 Petra Ehling
Petra Ehling Nico Melzer
Nico Melzer Thomas Budde
Thomas Budde Sven G. Meuth
Sven G. Meuth- 1Department of Neurology, Westfälische Wilhelms-University of Münster, Münster, Germany
- 2Institute of Physiology I – Neuropathophysiology, Westfälische Wilhelms-University, Münster, Germany
- 3Institute of Physiology I, Westfälische Wilhelms-University, Münster, Germany
Autoimmune inflammation of the limbic gray matter structures of the human brain has recently been identified as major cause of mesial temporal lobe epilepsy with interictal temporal epileptiform activity and slowing of the electroencephalogram, progressive memory disturbances, as well as a variety of other behavioral, emotional, and cognitive changes. Magnetic resonance imaging exhibits volume and signal changes of the amygdala and hippocampus, and specific anti-neuronal antibodies binding to either intracellular or plasma membrane neuronal antigens can be detected in serum and cerebrospinal fluid. While effects of plasma cell-derived antibodies on neuronal function and integrity are increasingly becoming characterized, potentially contributing effects of T cell-mediated immune mechanisms remain poorly understood. CD8+ T cells are known to directly interact with major histocompatibility complex class I-expressing neurons in an antigen-specific manner. Here, we summarize current knowledge on how such direct CD8+ T cell–neuron interactions may impact neuronal excitability, plasticity, and integrity on a single cell and network level and provide an overview on methods to further corroborate the in vivo relevance of these mechanisms mainly obtained from in vitro studies.
Role of Neuronal Antigen-Reactive CD8+ T Cells in Limbic Encephalitis – The Story So Far
Clinical Features of Limbic Encephalitis
Patients with limbic encephalitis (LE) (1–3) usually present with new onset mesial temporal lobe seizures, progressive memory disturbance, and a variety of other behavioral, emotional, and cognitive changes. Cerebrospinal fluid (CSF) exhibits inflammatory changes including lymphocytic pleocytosis, elevated protein, as well as intrathecal immunoglobulin (Ig)G synthesis or oligoclonal bands (OCB). The electroencephalogram (EEG) typically shows temporal epileptiform activity and slowing. Magnetic resonance imaging (MRI) exhibits volume and signal changes of the amygdala and hippocampus suggesting a sequence of acute inflammation followed by inflammation-driven neurodegeneration (4–7). Moreover, serum and CSF may contain specific auto-antibodies binding to either intracellular or plasma membrane-bound neuronal antigens (8–10) illustrating the presence of an adaptive neuron-directed autoimmune reaction.
Putative Immunopathogenesis of Limbic Encephalitis
Antigen-specific cellular and humoral immune responses directed toward central nervous system (CNS) neurons are believed to develop as a multi-step process (8, 11, 12). Soluble or cell-bound neuronal antigens or epitopes resembling them are engulfed and presented in the context of major histocompatibility complex (MHC) II molecules to CD4+ T cells by professional antigen-presenting cells (APCs) within secondary lymphatic organs (e.g., cervical lymph nodes). This in turn permits CD4+ T cells to license APCs to cross-present these antigens in the context of MHC I molecules to naïve CD8+ T cells, which then become activated and acquire cytotoxic effector functions (cellular effectors). Moreover, naïve B cells, which encounter, ingest, and present their cognate antigen in the context of MHC II molecules to CD4+ T cells, are in turn activated and become antibody-secreting plasma cells (humoral effectors). Following peripheral activation, both antibody-secreting plasma cells and cytotoxic CD8+ T cells (together with CD4+ T cells) may enter the CNS to attack neurons and cause functional and structural impairment (9, 10, 13). In general, both effector arms of the adaptive immune response may be activated irrespective of the cellular localization of the neuronal antigen or its antigenic epitope (plasma membrane vs. interior cellular compartments).
In terms of relevant effector mechanisms, plasma cell-derived antibodies bind to extracellular conformational epitopes of neuronal plasma membrane antigens and specifically impact their function, expression, and localization. Whether antibodies may also bind to and impact the function of intracellular neuronal antigens either by passive uptake into the neuron or by active binding to intracellular antigens, which are transiently exposed to the plasma membrane, is currently a matter of debate (14–17). In contrast, cytotoxic CD8+ T cells usually recognize linear peptides derived from antigens located in interior cell compartments following their MHC I-bound presentation on the neuronal surface (13). Whether peptides derived from neuronal surface membrane antigens are also presented to cytotoxic CD8+ T cells in the context of MHC I molecules is unclear at present (12).
Indeed, several findings suggest a pathogenic role of cytotoxic CD8+ T cells for neuronal damage in different forms of LE are as follows (18, 19): (i) neuronal damage often correlates with the number of CD8+ T cells, (ii) CD8+ T cells are found in close spatial proximity to neurons within the CNS, (iii) CD8+ T cells show an activated phenotype with substantial expression of the effector molecules (e.g., perforin and granzymes) in cytotoxic granules with a polar orientation toward neuronal cell membranes, (iv) some CD8+ T cells stain positive for CD107 indicating recent exocytosis of cytotoxic granules (i.e., degranulation), (v) neurons exhibit substantial cell surface expression of MHC I molecules allowing for cognate antigen-recognition by CD8+ T cells, and (vi) CD8+ T cells exhibit a restricted T cell receptor (TCR) repertoire (i.e., oligoclonal expansions), suggesting that they have expanded from a few precursors locally responding to distinct antigen epitopes in the CNS.
Hence, ongoing studies focus on the hypothesis that the encephalitides with antibodies against intracellular antigens [e.g., glutamic acid decarboxylase (GAD)] show neurodegeneration mediated by T cells, while encephalitides with antibodies against surface antigens [e.g., γ-amino butyric acid (GABA) receptor] are antibody mediated (Figure 1) (18, 20). These findings indicate that substances increasing GABA signaling may prevent tissue damage and seizures (21).
