anoxia does undoubtedly elicit an increase in [Ca2+]i, its effect could be beneficial for neurons because it contributes to decrease [Na+]i overload, a phenomenon which would otherwise lead to cell swelling and thus to sudden necrotic neuronal death. Conversely, in the later phase of neuronal anoxia, when [Ca2+]i overload takes place, NCX working in the forward mode of operation can contribute to the lowering of Ca2+ concentrations, and thus it can protect neurons from intracellular Ca2+overload, neurotoxicity, and subsequent cell death (74). As we recently demonstrated, NCX1, NCX2, and NCX3 may exert different roles during in vitro and in vivo anoxic conditions leading to a new paradigm in the pathogenesis of ischemic damage. Indeed, we provided evidences that in cells singly and stably transfected with NCX3, this isoform contributes more significantly to the maintenance of [Ca2+]i homeostasis during experimental conditions mimicking ischemia, thereby preventing mitochondrial ΔΨ collapse and cell death (76). This is due to a different sensitivity of the three NCX isoforms to the changes in ATP content occurring during anoxia. Indeed, NCX3 results more resistant to ATP changes compared to NCX1 and NCX2 (76). In addition, in in vivo experiments, the selective knocking down of NCX1 and NCX3, but not of NCX2, by antisense oligodeoxynucleotide strategy (57) or the disruption of the ncx3 gene, renders the brain more susceptible to the ischemic insult (77). Moreover, the induction of permanent middle cerebral artery occlusion in rats correlates with NCX1 mRNA upregulation in the peri-infarct area thus suggesting the possibility that this isoform could be a new druggable target for the treatment of cerebral ischemia. In line with this hypothesis, we recently demonstrated that NCX1 transcript and protein were upregulated by ischemic cerebral preconditioning. This effect was mediated by the transcriptional factor HIF1α and was accompanied by a relevant neuroprotection (71).
Role of NCX1 and NCX3 in the Ischemic Preconditioning
Recent findings demonstrated that the two isoforms of the Na+/Ca2+ exchanger, NCX1 and NCX3, which are involved in several pathophysiological aspects of cerebral ischemia, can be included among the members of the growing family of the mediators involved in the ischemic brain tolerance due to their ability to regulate neuronal calcium homeostasis (Figure 1). In particular, we provide evidence that neuroprotection observed in preconditioned neurons exposed to OGD/reoxygenation is correlated to the increase in NCX1 and NCX3 protein expression. Indeed, the treatment with siRNA against NCX1 and NCX3 prevents this effect. These data are in accordance with results recently observed in vivo in an animal model of IPC (50). Consistently with these results, the upregulation of NCX1 and NCX3 protein expression in neurons exposed to IPC was dependent on PI3K/Akt activation, since the treatment with LY294002 was able to abolish this increase. Interestingly, we demonstrated that NO plays a key role in the triggering PI3K/Akt pathway as the increase in the phosphorylated form of Akt observed within 30 min after IPC was completely abolished by the treatment with L-NAME. More importantly, the treatment with L-NAME was able to inhibit NCX3 but did not affect NCX1 protein expression (31).
