 Gregory Hook
Gregory Hook J. Steven Jacobsen
J. Steven Jacobsen Kenneth Grabstein
Kenneth Grabstein Mark Kindy4,5
Mark Kindy4,5 Vivian Hook
Vivian Hook- 1American Life Science Pharmaceuticals, Inc., San Diego, CA, USA
- 2AstraZeneca Neuroscience iMed, Cambridge, MA, USA
- 3Department of Chemical Engineering, University of Washington, Seattle, WA, USA
- 4Department of Neurosciences, Medical University of South Carolina, Charleston, SC, USA
- 5Ralph H. Johnson Veterans Administration Medical Center, Charleston, SC, USA
- 6Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA, USA
- 7Department of Neurosciences, Department of Pharmacology, University of California San Diego, La Jolla, CA, USA
There is currently no therapeutic drug treatment for traumatic brain injury (TBI) despite decades of experimental clinical trials. This may be because the mechanistic pathways for improving TBI outcomes have yet to be identified and exploited. As such, there remains a need to seek out new molecular targets and their drug candidates to find new treatments for TBI. This review presents supporting evidence for cathepsin B, a cysteine protease, as a potentially important drug target for TBI. Cathepsin B expression is greatly up-regulated in TBI animal models, as well as in trauma patients. Importantly, knockout of the cathepsin B gene in TBI mice results in substantial improvements of TBI-caused deficits in behavior, pathology, and biomarkers, as well as improvements in related injury models. During the process of TBI-induced injury, cathepsin B likely escapes the lysosome, its normal subcellular location, into the cytoplasm or extracellular matrix (ECM) where the unleashed proteolytic power causes destruction via necrotic, apoptotic, autophagic, and activated glia-induced cell death, together with ECM breakdown and inflammation. Significantly, chemical inhibitors of cathepsin B are effective for improving deficits in TBI and related injuries including ischemia, cerebral bleeding, cerebral aneurysm, edema, pain, infection, rheumatoid arthritis, epilepsy, Huntington’s disease, multiple sclerosis, and Alzheimer’s disease. The inhibitor E64d is unique among cathepsin B inhibitors in being the only compound to have demonstrated oral efficacy in a TBI model and prior safe use in man and as such it is an excellent tool compound for preclinical testing and clinical compound development. These data support the conclusion that drug development of cathepsin B inhibitors for TBI treatment should be accelerated.
Introduction
Traumatic brain injury (TBI) occurs when an external force, such as that due to a vehicular accident, a football collision, or a bullet, causes brain dysfunction and pathology. Unfortunately, TBI is all too common with over 10 million people worldwide afflicted each year (1) and at least 1.7 million cases annually in the United States where it is a leading cause of death among the young and the elderly (2, 3). Falls are the primary cause of TBI among the very young and old, whereas auto accidents and sports injuries are the main cause in 15- to 24-year-olds and most of those occur in males (2, 4). Military casualties add to these numbers with an overwhelming number of those due to blast injury and most of those are males (5).
TBI encompasses a continuum of injuries and pathologies and is symptomatically classified into mild, moderate, or severe based on the level of patient consciousness (6, 7). Much is known about the complex consequences of TBI (8, 9) including the necrotic and apoptotic neuronal cell death that occurs (10) and the TBI-related pathologies, which include, for example, inflammation (11), breakdown of vascular walls (12), ischemia (13), subarachnoid aneurysms and hemorrhages (14), brain edema (15), inflammatory pain (16), increased intracranial pressure (ICP) (17), infections (18), and neuroexcitatory toxicity (19). Moreover, sufficiently severe or repetitive TBI results in increased risks for many age-related neurological diseases, including amyotrophic lateral sclerosis (ALS) (20), Parkinson’s disease (PD) (21), Alzheimer’s disease (AD) (22), epilepsy (23), and possibly multiple sclerosis (MS) (24). As such, TBI can also be viewed as a model of neurodegenerative diseases generally (25).
But despite this knowledge, there is currently no pharmaceutically effective treatment for TBI even though many experimental clinical trials have been conducted over decades to find such (26–28). This may be because the drug targets, which affect outcomes, have yet to be identified and exploited and thus there is a continuing need for new TBI therapeutic targets.
Proteases and small molecule protease inhibitors are a proven target and means for successful pharmaceutical intervention as such inhibitors are approved for drug use to treat hypertension (29), HIV infection (30), and multiple myeloma cancer (31). The cysteine protease, cathepsin B, is a potential drug target for several diseases (32), including, for example, various cancers (33–36), pancreatitis (37), liver fibrosis (38), rheumatoid arthritis (RA) (39, 40) viral Ebola (41), bacterial Streptococcus pneumoniae meningitis (42), and parasitic Trypanosoma cruzi infections (43). While no cathepsin B inhibitor has yet been approved for drug use, one has completed Phase 1 trials for fatty liver disease (44, 45) and another is in late preclinical stage for treating Chagas disease (46).
However, cathepsin B’s potential as a TBI drug target has received relatively little attention with the last review to discuss this protease in a TBI context published a decade ago (47). Since then, cathepsin B gene knockout data have clearly demonstrated that cathepsin B is a significant cause of the behavioral dysfunction and pathology that occurs in animal models as a result of TBI (48). This review summarizes the evidence showing that genetically eliminating or pharmaceutically reducing cathepsin B activity produces improved outcomes in animal models of TBI, other types of trauma, and the many TBI-related pathologies mentioned above. The review also focuses on the small molecule, E64d and its derivatives, as a tool compound for developing a TBI lead therapeutic because of its demonstrated efficacy by many groups by many routes of administration in many TBI and TBI-related animal models and its prior safe use in man. The paper includes a basic introduction to the enzymology, biology, distribution, regulation, and function of cathepsin B and how increased expression and redistribution of cathepsin B from lysosomes to the cell cytosol and extracellular matrix (ECM) likely causes the cathepsin B-induced pathology. The overall conclusion drawn is that cathepsin B is an important target for treatment of TBI and that E64d and its derivatives have many of the preclinical properties needed for a successful TBI therapeutic agent (49–51) and should be developed for such.
Cathepsin B Properties: Enzymology, Genetics, Transcription, and Translation
Cathepsin B Enzymology
Cathepsin B is among the most studied proteases as there are numerous reports written over the last 76 years. Its proteolytic activity was first identified in beef tissue (52). Originally called cathepsin II (53), it was renamed cathepsin B 63 years ago (54) and was purified 5 years after that (55). The first amino acid sequences were determined 32 years ago (56), and the first human, rat, and mouse genes were cloned 3 years later (57). The first X-ray crystal structure was resolved almost 25 years ago (58). While the first report on cathepsin B gene-deficient mice was made 17 years ago (59), it was not until last year that such animals were evaluated for improving TBI deficits (48).
Endopeptidase and Exopeptidase Activity
Most proteases have either endopeptidase or exopeptidase activity. Cathepsin B is unusual in having endopeptidase activities (60) as well as peptidyl-dipeptidase (61) and carboxypeptidase (62) exopeptidase activities. Its multi-enzymatic capability is due to a unique structural element of the protein called the “occluding loop” (58). At low pH, as occurs in the lysosome, the loop closes on the enzymatic active site and does not allow polypeptide binding and thereby reduces endopeptidase activity but permits peptidyl-dipeptidase and carboxypeptidase activities. At higher pH, the loop opens from the active site and allows more endopeptidase activity (63). The multiple activities of cathepsin B make it particularly well suited among proteases for digesting unwanted proteins, but also make it potentially more destructive in TBI brain damage and pathology.
Selectivity of Peptide Cleavage Sites
Endopeptidases recognize specific amino acid sequences flanking the scissile peptide bond of the cleavage site (64) (see Figure 1). A small peptide substrate containing the recognition sequence can mimic the recognition site in proteins and can be used to monitor protease activity. Using such substrates, cathepsin B has been shown to prefer positively charged arginine (Arg) or lysine (Lys) at the P1 position, which is an unusual recognition signal among proteases, and a bulky hydrophobic amino acid residue at P2 (65, 66). Cathepsin B will also accept a basic residue, Arg, at the substrate P2 position (67). Thus, the substrate Z-Arg-Arg-AMC (where Z = benzyloxycarbonyl) with AMC (amido-4-methylcoumarin hydrochloride) is commonly used to assay cathepsin B activity in vitro as its cleavage liberates fluorescent AMC that monitors protease activity. While the recognition motif is not absolute, significant changes in the types of amino acids adjacent to the cleavage site can drastically affect endopeptidase binding and cleavage.
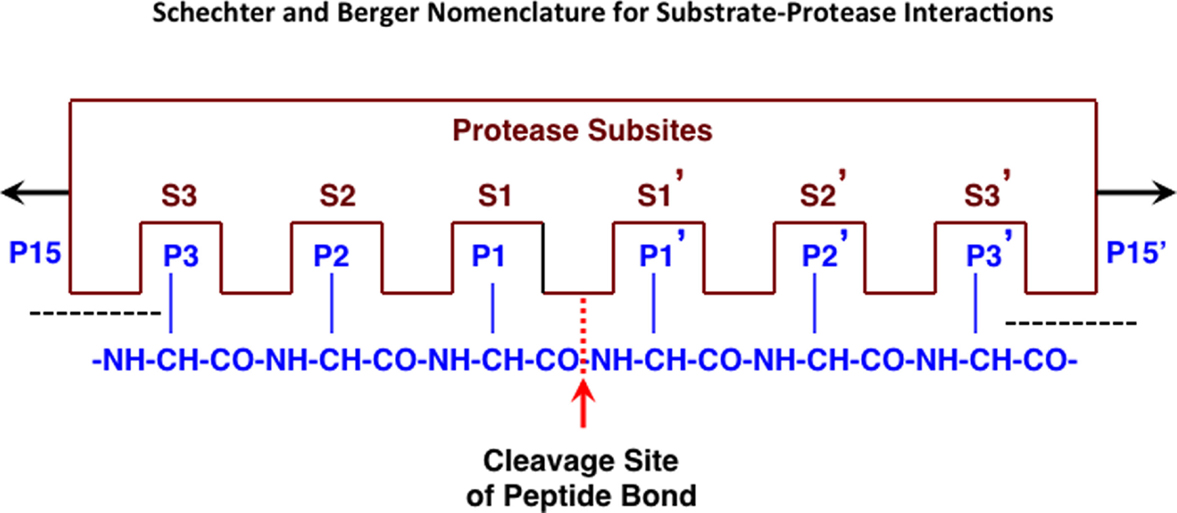
Figure 1. Protease interactions with polypeptide substrates for proteolysis. This figure illustrates protease and polypeptide substrate interactions utilizing the Schechter and Berger nomenclature (68). The active site of a protease is composed of several subsites. The scheme shows an active site of six subsites, termed S1–S3 and S1′–S3′. Subsites are located on both sides of the catalytic site and are numbered from there in either direction. The positions of the amino acid residues of the peptide substrate represent their locations from the cleaved peptide bonds and have the same numbering as the subsites they occupy (P1–P3 and P1′–P3′). Cleavage occurs between P1 and P1′ residues. Figure revised from Ref. (69).
Classification
Proteases are classified by catalytic type, which are composed of aspartic, cysteine, metallo, serine, and threonine, and a small group of uncertain catalytic type. Cathepsin B has a reactive cysteine residue within its catalytic domain and belongs to the cysteine catalytic class. Proteases are further classified according to their clan, which is based on the homology of the three-dimensional structure, the arrangement of catalytic residues in the active site, and the amino acid sequence around the catalytic site. Cathepsin B is a member of the CA clan and shares a common structure with the dyad reactive cysteine and histidine residues at specific positions within the catalytic site (70). They are further classified into families in which they are grouped together with proteases having a statistically significant relationship in the amino acid sequence to a representative member type. The cathepsin B sequence is highly homologous to the plant cysteine protease papain (56), which is the reference protease of the papain-like family. Thus, cathepsin B is a cysteine protease belonging to the CA clan and the papain-like family, C1A. The other papain-like cysteine proteases in man are cathepsins F, H, C, K, L, O, S, V, W, and X. Enzymes as a whole are also systematically classified according to the Enzyme Commission (EC) based on a numerical code related to the type of activity catalyzed by the enzyme but not on homology and the enzyme code for cathepsin B is EC3.4.22.1.
Common protease names, which are often based on their discovery and not on catalytic type or homology, can cause confusion. For example, the name “cathepsin,” which is derived from the Greek katehepsin (to digest), was given to the protease activity in an acidic environment. Those proteases were subsequently found to be of different catalytic types and include the serine proteases cathepsins A and G, the aspartic proteases cathepsins D and E, and the lysosomal papain-like cysteine proteases (70, 71).
Cathepsin B Genetics
In man, cathepsin B is encoded as a single gene on chromosome 8 at position p22-23.1 (72), spans 27 kilobases, and contains 13 exons (73, 74). Interestingly, the gene location of cathepsin B on chromosome 8p is near a potential hub for development of neuropsychiatric disorders and an area of high divergence between chimpanzee and man (75). Human cathepsin B gene polymorphisms are associated with the disease tropical calcific pancreatitis (76). In mouse, cathepsin B is also encoded as a single gene on chromosome 14 at position 33.24, spans 20 kilobases, and contains 10 exons and 9 introns (77, 78). The human and mouse nucleic acid sequences encoding the proteolytically active form have 82% homology (57).
Cathepsin B Transcription
Human cathepsin B messenger ribonucleic acid (mRNA) consists of multiple messages that differ in their 5′ and 3′ untranslated regions (UTRs) and arise by alternative splicing. Most tissues express 2.3 and 4.0 kilobase (kb) transcripts at a ratio of 2:1; but the ratio of mRNAs with variant 5′ UTRs differs widely (74). Normal human brain contains both cathepsin B mRNA transcripts but most is the 2.3 kb form (79).
Small, non-coding microRNA (miRNA) inhibit the expression and function of endogenously encoded proteins (80). The miRNA expression patterns vary among organs (81), are altered by central nervous system (CNS) injuries, and the changes vary among spinal cord contusion (SCC), stroke, and TBI injuries (82). TBI causes at least 35 and 50 miRNAs to be up- and down-regulated, respectively, and their predicted targets involve signal transduction, transcription, proliferation, and differentiation (83). miR-218 and miR-128 down-regulate cathepsin B expression when they are up-regulated in medulloblastoma cell lines (84) and AD monocytes and lymphocytes (85) and are expressed in brain neurons (81, 86).
Cathepsin B Translation
Cathepsin B post-translational processing is well understood and has been the subject of many reviews (60, 70, 87). Human cathepsin B is initially transcribed as a preproenzyme of 339 amino acids. The 17 amino acid pre-signal sequence is co-translationally removed and the resultant proenzyme is translocated to the Golgi-apparatus where aspargine becomes glycosylated by a high-mannose-type sugar. The mannose-6-phosphate residues target the proenzyme to the lysosomes. The preproenzye or proenzyme forms are not enzymatically active. Activation of the proenzyme occurs with cleavage of the 62-residue pro region, resulting in mature cathepsin B. The mature form (30 kDa) is enzymatically active and can be further processed by removal of the C-terminal 6 amino acids and the excision of a dipeptide resulting a heavy chain (25 kDa) and a light chain (5 kDa). The heavy and light chains form a 2-chain form (30 kDa) linked together by a cysteine–cysteine bond. The heavy and 2-chain forms are enzymatically active. In some cells, such as neuroendocrine cells, cathepsin B is also trafficked to the regulated secretory pathway (RSP) but how that occurs is not known. The cathepsin B prepro, pro, and mature amino acid sequences of mouse and man are 65%, 74%, and 82% homologous, respectively (57). Normal human brain contains inactive precursor cathepsin B and proteolytically active forms (79).
Proteases are stored in the cell as inactive zymogens that require hydrolysis for activation, and allow cells to rapidly deploy active protease as needed (88). In the case of cathepsin B, the inactive proenzyme is hydrolyzed by a variety of proteases, including the aspartic protease cathepsin D, other cysteine proteases, and cathepsin B self-cleavage to liberate the mature active form (89–91). Maximum cathepsin B zymogen activation occurs at an acidic pH and very little occurs at a neutral pH (91), but cathepsin B zymogen can endure a neutral pH and be subsequently activated in an acidic environment (60). Mature cathepsin B is degraded by cysteine protease nicking (87, 92).
Traumatic Brain Injury and TBI-Related Injuries Activate Cathepsin B
PreClinical Data: Cathepsin B in TBI and TBI-Related Animal Models
TBI Increases Cathepsin B Expression in Animal Models
As shown in Table 1, cathepsin B mRNA, protein, and activity are increased following trauma in TBI animal models. Trauma significantly up-regulates brain cathepsin B mRNA expression about 2.5-fold relative to controls at 3 days after TBI in fluid percussion injury rat and controlled cortical impact (CCI) mouse models (93). Trauma elevated cathepsin B protein levels and activity, using a weight drop mouse model, in the cortex at the injury site and in the hippocampus distal to the injury site beginning as soon as 6 h; the elevated brain cathepsin B continued for at least 1 week with a 75% maximum increase occurring at about two days post-trauma (94, 95). In a severe TBI rat model, brain trauma elevated brain cathepsin B protein levels within 1 h and those levels remain elevated for up to 32 days after injury with a 300% maximum increase at 8 days post injury (96). Our data in a severe CCI mouse model show a 100% and 380% increase in brain cathepsin B activity and protein levels at 2 and 24 h post-trauma, respectively (48). Trauma elevates cathepsin B protein levels 1 day to 3 days post injury in a moderate closed-skull TBI rat model and this increase varied with injury location (97). The prolonged elevation of cathepsin B after trauma suggests that it may be possible to therapeutically intervene clinically, as treatment often does not begin until several hours after injury. The fact that different groups using different moderate and severe TBI animal models find increased cathepsin B following injury shows that higher cathepsin B expression is a consistent response in rodent TBI models.