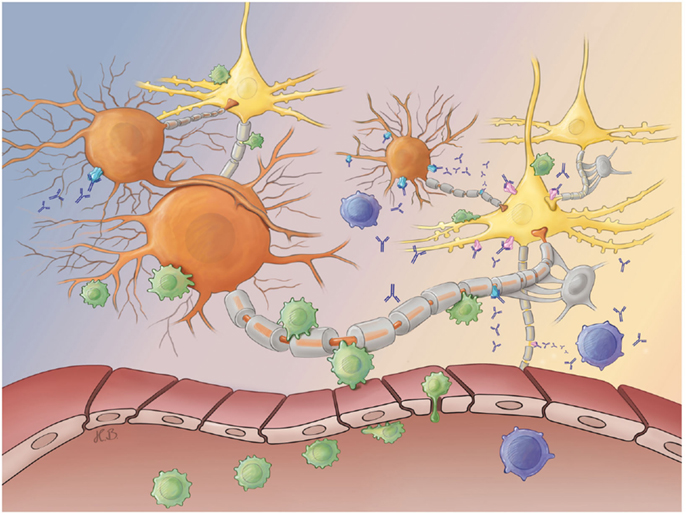
Figure 1. Antibody- and T cell-mediated neurotoxicity. After their peripheral activation, antibody-releasing plasma (blue) and activated CD8+ T cells (green) cross the blood–brain barrier and migrate into the brain parenchyma. Both effector arms of the adaptive immune system may selectively attack either inhibitory GABAergic interneurons (orange) or excitatory glutamatergic principal neurons (yellow) at synaptic as well as extra-synaptic sites. While plasma cell-derived antibodies may bind to neuronal surface antigens [e.g., GABA receptors (blue) or glutamate receptors (pink)], pathogenic CD8+ T cells recognize peptides derived from intracellular antigens (e.g., GAD in inhibitory interneurons or Hu in excitatory principal neurons) presented in the context of a MHC I molecule on the neuronal surface membrane. Both antibody- and T cell-mediated attacks finally cause neuronal dysfunction and degeneration. Abbreviations: GABA, γ-amino butyric acid; GAD, glutamic acid decarboxylase; MHC, major histocompatibility complex.
In contrast to antibodies, CD8+ T cells cannot directly impact the function or expression of their cognate antigens. Instead, cytotoxic T cells recognize their specific antigen only when presented on MHC I molecules on the surface of respective neuronal subtypes. This enables them to contribute to neuronal dysfunction and cell death by antigen-dependent release of effector molecules (13), as discussed below. Mouse data suggest that neuronal vulnerability against CD8+ T cell attacks might vary owing to their propensity to upregulate MHC class I molecules in response to inflammatory IFN-γ release (22). Moreover, catecholaminergic neurons in mice and humans have been shown to be particularly susceptible to T cell-mediated cytotoxic attacks (23). Thus, cellular immune responses may be restricted to distinct neuronal populations and networks due to their differential antigen expression pattern and capability of MHC I-mediated antigen presentation (22, 23). Moreover, distinct neuronal populations may take up soluble protein antigens released from other neural cell populations, process and present them in the context of MHC I molecules, thereby triggering a CD8+ T cell attack (23).
Effector Mechanisms during CD8+ T Cell–Neuron Interactions In Vitro
It has generally been presumed that the CNS is an immune-privileged organ, and that MHC I molecules are not expressed on neurons (24). However, CD8+ T cell-derived IFN-γ has recently been shown to cause immediate loss of dendrites and synapses, i.e., deafferentiation of neurons followed by delayed loss of neuronal somata in CNS gray matter areas (25). Reduced synaptic input through neuronal deafferentiation together with proinflammatory cytokine release may, in turn, reduce neuronal electrical activity below a critical threshold. Consequently, MHC I expression (22, 23) and endogenous or even exogenous antigen presentation are promoted and may thus render neurons susceptible for an antigen-dependent CD8+ T cell attack (26, 27). Consistently, it has recently been shown that MHC I expression per se in turn exerts profound effects on neuronal long-term plasticity in mice (28–33).
After encountering such neurons that present cognate antigens in the context of MHC I molecules, CD8+ T cells arrest and undergo stable long-term interactions (13, 34). TCR-signaling upon recognition of the appropriate antigen in the context of MHC I molecules leads to redistribution and accumulation of cytoskeletal, adhesion, co-stimulatory, and signal transduction molecules of the CD8+ T cell toward the cell–cell interface, resulting in the formation of the immunological synapse (18, 35). Similar to those formed by CD4+ T cells, the synapses formed by cytotoxic T cells during killing of their target consists of a ring of adhesion proteins surrounding a central core containing the TCR and downstream signaling proteins. However, synapses in CD8+ cells additionally possess a secretory domain for the exocytosis of effector molecules and reveal a shorter lifespan compared to CD4+ cell synapses (35).
CD8+ T cell-mediated cytotoxicity is predominantly mediated via two largely independent pathways (36, 37): (i) Granule cytotoxicity occurs by release of perforin together with a variety of granzymes. Perforin alone can lead to rapid necrosis of the target cell within minutes through the formation of large unselective transmembrane pores leading to rapid swelling and rupture of the cell membrane (38). Alternatively, perforin mediates the trafficking of granzymes into the target cell promoting apoptosis within a few hours. The exact mechanisms remain somewhat elusive (38, 39). (ii) Target cell apoptosis may also occur through the ligation of cell death receptors [e.g., FasL/Fas; (40)]. Together, Fas-induced apoptosis and the perforin pathway are the two main mechanisms by which cytotoxic T lymphocytes induce cell death in cells expressing foreign antigens (41).
The use of either the FasL–Fas or the perforin–granzyme pathway of CD8+ T cells depends on the strength of the antigen-signal delivered to the CD8+ T cell [i.e., the number of peptide (p) MHC I (pMHC I) complexes and the affinity of the TCR complex including co-receptors to the pMHC I complex]. This eventually results in different intracellular Ca2+ signals in T cells. Weak antigen-signals favor killing via the FasL–Fas pathway, whereas strong antigen-signals promote killing via perforin–granzyme exocytosis (42–44). Notably, 1–3 pMHC I-complexes per neuron are shown to be sufficient to elicit a cytotoxic T cell response when the TCR–pMHC I-affinity is high (44, 45). However, in case of low TCR–pMHC I-affinity, several thousand pMHC I complexes per target cell are needed to elicit an equal response (46).
Impact of CD8+ T Cells on Neuronal Excitability and Neuronal Network Activity
Besides the induction of cell death, effector molecules of cytotoxic CD8+ T cells are capable of disturbing electrical signaling in excitable target cells. The impact of these molecules on the electrical excitability has been extensively studied in ventricular cardiomyocytes but not neurons (47, 48).