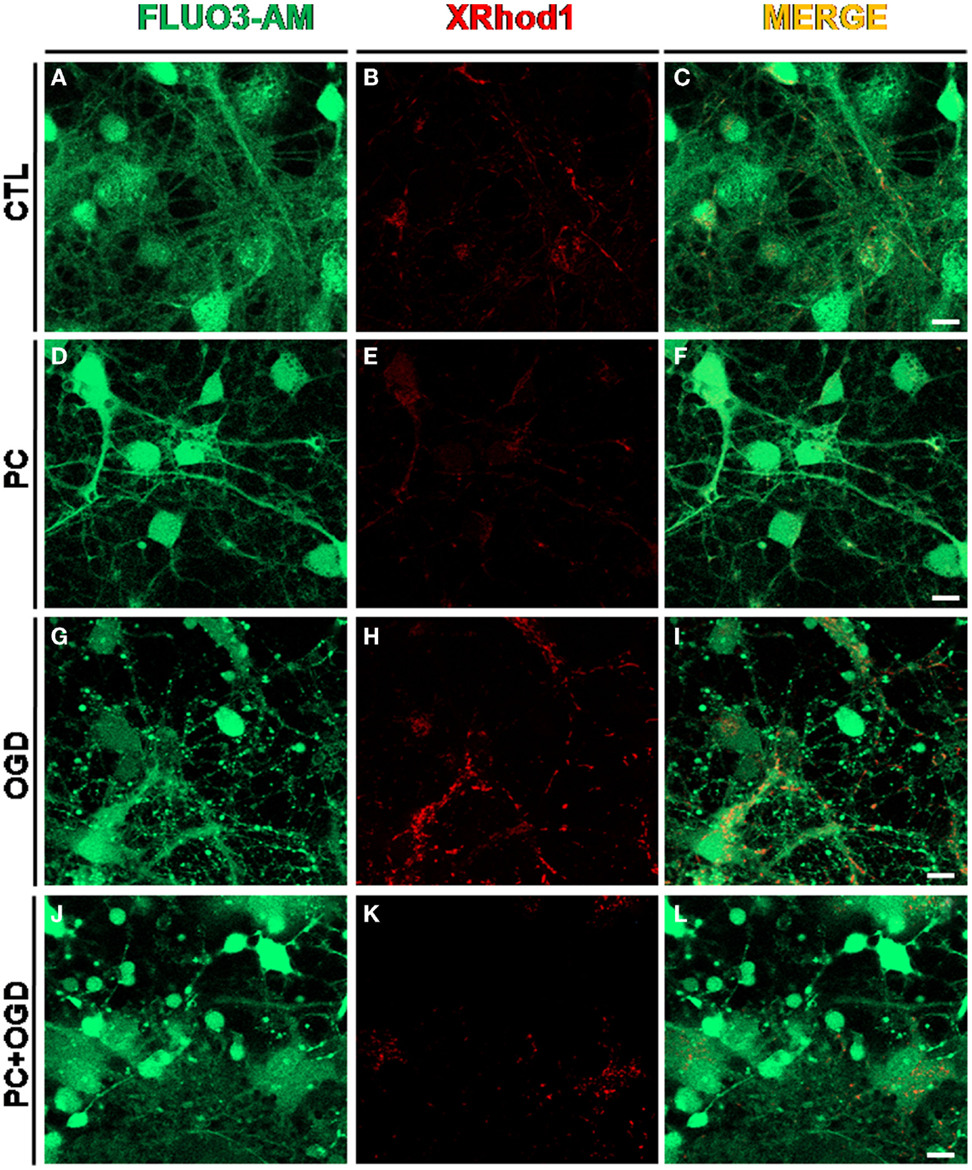
Figure 1. Effect of ischemic preconditioning on cytosolic and mitochondrial calcium content in cortical neurons exposed to OGD. Confocal images showing in red mitochondrial Ca2+ content measured by XRhod-1 probe (200 nM, 15 min at RT) and in green cytosolic calcium concentration measured by Fluo 3 AM probe (5 μM, 30 min at RT). (A–C): cortical neurons under control conditions (CTL); (D–F): cortical neurons exposed to 30 min of oxygen and glucose deprivation (OGD), preconditioning stimulus (PC); (G–I): cortical neurons exposed to 3 h OGD; (J-L): preconditioned neurons exposed to OGD (PC + OGD). Scale bars: 1 μm.