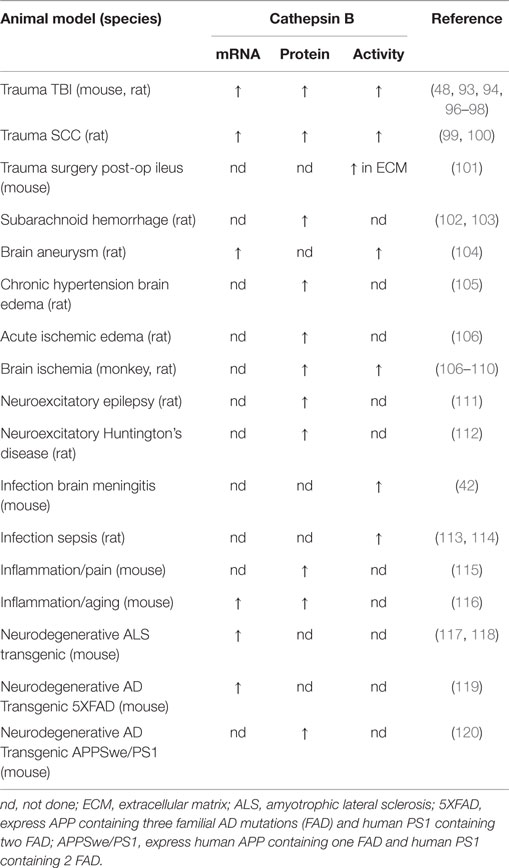
Table 1. Cathepsin B is activated in TBI and TBI-related animal models.
TBI-Related Animal Models
Table 1 shows that cathepsin B expression is increased in several TBI-related injuries in animal models. SCC is related to TBI in that both are trauma to the CNS and in a SCC rat model, cathepsin B mRNA, 37 kDa proprotein, 30 kDa mature protein, 25 kDa mature protein, and activity increased 20-, 3.5-, 4.5-, 10-, and 7-fold, respectively, at the injury site 7 days after trauma relative to sham animals (99, 100). Surgery is a form of trauma and in a surgical post-operative ileus mouse model, cathepsin B activity increased twofold in the ECM on the day after trauma relative to control (101) and was accompanied by a loss of the ECM microvascular basal lamina and collagen-type IV as is characteristically observed in TBI (12). Thus, other trauma models also show a similar increase in cathepsin B expression as seen in TBI models.
TBI causes many vascular pathologies including ischemia, aneurysms, subarachnoid hemorrhages, and edema (13–15). Increased cathepsin B expression has long been known to result from ischemia with brain activity increasing about two- and fivefold relative to controls at 2 and 5 days after injury in ischemic rat and monkey models, respectively (107–109). Similarly, in an aneurysm model, cathepsin B mRNA increased about fivefold and activity increased about twofold 3 months after aneurysm induction relative to controls (104). In a subarachnoid hemorrhage model, cathepsin B protein increased about 3.5-fold at 2 and 3 days after hemorrhage induction relative to controls (102, 103). Cathepsin B protein levels are increased in chronic hypertension brain edema (105) and acute ischemic edema models (106). Thus, cathepsin B expression is also increased in important vascular pathologies caused by TBI.
Brain infections are a common TBI complication (18) and cathepsin B levels are greatly increased in meningitis and sepsis animal models (42, 113, 114). Moreover, aseptic inflammation and pain are major factors in TBI (16), and cathepsin B is elevated in aseptic inflammatory and pain animal models (115, 116).
Finally, as discussed above, TBI is a risk factor for several neurodegenerative diseases. Cathepsin B mRNA is elevated 3.5-fold relative to controls in spinal cord samples from an ALS mouse model (117). Moreover, an ALS meta-analysis of the literature identified cathepsin B as repeatedly up-regulated in ALS patients and mouse models (121). TBI causes excitotoxicity (19) and cathepsin B protein levels are elevated in excitotoxicity animal models of recurring epilepsy and Huntington’s chorea (112, 122). Furthermore, cathepsin B mRNA is increased in the transgenic AD 5Xfamilial AD (FAD) mouse model, which expresses human amyloid precursor protein (APP) containing three familial AD mutations and human presenilin 1 (PSN1) containing two familial mutations (119). Cathepsin B protein is also increased by 50% in the cortex and hippocampus of the APPSwe/PS1 model, which expresses human APP containing the Swedish (Swe) FAD mutation and PS1 with FAD mutations, relative to controls (120). Thus, cathepsin B is elevated in several neurodegenerative animal models associated with TBI.
Clinical Data: Cathepsin B Regulation in TBI-Related Injuries
Short-Term Changes in Cathepsin B Observed in Polytrauma and Aneurysm
We are not aware of any clinical studies on acute cathepsin B regulation due to TBI, but non-brain polytrauma patients show increases in plasma cathepsin B activity during the first day after trauma, which subsequently falls to moderately elevated levels by the third day, and remains roughly at that level for up to 2 weeks. Importantly, the increase in plasma cathepsin B activity correlates with the severity of injury. Patients with a sixfold increase in plasma cathepsin B activity 1 day post-trauma subsequently developed fatal or reversible multiple organ failure, whereas those who had only a threefold increase, at the same day, did not display organ failure (123, 124).
Moreover, in human cerebral aneurysm tissue, cathepsin B is highly expressed in the endothelial cell layer and the media in the aneurysmal walls in contrast to control artery tissue where it is barely expressed at all (104). Thus, cathepsin B expression appears to be significantly increased as a result of TBI type injuries in man.
Long-Term Changes in Cathepsin B in Chronic Inflammatory Conditions
Patients having the chronic inflammatory neurological diseases Guillain–Barre syndrome, chronic demyelinating polyneuropathy, or MS have higher cerebrospinal fluid (CSF) cathepsin B activity levels than controls (125, 126). Cathepsin B mRNA levels in ALS postmortem spinal cord tissue has been shown to be about 2- and 3.3-fold higher than that of non-neurological age-matched controls (117, 127) and cathepsin B protein expression is increased and has an abnormal distribution, especially in the anterior horn, relative to controls (128). AD brain autopsy samples show a high cathepsin B protein expression, especially near pathological amyloid plaque brain deposits, relative to age-matched control samples (129). Moreover, AD patients have a significant 50% higher plasma and serum cathepsin B protein level than age-matched control samples (120, 130) and higher serum cathepsin B levels in AD patients strongly correlates with reduced cognitive ability (120). Interestingly, cathepsin B protein levels in peripheral blood lymphocytes and monocytes from AD patients are about 50% lower than those from controls (85) and that, taken together with the plasma and serum data, suggest that cathepsin B may redistribute from peripheral blood cells to the serum/plasma compartment in AD patients. CSF cathepsin B studies in AD patients have shown a significant increase (131) by proteomic analysis and no significant difference but a trend toward higher levels by ELISA and Western blot analysis relative to controls (130, 132).
In the peripheral inflammatory conditions of RA and osteoarthritis (OA) synovial cells and chondrocytes have increased cathepsin B mRNA levels and cathepsin B protein secretion relative to controls (133, 134). Furthermore, in patients with inflammatory bowel disease, cathepsin B is up-regulated in areas of tissue damage and mucosal ulceration (135).
Cathepsin B Gene Knockout Improves Deficits in TBI and TBI-Related Animal Models
Cathepsin B Knockout Mice are Healthy
The health of cathepsin B gene knockout mice is maintained and generally indistinguishable from normal littermates in behavior, histology, and fertility (136), as shown by data summarized in Table 2. The only reported difference is a decrease in thyroglobulin (Tg) solubilization (137) but this does not appear to cause a significant phenotypic or behavioral effect. The normal health of mice lacking cathepsin B implicates that pharmacologic inhibition of cathepsin B will likely be generally safe.

Table 2. Animals lacking the cathepsin B gene are healthy.
TBI Mice with Cathepsin B Knockout: Improved Deficits and Pathology
Table 3 summarizes the data on the effect of deleting the cathepsin B gene in TBI models and shows that the deletion improves behavior, pathology, and neuronal cell survival (48). Figure 2 shows that deleting the cathepsin B gene reduces the severity and duration of the neuromotor dysfunction that occurs during the week following trauma with the deficient mice suffering about half the dysfunction of wild-type (wt) mice 1 day after trauma and regaining normal function, whereas the wt mice remain significantly impaired 1 week after trauma. Figures 3 and 4 show that 1 week after trauma, cathepsin B gene-deficient animals have about one-third the brain lesion volume of wt animals and no hippocampal neuronal cell loss, whereas wt mice suffered a 60% loss relative to the sham animals. As discussed below, apoptosis contributes to TBI-induced cell death and the pro-apoptotic cell death biomarker, Bax, is elevated 1 day post-trauma in wt mice, but not in cathepsin B gene knockout mice relative to sham controls. The clear conclusion is that cathepsin B is a significant contributor to the behavioral dysfunction and neuronal cell loss that follows TBI.
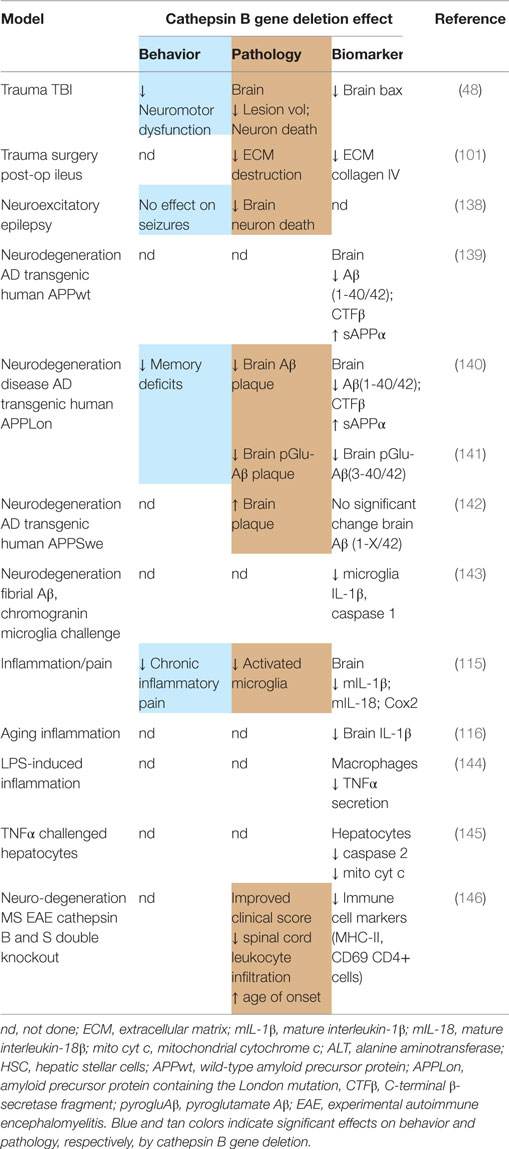
Table 3. Cathepsin B gene deletion improves deficits of TBI and TBI-related animal models.
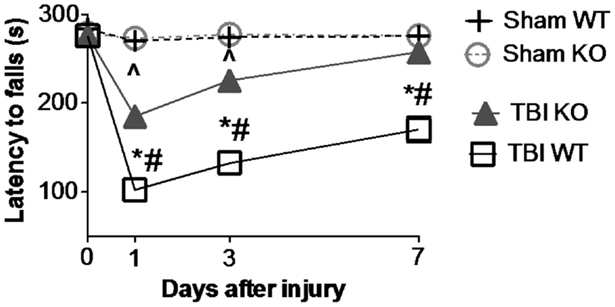
Figure 2. Cathepsin B gene deletion improves neuromotor deficits caused by TBI. Mice with knockout of the cathepsin B gene were assessed for TBI-caused neuromotor deficits. Four groups of mice were assessed: sham wild-type (Sham WT), sham cathepsin B gene knockout (Sham KO), TBI WT, and TBI cathepsin B gene knockout (TBI KO). TBI was modeled by controlled cortical impact (CCI) and mice were subjected to rotarod behavioral neuromotor evaluations before and 7 days after TBI trauma. Longer latency times indicate better neuromotor function. Sham WT and Sham KO animals were not surgically treated the same as TBI animals and were not traumatized. Significantly, knockout of the cathepsin B gene resulted in improved neuromotor defictis and a shorter recovery period compared to TBI WT mice (mean ± SEM, Bonferroni’s multiple comparison test P < 0.05, N = 10 animals/group, *TBI WT vs. Sham WT, ^TBI KO vs. Sham KO, and #TBI WT vs. TBI KO) (48). Data from cited publication adapted for graphic display.
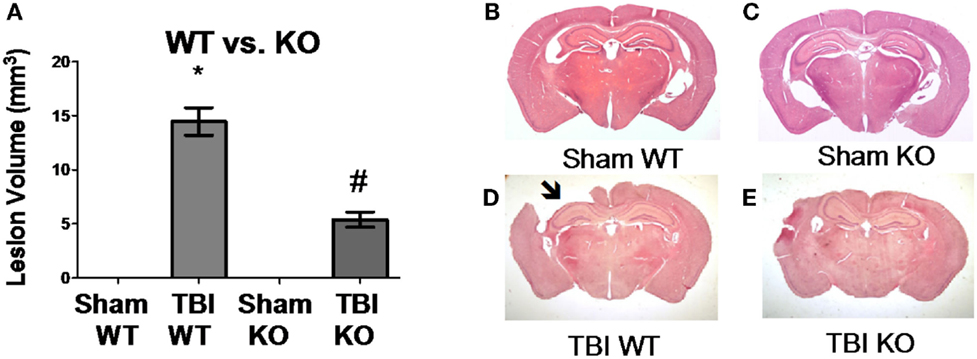
Figure 3. Cathepsin B gene deletion reduces brain tissue lesions caused by TBI. At 7 days post-TBI (mice receiving CCI TBI), mouse brains were evaluated to determine the brain volume loss (Lesion Volume) at the impact site. (A) Quantitative image analysis of brain sections showed that cathepsin B gene deletion minimized brain tissue loss. The Sham WT and Sham KO animals had no loss whereas the TBI WT mice had significant loss, but the TBI KO mice had roughly one-third the loss suffered by the TBI WT mice. Representative micrographs from the brains of Sham WT, Sham KO, TBI WT, and TBI KO animals are shown in (B–E), respectively. (mean ± SEM, Bonferroni’s multiple comparison test P < 0.05, N = 10 animals/group, *TBI WT vs. Sham WT, Sham KO, and TBI KO, and #TBI KO vs. Sham WT and Sham KO) (48). Data from cited publication adapted for graphic display.
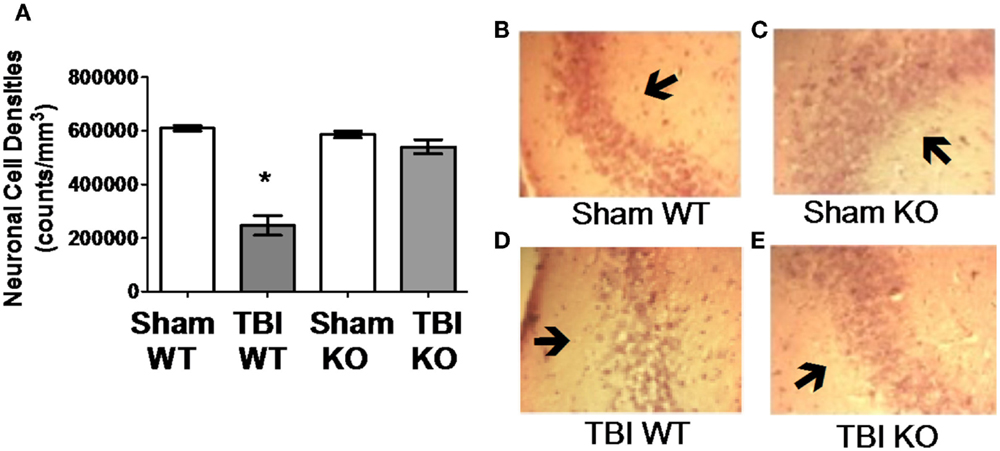
Figure 4. Cathepsin B gene deletion reduces neuronal loss after TBI. Quantitative image analyses of brain sections evaluated for Lesion Volume were also analyzed for neuronal cell density in the CA3 region of the hippocampus, which is distal to the impact site. (A) TBI WT, but not TBI KO, mice had lower neuronal density than Sham WT and Sham KO animals. Thus, cathepsin B knockout resulted in reduced neuronal loss. Representative micrographs from the brains of Sham WT, Sham KO, TBI WT, and TBI KO animals are shown in (B–E), respectively (mean ± SEM, Bonferroni’s multiple comparison test P < 0.05, N = 10 animals/group, * TBI WT vs. Sham WT, Sham KO and TBI KO) (48). Data from cited publication adapted for graphic display.
TBI-Related Models with Cathepsin B Knockout Show Improved Outcomes
Deleting the cathepsin B gene produces significant beneficial outcomes in TBI-related pathological animal models and includes surgery, epilepsy, AD, inflammation, pain, and cytokine cell death models. Deletion of both cathepsin B and cathepsin S improves the outcomes in an MS model (see Table 3).
Surgery Model
In the traumatic post-operative ileus surgery model, deleting the cathepsin B gene results in significantly less ECM breakdown and collagen-type IV loss than occurs in wt animals in that model (101). Cathepsin B, therefore, is likely a key target to preventing ECM breakdown resulting from TBI.
Epilepsy Model
TBI increases the risk of epilepsy (23), which can result in neuroexcitotoxicity. Deleting the cathepsin B gene in the Unverricht–Lundborg progressive myoclonus epilepsy model prevents about 90% of the apoptotic neuronal cell death that usually occurs in these animals. (138). Thus, cathepsin B is also likely to be an important target for preventing the TBI neuroexcitotoxity-induced cell death.
Alzheimer’s Disease Model
TBI increases the risk of AD (22), which is thought to result from the abnormal accumulation of brain amyloid-β (Aβ). In transgenic AD mice expressing human APP containing the human wt β-secretase site, which is what most AD patients have, deleting the cathepsin B gene improves memory deficits and reduces the brain amyloid plaque, which is a neuropathological hallmark of AD, that develop in these animals (140, 141). That deletion in such transgenic animals also reduces brain Aβ(1-40/42) and the pernicious post-translationally modified pyroglutamate (pGlu) Aβ(3-40/42) forms (139–141), which are thought to be particularly neurotoxic among Aβ peptide species (147). These and other data (148–150) show that cathepsin B has wt β-secretase activity, which cleaves APP and can produce Aβ species.