Within minutes, purified perforin or lytic granules exposed to ventricular cardiomyocytes cause membrane depolarization as well as changes in amplitude and duration of action potentials. These effects are mediated by perforin per se and cannot be induced by granzymes alone (49, 50). Perforin monomers assemble to form large, unselective voltage-independent polyperforin channels in the target cell membrane (49, 50). This allows large non-selective ion fluxes over the plasma membrane, as also shown in lipid bilayer membranes and other intact cells (51, 52). After 2 h, affected cells exhibit an intracellular Ca2+ concentration in the micromolar range compared to low-nanomolar concentrations under physiological resting conditions. This is most likely due to transmembrane Ca2+ entry through perforin pores rather than through voltage-gated Ca2+ channels or by Ca2+ release from intracellular stores (53). Most importantly, these ion fluxes lead to the abolishment of transmembrane electrochemical ion gradients, an intracellular Ca2+ overload, and finally result in total electrical silence and collapse of the target cell.
Similarly, exposition with activating anti-Fas-receptor antibodies as well as conjugation with perforin-deficient CD8+ T cells also induced pronounced perturbation of electrical signaling in ventricular cardiomyocytes. The cells’ resting membrane potentials depolarize and their action potential amplitudes are reduced. Notably, the action potential duration is prolonged, a finding in marked contrast to the effects of perforin (54). Fas-receptor activation results in generation of 1,4,5-inositol-trisphosphate (IP3), which in turn triggers the release of Ca2+ from intracellular stores. Fas activation further causes a pronounced attenuation of transient outward K+ currents and an enhancement of L-type Ca2+ currents, and thus, prolongs the action potential duration. Electrophysiological effects of Fas-receptor activation can be mimicked by intracellular application of IP3 and can be abrogated by blocking phospholipase C, the IP3 receptor channel or store depletion (47, 54). Together, similar to perforin, Fas activation results in an intracellular Ca2+ overload of ventricular cardiomyocytes within a few hours.
In neurons, Ca2+ overload is associated with long-lasting changes in neuronal Ca2+ homeostasis and disturbed functioning of several Ca2+-dependent proteins (55–58). Furthermore, provided that most of the subcellular elements required for CD8+ T cell-mediated impairment of electrical excitability in cardiomyocytes are also present in neurons (59), it seems conceivable to assume that similar mechanisms will also lead to perturbation of neuronal excitability and Ca2+ homeostasis upon direct and indirect CD8+ T cell-neuron interactions.
In a previous study from our group, we used whole-cell patch clamp recordings and Ca2+ imaging from MHC I-expressing, ovalbumin (OVA)–peptide loaded cultured hippocampal neurons (60). Thereby, we demonstrated an immediate increase of the whole-cell membrane conductance upon direct cell–cell contact with activated antigen-specific CD8+ T cells leading to an impairment of electrical signaling (silencing). This was due to shunting of the membrane capacitance following insertion of CD8+ T cell-derived channel-forming perforin, which was paralleled by an increase of intracellular Ca2+ levels. Thus, perforin-dependent neuronal silencing is an immediate consequence of MHC I-restricted interaction of CD8+ T cells with cultured neurons much like the effects observed in cardiomyocytes (13, 60).
Importantly, an increase of the intracellular Ca2+ concentration could not only be detected in the neuron directly engaged with the CD8+ T cell but also in neighboring neurons without direct T cell contact (60). The nature of this “spill over-mechanism” explaining the intracellular Ca2+ accumulation in neighboring neurons has not been studied in detail. Possible mechanisms include that either membrane depolarization or Ca2+ accumulation causes intense synaptic signaling within the network of cultured neurons. However, glutamate toxicity is unlikely because glutamate could not be detected in culture supernatants after granule-induced cytotoxicity (61). Thus, these in vitro data indicate that only spatially confined trans-synaptic neuronal excitotoxicity involving activation of the respective ionotropic glutamate receptors might further promote neuronal damage. Alternatively, spatially non-confined release of cytotoxic effector molecules from the CD8+ T cell in contact with the neuron could also explain collateral effects in surrounding neurons (13).
Moreover, CD8+ (and CD4+) T cells have been shown to release glutamate that may contribute to effects on remote neurons within a distinct network (62, 63). Also, cytokines are released from CD8+ T cells and might affect the excitability and viability of neuronal networks. Thus, INF-γ has been shown to enhance glutamate excitotoxicity by direct intracellular trans-signaling between its INF-γ and AMPA/kainate receptors (64). TNF-α is shown to have an intrinsic ion channel-forming activity. Similar to polyperforin molecules, TNF-α trimers form largely unselective, high-conductance ion channels that may insert into lipid bilayer or cell membranes promoted under low pH values (65–67). Moreover, other inflammatory mediators are also likely to contribute to the perturbation of excitability and structure of neuronal networks [e.g., IFN-α and IL-1β in hypothalamic slice preparations, intraventricular IL-2 injections; reviewed in Ref. (68)].
These findings suggest a profound disturbance of neuronal function in close vicinity to as well as remote from the site of direct CD8+ T cell–neuron interaction and thus a significant impact of CD8+ T cells on structure and function of distinct neuronal networks (60). Significant limitations of this study, however, include that it was performed using neurons loaded with non-limiting amounts of exogenous OVA peptides following IFN-γ-induced MHC I expression. Moreover, CD8+ T cells had a transgenic TCR specific for the respective OVA–peptide and were strongly pre-activated in culture before experimentation.
These limitations have been partially overcome by the use of virus-infected neurons incubated with virus-specific CD8+ T cells isolated from brains of infected rodents (34). In this experimental setting, CD8+ T cells caused an immediate profound increase of neuronal network activity in multi-electrode recordings (34), suggesting that depending on the experimental conditions, direct CD8+ T cell–neuron interactions may effectively modulate electrical signaling within neuronal networks.
However, as most of these findings have been obtained from in vitro studies, the in vivo relevance of these results awaits demonstration.