Calcium Homeostasis in ER and Mitochondria during Ischemic Preconditioning
It has recently demonstrated that in neurons NCX3, apart its localization on the plasma membrane, is also expressed on the outer mitochondrial membrane where it contributes to the extrusion of calcium from mitochondria (78). It is well known that mitochondria, in addition to the generation of cellular energy, play an important role in regulating cellular calcium homeostasis (79–82) in concert with the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), the plasma membrane Ca2+-ATPase, and Na+/Ca2+ exchanger (83). On the other hand, the maintenance of mitochondrial calcium homeostasis is an important requirement ensuring mitochondrial function. In fact, Ca2+-sensitive dehydrogenases can regulate oxidative phosphorylation and ATP synthesis during times of high cellular demand (43). Therefore, it is possible to hypothesize that the increased expression of NCX3, we observed within 48 h from the sub-lethal insult, might exert neuroprotective effects regulating calcium handling and improving mitochondrial oxidative capacity. This finding is in line with the results previously obtained in an in vitro model of IPC and demonstrating that the increase in nNOS expression and NO• production through the activation of Ras/ERK1/2 pathway stimulated mitochondrial MnSOD. These effects associate with a reduction in free radical production and cytocrome c release from mitochondria to cytosol, and in turn with an improvement of neuronal survival (24). The demonstration that NCX3 might represent a target of IPC-induced neuroprotection (31) adds new insight into the molecular mechanisms involved in the ischemic brain preconditioning. Indeed, the over-expression of NCX3 might help mitochondria to preserve their energetic capacity making them less vulnerable to the subsequent lethal insult represented by OGD/reoxygenation. This hypothesis was strongly supported by the results obtained measuring the activity of NCX during IPC. In fact, we demonstrated that IPC induced an increase of NCX activity in the reverse mode of operation that was still observed in preconditioned neurons exposed to OGD/Reoxygenation. This is an early event since it occurred within 30 min after IPC stimulus was due to the contribution of NCX1 and NCX3 isoforms and was promoted by NO because it was abolished by the treatment with L-NAME. Intriguingly, this effect was associated with an increase in ER calcium content. Consistently with data previously published in our Laboratory (84), we speculated that NCX1, working in the reverse mode of operation, plays a key role in the regulation of ER calcium refilling in the early phase of IPC (Figure 2). Indeed, the treatment with siNCX1, but not siNCX3, was able to prevent this phenomenon. The novel aspect of the study is the demonstration that NO promoted NCX1-induced ER refilling that was hampered by L-NAME pretreatment. This finding is in accordance with data recently published by Secondo et al. and demonstrating that NO was able to stimulate NCX1 to work in the reverse mode of operation, whereas NO did not affect NCX3 activity (85). The possibility that NCX1 activation in the early phase of IPC could affect mitochondrial calcium content promoting mitochondrial calcium uptake could not be excluded. However, in the late phase of IPC mitochondrial calcium handling is mainly regulated by NCX3 that is able to promote mitochondrial calcium extrusion (Figure 2). These data are supported by the finding that NCX3 expression increased 48 h after the IPC insult (31). We have previously demonstrated that NCX3 is distributed also at mitochondrial level; therefore, it is possible to speculate that during IPC, the increased