On the other hand, cathepsin B gene deletion in transgenic models expressing APP containing the Swe FAD mutation does not significantly affect Aβ and increases amyloid plaque (142). The Swe mutation alters the amino acid sequence at the β-secretase site, which destroys the cathepsin B target sequence and thus cathespsin B does not cleave this form of APP (151). However, the Swe FAD mutation occurs in only one extended family, whereas most people express normal APPwt. Thus, cathepsin B gene deletion reduces Aβ in models mimicking the β-secretase activity occurring in most humans. Given that, the inhibition of cathepsin B may be able to prevent the increased brain Aβ, which also occurs after TBI (152, 153).
Inflammation and Inflammatory Pain Models
TBI induces microglia activation and inflammation, which can occur for a prolonged period after trauma and can cause neuronal cell death (154). Moreover, TBI causes inflammatory pain (16) and increases the proinflammatory cytokines interleukin-1β (IL-1β), and interleukin-18 (IL-18), which are related to pain (155, 156). These cytokines are controlled by caspase 1 in protein complexes known as inflammasomes, which are also activated by TBI (157). Administering Freund’s adjuvant to animals, in the peripheral tissues, induces inflammatory pain and inflammation, but doing so to cathepsin B gene-deficient mice results in significantly less pain (tactile allodynia), less IL-1β, IL-18, caspase 1 activation, and less inflammasome activation than occurs in wt animals (115, 143). Microglia phagocytosis of fibrillar Aβ or chromogranin A (CGA) causes microglia activation and cathepsin B expression leading to inflammasome formation and production of IL-1β, and activated caspase 1. These microglia responses do not occur in cathepsin B-deficient mice (116, 158). These data suggest that cathepsin B is also a critical target for reducing CNS inflammation and inflammatory pain caused by TBI.
TNFα Models
Tumor necrosis factor alpha (TNFα) is a cytokine that induces a cytoplasmic molecular cascade including cathepsin B release from lysosomes and apoptotic cell death (159) and TBI increases brain TNFα (160). Cathepsin B gene-deficient mice are resistant to TNFα-induced liver damage and hepatocyte apoptotic cell death and have reduced TNFα-induced caspase activation and mitochondrial cytochrome c (cyt c) release, which are key apoptotic proteins (145, 161). More recently, macrophages from cathepsin B-deficient mice treated with lipopolysaccharide (LPS), which induces an intense TNFα response, were shown to secrete 50% less TNFα than wt macrophages (144). Thus, cathepsin B is an important target for preventing the TNFα-induced cell death that occurs as a result of TBI.
Cathepsin B and Cathepsin S Knockouts have Improved Outcomes in Multiple Sclerosis Model
TBI has been associated with an increased risk of subsequent MS development in the Chinese population (24). In an experimental autoimmune encephalomyelitis (EAE) mouse model of MS, deletion of cathepsin B or cathepsin S gene alone had no effect but deletion of both cathepsin B and S genes improved clinical scores and significantly delayed age of disease onset relative to sufficient animals (146).
Summary: Cathepsin B Knockout has Little Effect in Development but has Major Neuroprotective Effects on TBI and TBI-Related Injuries
The cathepsin B gene knockout mice show little or no adverse impact on normal functions, but have major beneficial effects in TBI, including reductions in neuromotor deficits, brain pathology, and neuronal cell death. Cathepsin B deficiency produces substantial improvement in important pathologies related to TBI, including reductions in ECM breakdown, neuroexcitatory-induced cell death, inflammation, inflammatory pain, TNFα-induced cell death, Aβ levels, and memory deficits.
It is likely that the absence of developmental defects associated with cathepsin B deficiency is due to redundant protease specificity of the closely related protease, cathepsin L, which can substitute for the normal function of cathepsin B. Evidence in support of this hypothesis is that cathepsin B and L double knockouts are lethal and have profound neurological abnormalities (162, 163). Thus, some cysteine cathepsin proteolytic activity is required for fetal development. This has implications, which are discussed below (7.5), for the therapeutic development of compounds.
The large volume of data from the cathepsin B knockout mice indicates that this protease is central to many different pathological processes. Many benefits occur from this deficiency, leading to the compelling conclusion that cathepsin B inhibition is a promising therapeutic approach for TBI.
Mechanisms of Cathepsin B Regulation in Normal Compared to TBI Injury Conditions
Neurobiology of Normal Cathepsin B Regulation
Cathepsin B Distribution
Tissue distribution of cathepsin B. Early studies demonstrated that cathepsin B is present in human tissues throughout the body (164), but that the concentration varied as exemplified in Figure 5, which shows the cathepsin B concentrations in selected rat tissues (165). Moreover, these studies also established that cathepsin B concentrations varied among cell types with rat peripheral macrophages having cathepsin B concentrations that are 33, 50, and 400 times higher than lymphocytes, neutrophils, and erythrocytes, respectively, and that immunologically activating macrophages causes a further sixfold increase in cathepsin B levels (165). The cathepsin B variation among the different tissues and cells suggests that cathepsin B is differentially expressed among the tissues and has specialized functions in addition to lysosomal protein degradation (165, 166).
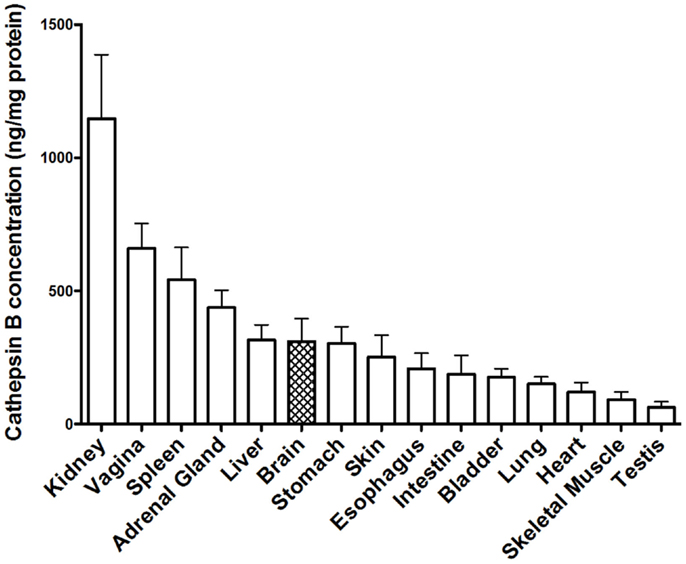
Figure 5. Cathepsin B protein concentration vary among tissues. The levels of mature cathepsin B concentrations are shown from rat tissues (mean ± SD displayed, N = 4 analyses, means significantly different, ANOVA, p < 0.0001) (165). Data from the cited publication and adapted to graphic display and analyzed for variance.
An on-line human protein tissue atlas (167) also shows that cathepsin B is widely expressed in the body with cathepsin B mRNA found in all and protein detected in 80% of the tissues (http://www.proteinatlas.org/). Moreover, the amount of cathepsin B expression varies among different tissues and cell types.
Brain cathepsin B. Early immuno-microscopic analysis showed that human brain contains cathepsin B and it is concentrated in neuronal cell types, especially in the hippocampus (164, 168–171). By contrast, only a few glia cells in normal brain were seen to contain significant amounts of cathepsin B (168). A similar intense neuronal cell staining was also seen in rodent brain, especially in the pyramidal cells of the cortex, large neurocytes of the septal region, many hippocampal neurons, and magnocellular nerve cells of the hypothalamus (97, 168, 172, 173).
Early cathepsin B mRNA analysis of rat brain also showed cathepsin B expression concentrated in neurons (174) and more recent data confirm this distribution. Figure 6 shows cathepsin B mRNA expression and histological images in a normal mouse brain section. Cathepsin B is selectively and intensely expressed in the hippocampal neuronal cell layer and in the cortex. The images were obtained from the Allen Brain Institute web site. A comparison of the papain-like cysteine protease mRNA expression levels in that section shows that cathepsin B is expressed the most followed by cathepsin L with all others expressed at very low levels or not at all.
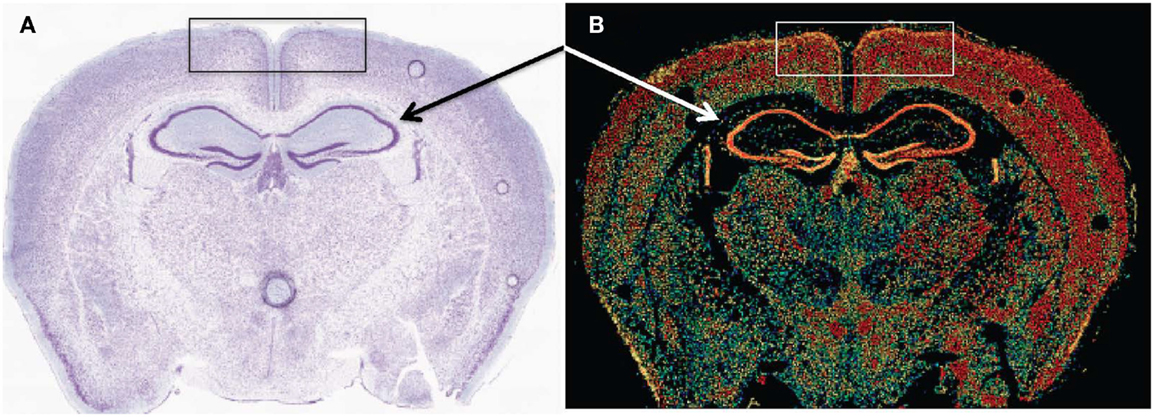
Figure 6. Cathepsin B expression occurs in selected regions of the brain. (A,B) are micrographs of the same coronal mouse brain section and show tissue structure and cathepsin B mRNA expression, respectively. In (A), the section is nissl stained, which highlights neurons as dark blue. In (B), in situ hybridization of sections with antisense mRNA to cathepsin B illustrates the brain regions of cathepsin B mRNA expression. Hotter colors, such as yellow and red, signify high expression, cooler colors, such as green and blue, indicate low expression, and black indicates undetectable expression. A comparison of the two micrographs shows that cathepsin B is intensely expressed in the hippocampal neuronal cell layer (arrows) and in the cortex (box). Figures taken from the Allen Brain Institute web site http://www.brain-map.org/.
CSF and plasma cathepsin B. Table 4 shows the human CSF and plasma cathepsin B concentrations (130). The plasma concentration is about 14 times greater than that in the CSF. Human and rat CSF cathepsin B concentrations increase with age (175, 176).
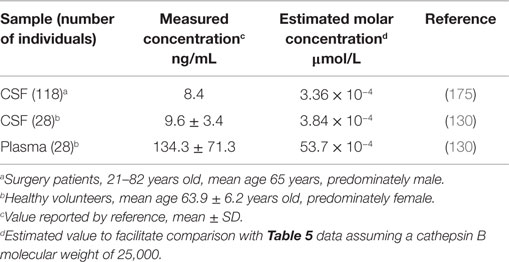
Table 4. Human CSF and plasma cathepsin B concentrations.
Subcellular Organelle Compartments and Functions
Lysosomes. A major destination for cathepsin B is the lysosome, the name of which is from the Greek lysis: loosen and soma: body. Christian de Duve discovered that these subcellular structures were recycling centers in which unwanted proteins are dismantled to their amino acid building blocks for recycling into new proteins for which he received the 1974 Nobel Prize in Physiology or Medicine (177). Lysosomes are membrane-bound compartments present within neurons and cells in the cytosol but outside the nucleus. Lysosomes sequester cytosolic proteins at a rate of no more than 4% per hour but rapidly digest those with a half-life of less than 10 min, which means that the proteolytic capability is at least 20 times that of the highest substrate uptake (178, 179). This rapid hydrolysis is due to the high lysosomal protease concentration, which can be more than 1 mM (180), present at a pH significantly more acidic than the cytosol of the cell (discussed in the section below on pH and cystatin). Lysosomal cathepsins L, B, and S account for roughly 50% of the total bulk protein degradation (181) and the lysosomal cathepsin B concentration is about 330 ng/mg protein (87). Thus, cathepsin B is a major and key component of lysosomes, which are powerful proteolytic machines containing high concentrations of proteases capable of rapid digestion of large amounts of proteins.
In addition to digestive lysosomes, cathepsin B is also trafficked to secretory lysosomes, which release their contents to the extracellular environment in response to a triggering event. These lysosomes are primarily found in immunological cell types and brain astrocytes (182, 183). Containment in and release from secretory lysosomes to the extracellular space allows the cell to safely export the proteolytic capability of cathepsin B and other lysosomal proteases as needed. For example, glia cells secrete cathepsin B in response to neurotoxin exposure via secretory lysosomes (184).
But lysosomal cathepsin B can also cleave proteins at specific recognition sites to produce protein fragments having important biological activities. For example, cathepsin B cleaves in the lysosome non-phosphorylated myristoylated alanine-rich C kinase substrate (MARCKS), which is the primary substrate for protein kinase C (PKC), to prevent phosphorylation and thereby control cell morphology and motility (185–187). In the thyroid gland, cathepsin B traffics via secretory lysosomes to the extracellular lumen of the thyroid follicles (188) where it cleaves Tg and solubilizes it (137).
Autophagosomes. Autophagosomes are structures in which unnecessary or dysfunctional cellular components are degraded. Lysosomes fuse with autophagosomes to provide the proteases needed for that degradation and cathepsin B is a key protease in autophagy catabolism (189).
Regulated secretory vesicles. Lysosomes are not the only subcellular destination for cathepsin B (190). It is also found in regulated secretory vesicles of cells, which secrete large amounts of enzymes, hormones, or neurotransmitters to the extracellular space. Neurons and endocrine cells are such cells and are unique in having a RSP (regulated secretory pathway), in which material to be exported is densely stored in regulated secretory vesicles that discharge their contents out of the cell in response to a triggering signal (191). In pancreatic acinar cells, more cathepsin B is present in the RSP than lysosomes, and in the RSP cathepsin B cleaves trypsinogen to activate trypsin (192, 193). Cathepsin B is in the RSP of β-cells of mouse pancreatic islets where it co-localizes with insulin (194). Cathepsin B is found in dense secretory granules of kidney juxtaglomerular cells (195). In bovine adrenal chromaffin cells, cathepsin B is present in the RSP where it proteolytically cleaves wt APP to produce Aβ (148). As in the lysosome, cathepsin B is physically separated from the cytosol by membranes of secretory vesicles within the RSP.
Summary: Cathepsin B is Normally Packaged into Discrete Subcellular Organelles. From a TBI point of view, the important point is that cathepsin B is normally mostly contained within subcellular organelles and this compartmentalization allows the cell to safely utilize the proteolytic powers of cathepsin B without damaging the cell structure. As discussed below, that segregation is compromised after brain trauma leading to cathepsin B-induced injury and pathology.
pH and Cystatin Control of Cathepsin B and Cysteine Protease Activity
pH regulation. Cathepsin B zymogen is activated by cathepsins in the lysosome/endosome compartments where they have optimum efficiency in the acidic environment of about pH 5.0 and the papain-like cysteine proteases require a reducing environment (67). The lysosomal membrane contains molecular pumps, which create that environment inside the lysosome and maintain it against the neutral pH 7.4 of the cytosol.
Cathepsin B’s enzymatic activity is reduced at neutral pH (196). For example, cathepsin B cleaves trypsinogen at pH 4.0 to 5.2, but not at a higher pH (192). Nonetheless, cathepsin B retains significant activity at the neutral pH of the cell cytosol after translocation from the lysosomes (197). Cathepsin B has a complex pH dependency among its endopeptidase and exopeptidase activities. In vitro, cathepsin B exopeptidase activity predominates below pH of 5.5 and endopeptidase activity prevails above pH 5.5 (198). Glycosaminogycans bind to cathepsin B to stabilize its structure and preserve its endopeptidase activity in the neutral pH of the cytosol (199). In comparison, cathepsin B is much more stable than cathepsin L at neutral pH (200). The key point is that cathepsin B retains significant enzymatic activity at pH 7.4 and thus can inflict major damage when set loose in the cytosol or the extracellular environment.
Cystatin regulation. The endogenous cystatin proteins are probably the most important means of controlling cytosolic and extracellular cysteine protease activity (200); cystatins are emergency inhibitors that neutralize cysteine proteases, which escape from the lysosomes (70, 201) and are of high importance in TBI. Type 1 cystatins are primarily intracellular proteins and type 2 cystatins are found in extracellular fluids. Cystatin C, a type 2 cystatin, is the controlling inhibitor for cathepsin B in human extracellular fluids, including CSF, blood plasma, synovial fluid, milk, saliva, seminal fluid, amniotic fluid, and tears (202). The inhibitor concentrations vary among the fluids but cystatin C has the highest CSF concentration of any of the endogenous inhibitors and is the most potent cathepsin B inhibitor of the endogenous inhibitors in those fluids. The cathepsin B activity half-life (t1/2) when exposed to cystatin C at the concentrations in the extracellular fluids is less than a second (202).
Table 5 shows the human CSF and plasma cystatin C concentrations (203). The CSF cystatin C concentration is about 5.5 times higher than that in plasma. Comparing the molar concentrations with those for cathepsin B in Table 4 shows that the CSF and plasma cystatin C concentrations are 16,000 and 170-fold greater than that of CSF and plasma cathepsin B concentrations, respectively. The high cystatin C to cathepsin B ratios show that inhibiting cathepsin B activity in these compartments is critical, especially in the brain. TBI increases cystatin C and an increase in cystatin C soon after trauma is associated with reduced neuronal cell damage (97).

Table 5. Human CSF and plasma cystatin C concentrations.
Lysosomal Leakage of Cathepsin B
Lysosomal release of cathepsin B to the cytoplasm can overwhelm the normal controls and cause significant damage. The potential of the lysosome as a “suicide bag” capable of killing the cell if its protease contents escape to cytosol and other compartments has long been recognized (204).
TBI Causes Lysosomal Leakage of Cathepsin B
Trauma causes lysosomal cathepsin B leakage to the cytosol in animal models. In normal brain, immunostaining shows that all neurons have a punctate cathepsin B distribution, which reflects its lysosomal location, but after TBI injury many neurons display diffuse staining throughout the neuron cell body showing that cathepsin B has escaped the lysosome containment (97, 205).