Lack of Tools for the Study of CD8+ T Cell-Neuron Interactions and Potential Methods of Resolution
Despite the clear evidence for a pathogenic role of CD8+ T cells in autoimmune inflammation of the limbic gray matter structures constituting LE and other conditions, it has been difficult to develop an adequate in vivo model of autoimmune CD8+ T cell-mediated CNS inflammation directed against an endogenous neuronal antigen in rodents. In adoptive transfer experiments with myelin-reactive encephalitogenic CD8+ T cells, analogous to those performed with encephalitogenic CD4+ T cells, it was extremely difficult to induce clinically apparent disease in rodents. For example, transfer of very high numbers (3 × 107, corresponding to about 10% of the endogenous CD8+ T cell population) of in vitro pre-activated hemagglutinin (HA)-specific CD8+ T cells induces experimental autoimmune encephalitis (EAE) in less than half of recipient mice expressing HA as a neo-self antigen in oligodendrocytes (69). In another EAE-model employing TCR-transgenic myelin basic protein (MBP)-specific CD8+ T cells, disease induction requires transfer of 2 × 107 pre-activated cells (70). In contrast, as few as 1 × 105 MBP-specific CD4+ T cells suffice to induce EAE in mice (71), indicating that the pathogenic potency of activated CNS–antigen reactive CD4+ T cells exceeds that of their CD8+ counterparts by more than two orders of magnitude. Similarly, strong tolerance also exists for CD8+ T cell reactions toward endogenous neuronal antigens (72, 73). This is most likely to be distinct but poorly understood central and peripheral (74) as well as CNS-specific (75) tolerance mechanisms (13). These physiological mechanisms might establish a strict regulation of such immune responses given their strong destructive potential, especially in target organs with limited regenerative capacity, such as the CNS.
However, in case of distinct foreign neuronal antigens, CD8+ T cell–neuron interactions could be studied in vivo without requiring the break of strong self-tolerance to endogenous neuronal antigens (25, 34, 76).
Non-neuronal cells play a central role for the maintenance and functionality of neurons and networks in the CNS and may become targets of auto-reactive CNS-invading CD8+ T cells. For instance, in addition to neurons, astrocytes have been identified as targets for CD8+ T cells in Rasmussen’s encephalitis (RE) (77), a paradigmatic model of CD8+ T cell-driven neurodegeneration and epilepsy (78). Moreover, intracellular antigens expressed in oligodendrocytes have been described as targets of cerebral autoimmunity in CV2-antibody-associated encephalitis (79–81). In the context of several other inflammation-driven neurodegenerative diseases, it has already been shown that death of non-neuronal cells promotes subsequent neuronal loss [e.g., dying of oligodendrocytes promotes axonal degeneration in multiple sclerosis, Ref. (82)]. Further, it has been shown that cytotoxic effector CD8+ T cells selectively directed toward an oligodendrocyte-related neo-self-antigen are capable of inducing caspase-dependent apoptosis upon recognition of their cognate antigen, which is accompanied by simultaneous apoptosis of gray matter neurons (83, 84). Hence, impairment of neuronal structure and function may also occur as a collateral effect of an attack by CNS-invasive CD8+ T cells.
Methodological Repertoire for the Study of Neuronal Effects of CD8+ T Cell Attacks: Possible Solutions to Pending Problems
Compared to other disease entities research in the field of LE pathogenesis is still in its early stages. Meanwhile, pathogenic effects of plasma cell-derived antibodies on neuronal function and integrity are increasingly becoming characterized (85–90), potentially contributing effects of T cell-mediated immune mechanisms are highly likely to be of pivotal importance but remain poorly understood.
In this respect, the following issues are of primary importance: (i) how do cytotoxic CD8+ T cells influence the functionality of neurons or neuronal networks?; (ii) it remains to be demonstrated that an attack of cytotoxic CD8+ T cells is a conclusive mechanism to provoke pathological hyperexcitability in a neuronal network that finally leads to temporal lobe epileptic seizures; and (iii) are non-neuronal cells affected by the immune attack in a way that involves collateral neuronal damage?
Very recently, a new mouse model for anti-NMDA encephalitis was described in which patient CSF (containing NMDA receptor specific auto-antibodies) was delivered directly into mouse brains via osmotic pumps thereby inducing behavioral and memory deficits in mice (86). However, the establishment of an animal model specifically reflecting LE-specific pathophysiology, namely, primary CNS-specific and CD8+ T cell-driven autoimmunity against neuronal targets, is still one of the most pressing tasks in LE research. Indeed, it has been demonstrated that several weeks following intrahippocampal kainate administration accompanied by acute symptomatic seizures, mainly CD8+ T cells accumulate in the parenchyma of lesioned hippocampi and seem to strongly modulate neuronal degeneration and reorganization leading to an attenuated neuronal network excitability and generation of spontaneous epileptic seizures implying a profound impact of secondary adaptive neuron-directed immunity on epileptogenesis in this model (91, 92). However, to date, no animal model exists, in which a primary CD8+ T cell response targets distinct neuronal populations based on an antigen-specific direct interaction. Such a disease model, however, would give insights into pathophysiological mechanisms in vivo and ex vivo as well as it would be beneficial for any kind of treatment studies addressing primarily immune-mediated epilepsies. Despite the lack of an adequate LE animal model, antigen-directed CD8+ T cell-mediated neuronal attacks can be mimicked in different experimental settings. For instance, in vitro activated MHC I-restricted, OVA-reactive T cells from OT-I mice (93) can be used in combination with neurons/neuronal tissue from NSE-OVA mice expressing OVA as a neuronal auto-antigen under control of the neuron-specific enolase promoter (94).
Our group published one of the first in vitro studies focusing on immediate electrophysiological consequences on a single neuron upon direct antigen-directed CD8+ T cell attack. Real-time patch clamp recordings from OVA-loaded cultured hippocampal neurons (s. above) were performed during an attack from an OVA-directed activated CD8+ T cell. Analyses of different basic electrophysiological parameters showed that the cytotoxic attack against OVA-presenting neurons led to cellular shrinking and significant reduction of the membrane resistance compared to recordings with control peptide. Increased neuronal membrane conductance during antigen-directed T cell–neuron interaction was attributable to incorporation of perforin into the neuronal surface membrane. Impairment of electrical signaling paralleled by Ca2+ overload occurred within minutes. However, antigen-dependent apoptosis was not necessarily dependent on perforin and granzymes indicating other, so far unknown CD8+ T cell-mediated mechanisms promoting cell death (60).