expression of NCX3 on mitochondria might contribute to the efflux of calcium from the organelle thus protecting neurons by the subsequent mitochondrial calcium overload induced by lethal OGD/reoxygenation exposure. The finding that the treatment with CGP and siNCX3 counteracted the effect of IPC on mitochondrial calcium content, leading to the lack of IPC-neuroprotection, further supports this hypothesis (31). Collectively, we can conclude that a functional interplay between NCX1 and NCX3 occurs during IPC. This phenomenon is tightly dependent on NO and Akt activation, and by contributing to the modulation of intracellular ionic homeostasis, could represent one of the mechanisms responsible for neuroprotection induced by IPC (Figure 2).
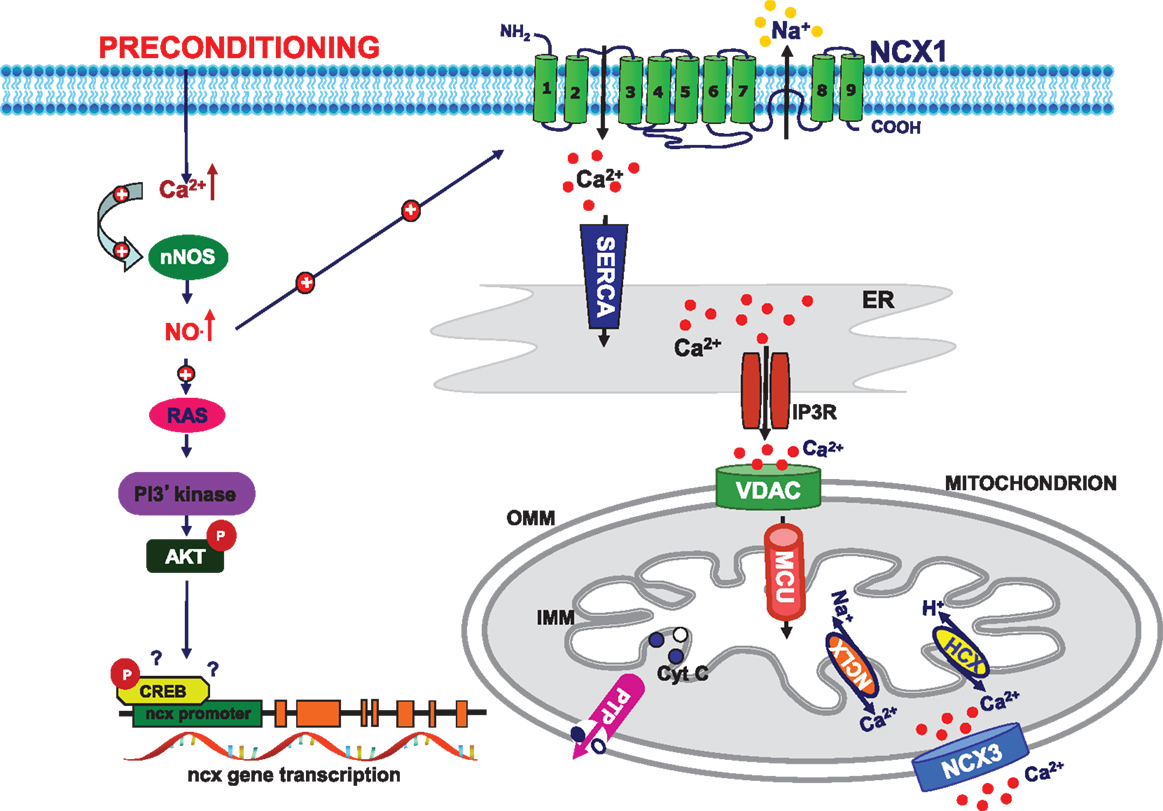
Figure 2. Schematic model of preconditioning-induced neuroprotection. Preconditioning stimulus activates neuronal nitric oxide synthase (nNOS) to produce NO which in turn stimulates plasma membrane NCX1 activity in the reverse mode of operation and promotes endoplasmic reticulum (ER) refilling in the early phase of PC. NO activate PI3K/Akt pathway, which increases the expression and activity of plasma membrane and mitochondrial NCX3, thus promoting mitochondrial calcium efflux in the late phase of preconditioning. These mechanisms working in concert promote neuronal survival. PI3K: phosphatidylinositol-3-kinases; CREB: cAMP response element-binding protein; SERCA: sarco/ER Ca2+-ATPase; IP3R: inositol trisphosphate receptor; MCU: mitochondrial calcium uniporter; VDAC: voltage-dependent anionic channel; NCLX: Na+/Ca2+ exchanger Li-dependent; HCX: Ca2+/H+ exchanger; PTP: permeability transition pore; OMM: outer mitochondrial membrane; IMM: inner mitochondrial membrane.
Conclusion
The experimental data described in this review support the hypothesis that mitochondria play a pivotal role in neuroprotective events provided by preconditioning. Indeed, both the mitochondrial antioxidant enzyme MnSOD and the proteins involved in the regulation of mitochondrial calcium homeostasis, such as mNCX3, represent new targets and mediators involved in preconditioning-induced neuroprotective responses. This is in line with the results reported in the literature that “mitochondrial preconditioning” has many neuroprotective effects, both during and following neurotoxic insults, including an improvement of neuronal viability, the attenuation of the intracellular Ca2+ influx, the suppression of ROS generation, the inhibition of apoptosis, and the maintenance of ATP levels (86). Therefore, understanding the precise mitochondrial mechanisms involved in preconditioning will provide important information necessary to develop new and more effective therapeutic strategies for neurodegeneration occurring in brain ischemia. In this regards, the identification of NCX3 as new player in the regulation of mitochondrial calcium efflux and the demonstration that an increase in its expression occurs during preconditioning, promoting mitochondrial calcium homeostasis, might help to draw new therapeutic strategy potentially able to delay neuronal loss occurring in ischemia. More interestingly, the demonstration that an increase in NCX1 and NCX3 activity is responsible for Ca2+ cycling from ER and mitochondria, and proof that it activates intracellular events leading to neuroprotection observed in IPC support this hypothesis (31). In this scenario, the identification of a compound able to selectively stimulate the activity of NCX1 and to prevent neuronal degeneration in in vitro and in vivo models of ischemia has been recently synthesized (77). Moreover, compounds able to stimulate NCX3 activity are in progress of development in our Laboratory.