Controlled cell shear of primary neuronal cell cultures mimics TBI damage and causes neuronal cell death (206). Cytosolic and lysosome fractions from sheared cells have elevated and reduced cathepsin B, respectively, which is consistent with lysosomal cathepsin B leakage. Sheared neurons are more likely to die than unsheared neurons and treatment with the membrane sealant Poloxamer 188 prevents leakage and reduces cell death in sheared cells (206). Moreover, Poloxamer 188 treatment in TBI CCI and ischemic mouse models reduces neuronal cell death (207, 208).
TBI-Related Injuries also Cause Lysosomal Leakage of Cathepsin B
Ischemia causes the rearrangement of cathepsin B from the lysosomes to the cytoplasm in non-human primate brain neurons (108). Niemann–Pick disease Type C is a genetic lysosomal storage disease, and in mouse models of this disease, cathepsin B leaks out of lysosomes in neurons of the cerebellum (209). In epilepsy animal models, seizures result in translocation of lysosomal cathepsin B to the cell body and nucleus (210). ALS autopsy samples show that cathepsin B is diffusely distributed within degenerative neurons (128). In nerve cells of brain autopsy tissue from AD and Parkinson’s dementia of Guam and senile dementia, but not age-matched controls, cathepsin B is found in neurites and dendrites and in the pathological neurofibrillary tangles and plaque structures (129, 170, 171). And in AD and Parkinson’s cell culture models, Aβ(1-42) and α-synuclein cause lysosomal leakage of cathepsin B into the cytoplasm (211–213).
Mechanisms of Lysosomal Leakage of Cathepsin B
Molecular mechanisms of lysosomal leakage have been summarized in an excellent recent review (214). Many of the mechanisms were discovered in abnormal cancer cells and those are not discussed because it is not clear how relevant they are to normal neurons.
Unique to trauma is that mechanical force directly breaks membranes and is likely the primary cause of lysosomal rupture at the site of injury. These forces can rapidly damage large amounts of tissue and cause the release of significant amounts of proteases, which autodigest the brain tissue.
Ischemic monkey and rat animal model data generated the “calpain/cathepsin” neuronal cell death hypothesis, which is based on lysosomal cathepsin B leakage (215, 216). Injury induces calcium ion entry into neurons causing μ-calpain activation, which indirectly permeabilizes lysosomal membranes. Heat shock protein 70.1 (HSP 70.1) normally stabilizes lysosomal membranes by binding to them via endolysosomal phospholipid bis(monoacylglycero)phosphate (BMP) and enhances membrane stabilizing acid sphingomylinase (ASM) activity. Ischemia decreases HSP 70.1 and BMP and increases oxidized HSP 70.1, which is cleaved by activated μ-calpain, and those changes decrease ASM and together cause lysosomal membrane permeability, the release of lysosomal cysteine proteases, and cell death (217).
Cathepsin B also contributes to lysosomal leakage as illustrated by the reduced lysosomal leakage that occurs in cathepsin B-deficient hepatocytes vs. wt cells responding to TNFα toxicity (218). The fact that trauma causes increased cathepsin B and TNFα suggests that cathepsin B may contribute to lysosomal leakage in TBI.
Free radical formation can also contribute to lysosomal membrane breakdown. TBI pathology causes a pronounced increase in free radicals and oxidative brain damage (219, 220). The free radicals enter lysosomes, react with iron to form hydroxyl radicals, which react with lysosomal membrane components thereby destabilize the lysosomal membrane and cause it to leak (221). Treating subarachnoid hemorrhage models with iron chelators deferoxamine or α-lipoic acid protected lysosomal membranes and prevented cathepsin B leakage, which correlated with improved outcomes, such as reductions in brain edema, blood–brain-barrier impairment, and neuronal cell death, and also improved behavioral deficits (102, 103).
TBI-Induced Lysosomal Leakage Causes Necrotic and Apoptotic Cell Death, Inflammation, and Axonal Damage
Necrotic Cell Death
The extent to which the lysosomal membrane is disrupted by an injury is a key driver in determining the resulting pathology. A frank rupture is generally thought to result in necrotic cell death whereas a lesser amount of leakage causes apoptotic cell death (222, 223). The molecular machinery causing necrotic and apoptotic death is complex and diverse and has been the subject of several reviews (214, 222, 224).
The molecular mechanisms by which cathepsin B participates in necrotic cell death are yet to be fully understood presumably because of the difficultly in studying the rapidly occurring multiplicity of reactions that lead to necrotic death. But one mechanism by which lysosomal rupture causes necrotic cell death is through activation of the inflammasome receptor NOD-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) by cathepsin release into the cytoplasm (225). As discussed below, NLRP3 activation by cytosolic cathepsin B is also central to the inflammatory response caused by lysosome leakage.
Apoptotic Cell Death
Apoptotic cell death cascade occurs following trauma and cathepsin B is a key component of that cascade (94, 98). TNFα is elevated after trauma and it is an extracellular cell death signal that sets into motion intracellular molecular events that ultimately lead to cell death. The pathway includes TNFα binding to tumor necrosis factor receptor 1 (TNFR1) causing cytosolic activation of caspase 8, Bid cleavage to t-Bid, and polymerization of BAX, which forms mitochondrial membrane pores causing mitochondrial leakage to the cytoplasm of cytochrome C (cyt c), which activates the apoptotic initiator caspase 9 causing activation of the executioner caspase 3 (226). Direct evidence that cathepsin B contributes to this pathway in TBI comes from data showing that deleting the cathepsin B gene in a TBI mouse model blocks BAX activation following trauma (48). E64d, which is an inhibitor of cathepsin B and other papain-like cysteine proteases and calpains 1 and 2 discussed below (see section on ‘E64, E64d, and E64c cysteine protease inhibitors’), administered to TBI mouse models reduced proapoptotic proteins t-Bid, BAX, and cytosolic cyt c, and activated caspase 3 and antiapoptotic protein Bcl-2 (94). The effects of E64d on reducing apoptotic cell death are likely primarily due to cathepsin B inhibition because E64d given to cathepsin B knockout mice produced no additional reduction on BAX levels after trauma than that obtained in cathepsin B gene deletion mice given the vehicle solution (48). Thus, cathepsin B is an important component to TBI-induced TNFα cell death.
Studies in hepatocytes from cathepsin B knockout animals that show the TNFα-induced lysosomal leakage of cathepsin B to the cytosol cleaves Bid and activates caspase 2, which facilitate mitochondrial release of cyt c to the cytosol and that triggers caspase apoptotic cell death (145, 161). Treatment of normal human blood lymphocytes with antithymocyte antibodies also results in lysosomal cathepsin B leakage to the cytosol and apoptotic cell death by an undefined mechanism that does not involve the intrinsic mitochondrial cyt c pathway (227). Unraveling the exact means by which cathepsin B functions in the TBI-induced death pathway will be an exciting area of future research.
Inflammation
Cathepsin B is secreted by inflamed microglia and induces apoptosis. Trauma to the CNS causes diverse inflammatory cells expressing high levels of cathepsin B, which include microglia and macrophages, to gather at a site of injury (100). Inflammation induced by CGA in primary microglia cultures causes those cells to secrete cathepsin B, which induces apoptotic cell death in primary granule neuronal cultures possibly by executioner caspase 3 activation (228). Thus, cathepsin B in the extracellular space can induce neuronal apoptotic cell death.
TBI can cause massive cellular and cytokine inflammatory responses that focus on cathepsin B as the center of TBI-caused damages. As discussed above, cathepsin B is clearly involved in the production of IL-1β by activating caspase 1, which in turn cleaves pro-IL-1β to produce active IL-1β (116) as has been shown in microglia inflamed by fibrillar Aβ or CGA or in alveolar macrophages inflamed by silica. Interestingly, lysosomal permeabilization and NLRP3 activation are required for fibrillar Aβ- (158) and silica- (229) induced inflammations, but not in the CGA-induced inflammation (115). Alternatively, cathepsin B can also activate IL-1β by cleaving pro-caspase 11 to activate proinflammatory caspase 11 (230), which in turn can cleave pro-caspase 1 (231). As such, cathepsin B can control IL-1β activation by multiple mechanisms. Moreover, cathepsin B also increases TNFα secretion in response to LPS-induced inflammation by functioning in the trafficking of vesicles containing TNFα (144). Thus, cathepsin B both contributes to TNFα levels and transduction of the TNFα apoptotic cell death signal. Cathepsin B also activates IL-18 levels in response to CGA-induced inflammation (115).
Peroxisome proliferator-activated receptor (PPAR) receptors are key regulators of neuroinflammation after CNS injury, including TBI (232). PPARα agonist fenofibrate is effective in TBI models (233, 234). PPARδ agonists suppress cathepsin B levels in human endothelial cells in a PPARδ-dependent manner (235). Thus, PPARδ agonists target cathepsin B and may be beneficial in TBI because they reduce cathepsin B.
Axonal Damage
Pathological axonal damage is a predictor of outcome in CNS diseases (236) and cathepsin B may be involved in causing that damage. As mentioned above, cathepsin B cleaves MARCKS, which is the PKC substrate that controls cell morphology and motility by regulating actin dynamics near the cell surface. N-Methyl-d-aspartate (NMDA) treatment of primary hippocampal neuronal cell cultures mimics excitotoxicity, increases cathepsin B, reduces MARCKS at synapses, and causes dendritic spine collapse. Treating with CA-074Me, which is a cathepsin B inhibitor discussed below, prevents spine collapse in the NMDA-treated neurons. (186, 237). Thus, activated cathepsin B cleavage of MARCKS may be a mechanism that contributes to axonal swelling.
TBI-Induced Autophagy
TBI increased autophagy activation within 1 h and up to at least 3 days after trauma in the brain relative to non-traumatized controls as measured by lipidated microtubule-associated protein light chain 3 (LC3II) (94, 238–241) with higher LC3II levels suggesting autophagy activation (242). Moreover, beclin 1, which interacts with phosphatidylinositol-3-kinase (Class III PI3K) to induce autophagy (243), was also elevated 1 h and up to at least 3 days post-TBI in the cortex and hippocampus relative to controls (239–241). Furthermore, microscopic examination shows more cellular autophagy structures after trauma than before (238, 239). Autophagy proteolysis was also increased post-TBI as measured by P62 with less P62 generally reflecting more autophagy proteolysis (244), as P62 was reduced within 1 h and up to at least 2 days post-trauma in the cortex and hippocampus relative to controls (240, 241).
Inhibiting Autophagy Improves TBI Outcomes
While it has been suggested that autophagy might provide neuroprotection after TBI (239), administration of the autophagy inhibitor 3-methyladenine (3-MA) to TBI animal models reduced the TBI-increased LC3II and Beclin 1 levels and restored the reduced P62 levels that resulted from trauma, while improving memory and neuromotor defects and reducing brain lesion volume, neuronal cell death, cathepsin B activity, and caspase 3 activity (241). 3-MA reduces autophagy by inhibiting Class III PI3K and does not inhibit cathepsin B activity (245). Moreover, treating animals with the gamma-glutamylcysteinyl ether ester, which is a prodrug of glutathione and an antioxidant, also reduced autophagy and improved TBI outcomes (238). These data suggest that reducing autophagy improves TBI outcomes. Cathepsin B inhibitors inhibit lysosomal cysteine proteases, which reduces autophagy function (246) and thereby may also improve TBI outcomes.
Small Molecule Inhibitors of Cathepsin B Improve Behavioral and Pathological Deficits in TBI and TBI-Related Animal Models
Inhibitors of Cathepsin B Improve Deficits of TBI Animal Models
Table 6 summarizes the data showing the behavioral, pathological, and biomarker effects of administering small molecule inhibitors of cathepsin B to TBI and TBI-related injury animal models. These include models of ischemia, subarachnoid and cerebral hemorrhage, meningitis, pain, and neurodegenerative conditions including epilepsy, AD, MS, and Huntington’s disease. Table 7 summarizes the protease inhibition profiles of the small molecules cited in Table 6.
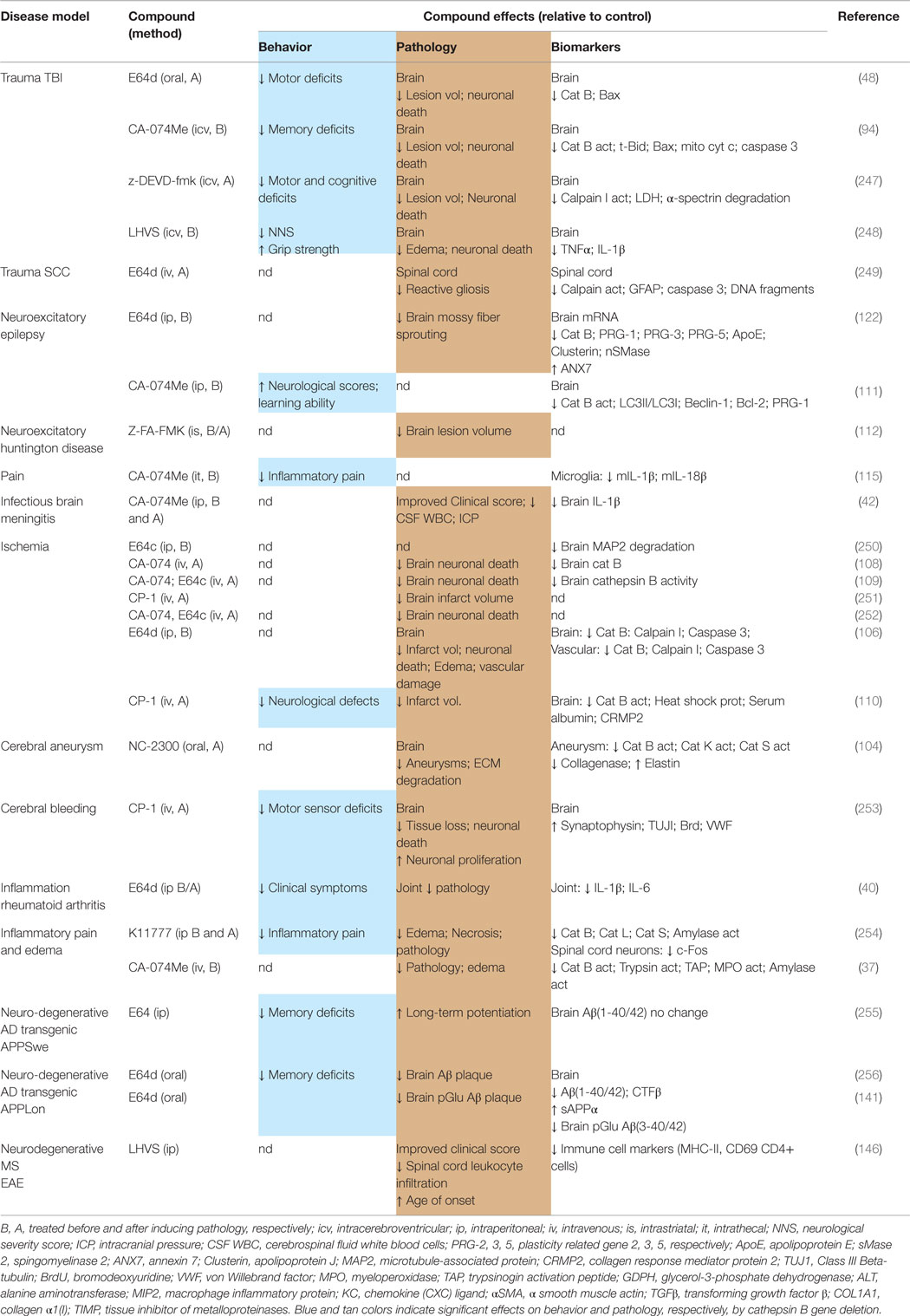
Table 6. Small molecule inhibitors of cathepsin B are efficacious for improving behavioral and pathological deficits in TBI and TBI-related animal models.
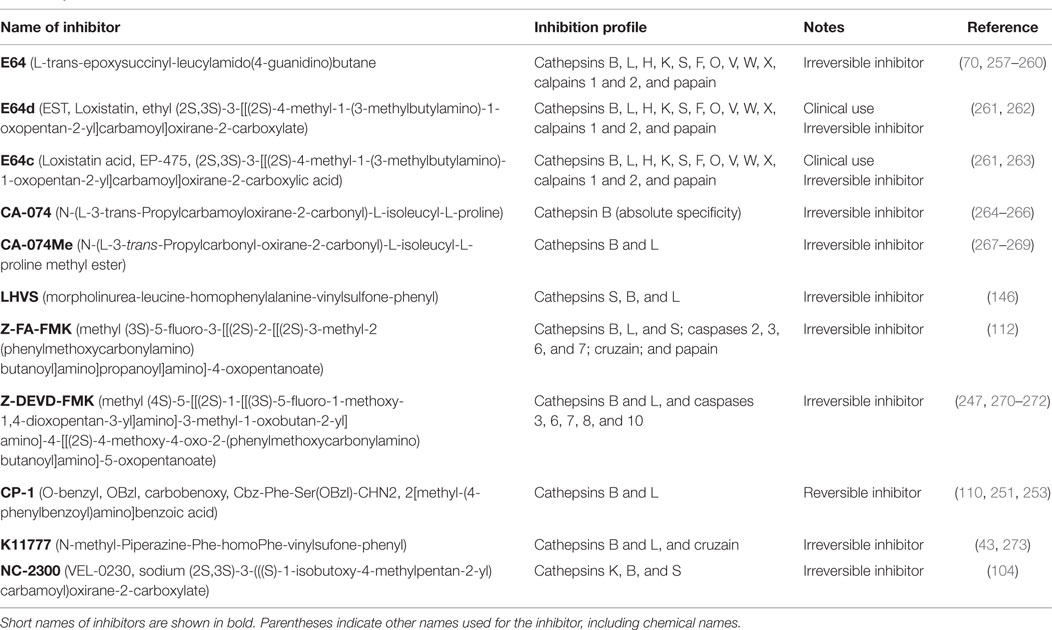
Table 7. Protease inhibition profiles of small molecule inhibitors of cathepsin B, from studies of TBI and TBI-related animals.