Besides our sparse knowledge about consequences on single neuron functions due to direct immune cell attacks, we can only speculate on changes on the network level. At this point, an early imaging technique, whose widespread usage has been limited due to the sophisticated technical effort necessary to run successful experiments (95), may prove useful. It makes use of living brain slices loaded with voltage-sensitive dyes that change their fluorescent properties upon alterations of the membrane potential. Upon a defined depolarizing stimulus, the strength and the spreading of a network response can be visualized with high-temporal–spatial resolution. Conclusions can be drawn about the network’s functionality and intra-connectivity (96). An alternative in vitro technique to record network activity uses multi-electrode arrays, which offer the capability of recording and stimulating at multiple sites simultaneously (97). This technique has already been used to investigate pharmacologically induced epileptiform network activity (98). However, to obtain insights into immune attack-induced changes of neuronal functioning in vivo, electrophysiological recordings from freely behaving animals need to be done. This advanced technique permits the observation of cross-talk between single or multiple neurons in interconnected brain areas in combination with different behavioral contexts [e.g., Ref. (99)].
Further classical electrophysiological techniques may be suitable and informative. Long-term plasticity measurements in living hippocampal slices from rodents could reveal direct neuronal effects of a cellular immune response on synaptic plasticity as a basis for learning and memory processes. Especially, since memory disturbances are one of the prominent LE symptoms, this technique can be used to approach this aspect of the disease.
Conclusion
In recent years, an ever increasing number of endogenous neuronal plasma membrane auto-antigens have been identified as targets of specific auto-antibodies in LE and other autoimmune encephalitides. Pathogenic effects of auto-antibodies on neuronal excitability and integrity are increasingly becoming recognized on the single cell, network, and systems level. Auto-reactive T cells have been suggested to target neurons in autoimmune encephalitides associated with antibodies to endogenous intracellular neuronal antigens. However, the consequences of such direct T cell–neuron interactions remain poorly understood.
CD8+ T cell-derived IFN-γ may first contribute to neuronal MHC I up-regulation and antigen presentation. By a variety of molecular mechanisms, neuron-directed CD8+ T cell attacks may immediately modulate neuronal excitability and network activity over a wide range of functional states and trigger acute symptomatic seizures as well as neuropsychiatric symptoms. Subsequent inflammation-driven neuronal degeneration together with thus far poorly defined processes of neuronal reorganization may permanently alter structure and excitability in distinct neuronal networks targeted due to their specific antigen expression pattern and thus promoting chronic spontaneous seizures and epilepsy together with other neuropsychiatric symptoms. To corroborate these largely speculative mechanisms, effects of T cell–neuron interactions need to be studied on single cell, network, and systems level ideally in a suitable animal model using advance electrophysiological and imaging methods.
Conflict of Interest Statement
All authors declare no relevant conflicts of interest. Petra Ehling has received honoria for lecturing, travel expenses for attending meetings, and financial research support from Merck Serono and Novartis. Nico Melzer has received honoria for lecturing and travel expenses for attending meetings from Biogen Idec, GlaxoSmith Kline, Teva, and Fresenius Medical Care and has received financial research support from Fresenius Medical Care. Thomas Budde has received financial research support from Bayer, Biogen Idec, and Novartis. Sven G. Meuth has received honoraria for lecturing and travel expenses for attending meetings and has received financial research support from Bayer, Bayer Schering, Biogen Idec, Genzyme, Merck Serono, MSD, Novartis, Novo Nordisk, Sanofi-Aventis, and Teva.
Acknowledgments
We gratefully thank Heike Blum for excellent graphical illustration. This work was supported by the German research foundation (DFG; EH 469/1-1), the IMF Münster (AL 12 11 08), and the DFG funded excellence cluster “CiM – Cells in Motion.”
References
1. Tuzun E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol (2009) 118(6):737–43. doi: 10.1007/s00401-009-0582-4
2. Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life. Mainly affecting the limbic areas. Brain (1960) 83:357–68. doi:10.1093/brain/83.3.357
3. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol (2010) 67(4):470–8. doi:10.1002/ana.21917
4. Bien CG, Schulze-Bonhage A, Deckert M, Urbach H, Helmstaedter C, Grunwald T, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology (2000) 55(12):1823–8. doi:10.1212/WNL.55.12.1823
5. Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R, et al. Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology (2007) 69(12):1236–44. doi:10.1212/01.wnl.0000276946.08412.ef
6. Wagner J, Schoene-Bake JC, Malter MP, Urbach H, Huppertz HJ, Elger CE, et al. Quantitative FLAIR analysis indicates predominant affection of the amygdala in antibody-associated limbic encephalitis. Epilepsia (2013) 54(9):1679–87. doi:10.1111/epi.12320
7. Wagner J, Witt JA, Helmstaedter C, Malter MP, Weber B, Elger CE. Automated volumetry of the mesiotemporal structures in antibody-associated limbic encephalitis. J Neurol Neurosurg Psychiatry (2015) 86:735–42. doi:10.1136/jnnp-2014-307875
8. Melzer N, Meuth SG, Wiendl H. Paraneoplastic and non-paraneoplastic autoimmunity to neurons in the central nervous system. J Neurol (2013) 260(5):1215–33. doi:10.1007/s00415-012-6657-5
9. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol (2008) 7(4):327–40. doi:10.1016/S1474-4422(08)70060-7
10. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol (2011) 10(8):759–72. doi:10.1016/S1474-4422(11)70096-5
11. Pedemonte E, Mancardi G, Giunti D, Corcione A, Benvenuto F, Pistoia V, et al. Mechanisms of the adaptive immune response inside the central nervous system during inflammatory and autoimmune diseases. Pharmacol Ther (2006) 111(3):555–66. doi:10.1016/j.pharmthera.2005.11.007
12. Melzer N, Meuth SG, Wiendl H. Neuron-directed autoimmunity in the central nervous system: entities, mechanisms, diagnostic clues, and therapeutic options. Curr Opin Neurol (2012) 25(3):341–8. doi:10.1097/WCO.0b013e3283531efb
13. Melzer N, Meuth S, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J (2009) 23(11):3659–73. doi:10.1096/fj.09-136200
14. Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T, et al. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain (2010) 133(11):3166–80. doi:10.1093/brain/awq253
15. Greenlee JE, Clawson SA, Hill KE, Wood B, Clardy SL, Tsunoda I, et al. Neuronal uptake of anti-Hu antibody, but not anti-Ri antibody, leads to cell death in brain slice cultures. J Neuroinflammation (2014) 11(1):160. doi:10.1186/s12974-014-0160-0
16. Greenlee JE, Clawson SA, Hill KE, Wood BL, Tsunoda I, Carlson NG. Purkinje cell death after uptake of anti-Yo antibodies in cerebellar slice cultures. J Neuropathol Exp Neurol (2010) 69(10):997–1007. doi:10.1097/NEN.0b013e3181f0c82b
17. Hill KE, Clawson SA, Rose JW, Carlson NG, Greenlee JE. Cerebellar Purkinje cells incorporate immunoglobulins and immunotoxins in vitro: implications for human neurological disease and immunotherapeutics. J Neuroinflammation (2009) 6:31. doi:10.1186/1742-2094-6-31
18. Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain (2012) 135(Pt 5):1622–38. doi:10.1093/brain/aws082
19. McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol (2011) 122(4):381–400. doi:10.1007/s00401-011-0876-1
20. Bien CG, Bauer J. T-cells in human encephalitis. Neuromolecular Med (2005) 7(3):243–53. doi:10.1385/NMM:7:3:243
21. Sajadian A, Esteghamat S, Karimzadeh F, Eshaghabadi A, Sieg F, Speckmann EJ, et al. Anticonvulsant effect of neural regeneration peptide 2945 on pentylenetetrazol-induced seizures in rats. Neuropeptides (2015) 49:15–23. doi:10.1016/j.npep.2014.11.002
22. Scheikl T, Pignolet B, Dalard C, Desbois S, Raison D, Yamazaki M, et al. Cutting edge: neuronal recognition by CD8 T cells elicits central diabetes insipidus. J Immunol (2012) 188(10):4731–5. doi:10.4049/jimmunol.1102998
23. Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun (2014) 5:3633. doi:10.1038/ncomms4633
24. Lampson LA. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: reconciling divergent findings. Microsc Res Tech (1995) 32(4):267–85. doi:10.1002/jemt.1070320402
25. Kreutzfeldt M, Bergthaler A, Fernandez M, Bruck W, Steinbach K, Vorm M, et al. Neuroprotective intervention by interferon-gamma blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J Exp Med (2013) 210(10):2087–103. doi:10.1084/jem.20122143
26. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science (1995) 269(5223):549–52. doi:10.1126/science.7624779
27. Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med (1997) 185(2):305–16. doi:10.1084/jem.185.2.305
28. Edamura M, Murakami G, Meng H, Itakura M, Shigemoto R, Fukuda A, et al. Functional deficiency of MHC class I enhances LTP and abolishes LTD in the nucleus accumbens of mice. PLoS One (2014) 9(9):e107099. doi:10.1371/journal.pone.0107099
29. Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron (1998) 21(3):505–20. doi:10.1016/S0896-6273(00)80562-0
30. Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science (2000) 290(5499):2155–9. doi:10.1126/science.290.5499.2155
31. Oliveira AL, Thams S, Lidman O, Piehl F, Hokfelt T, Karre K, et al. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc Natl Acad Sci U S A (2004) 101(51):17843–8. doi:10.1073/pnas.0408154101
32. Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A (2007) 104(16):6828–33. doi:10.1073/pnas.0702023104
33. Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron (2009) 64(1):40–5. doi:10.1016/j.neuron.2009.09.044
34. Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, Farrugia F, et al. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog (2011) 7(11):e1002393. doi:10.1371/journal.ppat.1002393
35. Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol (2007) 23:495–517. doi:10.1146/annurev.cellbio.23.090506.123521
36. Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, et al. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science (1994) 265(5171):528–30. doi:10.1126/science.7518614
37. Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature (1994) 370(6491):650–2. doi:10.1038/370650a0
38. Waterhouse NJ, Sutton VR, Sedelies KA, Ciccone A, Jenkins M, Turner SJ, et al. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J Cell Biol (2006) 173(1):133–44. doi:10.1083/jcb.200510072
39. Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol (2007) 19(3):301–8. doi:10.1016/j.coi.2007.04.011
40. Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev (2004) 44(1):65–81. doi:10.1016/j.brainresrev.2003.08.007
41. Andersen MH, Schrama D, Thor Straten P, Becker JC. Cytotoxic T cells. J Invest Dermatol (2006) 126(1):32–41. doi:10.1038/sj.jid.5700001
42. Esser MT, Haverstick DM, Fuller CL, Gullo CA, Braciale VL. Ca2+ signaling modulates cytolytic T lymphocyte effector functions. J Exp Med (1998) 187(7):1057–67. doi:10.1084/jem.187.7.1057
43. Kessler B, Hudrisier D, Schroeter M, Tschopp J, Cerottini JC, Luescher IF. Peptide modification or blocking of CD8, resulting in weak TCR signaling, can activate CTL for Fas- but not perforin-dependent cytotoxicity or cytokine production. J Immunol (1998) 161(12):6939–46.
44. Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol (2004) 5(5):524–30. doi:10.1038/ni0604-658a
45. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity (1996) 4(6):565–71. doi:10.1016/S1074-7613(00)80483-5
46. Kageyama S, Tsomides TJ, Sykulev Y, Eisen HN. Variations in the number of peptide-MHC class I complexes required to activate cytotoxic T cell responses. J Immunol (1995) 154(2):567–76.