Therefore, based on this early observation, these studies have potentially revealed new molecular targets in cerebral ischemia and neurodegenerative diseases pathogenesis, which ultimately may open up alternative avenues for future therapeutic intervention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Regione Campania #PON01_01602 and PON03_00785 to Lucio Annunziato.
References
1. Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics: 2008 update. A report from the American heart association statistics committee and stroke statistics subcommittee. Circulation (2008) 117:E25–46. doi:10.1161/CIRCULATIONAHA.107.187998
2. Dichgans M. Genetics of ischaemic stroke. Lancet Neurol (2007) 6:149–61. doi:10.1016/S1474-4422(07)70028-5
3. Siesjö BK. A new perspective on ischemic brain damage? Prog Brain Res (1993) 96:1–9. doi:10.1016/S0079-6123(08)63255-0
4. Livnat A, Barbiro-Michaely E, Mayevsky A. Real-time monitoring of spatial and temporal metabolic changes during focal cerebral ischemia in rats. Brain Res (2011) 1389:125–32. doi:10.1016/j.brainres.2011.03.008
5. Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta (2010) 1802:80–91. doi:10.1016/j.bbadis.2009.09.003
6. Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, et al. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res (1991) 561:203–11. doi:10.1016/0006-8993(91)91596-S
7. Dawson VL, Dawson TM. Neuronal ischemic preconditioning. Trends Pharmacol Sci (2000) 21:423–4. doi:10.1016/S0165-6147(00)01560-1
8. Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab (2002) 22:1283–96. doi:10.1097/00004647-200211000-00001
9. Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol (2002) 249:1503–11. doi:10.1007/s00415-002-0933-8
10. Cho S, Park EM, Zhou P, Frys K, Ross ME, Iadecola C. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab (2005) 25:493–501. doi:10.1038/sj.jcbfm.9600058
11. Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, et al. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab (2005) 25:234–46. doi:10.1038/sj.jcbfm.9600024
12. Weih M, Kallenberg K, Bergk A, Dirnagl U, Harms L, Wernecke KD, et al. Attenuated stroke severity after prodromal TIA: a role for ischemic tolerance in the brain. Stroke (1999) 30:1851–4. doi:10.1161/01.STR.30.9.1851
13. Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect. Neurology (2000) 54:2089–94. doi:10.1212/WNL.54.11.2089
14. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation (1986) 74:1124–36. doi:10.1161/01.CIR.74.5.1124
15. Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. “Ischemic tolerance” phenomen found in the brain. Brain Res (1990) 528:21–4. doi:10.1016/0006-8993(90)90189-I
16. Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke (2007) 38:680–5. doi:10.1161/01.STR.0000251444.56487.4c
17. Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci (2006) 7:437–48. doi:10.1038/nrn1927
18. Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab (2002) 22:393–403. doi:10.1097/00004647-200204000-00003
19. Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet (2003) 362:1028–37. doi:10.1016/S0140-6736(03)14412-1
20. Tang Y, Pacary E, Freret T, Divoux D, Petit E, Schumann-Bard P, et al. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identifi cation of potential neuroprotective candidates for stroke. Neurobiol Dis (2006) 21:18–28. doi:10.1016/j.nbd.2005.06.002
21. Kariko K, Weissmann D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab (2004) 24:1288–304. doi:10.1097/01.WCB.0000145666.68576.71
22. Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, et al. Requirement for nitric oxide activation of p21ras/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A (2000) 97:436–41. doi:10.1073/pnas.97.1.436
23. Danielisova V, Nemethova M, Gottlieb M, Burda J. Changes of endogenous antioxidant enzymes during ischemic tolerance acquisition. Neurochem Res (2005) 30:559–65. doi:10.1007/s11064-005-2690-4
24. Scorziello A, Santillo M, Adornetto A, Dell’aversano C, Sirabella R, Damiano S, et al. NO-induced neuroprotection in ischemic preconditioning stimulates mitochondrial Mn-SOD activity and expression via Ras/ERK1/2 pathway. J Neurochem (2007) 103:1472–80. doi:10.1111/j.1471-4159.2007.04845.x
25. Jones NM, Bergeron M. Hypoxia-induced ischemic tolerance in neonatal rat brain involves enhanced ERK1/2 signaling. J Neurochem (2004) 89:157–67. doi:10.1111/j.1471-4159.2004.02324.x
26. Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci U S A (1995) 92:4666–70. doi:10.1073/pnas.92.10.4666
27. Yoshida M, Nakakimura K, Cui YJ, Matsumoto M, Sakabe T. Adenosine A1 receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab (2004) 24:771–9. doi:10.1097/01.WCB.0000122742.72175.1B
28. Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, et al. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab (2001) 21:351–60. doi:10.1097/00004647-200104000-00004
29. Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci (2002) 22:6401–7.
30. Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, et al. Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab (2004) 24:271–9. doi:10.1097/01.WCB.0000110539.96047.FC
31. Sisalli MJ, Secondo A, Esposito A, Valsecchi V, Savoia C, Di Renzo GF, et al. Endoplasmic reticulum refilling and mitochondrial calcium extrusion promoted in neurons by NCX1 and NCX3 in ischemic preconditioning are determinant for neuroprotection. Cell Death Differ (2014) 21:1142–9. doi:10.1038/cdd.2014.32
32. Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Pérez-Pinzón MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci (2003) 23:384–91.
33. Wingrove JA, O’Farrell PH. Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell (1999) 98:105–14. doi:10.1016/S0092-8674(00)80610-8
34. Huang PL. Nitric oxide and cerebral ischemic preconditioning. Cell Calcium (2004) 36:323–9. doi:10.1016/j.ceca.2004.02.007
35. Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Perez-Pinzon MA. Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. J Cereb Blood Flow Metab (2001) 21:1401–10. doi:10.1097/00004647-200112000-00004
36. Zhang HX, Du GH, Zhang JT. Ischemic pre-conditioning preserves brain mitochondrial functions during the middle cerebral artery occlusion in rat. Neurol Res (2003) 25:471–6. doi:10.1179/016164103101201878
37. Puisieux F, Deplanque D, Bulckaen H, Maboudou P, Gelé P, Lhermitte M, et al. Brain ischemic preconditioning is abolished by antioxidant drugs but does not up-regulate superoxide dismutase and glutathione peroxidase. Brain Res (2004) 1027:30–7. doi:10.1016/j.brainres.2004.08.067
38. Zhang X, Xiong L, Hu W, Zheng Y, Zhu Z, Liu Y, et al. Preconditioning with prolonged oxygen exposure induces ischemic tolerance in the brain via oxygen free radical formation. Can J Anaesth (2004) 51:258–63. doi:10.1007/BF03019107
39. Ravati A, Ahlemeyer B, Becker A, Klumpp S, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factorkappaB. J Neurochem (2001) 78:909–19. doi:10.1046/j.1471-4159.2001.00463.x
40. Plamondon H, Blondeau N, Heurteaux C, Lazdunski M. Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and KATP channels. J Cereb Blood Flow Metab (1999) 19:1296–308. doi:10.1097/00004647-199912000-00002
41. Nakamura M, Nakakimura K, Matsumoto M, Sakabe T. Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J Cereb Blood Flow Metab (2002) 22:161–70. doi:10.1097/00004647-200202000-00004
42. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev (2006) 86:369–408. doi:10.1152/physrev.00004.2005
43. Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett (1980) 119:1–8. doi:10.1016/0014-5793(80)80986-0
44. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1:11–21. doi:10.1038/35036191
45. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol (2003) 4:517–29. doi:10.1038/nrm1155
46. Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, et al. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell (2010) 38:280–90. doi:10.1016/j.molcel.2010.04.003
47. Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev (2008) 88:211–47. doi:10.1152/physrev.00039.2006
48. Pignataro G, Scorziello A, Di Renzo G, Annunziato L. Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J (2009) 276:46–57. doi:10.1111/j.1742-4658.2008.06769.x
49. Gidday JM. Pharmacologic preconditioning: translating the promise. Transl Stroke Res (2010) 1:19–30. doi:10.1007/s12975-010-0011-y
50. Pignataro G, Boscia F, Esposito E, Sirabella R, Cuomo O, Vinciguerra A, et al. NCX1 and NCX3: two new effectors of delayed preconditioning in brain ischemia. Neurobiol Dis (2012) 45:616–23. doi:10.1016/j.nbd.2011.10.007
51. Cantarella G, Pignataro G, Di Benedetto G, Anzilotti S, Vinciguerra A, Cuomo O, et al. Ischemic tolerance modulates TRAIL expression and its receptors and generates a neuroprotected phenotype. Cell Death Dis (2014) 5:e1331. doi:10.1038/cddis.2014.286