The important conclusion from Table 6 is that different compounds, which inhibit cathepsin B, used by different groups in different TBI animal models resulted in improvements of neuromotor and cognitive defects, brain lesion volume, brain edema, neuronal cell death, and key brain biomarkers (48, 94, 247, 248). A particularly dramatic result is the significant reduction in lesion volume as the inhibitors protected against brain tissue loss much of which was likely due to necrotic cell death. Another major result was the prevention of neuronal cell death in the hippocampus as the inhibitors arrested nearly all the cell death that occurs in that region as a result of TBI. Behavioral dysfunction was also greatly improved by treatment with these compounds suggesting that it may be possible to also observe improved clinical outcomes with cathepsin B inhibitor treatment of TBI patients.
Inhibitors of Cathepsin B Improve Deficits in TBI-Related Conditions
Compounds, which inhibit cathepsin B, also produced substantial beneficial effects in TBI-related injuries in animal models. For example, neuromotor, neurological learning and cognitive defects were improved by cathepsin B inhibition in cerebral bleeding (253), ischemia (110), epilepsy (111), and AD neurodegeneration (256) animal models. Inflammatory pain is a major problem in TBI and was alleviated by cathepsin B inhibitors in inflammatory pain models (115, 254). Moreover, E64d treatment essentially eliminated the clinical symptoms of chronic inflammation due to RA (40) and greatly reduced the reactive inflammatory gliosis caused by the trauma of SCC (249).
Edema is often a major complication of TBI and compounds, which inhibit cathepsin B, reduced edema in ischemic (37, 106) and pancreatitis models (254). Excessive ICP and infections also often accompany TBI and treatment with a cathepsin B inhibitor reduced ICP, improved clinical scores, and lowered CSF white blood cell counts in a brain meningitis animal model (42). Brain aneurysms and vascular ECM degradation occur as a result of TBI, and cathepsin B inhibition stopped aneurysm progression and vascular ECM degradation in brain aneurysm (104) and ischemic (106) animal models. Brain tissue loss was significantly prevented by cathepsin B inhibitor treatment of cerebral bleeding (253), ischemia (106, 110, 251), and kainic acid-induced Huntington’s chorea animal models (112). Cathepsin B inhibitors have also been shown to prevent the vast majority of the neuronal cell death that occurs in ischemia (106, 108, 109, 252) and a great deal of the death resulting from cerebral bleeding animal models (253).
Inhibition of Cathepsin B Modulates TBI Biomarkers
The data in Table 6 also show that the inhibitors affected many TBI relevant biomarkers. TBI animal models treated with the cathepsin B inhibitors had reduced brain pro-apoptotic biomarkers including t-Bid, Bcl-2, Bax, cyt c, and caspase 3 (48, 94) and inflammatory proteins IL-1β and TNFα (248). The biomarker data show that cathepsin B inhibitors improve TBI outcomes in part by reducing apoptosis and inflammation.
Glia fibrillary acidic protein (GFAP), which is biomarker of CNS inflammation and a potential TBI diagnostic marker (274), is reduced by E64d treatment of SCC animals (249). Cathepsin B inhibitors also reduced cytokine IL-1β in pain, meningitis, and RA animal models (40, 42, 115), IL-18 in a pain animal model (115), and IL-6 in a RA model (40). As in the cathepsin B knockout data, cathepsin B inhibitors reduced brain Aβ(1-40/42) and pGlu Aβ(3-40/42) in transgenic AD mice expressing human APP containing the wt β-secretase site but not in those expressing the Swe mutant β-secretase site (141, 256).
These data support the conclusion that compounds, which inhibit cathepsin B, are effective in many TBI relevant animal models at improving behavior and pathology. As such, these data provide strong motivation to explore cathepsin B inhibitors as potential TBI therapeutic agents.
E64d is a Promising Drug Inhibitor of Cathepsin B for Preclinical and TBI Therapeutic Drug Development
E64d Improves Deficits of TBI and Related Injuries
TBI and Multiple Injury Conditions are Improved by E64d
Most of the preclinical and essentially all the clinical studies on small molecule inhibitors of cathepsin B investigated E64 and its derivatives E64d, E64c, CA-074Me, and CA-074. Table 6 shows that these compounds were efficacious in at least 17 different preclinical pathological models including two TBI models as well as one SCC, two epilepsy, two pain, one infectious, five ischemic, one arthritis, and three neurodegeneration (AD) models. Different dosing regimens, including administering the drug before or after the pathology onset, have been effective, as have different routes of administration, including oral, intraperitoneal (ip), intravenous (iv), intracerebroventricular (icv), and spinal intrathecal (it). Thus, there are robust data showing that E64 and its related compounds are efficacious in a wide range of TBI-related models using different treatment regimens and routes of administration. This is important because it is thought that potential TBI therapeutics must demonstrate efficacy in diverse animal models in order to have hope of success in treating the heterogeneity of TBI injuries in a clinical setting (275).
E64d TBI Efficacy is Primarily Due to Cathepsin B Inhibition, but Inhibition of Other Proteases Provides Added Benefits
Without additional data, it is difficult to conclude that the beneficial TBI effects of E64d are due to cathepsin B inhibition because the biologically active acid form of E64d, which is E64c, inhibits proteases in addition to cathepsin B, which are the papain-like cysteine proteases and calpains 1 and 2 (see section below on E64, E64d, and E64c cysteine protease inhibitors). However, the relative importance of cathepsin B inhibition can be resolved by evaluating E64d in cathepsin B gene-deficient animals. Those studies show that E64d treatment produced, with one exception, similar improvement in neuromotor deficits after trauma as did vehicle controlled treatment of cathepsin B knockout animals (48). Also, the E64d-treated cathepsin B knockout animals tended to have smaller brain lesion volumes and higher neuronal cell densities than vehicle carrier-treated knockout animals but the differences were not statistically significant. As such, those data argue that E64d acts primarily, but perhaps not exclusively, through inhibiting cathepsin B in this TBI model.
The E64d-treated cathepsin B knockout animals performed significantly (20%) better in the neuromotor assay than carrier-treated cathepsin B knockout animals on day 1 post-TBI (48). These results suggest that E64d produced an additional benefit in behavior from inhibiting other proteases in addition to cathepsin B. While it is impossible to say from the data what those other protease(s) are, it is likely that E64c inhibition of calpain contributed to the additional benefit. That is because calpain activity spikes in the first day after TBI (276). Calpain 1 gene-deficient animals have reduced brain lesion volume and neuronal cell death following TBI (277), and E64d has been shown to inhibit calpain activity and provide neuroprotection after trauma (249). Thus, while E64c inhibition of cathepsin B is primarily responsible for the improvements in the TBI models, its ability to inhibit other proteases produces even better outcomes compared to inhibition of only cathepsin B.
Primary benefits due to inhibiting cathepsin B and the additional benefits from inhibiting related proteases have also been demonstrated in ischemic non-human primate models. CA-074, which has absolute specificity for cathepsin B (see below, section on CA-074, selective inhibitor of cathepsin B), given iv immediately after ischemia significantly inhibited brain cathepsin B by about 75% in the CA1 region of the hippocampus and caused substantial neuronal protection by saving about 67% of the neurons in that region relative to controls (108). Studies comparing the effects of CA-074 with that of E64c in the non-human primate model showed that even better outcomes resulted from using E64c (109, 252). For example, E64c and CA-074 treatments resulted in an 84% and 67% CA1 neuronal cell survival, respectively (109), and a range of cell protection from 75% (cortical layer 5) to 91.6% (cerebellum) vs. 47.4% (CA1) to 89.9% (caudate nucleus), respectively, relative to controls (252). The authors attributed the increased neuronal cell survival obtained with E64c over that of CA-074 to E64c inhibition of cathepsin L and calpains in addition to cathepsin B, whereas CA-074 only inhibited cathepsin B.
Another advantage of multiple protease inhibition by E64d is the indirect reduction in matrix metallopeptidase-9 (MM-9), which is a protease that contributes to TBI dysfunction (278). Up-regulation of both cathepsin B and calpain causes up-regulation of MMP-9 (279) and since E64c inhibits both cathepsin B and calpain, E64d treatment also indirectly down-regulates MMP-9 as E64d has been shown to do in an ischemic animal model (280).
The cysteine protease inhibitor, morpholinurea-leucine-homophenylalanine-vinylsulfone-phenyl (LHVS), provides significant improvements in behavioral deficits, brain edema, and cell survival following TBI (248). LHVS was thought to specifically inhibit cathepsin S, which is up-regulated following TBI (248), but LHVS has subsequently been shown to inhibit cathepsin B, S, and L (146). Thus, the beneficial results in the TBI model may be the result of LVHS inhibiting these several proteases. That is the case for the improved outcomes seen in an MS model given LHVS in which the compound has been shown to act by inhibiting both cathepsins B and S (146). E64 has been reported to inhibit cathepsin S (260) and thus E64d treatment may also provide beneficial TBI effects through inhibiting cathepsin S in addition to that achieved by cathepsin B inhibition.
Thus, cathepsin B inhibition is a primary means by which E64d provides beneficial outcomes in TBI and ischemic models, but additional benefits are obtained by the compound’s inhibition of other related proteases, especially calpains, cathepsin L, and cathepsin S. The ability of E64d to inhibit multiple related cysteine proteases is thus an advantage and may be required to “move the needle” and affect outcomes in the complex condition of TBI.
Biological Effects of Irreversible Cathepsin B Inhibition
E64 and its related compounds as well as many other cathepsin B inhibitors are irreversible inhibitors meaning that they covalently bind to the traget protease and permanently stop that protease’s activity. As a result, cathepsin B activity lost due to these inhibitors can only be restored through production of new mature cathepsin B protein. Normally, cathepsin B turnover (net synthesis and degradation) as measured by the half-life is about 14 h and results in a lysosomal content of about 330 mg/g in rat macrophages (87, 92). E64 treatment of those cells reduces cathepsin B turnover as the cathepsin B half-life is increased 3.6-fold (50 h) and increases lysosomal content threefold higher (990 mg/g) relative to untreated cells (87, 92). The reduced cathepsin B turnover and increased lysosomal content are due to E64 inhibiting the cysteine proteases which degrade cathepsin B (87). Thus, irreversible inhibition of cathepsin B results in its inactivation as well as increased half-life. New synthesis of cathepsin B will be needed to replenish cellular levels of active cathepsin B.
Discovery of E64d and Related Compounds
E64, E64d, and E64c Cysteine Protease Inhibitors
The discovery of the epoxysuccinyl-based inhibitor E64 was seminal in the development of cathepsin B inhibitors (257). This natural product of Aspergillus japonicas selectively inhibits the papain-like cathepsin proteases (except cathepsin C) and calpains 1 and 2. But E64 does not affect cysteine proteases belonging to the CD clan, which includes caspases, or proteases belonging to the aspartyl, serine or metalloprotease classes or thiol enzymes (70, 257–260). For over 30 years, this scaffold has been used to develop new compounds having medicinal properties and that use continues today (281).
The most important derivative of E64 is E64d (aka EST and Loxistatin), which was developed by Taisho Drug Company, Japan, and the Japanese Ministry of Health in the 1980s as an experimental therapeutic agent for treating muscular dystrophy (262). The clinical trials in pediatric patients were initiated based on studies in which E64d had some, but not a great deal, of effect in a dystrophic hamster model (282). The trials completed through Phase 3, but did not show sufficient efficacy for muscular dystrophy and the compound did not advance (283).
But importantly, extensive E64d pharmaceutical data were published as a result of this effort in many peer-reviewed Japanese scientific articles showing that the compounds could be safely used in man. Some of those data are summarized below (section on pharmaceutical properties of E64d and safety).
E64d is an ethyl ester prodrug, which is hydrolyzed in the gut to its active acid form E64c, which systemically circulates and is the biologically active inhibitor form (284). E64c has the same inhibition profile as E64, but is more potent (285). For example, the potency (IC50) of E64c against rat liver cathepsins L, B, H, and porcine kidney calpain 2 is 0.09, 3.36, 1,640, and 3,000 nM, respectively, with the substrates Z-Phe-Arg-MCA, Z-Arg-Arg-MCA, Arg-MCA, and casein, respectively (264), indicating varying potencies of E64c for inhibition of these cysteine proteases. E64c inhibition of calpain 2 is dependent on pH and calcium ion concentration and pH (286). Thus, oral administration of E64d results in systemic circulation of E64c, which can inhibit cathepsins B, L, K, H, O, S, V, W, and X, and calpains 1 and 2, but not other proteases or thiol-containing enzymes.
CA-074, Selective Inhibitor of Cathepsin B, and CA-074Me
A specific inhibitor of cathepsin B was developed from the E64 template, known as CA-074, which irreversibly inhibits with high potency (264, 265). CA-074Me was developed from CA-074 to have better membrane permeability (269) but inhibits both cathepsin B and L (267, 268).
E64 Activity-Based Probes (ABPs)
E64 has given rise to activity-based probes (ABPs), which bind to the active site of proteases and report on activities via a biotin or quencher-fluorophore tag (287–289). ABPs have been used to affinity purify cathepsin B activity from regulated secretory vesicles of chromaffin cells to discover its β-secretase activity in those vesicles (148). ABPs can image proteases in vivo (254, 290, 291) and would provide powerful visual data on cathepsin B distribution in TBI. ABPs can evaluate a drug’s engagement with cathepsin B (290). ABPs could be adapted to evaluate inhibitor target engagement with brain cathepsin B in animal models and in the clinic.
Pharmaceutical Properties of E64d
Oral and Peripheral E64d Administration is Efficacious in Animal Models
An important result from a TBI translational point of view is that oral administration of E64d (one 10 mg/kg dose) has been shown to be efficacious in a TBI animal model when given up to 8 h after trauma (48). That is a key milestone because 8 h represents a time frame in the TBI animal model for drug administration that could be applied post injury in the clinic. In most clinical TBI therapeutic applications, the earliest time when therapy can begin is about 6 h after injury and, thus, it is critical that a compound is effective at times after the TBI injury event in the animal model. Moreover, the data discussed above show that cathepsin B remains elevated for days after injury suggesting that an even longer effective treatment window is likely.
Furthermore, administration of E64d by oral gavage to guinea pigs (1–10 mg/kg/day, 7 days) resulted in dose–response reductions in brain cathepsin B activity and Aβ (256). And feeding E64d (10 mg/kg/day, 1 to 2 months) in chow to transgenic mice also reduced brain Aβ and pGluAβ (141, 256). Moreover, icv E64c (4 mg/kg) administration reduced brain cathepsin B activity in ischemic monkey models (109, 252) and ip E64d administration inhibited brain cathepsin B activity in ischemic rat (5 mg/kg) (106) and epileptic rat (4 μg dose) models (122). Thus, oral and other peripheral routes of E64d administration at doses comparable to that used in man (5 mg/kg/day) have been shown to inhibit cathepsin B and affect brain biomarkers in animal models.
E64d/E64c Biodistribution
Radioactive E64d biodistribution studies show that about 60% of an oral E64d dose is taken up in rats and hamsters and most of that occurs in the small intestine. Blood levels quickly rise and the maximum blood concentration varies among animal species with rabbits having the highest, and hamsters, dogs, and rats having 75%, 33%, and 25% that of rabbits, respectively. The excretion also varies among species, with most occuring via the urine in rabbits and hamsters whereas the feces was the primary route in dogs and rats. Biliary excretion was greater than 50% and 13% of the dose in rats and hamsters, respectively. Respiration only accounted for about 7% of the dose (292).
The tissue distribution was similar for rats, rabbits, and hamsters, but the highest drug concentration was in the gastrointestinal track followed by the kidney and liver which had about 25- and 12-fold higher concentrations than the plasma concentration, respectively. But the brain concentration was one-tenth that of the plasma concentration. However, the liver drug concentration in rats was about twice that of hamsters (293).
At a cellular level, subcutaneous radioactive E64c injection of rats resulted in a muscle cytoplasm to lysosome-specific activity ratio of about 5:1 (294). That distribution is advantageous for TBI treatment because the drug is concentrated in the cytoplasm where it is needed in the injury condition and not in the lysosomes where it could interfere with normal proteolysis.
E64d Pharmacokinetics and Metabolites
Pharmacokinetic properties of E64d have been determined in animals and man (284, 295, 296, 297). In man, an oral E64d dose is completely hydrolyzed in the gut to the acid form of E64c which systemically circulates and about half of that is metabolized to two hydroxylated E64c metabolites (M4a and M4b). E64c and M4 have about the same area under the curve (AUC) values (5.08 μg-hr/mL), serum half-lives of about 1.34 and 2.5 h, respectively, and urinary excretion rates of 28.9% and 18.5%, respectively. Single and multi-dose pharmacokinetics are the same, the drug does not accumulate and is completely eliminated within 24 h of the last dose in the urine at a recovery rate is about 30%. The M4 metabolites have the same inhibitory profile as E64c. Once a day dosing is effective despite the relatively short half-lives because the compounds are potent irreversible inhibitors.
E64d Mutagenesis
In vitro E64d mutagenic studies using the reverse mutation (Ames bacterial cell test) and chromosome aberration test (Chinese hamster lung fibroblast cells) showed no mutations or chromosome aberrations. In vivo mutagenic studies using the mouse bone marrow chromosome aberration test showed that E64d did not have any effects in that assay. The conclusion was that E64d is unlikely to have mutagenic effects (298).