47. Shilkrut M, Gealekman O, Rosen D, Berke G, Woodcock E, Binah O. Electrophysiologic perturbations and arrhythmogenic activity caused by activation of the Fas receptor in murine ventricular myocytes: role of the inositol trisphosphate pathway. J Cardiovasc Electrophysiol (2001) 12(2):185–95. doi:10.1046/j.1540-8167.2001.00185.x
48. Binah O. Cytotoxic lymphocytes and cardiac electrophysiology. J Mol Cell Cardiol (2002) 34(9):1147–61. doi:10.1006/jmcc.2002.2056
49. Binah O, Marom S, Rubinstein I, Robinson RB, Berke G, Hoffman BF. Immunological rejection of heart transplant: how lytic granules from cytotoxic T lymphocytes damage guinea pig ventricular myocytes. Pflugers Arch (1992) 420(2):172–9. doi:10.1007/BF00374987
50. Felzen B, Berke G, Rosen D, Coleman R, Tschopp J, Young JD, et al. Effects of purified perforin and granzyme A from cytotoxic T lymphocytes on guinea pig ventricular myocytes. Cardiovasc Res (1994) 28(5):643–9. doi:10.1093/cvr/28.5.643
51. Persechini PM, Young JD, Almers W. Membrane channel formation by the lymphocyte pore-forming protein: comparison between susceptible and resistant target cells. J Cell Biol (1990) 110(6):2109–16. doi:10.1083/jcb.110.6.2109
52. Young JD, Nathan CF, Podack ER, Palladino MA, Cohn ZA. Functional channel formation associated with cytotoxic T-cell granules. Proc Natl Acad Sci U S A (1986) 83(1):150–4. doi:10.1073/pnas.83.1.150
53. Binah O, Berke G, Rosen D, Hoffman BF. Calcium channel blockers modify electrophysiological effects induced by lytic granules from cytotoxic T lymphocytes in guinea pig ventricular myocytes. J Pharmacol Exp Ther (1994) 268(3):1581–7.
54. Felzen B, Shilkrut M, Less H, Sarapov I, Maor G, Coleman R, et al. Fas (CD95/Apo-1)-mediated damage to ventricular myocytes induced by cytotoxic T lymphocytes from perforin-deficient mice: a major role for inositol 1,4,5-trisphosphate. Circ Res (1998) 82(4):438–50. doi:10.1161/01.RES.82.4.438
55. Beck H, Steffens R, Heinemann U, Elger CE. Ca(2+)-dependent inactivation of high-threshold Ca(2+) currents in hippocampal granule cells of patients with chronic temporal lobe epilepsy. J Neurophysiol (1999) 82(2):946–54.
56. Lee J, Park K, Lee S, Whang K, Kang M, Park C, et al. Differential changes of calcium binding proteins in the rat striatum after kainic acid-induced seizure. Neurosci Lett (2002) 333(2):87–90. doi:10.1016/S0304-3940(02)00987-4
57. Otmakhov N, Gorbacheva EV, Regmi S, Yasuda R, Hudmon A, Lisman J. Excitotoxic insult results in a long-lasting activation of CaMKIIalpha and mitochondrial damage in living hippocampal neurons. PLoS One (2015) 10(3):e0120881. doi:10.1371/journal.pone.0120881
58. Meuth S, Kanyshkova T, Landgraf P, Pape H-C, Budde T. Influence of Ca2+-binding proteins and the cytoskeleton on Ca2+-dependent inactivation of high-voltage activated Ca2+ currents in thalamocortical relay neurons. Pflugers Arch (2005) 450(2):111–22. doi:10.1007/s00424-004-1377-z
59. Berridge MJ. Neuronal calcium signaling. Neuron (1998) 21(1):13–26. doi:10.1016/S0896-6273(00)80510-3
60. Meuth S, Herrmann A, Simon O, Siffrin V, Melzer N, Bittner S, et al. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci (2009) 29(49):15397–409. doi:10.1523/jneurosci.4339-09.2009
61. Malipiero U, Heuss C, Schlapbach R, Tschopp J, Gerber U, Fontana A. Involvement of the N-methyl-D-aspartate receptor in neuronal cell death induced by cytotoxic T cell-derived secretory granules. Eur J Immunol (1999) 29(10):3053–62. doi:10.1002/(SICI)1521-4141(199910)29:10<3053::AID-IMMU3053>3.0.CO;2-I
62. Melzer N, Hicking G, Bittner S, Bobak N, Göbel K, Herrmann AM, et al. Excitotoxic neuronal cell death during an oligodendrocyte-directed CD8+ T cell attack in the CNS gray matter. J Neuroinflammation (2013) 10:121. doi:10.1186/1742-2094-10-121
63. Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol (2008) 180(6):3866–73. doi:10.4049/jimmunol.180.6.3866
64. Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J (2008) 22(6):1797–806. doi:10.1096/fj.07-099499
65. Baldwin RL, Stolowitz ML, Hood L, Wisnieski BJ. Structural changes of tumor necrosis factor alpha associated with membrane insertion and channel formation. Proc Natl Acad Sci U S A (1996) 93(3):1021–6. doi:10.1073/pnas.93.3.1021
66. Kagan BL, Baldwin RL, Munoz D, Wisnieski BJ. Formation of ion-permeable channels by tumor necrosis factor-alpha. Science (1992) 255(5050):1427–30. doi:10.1126/science.1371890
67. Kagan BL, Mirzabekov T, Munoz D, Baldwin RL, Wisnieski B. The role of channel formation in the mechanism of action of tumor necrosis factors. Ann N Y Acad Sci (1993) 707:317–27. doi:10.1111/j.1749-6632.1993.tb38062.x
68. Köller H, Siebler M, Hartung HP. Immunologically induced electrophysiological dysfunction: implications for inflammatory diseases of the CNS and PNS. Prog Neurobiol (1997) 52(1):1–26. doi:10.1016/S0301-0082(96)00065-2
69. Saxena A, Bauer J, Scheikl T, Zappulla J, Audebert M, Desbois S, et al. Cutting edge: multiple sclerosis-like lesions induced by effector CD8 T cells recognizing a sequestered antigen on oligodendrocytes. J Immunol (2008) 181(3):1617–21. doi:10.4049/jimmunol.181.3.1617
70. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med (2001) 194(5):669–76. doi:10.1084/jem.194.5.669
71. Zamvil S, Nelson P, Trotter J, Mitchell D, Knobler R, Fritz R, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature (1985) 317(6035):355–8. doi:10.1038/317355a0
72. DeLuca I, Blachere NE, Santomasso B, Darnell RB. Tolerance to the neuron-specific paraneoplastic HuD antigen. PLoS One (2009) 4(6):e5739. doi:10.1371/journal.pone.0005739