52. Boscia F, Casamassa A, Secondo A, Esposito A, Pannaccione A, Sirabella R, et al. NCX1 exchanger cooperates with calretinin to confer preconditioning-induced tolerance against cerebral ischemia in the striatum. Mol Neurobiol (2015). doi:10.1007/s12035-015-9095-4
53. Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology (2008) 55:334–44. doi:10.1016/j.neuropharm.2008.02.017
54. Calderone V, Testai L, Martelli A, Rapposelli S, Digiacomo M, Balsamo A, et al. Anti-ischemic properties of a new spiro-cyclic benzopyran activator of the cardiac mito-KATP channel. Biochem Pharmacol (2010) 79:39–47. doi:10.1016/j.bcp.2009.07.017
55. Shah NH, Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res (2014) 5:38–58. doi:10.1007/s12975-013-0297-7
56. De Souza Wyse AT, Streck EL, Worm P, Wajner A, Ritter F, Netto CA. Preconditioning prevents the inhibition of Na+,K+-ATPase activity after brain ischemia. Neurochem Res (2000) 25:971–5. doi:10.1023/A:1007504525301
57. Pignataro G, Gala R, Cuomo O, Tortiglione A, Giaccio L, Castaldo P, et al. Two sodium/calcium exchanger gene products, ncx1 and ncx3, play a major role in the development of permanent focal cerebral ischemia. Stroke (2004) 35:2566–70. doi:10.1161/01.STR.0000143730.29964.93
58. Shimazaki K, Nakamura T, Nakamura K, Oguro K, Masuzawa T, Kudo Y, et al. Reduced calcium elevation in hippocampal CA1 neurons of ischemia-tolerant gerbils. Neuroreport (1998) 9:1875–8. doi:10.1097/00001756-199806010-00038
59. Kato K, Shimazaki K, Kamiya T, Amemiya S, Inaba T, Oguro K, et al. Differential effects of sublethal ischemia and chemical preconditioning with 3-nitropropionic acid on protein expression in gerbil hippocampus. Life Sci (2005) 77:2867–78. doi:10.1016/j.lfs.2005.01.037
60. Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta (2007) 1767:1007–31. doi:10.1016/j.bbabio.2007.05.008
61. Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol (2006) 34:249–70. doi:10.1385/MN:34:3:249
62. Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med (2007) 204:2089–102. doi:10.1084/jem.20070198
63. Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J Biol Chem (2002) 277:39728–38. doi:10.1074/jbc.M204619200
64. Stetler RA, Zhang F, Liu C, Chen J. Ischemic tolerance as an active and intrinsic neuroprotective mechanism. Handb Clin Neurol (2009) 92:171–95. doi:10.1016/S0072-9752(08)01909-X
65. Zhan RZ, Fujihara H, Baba H, Yamakura T, Shimoji K. Ischemic preconditioning is capable of inducing mitochondrial tolerance in the rat brain. Anesthesiology (2002) 97:896–901. doi:10.1097/00000542-200210000-00022
66. Liu D, Lu C, Wan R, Auyeung WW, Mattson MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J Cereb Blood Flow Metab (2002) 22:431–43. doi:10.1097/00004647-200204000-00007
67. Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab (2001) 21:1105–14. doi:10.1097/00004647-200109000-00008
68. Shamloo M, Wieloch T. Changes in protein tyrosine phosphorylation in the rat brain after cerebral ischemia in a model of ischemic tolerance. J Cereb Blood Flow Metab (1999) 19:173–83. doi:10.1097/00004647-199902000-00009
69. Shamloo M, Kamme F, Wieloch T. Subcellular distribution and autophosphorylation of calcium/calmodulin-dependent protein kinase II-alpha in rat hippocampus in a model of ischemic tolerance. Neuroscience (2000) 96:665–74. doi:10.1016/S0306-4522(99)00586-2
70. Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1region in a rat model of global cerebral ischemic preconditioning. Neuroscience (1999) 93:81–8. doi:10.1016/S0306-4522(99)00137-2
71. Valsecchi V, Pignataro G, Del Prete A, Sirabella R, Matrone C, Boscia F, et al. NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke (2010) 42:754–63. doi:10.1161/STROKEAHA.110.597583
72. Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev (1999) 79:763–854.