E64d Toxicology
Toxicity studies show that E64d has a wide therapeutic window. Extensive E64d toxicology studies have been published. For example, acute oral E64d toxicity studies showed that high lethal doses to be over 10,000 and 5,000 mg/kg for rats and dogs, respectively, which killed 50% of the animals (LD50), with the main clinical effects being depression of voluntary movements, closed eye, and loss of righting reflex, from which it was concluded that acute toxicity is not an issue (299). Longer-term toxicity studies showed the kidneys and liver to be the primary target organs for toxicity and the toxic effects increased with increasing dose and generally abated upon discontinuing the drug. Sub-acute, 1 month oral E64d toxicity studies in dogs and rats found no-effect doses of 40 and 80 mg/kg/day, respectively, with slight renal tubular epithelia degeneration but without an increase in renal weight or impairment of function (300, 301). Six-month chronic oral E64d studies in dogs found a no-dose effect level at 5 mg/kg/day, with similar renal results as in the sub-acute studies (302). Six-month chronic oral E64d studies in rats, on the other hand, found a no-effect dose of 2 mg/kg/day for liver toxicity and10 mg/kg/day for renal toxicity and that the rat was unusually sensitive to liver toxicity (303). Subsequent studies confirmed the rat species-specific hepatotoxicity as a single oral E64d dose produced rat liver toxicity at 60 mg/kg, but in a hamster liver toxicity required a 500 mg/kg dose (297, 304). The rat liver toxicity was attributed to the rat metabolizing the drug differently than rabbits, hamsters, and man because the rat mostly excreted the drug as glutathione conjugates via bile whereas in man and other animals the free drug was mostly excreted via urine (284). Rats have much higher liver glutathione levels, which E64c and M4 metabolites bind to, and thus the rat liver dose is proportionally greater than that in other species (304).
E64d Teratogenic Effects
Teratogenic effects have also been studied and also show differences among species. In rabbits, the no teratogenic effect for oral E64d doses was found to be 100 and 500 mg/kg/day for fetuses and mothers, respectively, when administered from the 6th through the 18th day of gestation (305). By contrast, the no teratogenic effect oral E64d dose in rats was 20 mg/kg/day for fetuses and mothers when given at about the same time frame (7th through the 17th day of gestation) (306). Thus, the rat is much more susceptible to E64d teratogenic effects than rabbits. Rat studies using oral dosing before pregnancy and during mating and late-term and post-natal showed the lowest no-effect dose for the fetuses and parents to be about 50 and 80 mg/kg/day (307, 308). No effect was seen in the subsequent generation (F2) in these studies.
E64d Safely Used in Man
As mentioned above, E64d was developed for treating muscular dystrophy. Phase 1 single (oral 3.5 mg/kg maximum dose) and multi-dose (oral, 5 mg/kg/day for 7 days) studies in normal volunteers and showed no effect on observed clinical symptoms, including electrocardiograms, body temperature, blood pressure, grip strength, and physician interviews, or clinical tests, including hematological tests, blood chemistry tests, and urinalysis (295, 296). These E64d doses used in patients are below the range of doses evaluated in animals for toxicology and teratogenic studies.
Open trials on 73 Duchene’s muscular dystrophy patients were conducted for 3 years at four Japanese national sanatoriums and resulted in some muscle strength improvement but the results were inconclusive and final double-blind studies did not confirm the results (283). The fact that these trials were completed shows that no serious adverse event occurred in the pediatric patient population with long-term chronic administration of E64d.
Issues with E64d TBI Therapy
The gross abnormalities seen in offspring of double cathepsin B and L knockout mice clearly show that some cysteine cathepsin activity is required for normal embryogenesis. Possibly, the loss of cysteine cathepsin protease protein degradation in lysosomes and autophagosomes results in nutritional deficiencies, which cause the abnormalities. As discussed above, sufficiently high E64d doses can also cause teratogenic abnormalities when administered to pregnant animals.
This has consequences for the therapeutic use of E64d or its derivative, the most obvious being that very careful consideration will have to be given to its use in pregnant women, women of child-bearing age, and infants. But as most young civilian and military TBI patients are adolescent or young adult males, the teratogenic danger may not be as much of a risk for the majority of that TBI patient population. Moreover, the teratogenic risk is not likely an issue for a large and growing segment of the TBI population, which are elderly who are beyond the reproductive age.
These data also suggest that minimizing inhibition of lysosomal cathepsin B and cathepsin L activities is desirable in adults. To that end, E64c is unique among cysteine protease inhibitors in that it concentrates in the cytosol and not in the lysosome (see above section on E64d/E64c biodistribution), thus preferentially spares inhibition of lysosomal cathepsin B and L activities. The fact that E64d has been safely administered to pediatric patients for many years shows that, if lysosomal protease inhibition is a toxicity issue, it is not significant with an appropriate dose.
The low brain dose is the primary issue with E64d as a TBI therapeutic because a small portion of an oral dose gets into the brain as discussed above (section on biodistribution). Even though a low brain dose can effectively inhibit brain cathepsin B activity, it would be desirable to have a derivative compound, which delivers a higher brain dose than does E64d so that efficacy could be achieved with reduced systemic exposure.
E64d is a Promising Tool Compound for Preclinical TBI Testing and TBI Clinical Compound Development
E64d is unique among the small molecule cathepsin B inhibitors because of the extraordinary amount of data on the compound. No other compound has the extensive information known for E64d on its efficacy, toxicology, and safety. These data greatly reduce the risk of using E64d vs. other small molecule compounds for testing in TBI and related models. Therefore, E64d is a promising tool compound for preclinical TBI testing.
The extraordinary amount of data makes the E64d scaffold extremely useful for developing clinical derivatives. Only the E64d compound has data on pharmaceutical properties that provides an expectation roadmap for the behavior of a clinical compound that is derived from it. Other small molecule scaffolds lack that information and thus all the data on clinical candidates derived from them will be without information as to the likelihood of pharmaceutical success. Thus, the E64d data greatly reduce the inherently high risk of drug development.
One might ask why not use a cathepsin B-specific inhibitor rather than the multi-protease inhibitor E64d since cathepsin B knockouts have no pathology? A druggable cathepsin B-specific inhibitor may be useful as a follow-on therapeutic. However, no orally bioavailable, brain effecting, cathepsin B-specific inhibitor is currently available and developing such will be difficult and will take considerable resources and time. For example, a high throughput screen of the National Institutes of Health (NIH) small molecule library for a cathepsin B-specific inhibitor did not find a druggable hit (309). The known cathepsin B-specific inhibitor, CA-074, is not orally druggable and when made more so with the addition of a methyl group, it looses specificity. Thus, an E64d derivative will likely reach clinical testing much sooner than a cathepsin B-specific compound.
TBI is an urgent unmet need that cannot wait. Furthermore, as discussed above, the multi-cysteine protease inhibition by E64c is more efficacious than selective cathepsin B inhibition and thus E64d derivatives offer a better chance of obtaining positive outcomes in clinical trials.
Conclusion
Cathepsin B gene deletion studies show that cathepsin B is a key player in the pathology of TBI. Cathepsin B is elevated following TBI and its removal or inhibition dramatically improves outcomes following trauma in animal models. Moreover, deleting or inhibiting cathepsin B improves outcomes in injury models related to TBI including epilepsy, aneurysm, ischemia, pain, surgical trauma, spinal cord trauma, infectious disease, and neurodegeneration. E64d is a promising compound for further preclinical testing as a candidate therapeutic agent for TBI because it has been shown to be orally effective when administered after trauma. E64d is the best scaffold upon which a TBI therapeutic can be developed because only E64d has extensive clinical and preclinical data available. It will also be beneficial to consider that inhibition of cathepsin B targets several mechanisms involved in TBI, including calpain and matrix metalloproteinases that contribute to TBI, as explained in this review. In conclusion, basic and translational research on cathepsin B for improving TBI-caused deficits should be accelerated as it is highly likely to generate an effective new TBI therapeutic.
Conflict of Interest Statement
Gregory Hook is an employee and has equity in ALSP, Inc., which is developing drugs to treat neurological conditions, including TBI. Vivian Hook is the chair of ALSP’s Scientific Advisory Board and J. Steven Jacobsen and Kenneth Grabstein are members of that Board and all hold equity in ALSP. J. Steven Jacobsen is an employee of AstraZeneca, a major pharmaceutical company. Vivian Hook and J. Steven Jacobsen have disclosed their relationship with ALSP to the University of California, San Diego (La Jolla, CA, USA) and AstraZeneca, respectively. Mark Kindy has no conflict of interest to declare.
Acknowledgments
The authors wish to thank Professor Tetsumori Yamashima at the Kanazawa University Graduate School of Medical Science, Kanazawa, Japan, for his insightful discussions. This work was supported in part by the United States National Institutes of Health (NIH) grants R44AG032784 (to American Life Science Pharmaceuticals, ALSP), NIH R21AG042828 (to VH), and the United States Veterans Administration (VA) Merit Reviews 1I01RX001450 and 1I01RX000331, NIH grants ES016774-01, R21AG043718, 1P20GM109091, 2P20GM103444, and 5P30GM103342, and NSF grants IIP-0903795 (to MK). MK is a Senior Research Career Scientist in the VA.
References
1. Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation (2007) 22:341–53.
2. Faul M, Xu LX, Wald M, Coronado V. Traumatic Brain Injury in the United States: Emergency Department visits, Hospitalizations, and Deaths. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Intjury Prevention and Control (2010).
3. Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil (2006) 21:544–8. doi:10.1097/00001199-200611000-00009
4. Gessel LM, Fields SK, Collins CL, Dick RW, Comstock RD. Concussions among United States high school and collegiate athletes. J Athl Train (2007) 42:495–503.
5. Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil (2006) 21:398–402. doi:10.1097/00001199-200609000-00004
6. Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT, et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma (2008) 25:719–38. doi:10.1089/neu.2008.0586
7. Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ (2008) 178:1163–70. doi:10.1503/cmaj.080282
8. McAllister TW. Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci (2011) 13:287–300.
9. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth (2007) 99:4–9. doi:10.1093/bja/aem131
10. Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res (2007) 161:43–59. doi:10.1016/S0079-6123(06)61004-2
11. Corps KN, Roth TL, Mcgavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol (2015) 72:355–62. doi:10.1001/jamaneurol.2014.3558
12. Muellner A, Benz M, Kloss CU, Mautes A, Burggraf D, Hamann GF. Microvascular basal lamina antigen loss after traumatic brain injury in the rat. J Neurotrauma (2003) 20:745–54. doi:10.1089/089771503767869971
13. Coles JP, Fryer TD, Smielewski P, Rice K, Clark JC, Pickard JD, et al. Defining ischemic burden after traumatic brain injury using 15O PET imaging of cerebral physiology. J Cereb Blood Flow Metab (2004) 24:191–201. doi:10.1097/01.WCB.0000100045.07481.DE
14. Miley JT, Rodriguez GJ, Qureshi AI. Traumatic intracranial aneurysm formation following closed head injury. J Vasc Interv Neurol (2008) 1:79–82.
15. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience (2004) 129:1021–9. doi:10.1016/j.neuroscience.2004.06.046
16. Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA (2008) 300:711–9. doi:10.1001/jama.300.6.711
17. Grande DJ, Koranda FC, Guthrie D. Monitoring respirations for outpatient surgery. J Dermatol Surg Oncol (1983) 9:338–9. doi:10.1111/j.1524-4725.1983.tb00811.x
18. Scott BN, Roberts DJ, Robertson HL, Kramer AH, Laupland KB, Ousman SS, et al. Incidence, prevalence, and occurrence rate of infection among adults hospitalized after traumatic brain injury: study protocol for a systematic review and meta-analysis. Syst Rev (2013) 2:68. doi:10.1186/2046-4053-2-68
19. Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, Dekosky ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem (1993) 61:2015–24. doi:10.1111/j.1471-4159.1993.tb07437.x
20. Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol (2007) 166:810–6. doi:10.1093/aje/kwm153
21. Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. Traumatic brain injury in later life increases risk for Parkinson’s disease. Ann Neurol (2015) 77(6):987–95. doi:10.1002/ana.24396
22. Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry (2003) 74:857–62. doi:10.1136/jnnp.74.7.857
23. Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med (1998) 338:20–4. doi:10.1056/NEJM199801013380104
24. Kang JH, Lin HC. Increased risk of multiple sclerosis after traumatic brain injury: a nationwide population-based study. J Neurotrauma (2012) 29:90–5. doi:10.1089/neu.2011.1936
25. Daneshvar DH, Goldstein LE, Kiernan PT, Stein TD, McKee AC. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci (2015) 66(Pt B):81–90. doi:10.1016/j.mcn.2015.03.007
26. Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma (2002) 19:503–57. doi:10.1089/089771502753754037
27. Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med (2008) 14:731–40. doi:10.2119/2008-00050.Beauchamp
28. Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med (2014) 371:2457–66. doi:10.1056/NEJMoa1404304
29. Powers BJ, Coeytaux RR, Dolor RJ, Hasselblad V, Patel UD, Yancy WS Jr, et al. Updated report on comparative effectiveness of ACE inhibitors, ARBs, and direct renin inhibitors for patients with essential hypertension: much more data, little new information. J Gen Intern Med (2012) 27:716–29. doi:10.1007/s11606-011-1938-8
30. Cruciani M, Malena M. Combination dolutegravir-abacavir-lamivudine in the management of HIV/AIDS: clinical utility and patient considerations. Patient Prefer Adherence (2015) 9:299–310. doi:10.2147/PPA.S65199
31. Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets (2011) 11:239–53. doi:10.2174/156800911794519752
32. Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des (2007) 13:387–403. doi:10.2174/138161207779313542
33. Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets (2013) 17:281–91. doi:10.1517/14728222.2013.740461
34. Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci (2008) 29:22–8. doi:10.1016/j.tips.2007.10.011
35. Yan S, Sloane BF. Molecular regulation of human cathepsin B: implication in pathologies. Biol Chem (2003) 384:845–54. doi:10.1515/BC.2003.095
36. Kos J, Mitrovic A, Mirkovic B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med Chem (2014) 6:1355–71. doi:10.4155/fmc.14.73
37. Van Acker GJ, Saluja AK, Bhagat L, Singh VP, Song AM, Steer ML. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am J Physiol Gastrointest Liver Physiol (2002) 283:G794–800. doi:10.1152/ajpgi.00363.2001
38. Canbay A, Guicciardi ME, Higuchi H, Feldstein A, Bronk SF, Rydzewski R, et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest (2003) 112:152–9. doi:10.1172/JCI17740
39. Hashimoto Y, Kakegawa H, Narita Y, Hachiya Y, Hayakawa T, Kos J, et al. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem Biophys Res Commun (2001) 283:334–9. doi:10.1006/bbrc.2001.4787
40. Yoshifuji H, Umehara H, Maruyama H, Itoh M, Tanaka M, Kawabata D, et al. Amelioration of experimental arthritis by a calpain-inhibitory compound: regulation of cytokine production by E-64-d in vivo and in vitro. Int Immunol (2005) 17:1327–36. doi:10.1093/intimm/dxh311
41. Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science (2005) 308:1643–5. doi:10.1126/science.1110656
42. Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, et al. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol (2011) 187:5440–51. doi:10.4049/jimmunol.1100790
43. McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. Development of protease inhibitors for protozoan infections. Curr Opin Infect Dis (2008) 21:668–72. doi:10.1097/QCO.0b013e328315cca9
44. Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol (2011) 5:201–12. doi:10.1586/egh.11.6
45. Holsinger LDC, Dener J, Green M, Booth R, Dalrymple S. Efficacy of a reversible cathepsin B inhibitor in a rodent model of liver fibrosis and human pharmacokinetic profile. Hepatology (2010) 52:1128A.
46. Doyle PS, Zhou YM, Engel JC, Mckerrow JH. A cysteine protease inhibitor cures Chagas’ disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother (2007) 51:3932–9. doi:10.1128/AAC.00436-07
47. Knoblach SM, Faden AI. Proteases in traumatic brain injury. In: Lendeckel U, Hooper NM, editors. Proteases in the Brain. Springer (2005).
48. Hook GR, Yu J, Sipes N, Pierschbacher MD, Hook V, Kindy MS. The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury. J Neurotrauma (2014) 31:515–29. doi:10.1089/neu.2013.2944
49. Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx C, Grimes J, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma (2013) 31(2):135–58. doi:10.1089/neu.2013.3019
50. Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci (2014) 15:1216–36. doi:10.3390/ijms15011216
51. Stocchetti N, Taccone FS, Citerio G, Pepe PE, Le Roux PD, Oddo M, et al. Neuroprotection in acute brain injury: an up-to-date review. Crit Care (2015) 19:186. doi:10.1186/s13054-015-0887-8
52. Fruton JS, Bergmann M. On the proteolytic enzymes of animal tissues: beef spleen. J Biol Chem (1939) 130:19–27.
53. Fruton JS, Irving GW, Bergmann M. One the proteolytic enzymes of beef spleen, beef kidney, and swine kidney. Classification of the cathepsins. J Biol Chem (1941) 138:249–62.
54. Tallan HH, Jones ME, Fruton JS. On the proteolytic enzymes of animal tissues. X. Beef spleen cathepsin C. J Biol Chem (1952) 194:793–805.
55. Greenbaum LM, Fruton JS. Purification and properties of beef spleen cathepsin B. J Biol Chem (1957) 226:173–80.
56. Takio K, Towatari T, Katunuma N, Teller DC, Titani K. Homology of amino acid sequences of rat liver cathepsins B and H with that of papain. Proc Natl Acad Sci U S A (1983) 80:3666–70. doi:10.1073/pnas.80.12.3666
57. Chan SJ, San Segundo B, Mccormick MB, Steiner DF. Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc Natl Acad Sci U S A (1986) 83:7721–5. doi:10.1073/pnas.83.20.7721
58. Musil D, Zucic D, Turk D, Engh RA, Mayr I, Huber R, et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J (1991) 10:2321–30.
59. Deussing J, Roth W, Saftig P, Peters C, Ploegh HL, Villadangos JA. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc Natl Acad Sci U S A (1998) 95:4516–21. doi:10.1073/pnas.95.8.4516
60. Mort JS. 333. Cathepsin B. Second ed. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Amsterdam: Elsevier Academic Press (2004) 1079–86.
61. Aronson NN Jr, Barrett AJ. The specificity of cathepsin B. Hydrolysis of glucagon at the C-terminus by a peptidyldipeptidase mechanism. Biochem J (1978) 171:759–65.
62. Takahashi T, Dehdarani AH, Yonezawa S, Tang J. Porcine spleen cathepsin B is an exopeptidase. J Biol Chem (1986) 261:9375–81.