73. Blachere NE, Orange DE, Santomasso BD, Doerner J, Foo PK, Herre M, et al. T cells targeting a neuronal paraneoplastic antigen mediate tumor rejection and trigger CNS autoimmunity with humoral activation. Eur J Immunol (2014) 44(11):3240–51. doi:10.1002/eji.201444624
74. Srinivasan M, Frauwirth KA. Peripheral tolerance in CD8+ T cells. Cytokine (2009) 46(2):147–59. doi:10.1016/j.cyto.2009.01.010
75. Na SY, Hermann A, Sanchez-Ruiz M, Storch A, Deckert M, Hunig T. Oligodendrocytes enforce immune tolerance of the uninfected brain by purging the peripheral repertoire of autoreactive CD8+ T cells. Immunity (2012) 37(1):134–46. doi:10.1016/j.immuni.2012.04.009
76. Merkler D, Horvath E, Bruck W, Zinkernagel RM, Del la Torre JC. Pinschewer DD. “Viral deja vu” elicits organ-specific immune disease independent of reactivity to self. J Clin Invest (2006) 116(5):1254–63. doi:10.1172/JCI27372
77. Bauer J, Elger CE, Hans VH, Schramm J, Urbach H, Lassmann H, et al. Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol (2007) 62(1):67–80. doi:10.1002/ana.21148
78. Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol (2014) 13(2):195–205. doi:10.1016/S1474-4422(13)70260-6
79. Honnorat J, Aguera M, Zalc B, Goujet C, Quach T, Antoine JC, et al. POP66, a paraneoplastic encephalomyelitis-related antigen, is a marker of adult oligodendrocytes. J Neuropathol Exp Neurol (1998) 57(4):311–22. doi:10.1097/00005072-199804000-00002
80. Honnorat J, Antoine JC, Derrington E, Aguera M, Belin MF. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry (1996) 61(3):270–8. doi:10.1136/jnnp.61.3.270
81. Honnorat J, Byk T, Kusters I, Aguera M, Ricard D, Rogemond V, et al. Ulip/CRMP proteins are recognized by autoantibodies in paraneoplastic neurological syndromes. Eur J Neurosci (1999) 11(12):4226–32. doi:10.1046/j.1460-9568.1999.00864.x
82. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain (2000) 123(Pt 6):1174–83. doi:10.1093/brain/123.6.1174
83. Göbel K, Melzer N, Herrmann A, Schuhmann M, Bittner S, Ip C, et al. Collateral neuronal apoptosis in CNS gray matter during an oligodendrocyte-directed CD8(+) T cell attack. Glia (2010) 58(4):469–80. doi:10.1002/glia.20938
84. Sobottka B, Harrer MD, Ziegler U, Fischer K, Wiendl H, Hunig T, et al. Collateral bystander damage by myelin-directed CD8+ T cells causes axonal loss. Am J Pathol (2009) 175(3):1160–6. doi:10.2353/ajpath.2009.090340
85. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol (2014) 76(1):108–19. doi:10.1002/ana.24195
86. Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain (2015) 138(Pt 1):94–109. doi:10.1093/brain/awu310
87. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30(17):5866–75. doi:10.1523/JNEUROSCI.0167-10.2010
88. Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol (2015) 77(3):381–98. doi:10.1002/ana.24293
89. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol (2009) 65(4):424–34. doi:10.1002/ana.21589
90. Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci (2013) 33(46):18161–74. doi:10.1523/JNEUROSCI.3506-13.2013
91. Deprez F, Zattoni M, Mura ML, Frei K, Fritschy JM. Adoptive transfer of T lymphocytes in immunodeficient mice influences epileptogenesis and neurodegeneration in a model of temporal lobe epilepsy. Neurobiol Dis (2011) 44(2):174–84. doi:10.1016/j.nbd.2011.06.011
92. Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci (2011) 31(11):4037–50. doi:10.1523/JNEUROSCI.6210-10.2011
93. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell (1994) 76(1):17–27. doi:10.1016/0092-8674(94)90169-4
94. Sanchez-Ruiz M, Wilden L, Muller W, Stenzel W, Brunn A, Miletic H, et al. Molecular mimicry between neurons and an intracerebral pathogen induces a CD8 T cell-mediated autoimmune disease. J Immunol (2008) 180(12):8421–33. doi:10.4049/jimmunol.180.12.8421
95. Carlson GC, Coulter DA. In vitro functional imaging in brain slices using fast voltage-sensitive dye imaging combined with whole-cell patch recording. Nat Protoc (2008) 3(2):249–55. doi:10.1038/nprot.2007.539
96. Broicher T, Bidmon HJ, Kamuf B, Coulon P, Gorji A, Pape HC, et al. Thalamic afferent activation of supragranular layers in auditory cortex in vitro: a voltage sensitive dye study. Neuroscience (2010) 165(2):371–85. doi:10.1016/j.neuroscience.2009.10.025
97. Obien ME, Deligkaris K, Bullmann T, Bakkum DJ, Frey U. Revealing neuronal function through microelectrode array recordings. Front Neurosci (2014) 8:423. doi:10.3389/fnins.2014.00423
98. Gonzalez-Sulser A, Wang J, Motamedi G, Avoli M, Vicini S, Dzakpasu R. The 4-aminopyridine in vitro epilepsy model analyzed with a perforated multi-electrode array. Neuropharmacology (2011) 60(7–8):1142–53. doi:10.1016/j.neuropharm.2010.10.007
Keywords: limbic encephalitis, cytotoxic CD8+ T cell, T cell–neuron interaction, future strategies, autoimmune neurological disease
Citation: Ehling P, Melzer N, Budde T and Meuth SG (2015) CD8+ T cell-mediated neuronal dysfunction and degeneration in limbic encephalitis. Front. Neurol. 6:163. doi: 10.3389/fneur.2015.00163
Received: 10 April 2015; Accepted: 02 July 2015;
Published: 15 July 2015
Edited by:
Christian E. Elger, University of Bonn, GermanyReviewed by:
Christopher French, University of Melbourne, AustraliaChristoph Kleinschnitz, University of Würzburg, Germany
Copyright: © 2015 Ehling, Melzer, Budde and Meuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Ehling and Nico Melzer, Department of Neurology, University of Münster, Albert-Schweitzer-Campus 1, Münster 48149, Germany,cGV0cmEuZWhsaW5nQHVrbXVlbnN0ZXIuZGU=;bmljby5tZWx6ZXJAdWttdWVuc3Rlci5kZQ==