73. Canitano A, Papa M, Boscia F, Castaldo P, Sellitti S, Taglialatela M, et al. Brain distribution of the Na+/Ca2+ exchanger-encoding genes NCX1, NCX2, and NCX3 and their related proteins in the central nervous system. Ann N Y Acad Sci (2002) 976:394–404. doi:10.1111/j.1749-6632.2002.tb04766.x
74. Annunziato L, Cataldi M, Pignataro G, Secondo A, Molinaro P. Glutamate-independent calcium toxicity: introduction. Stroke (2007) 38(2 Suppl):661–4. doi:10.1161/01.STR.0000247942.42349.37
75. Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev (2004) 56:633–54. doi:10.1124/pr.56.4.5
76. Secondo A, Staiano RI, Scorziello A, Sirabella R, Boscia F, Adornetto A, et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium (2007) 42:521–35. doi:10.1016/j.ceca.2007.01.006
77. Molinaro P, Cuomo O, Pignataro G, Boscia F, Sirabella R, Pannaccione A, et al. Targeted disruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemic brain damage. J Neurosci (2008) 28:1179–84. doi:10.1523/JNEUROSCI.4671-07.2008
78. Scorziello A, Savoia C, Sisalli MJ, Adornetto A, Secondo A, Boscia F, et al. NCX3 regulates mitochondrial Ca2+ handling through the AKAP121-anchored signaling complex and prevents hypoxia-induced neuronal death. J Cell Sci (2013) 126:5566–77. doi:10.1242/jcs.129668
79. Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol (1997) 136:833–44. doi:10.1083/jcb.136.4.833
80. Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem (1996) 67:2282–91. doi:10.1046/j.1471-4159.1996.67062282.x
81. Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci (1994) 14:348–56.
82. Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature (1995) 377:438–41. doi:10.1038/377438a0
83. Saris NE, Carafoli E. A historical review of cellular calcium handling, with emphasis on mitochondria. Biochemistry (Mosc) (2005) 70:187–94. doi:10.1007/s10541-005-0100-9
84. Sirabella R, Secondo A, Pannaccione A, Scorziello A, Valsecchi V, Adornetto A, et al. Anoxia-induced NF-kappaB-dependent upregulation of NCX1 contributes to Ca2+ refilling into endoplasmic reticulum in cortical neurons. Stroke (2009) 40:922–9. doi:10.1161/STROKEAHA.108.531962
85. Secondo A, Molinaro P, Pannaccione A, Esposito A, Cantile M, Lippiello P, et al. Nitric oxide stimulates NCX1 and NCX2 but inhibits NCX3 isoform by three distinct molecular determinants. Mol Pharmacol (2011) 79:558–68. doi:10.1124/mol.110.069658
Keywords: ischemic preconditioning, sodium calcium exchanger, mitochondria, neurons, calcium
Citation: Sisalli MJ, Annunziato L and Scorziello A (2015) Novel cellular mechanisms for neuroprotection in ischemic preconditioning: a view from inside organelles. Front. Neurol. 6:115. doi: 10.3389/fneur.2015.00115
Received: 02 February 2015; Accepted: 05 May 2015;
Published: 26 May 2015
Edited by:
Alexej Verkhratsky, University of Manchester, UKCopyright: © 2015 Sisalli, Annunziato and Scorziello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonella Scorziello, Department of Neuroscience, Reproductive and Odontostomatological Science, School of Medicine, Federico II University of Naples, Via S. Pansini 5, Naples 80131, Italy,c2NvcnppZWxAdW5pbmEuaXQ=