63. Illy C, Quraishi O, Wang J, Purisima E, Vernet T, Mort JS. Role of the occluding loop in cathepsin B activity. J Biol Chem (1997) 272:1197–202. doi:10.1074/jbc.272.2.1197
64. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun (1967) 27:157–62. doi:10.1016/S0006-291X(67)80055-X
65. Gosalia DN, Salisbury CM, Ellman JA, Diamond SL. High throughput substrate specificity profiling of serine and cysteine proteases using solution-phase fluorogenic peptide microarrays. Mol Cell Proteomics (2005) 4:626–36. doi:10.1074/mcp.M500004-MCP200
66. Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem (2006) 281:12824–32. doi:10.1074/jbc.M513331200
67. Barrett A, Rawlings N, Woessner J. Handbook of Proteolytic Enzymes. Amsterdam: Elsevier Academic Press (2004).
68. Schechter I, Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun (1968) 32:898–902. doi:10.1016/0006-291X(68)90326-4
69. Hook V, Schechter I, Demuth HU, Hook G. Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. Biol Chem (2008) 389:993–1006. doi:10.1515/BC.2008.124
70. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta (2012) 1824:68–88. doi:10.1016/j.bbapap.2011.10.002
71. Rossi A, Deveraux Q, Turk B, Sali A. Comprehensive search for cysteine cathepsins in the human genome. Biol Chem (2004) 385:363–72. doi:10.1515/BC.2004.040
72. Fong D, Chan MM, Hsieh WT, Menninger JC, Ward DC. Confirmation of the human cathepsin B gene (CTSB) assignment to chromosome 8. Hum Genet (1992) 89:10–2. doi:10.1007/BF00207033
73. Berquin IM, Cao L, Fong D, Sloane BF. Identification of two new exons and multiple transcription start points in the 5’-untranslated region of the human cathepsin-B-encoding gene. Gene (1995) 159:143–9. doi:10.1016/0378-1119(95)00072-E
74. Gong Q, Chan SJ, Bajkowski AS, Steiner DF, Frankfater A. Characterization of the cathepsin B gene and multiple mRNAs in human tissues: evidence for alternative splicing of cathepsin B pre-mRNA. DNA Cell Biol (1993) 12:299–309. doi:10.1089/dna.1993.12.299
75. Tabares-Seisdedos R, Rubenstein JL. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry (2009) 14:563–89. doi:10.1038/mp.2009.2
76. Mahurkar S, Idris MM, Reddy DN, Bhaskar S, Rao GV, Thomas V, et al. Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut (2006) 55:1270–5. doi:10.1136/gut.2005.087403
77. Qian F, Frankfater A, Chan SJ, Steiner DF. The structure of the mouse cathepsin B gene and its putative promoter. DNA Cell Biol (1991) 10:159–68. doi:10.1089/dna.1991.10.159
78. Deussing J, Roth W, Rommerskirch W, Wiederanders B, Von Figura K, Peters C. The genes of the lysosomal cysteine proteinases cathepsin B, H, L, and S map to different mouse chromosomes. Mamm Genome (1997) 8:241–5. doi:10.1007/s003359900401
79. Sivaparvathi M, Sawaya R, Wang SW, Rayford A, Yamamoto M, Liotta LA, et al. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin Exp Metastasis (1995) 13:49–56. doi:10.1007/BF00144018
80. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi:10.1016/j.cell.2009.01.035
81. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol (2004) 5:R13. doi:10.1186/gb-2004-5-3-r13
82. Bhalala OG, Srikanth M, Kessler JA. The emerging roles of microRNAs in CNS injuries. Nat Rev Neurol (2013) 9:328–39. doi:10.1038/nrneurol.2013.67
83. Redell JB, Liu Y, Dash PK. Traumatic brain injury alters expression of hippocampal microRNAs: potential regulators of multiple pathophysiological processes. J Neurosci Res (2009) 87:1435–48. doi:10.1002/jnr.21945
84. Venkataraman S, Birks DK, Balakrishnan I, Alimova I, Harris PS, Patel PR, et al. MicroRNA 218 acts as a tumor suppressor by targeting multiple cancer phenotype-associated genes in medulloblastoma. J Biol Chem (2013) 288:1918–28. doi:10.1074/jbc.M112.396762
85. Tiribuzi R, Crispoltoni L, Porcellati S, Di Lullo M, Florenzano F, Pirro M, et al. miR128 up-regulation correlates with impaired amyloid beta(1-42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol Aging (2014) 35:345–56. doi:10.1016/j.neurobiolaging.2013.08.003
86. Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, et al. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J (2007) 21:415–26. doi:10.1096/fj.06-6130com
87. Katunuma N. Posttranslational processing and modification of cathepsins and cystatins. J Signal Transduct (2010) 2010:375345. doi:10.1155/2010/375345
88. Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta (2000) 1477:98–111. doi:10.1016/S0167-4838(99)00263-0
89. Neurath H. Evolution of proteolytic enzymes. Science (1984) 224:350–7. doi:10.1126/science.6369538
90. Pungercar JR, Caglic D, Sajid M, Dolinar M, Vasiljeva O, Pozgan U, et al. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J (2009) 276:660–8. doi:10.1111/j.1742-4658.2008.06815.x
91. Mach L, Mort JS, Glossl J. Maturation of human procathepsin B. Proenzyme activation and proteolytic processing of the precursor to the mature proteinase, in vitro, are primarily unimolecular processes. J Biol Chem (1994) 269:13030–5.
92. Kominami E, Tsukahara T, Bando Y, Katunuma N. Autodegradation of lysosomal cysteine proteinases. Biochem Biophys Res Commun (1987) 144:749–56. doi:10.1016/S0006-291X(87)80028-1
93. Natale JE, Ahmed F, Cernak I, Stoica B, Faden AI. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J Neurotrauma (2003) 20:907–27. doi:10.1089/089771503770195777
94. Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, et al. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res (2010) 88:2847–58. doi:10.1002/jnr.22453
95. Zhang M, Shan H, Chang P, Wang T, Dong W, Chen X, et al. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS One (2014) 9:e87241. doi:10.1371/journal.pone.0087241
96. Zhang YB, Chen XP, Tao LY, Qin ZH, Li SX, Yang L, et al. Expression of cathepsin-B and -D in rat’s brain after traumatic brain injury. Fa Yi Xue Za Zhi (2006) 22(404–6):410.
97. Martinez-Vargas M, Soto-Nunez M, Tabla-Ramon E, Solis B, Gonzalez-Rivera R, Perez-Arredondo A, et al. Cystatin C has a dual role in post-traumatic brain injury recovery. Int J Mol Sci (2014) 15:5807–20. doi:10.3390/ijms15045807
98. Sun YX, Dai DK, Liu R, Wang T, Luo CL, Bao HJ, et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-kappaB, in treatment of TBI in mice. Neurol Sci (2013) 34:345–55. doi:10.1007/s10072-012-1007-z
99. Ellis RC, Earnhardt JN, Hayes RL, Wang KK, Anderson DK. Cathepsin B mRNA and protein expression following contusion spinal cord injury in rats. J Neurochem (2004) 88:689–97. doi:10.1046/j.1471-4159.2003.02197.x
100. Ellis RC, O’steen WA, Hayes RL, Nick HS, Wang KK, Anderson DK. Cellular localization and enzymatic activity of cathepsin B after spinal cord injury in the rat. Exp Neurol (2005) 193:19–28. doi:10.1016/j.expneurol.2004.11.034
101. Vreemann A, Qu H, Mayer K, Andersen LB, Stefana MI, Wehner S, et al. Cathepsin B release from rodent intestine mucosa due to mechanical injury results in extracellular matrix damage in early post-traumatic phases. Biol Chem (2009) 390:481–92. doi:10.1515/BC.2009.055
102. Yu ZQ, Jia Y, Chen G. Possible involvement of cathepsin B/D and caspase-3 in deferoxamine-related neuroprotection of early brain injury after subarachnoid haemorrhage in rats. Neuropathol Appl Neurobiol (2014) 40:270–83. doi:10.1111/nan.12091
103. Wang Y, Gao A, Xu X, Dang B, You W, Li H, et al. The neuroprotection of lysosomotropic agents in experimental subarachnoid hemorrhage probably involving the apoptosis pathway triggering by cathepsins via chelating intralysosomal iron. Mol Neurobiol (2014) 52(1):64–77. doi:10.1007/s12035-014-8846-y
104. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Cathepsin B, K, and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms. Stroke (2008) 39:2603–10. doi:10.1161/STROKEAHA.107.513648
105. Yamada E, Chue CH, Yukioka N, Hazama F. Causative role of lysosomal enzymes in the pathogenesis of cerebral lesions due to brain edema under chronic hypertension. Acta Neurochir Suppl (Wien) (1994) 60:83–5.
106. Tsubokawa T, Yamaguchi-Okada M, Calvert JW, Solaroglu I, Shimamura N, Yata K, et al. Neurovascular and neuronal protection by E64d after focal cerebral ischemia in rats. J Neurosci Res (2006) 84:832–40. doi:10.1002/jnr.20977
107. Seyfried D, Han Y, Zheng Z, Day N, Moin K, Rempel S, et al. Cathepsin B and middle cerebral artery occlusion in the rat. J Neurosurg (1997) 87:716–23. doi:10.3171/jns.1997.87.5.0716
108. Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, et al. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur J Neurosci (1998) 10:1723–33. doi:10.1046/j.1460-9568.1998.00184.x
109. Tsuchiya K, Kohda Y, Yoshida M, Zhao L, Ueno T, Yamashita J, et al. Postictal blockade of ischemic hippocampal neuronal death in primates using selective cathepsin inhibitors. Exp Neurol (1999) 155:187–94. doi:10.1006/exnr.1998.6988
110. Anagli J, Abounit K, Stemmer P, Han Y, Allred L, Weinsheimer S, et al. Effects of cathepsins B and L inhibition on postischemic protein alterations in the brain. Biochem Biophys Res Commun (2008) 366:86–91. doi:10.1016/j.bbrc.2007.11.104
111. Ni H, Yan JZ, Zhang LL, Feng X, Wu XR. Long-term effects of recurrent neonatal seizures on neurobehavioral function and related gene expression and its intervention by inhibitor of cathepsin B. Neurochem Res (2012) 37:31–9. doi:10.1007/s11064-011-0578-z
112. Wang Y, Gu ZL, Cao Y, Liang ZQ, Han R, Bennett MC, et al. Lysosomal enzyme cathepsin B is involved in kainic acid-induced excitotoxicity in rat striatum. Brain Res (2006) 1071:245–9. doi:10.1016/j.brainres.2005.10.074
113. Ruff RL, Secrist D. Inhibitors of prostaglandin synthesis or cathepsin B prevent muscle wasting due to sepsis in the rat. J Clin Invest (1984) 73:1483–6. doi:10.1172/JCI111352
114. Hummel RP III, James JH, Warner BW, Hasselgren PO, Fischer JE. Evidence that cathepsin B contributes to skeletal muscle protein breakdown during sepsis. Arch Surg (1988) 123:221–4. doi:10.1001/archsurg.1988.01400260105013
115. Sun L, Wu Z, Hayashi Y, Peters C, Tsuda M, Inoue K, et al. Microglial cathepsin B contributes to the initiation of peripheral inflammation-induced chronic pain. J Neurosci (2012) 32:11330–42. doi:10.1523/JNEUROSCI.0677-12.2012
116. Terada K, Yamada J, Hayashi Y, Wu Z, Uchiyama Y, Peters C, et al. Involvement of cathepsin B in the processing and secretion of interleukin-1beta in chromogranin A-stimulated microglia. Glia (2010) 58:114–24. doi:10.1002/glia.20906
117. Offen D, Barhum Y, Melamed E, Embacher N, Schindler C, Ransmayr G. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J Mol Neurosci (2009) 38:85–93. doi:10.1007/s12031-007-9004-z
118. Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci (2007) 27:9201–19. doi:10.1523/JNEUROSCI.1470-07.2007
119. Bouter Y, Kacprowski T, Weissmann R, Dietrich K, Borgers H, Brauss A, et al. Deciphering the molecular profile of plaques, memory decline and neuron loss in two mouse models for Alzheimer’s disease by deep sequencing. Front Aging Neurosci (2014) 6:75. doi:10.3389/fnagi.2014.00075
120. Sun Y, Rong X, Lu W, Peng Y, Li J, Xu S, et al. Translational study of Alzheimer’s disease (AD) biomarkers from brain tissues in AbetaPP/PS1 mice and serum of AD patients. J Alzheimers Dis (2015) 45:269–82. doi:10.3233/JAD-142805
121. Saris CG, Groen EJ, Koekkoek JA, Veldink JH, Van Den Berg LH. Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph Lateral Scler Frontotemporal Degener (2013) 14:177–89. doi:10.3109/21678421.2012.729842
122. Ni H, Ren SY, Zhang LL, Sun Q, Tian T, Feng X. Expression profiles of hippocampal regenerative sprouting-related genes and their regulation by E-64d in a developmental rat model of penicillin-induced recurrent epilepticus. Toxicol Lett (2013) 217:162–9. doi:10.1016/j.toxlet.2012.12.010
123. Assfalg-Machleidt I, Jochum M, Nast-Kolb D, Siebeck M, Billing A, Joka T, et al. Cathepsin B-indicator for the release of lysosomal cysteine proteinases in severe trauma and inflammation. Biol Chem Hoppe Seyler (1990) 371(Suppl):211–22.
124. Jochum M, Machleidt W, Fritz H. Phagocyte proteinases in multiple trauma and sepsi: pathomechanims and related therapeutic approaches. In: Neugebauer EA, Holaday JW, editors. Handbook of Mediators in Septic Shock. CRC Press (1993). p. 335–61.
125. Nagai A, Murakawa Y, Terashima M, Shimode K, Umegae N, Takeuchi H, et al. Cystatin C and cathepsin B in CSF from patients with inflammatory neurologic diseases. Neurology (2000) 55:1828–32. doi:10.1212/WNL.55.12.1828
126. Bever CT Jr, Garver DW. Increased cathepsin B activity in multiple sclerosis brain. J Neurol Sci (1995) 131:71–3. doi:10.1016/0022-510X(95)00039-5
127. Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, Stephanopoulos G, et al. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics (2004) 16:229–39. doi:10.1152/physiolgenomics.00087.2001
128. Kikuchi H, Yamada T, Furuya H, Doh-Ura K, Ohyagi Y, Iwaki T, et al. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol (2003) 105:462–8. doi:10.1007/s00401-002-0667-9
129. Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci U S A (1990) 87:3861–5. doi:10.1073/pnas.87.10.3861
130. Sundelof J, Sundstrom J, Hansson O, Eriksdotter-Jonhagen M, Giedraitis V, Larsson A, et al. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J Alzheimers Dis (2010) 22:1223–30. doi:10.3233/JAD-2010-101023
131. Zhang J, Goodlett DR, Quinn JF, Peskind E, Kaye JA, Zhou Y, et al. Quantitative proteomics of cerebrospinal fluid from patients with Alzheimer disease. J Alzheimers Dis (2005) 7:125–33.
132. Armstrong A, Mattsson N, Appelqvist H, Janefjord C, Sandin L, Agholme L, et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular Med (2014) 16:150–60. doi:10.1007/s12017-013-8269-3
133. Trabandt A, Gay RE, Fassbender HG, Gay S. Cathepsin B in synovial cells at the site of joint destruction in rheumatoid arthritis. Arthritis Rheum (1991) 34:1444–51. doi:10.1002/art.1780341116
134. Baici A, Lang A, Horler D, Kissling R, Merlin C. Cathepsin B in osteoarthritis: cytochemical and histochemical analysis of human femoral head cartilage. Ann Rheum Dis (1995) 54:289–97. doi:10.1136/ard.54.4.281
135. Menzel K, Hausmann M, Obermeier F, Schreiter K, Dunger N, Bataille F, et al. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin Exp Immunol (2006) 146:169–80. doi:10.1111/j.1365-2249.2006.03188.x
136. Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem (2001) 382:735–41. doi:10.1515/BC.2001.089
137. Friedrichs B, Tepel C, Reinheckel T, Deussing J, Von Figura K, Herzog V, et al. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest (2003) 111:1733–45. doi:10.1172/JCI15990
138. Houseweart MK, Pennacchio LA, Vilaythong A, Peters C, Noebels JL, Myers RM. Cathepsin B but not cathepsins L or S contributes to the pathogenesis of Unverricht-Lundborg progressive myoclonus epilepsy (EPM1). J Neurobiol (2003) 56:315–27. doi:10.1002/neu.10253
139. Hook VY, Kindy M, Reinheckel T, Peters C, Hook G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res Commun (2009) 386:284–8. doi:10.1016/j.bbrc.2009.05.131
140. Kindy MS, Yu J, Zhu H, El-Amouri SS, Hook V, Hook GR. Deletion of the cathepsin B gene improves memory deficits in a transgenic alzheimer’s disease mouse model expressing AbetaPP containing the wild-type beta-secretase site sequence. J Alzheimers Dis (2012) 29:827–40. doi:10.3233/JAD-2012-111604
141. Hook G, Yu J, Toneff T, Kindy M, Hook V. Brain pyroglutamate amyloid-beta is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimers Dis (2014) 41:129–49. doi:10.3233/JAD-131370
142. Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron (2006) 51:703–14. doi:10.1016/j.neuron.2006.07.027
143. Wu Z, Sun L, Hashioka S, Yu S, Schwab C, Okada R, et al. Differential pathways for interleukin-1beta production activated by chromogranin A and amyloid beta in microglia. Neurobiol Aging (2013) 34:2715–25. doi:10.1016/j.neurobiolaging.2013.05.018
144. Ha SD, Martins A, Khazaie K, Han J, Chan BM, Kim SO. Cathepsin B is involved in the trafficking of TNF-alpha-containing vesicles to the plasma membrane in macrophages. J Immunol (2008) 181:690–7. doi:10.4049/jimmunol.181.1.690
145. Guicciardi ME, Bronk SF, Werneburg NW, Yin XM, Gores GJ. Bid is upstream of lysosome-mediated caspase 2 activation in tumor necrosis factor alpha-induced hepatocyte apoptosis. Gastroenterology (2005) 129:269–84. doi:10.1053/j.gastro.2005.05.022
146. Allan ER, Yates RM. Redundancy between cysteine cathepsins in murine experimental autoimmune encephalomyelitis. PLoS One (2015) 10:e0128945. doi:10.1371/journal.pone.0128945
147. Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease. J Biol Chem (2011) 286:38825–32. doi:10.1074/jbc.R111.288308
148. Hook V, Toneff T, Bogyo M, Medzihradszky KF, Nevenu J, Lane W, et al. Inhibition of cathepsin B reduces β-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: evidence for cathepsin B as a candidate β-secretase of Alzheimer’s disease. Biol Chem (2005) 386:931–40. doi:10.1515/BC.2005.108
149. Hook V, Kindy M, Hook G. Cysteine protease inhibitors effectively reduce in vivo levels of brain beta-amyloid related to Alzheimer’s disease. Biol Chem (2007) 388:247–52. doi:10.1515/BC.2007.027
150. Hook G, Hook VY, Kindy M. Cysteine protease inhibitors reduce brain beta-amyloid and beta-secretase activity in vivo and are potential Alzheimer’s disease therapeutics. Biol Chem (2007) 388:979–83. doi:10.1515/BC.2007.027
151. Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce Abeta in transgenic Alzheimer’s Disease mice expressing the wild-type, but not the Swedish mutant, beta -secretase APP site. J Biol Chem (2008) 283:7745–53. doi:10.1074/jbc.M708362200
152. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry (1994) 57:419–25. doi:10.1136/jnnp.57.4.419
153. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol (2009) 19:214–23. doi:10.1111/j.1750-3639.2008.00176.x
154. Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Front Neurol (2013) 4:30. doi:10.3389/fneur.2013.00030
155. Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab (2002) 22:971–8. doi:10.1097/00004647-200208000-00008
156. Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J Trauma (2006) 60:152–7. doi:10.1097/01.ta.0000196345.81169.a1
157. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab (2014) 34:369–75. doi:10.1038/jcbfm.2013.227
158. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9:857–65. doi:10.1038/ni.1636
159. Salvesen GS. A lysosomal protease enters the death scene. J Clin Invest (2001) 107:21–2. doi:10.1172/JCI11829
160. Goodman JC, Robertson CS, Grossman RG, Narayan RK. Elevation of tumor necrosis factor in head injury. J Neuroimmunol (1990) 30:213–7. doi:10.1016/0165-5728(90)90105-V
161. Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest (2000) 106:1127–37. doi:10.1172/JCI9914
162. Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, et al. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A (2002) 99:7883–8. doi:10.1073/pnas.112632299
163. Sevenich L, Pennacchio LA, Peters C, Reinheckel T. Human cathepsin L rescues the neurodegeneration and lethality in cathepsin B/L double-deficient mice. Biol Chem (2006) 387:885–91. doi:10.1515/BC.2006.112
164. Howie AJ, Burnett D, Crocker J. The distribution of cathepsin B in human tissues. J Pathol (1985) 145:307–14. doi:10.1002/path.1711450404
165. Kominami E, Tsukahara T, Bando Y, Katunuma N. Distribution of cathepsins B and H in rat tissues and peripheral blood cells. J Biochem (1985) 98:87–93.
166. San Segundo B, Chan SJ, Steiner DF. Differences in cathepsin B mRNA levels in rat tissues suggest specialized functions. FEBS Lett (1986) 201:251–6. doi:10.1016/0014-5793(86)80618-4
167. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science (2015) 347:1260419. doi:10.1126/science.1260419
168. Bernstein HG, Kirschke H, Wiederanders B, Kloss P, Rinne A, Dorn A. Cathepsin B immunoreactivity is widely distributed in the rat brain. J Hirnforsch (1988) 29:17–9.
169. Bernstein HG, Kirschke H, Wiederanders B, Schmidt D, Rinne A. Antigenic expression of cathepsin B in aged human brain. Brain Res Bull (1990) 24:543–9. doi:10.1016/0361-9230(90)90157-U
170. Nakamura Y, Takeda M, Suzuki H, Hattori H, Tada K, Hariguchi S, et al. Abnormal distribution of cathepsins in the brain of patients with Alzheimer’s disease. Neurosci Lett (1991) 130:195–8. doi:10.1016/0304-3940(91)90395-A
171. Ii K, Ito H, Kominami E, Hirano A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch A Pathol Anat Histopathol (1993) 423:185–94. doi:10.1007/BF01614769
172. Bernstein HG, Sormunen R, Jarvinen M, Kloss P, Kirschke H, Rinne A. Cathepsin B immunoreactive neurons in rat brain. A combined light and electron microscopic study. J Hirnforsch (1989) 30:313–7.
173. Jung H, Lee EY, Lee SI. Age-related changes in ultrastructural features of cathepsin B- and D-containing neurons in rat cerebral cortex. Brain Res (1999) 844:43–54. doi:10.1016/S0006-8993(99)01888-0
174. Petanceska S, Burke S, Watson SJ, Devi L. Differential distribution of messenger RNAs for cathepsins B, L and S in adult rat brain: an in situ hybridization study. Neuroscience (1994) 59:729–38. doi:10.1016/0306-4522(94)90190-2
175. Nilsson E, Bodolea C, Gordh T, Larsson A. Cerebrospinal fluid cathepsin B and S. Neurol Sci (2013) 34:445–8. doi:10.1007/s10072-012-1022-0
176. Nakanishi H, Tominaga K, Amano T, Hirotsu I, Inoue T, Yamamoto K. Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp Neurol (1994) 126:119–28. doi:10.1006/exnr.1994.1048
177. de Duve C. Lysosomes revisited. Eur J Biochem (1983) 137:391–7. doi:10.1111/j.1432-1033.1983.tb07841.x
178. Mortimore GE, Lardeux BR, Adams CE. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J Biol Chem (1988) 263:2506–12.
179. Bohley P, Seglen PO. Proteases and proteolysis in the lysosome. Experientia (1992) 48:151–7. doi:10.1007/BF01923508
181. Bromme D, Bonneau PR, Lachance P, Storer AC. Engineering the S2 subsite specificity of human cathepsin S to a cathepsin L- and cathepsin B-like specificity. J Biol Chem (1994) 269:30238–42.
182. Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol (2002) 3:122–31. doi:10.1038/nrm732
183. Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, et al. Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol (2007) 9:945–53. doi:10.1038/ncb1620
184. Verderio C, Cagnoli C, Bergami M, Francolini M, Schenk U, Colombo A, et al. TI-VAMP/VAMP7 is the SNARE of secretory lysosomes contributing to ATP secretion from astrocytes. Biol Cell (2012) 104:213–28. doi:10.1111/boc.201100070
185. Spizz G, Blackshear PJ. Identification and characterization of cathepsin B as the cellular MARCKS cleaving enzyme. J Biol Chem (1997) 272:23833–42. doi:10.1074/jbc.272.38.23833
186. Graber S, Maiti S, Halpain S. Cathepsin B-like proteolysis and MARCKS degradation in sub-lethal NMDA-induced collapse of dendritic spines. Neuropharmacology (2004) 47:706–13. doi:10.1016/j.neuropharm.2004.08.004
187. Kopitar-Jerala N, Turk B. Cleavage of the myristoylated alanine-rich C kinase substrate (MARCKS) by cysteine cathepsins in cells and tissues of stefin B-deficient mice. Biol Chem (2007) 388:847–52. doi:10.1515/BC.2007.092
188. Linke M, Herzog V, Brix K. Trafficking of lysosomal cathepsin B-green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J Cell Sci (2002) 115:4877–89. doi:10.1242/jcs.00184
189. Pozuelo-Rubio M. 14-3-3zeta binds class III phosphatidylinositol-3-kinase and inhibits autophagy. Autophagy (2011) 7:240–2. doi:10.4161/auto.7.2.14286
190. Brix K, Mcinnes J, Al-Hashimi A, Rehders M, Tamhane T, Haugen MH. Proteolysis mediated by cysteine cathepsins and legumain-recent advances and cell biological challenges. Protoplasma (2015) 252:755–74. doi:10.1007/s00709-014-0730-0
191. Kelly RB. Pathways of protein secretion in eukaryotes. Science (1985) 230:25–32. doi:10.1126/science.2994224
192. Kukor Z, Mayerle J, Kruger B, Toth M, Steed PM, Halangk W, et al. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J Biol Chem (2002) 277:21389–96. doi:10.1074/jbc.M200878200
193. Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest (2000) 106:773–81. doi:10.1172/JCI9411
194. Kuliawat R, Klumperman J, Ludwig T, Arvan P. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic beta-cells. J Cell Biol (1997) 137:595–608. doi:10.1083/jcb.137.3.595
195. Shinagawa T, Nakayama K, Uchiyama Y, Kominami E, Doi Y, Hashiba K, et al. Role of cathepsin B as prorenin processing enzyme in human kidney. Hypertens Res (1995) 18:131–6. doi:10.1291/hypres.18.131
196. Turk B, Dolenc I, Zerovnik E, Turk D, Gubensek F, Turk V. Human cathepsin B is a metastable enzyme stabilized by specific ionic interactions associated with the active site. Biochemistry (1994) 33:14800–6. doi:10.1021/bi00253a019
197. Pratt MR, Sekedat MD, Chiang KP, Muir TW. Direct measurement of cathepsin B activity in the cytosol of apoptotic cells by an activity-based probe. Chem Biol (2009) 16:1001–12. doi:10.1016/j.chembiol.2009.07.011
198. Polgar L, Csoma C. Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B. J Biol Chem (1987) 262:14448–53.
199. Almeida PC, Nantes IL, Chagas JR, Rizzi CC, Faljoni-Alario A, Carmona E, et al. Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J Biol Chem (2001) 276:944–51. doi:10.1074/jbc.M003820200
200. Turk B, Dolenc I, Turk V, Bieth JG. Kinetics of the pH-induced inactivation of human cathepsin L. Biochemistry (1993) 32:375–80. doi:10.1021/bi00052a046
201. Turk B, Turk D, Salvesen GS. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr Pharm Des (2002) 8:1623–37. doi:10.2174/1381612023394124
202. Abrahamson M, Barrett AJ, Salvesen G, Grubb A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J Biol Chem (1986) 261:11282–9.
203. Sundelof J, Sundstrom J, Hansson O, Eriksdotter-Jonhagen M, Giedraitis V, Larsson A, et al. Cystatin C levels are positively correlated with both Abeta42 and tau levels in cerebrospinal fluid in persons with Alzheimer’s disease, mild cognitive impairment, and healthy controls. J Alzheimers Dis (2010) 21:471–8. doi:10.3233/JAD-2010-091594
204. de Duve C, De Reuck AVS, Cameron MP, Ciba Foundation. Ciba Foundation Symposium: Lysosomes; [Proceedings]. Boston, MA: Little, Brown (1963).
205. Lafrenaye AD, Mcginn MJ, Povlishock JT. Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poration but not axonal injury: evidence for primary intracranial pressure-induced neuronal perturbation. J Cereb Blood Flow Metab (2012) 32:1919–32. doi:10.1038/jcbfm.2012.95
206. Luo CL, Chen XP, Li LL, Li QQ, Li BX, Xue AM, et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J Neurotrauma (2013) 30:597–607. doi:10.1089/neu.2012.2425
207. Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, et al. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res (2012) 37:2856–67. doi:10.1007/s11064-012-0880-4
208. Gu JH, Ge JB, Li M, Xu HD, Wu F, Qin ZH. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS One (2013) 8:e61641. doi:10.1371/journal.pone.0061641
209. Amritraj A, Peake K, Kodam A, Salio C, Merighi A, Vance JE, et al. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am J Pathol (2009) 175:2540–56. doi:10.2353/ajpath.2009.081096
210. Zhao S, Aviles ER Jr, Fujikawa DG. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 minutes of generalized seizures. J Neurosci Res (2010) 88:1727–37. doi:10.1002/jnr.22338
211. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One (2013) 8:e55375. doi:10.1371/journal.pone.0055375
212. Freeman D, Cedillos R, Choyke S, Lukic Z, Mcguire K, Marvin S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One (2013) 8:e62143. doi:10.1371/journal.pone.0062143
213. Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis (2001) 8:19–31. doi:10.1006/nbdi.2000.0364
214. Aits S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci (2013) 126:1905–12. doi:10.1242/jcs.091181
215. Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol (2009) 89:343–58. doi:10.1016/j.pneurobio.2009.09.003
216. Yamashima T. Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’ – a perspective review. Prog Neurobiol (2013) 105:1–23. doi:10.1016/j.pneurobio.2013.02.004
217. Zhu H, Yoshimoto T, Yamashima T. Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem (2014) 289:27432–43. doi:10.1074/jbc.M114.560334
218. Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ. Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol (2002) 283:G947–56. doi:10.1152/ajpgi.00151.2002
219. Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med (2006) 41:77–85. doi:10.1016/j.freeradbiomed.2006.03.007
220. Bayir H, Kagan VE, Borisenko GG, Tyurina YY, Janesko KL, Vagni VA, et al. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab (2005) 25:673–84. doi:10.1038/sj.jcbfm.9600068
221. Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol (2008) 129:389–406. doi:10.1007/s00418-008-0394-y
222. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene (2008) 27:6434–51. doi:10.1038/onc.2008.310
223. Tontchev AB, Yamashima T. Ischemic delayed neuronal death: role of the cysteine proteases calpain and cathepsins. Neuropathology (1999) 19:356–65. doi:10.1046/j.1440-1789.1999.00259.x
224. Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene (2004) 23:2881–90. doi:10.1038/sj.onc.1207512
225. Lima H Jr, Jacobson LS, Goldberg MF, Chandran K, Diaz-Griffero F, Lisanti MP, et al. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle (2013) 12:1868–78. doi:10.4161/cc.24903
226. Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev (2000) 14:2060–71.
227. Michallet MC, Saltel F, Preville X, Flacher M, Revillard JP, Genestier L. Cathepsin-B-dependent apoptosis triggered by antithymocyte globulins: a novel mechanism of T-cell depletion. Blood (2003) 102:3719–26. doi:10.1182/blood-2003-04-1075
228. Kingham PJ, Pocock JM. Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem (2001) 76:1475–84. doi:10.1046/j.1471-4159.2001.00146.x
229. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi:10.1038/ni.1631
230. Schotte P, Van Criekinge W, Van De Craen M, Van Loo G, Desmedt M, Grooten J, et al. Cathepsin B-mediated activation of the proinflammatory caspase-11. Biochem Biophys Res Commun (1998) 251:379–87. doi:10.1006/bbrc.1998.9425
231. Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol (2000) 149:613–22. doi:10.1083/jcb.149.3.613
232. Stahel PF, Smith WR, Bruchis J, Rabb CH. Peroxisome proliferator-activated receptors: “key” regulators of neuroinflammation after traumatic brain injury. PPAR Res (2008) 2008:538141. doi:10.1155/2008/538141
233. Besson VC, Chen XR, Plotkine M, Marchand-Verrecchia C. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, exerts neuroprotective effects in traumatic brain injury. Neurosci Lett (2005) 388:7–12. doi:10.1016/j.neulet.2005.06.019
234. Chen XR, Besson VC, Palmier B, Garcia Y, Plotkine M, Marchand-Leroux C. Neurological recovery-promoting, anti-inflammatory, and anti-oxidative effects afforded by fenofibrate, a PPAR alpha agonist, in traumatic brain injury. J Neurotrauma (2007) 24:1119–31. doi:10.1089/neu.2006.0216
235. Reichenbach G, Starzinski-Powitz A, Doll M, Hrgovic I, Valesky EM, Kippenberger S, et al. Ligand activation of peroxisome proliferator-activated receptor delta suppresses cathepsin B expression in human endothelial cells in a posttranslational manner. Exp Dermatol (2012) 21:751–7. doi:10.1111/exd.12002
236. Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain (2003) 126:515–30. doi:10.1093/brain/awg061
237. Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron (2005) 48:77–90. doi:10.1016/j.neuron.2005.08.027
238. Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab (2008) 28:540–50. doi:10.1038/sj.jcbfm.9600551
239. Zhang YB, Li SX, Chen XP, Yang L, Zhang YG, Liu R, et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull (2008) 24:143–9. doi:10.1007/s12264-008-1108-0
240. Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ, Liu WL, et al. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem Res (2012) 37:1849–58. doi:10.1007/s11064-012-0791-4
241. Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience (2011) 184:54–63. doi:10.1016/j.neuroscience.2011.03.021
242. Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol (2009) 452:199–213. doi:10.1016/S0076-6879(08)03613-6
243. Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ (2011) 18:571–80. doi:10.1038/cdd.2010.191
244. Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol (2009) 452:181–97. doi:10.1016/S0076-6879(08)03612-4
245. Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem (2010) 285:10850–61. doi:10.1074/jbc.M109.080796
246. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy (2008) 4:151–75. doi:10.4161/auto.5338
247. Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab (2004) 24:1119–32. doi:10.1097/01.WCB.0000138664.17682.32
248. Xu J, Wang H, Ding K, Lu X, Li T, Wang J, et al. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm (2013) 2013:187873. doi:10.1155/2013/187873
249. Ray SK, Matzelle DC, Wilford GG, Hogan EL, Banik NL. E-64-d prevents both calpain upregulation and apoptosis in the lesion and penumbra following spinal cord injury in rats. Brain Res (2000) 867:80–9. doi:10.1016/S0006-8993(00)02260-5
250. Inuzuka T, Tamura A, Sato S, Kirino T, Toyoshima I, Miyatake T. Suppressive effect of E-64c on ischemic degradation of cerebral proteins following occlusion of the middle cerebral artery in rats. Brain Res (1990) 526:177–9. doi:10.1016/0006-8993(90)90269-H
251. Seyfried DM, Veyna R, Han Y, Li K, Tang N, Betts RL, et al. A selective cysteine protease inhibitor is non-toxic and cerebroprotective in rats undergoing transient middle cerebral artery ischemia. Brain Res (2001) 901:94–101. doi:10.1016/S0006-8993(01)02289-2
252. Yoshida M, Yamashima T, Zhao L, Tsuchiya K, Kohda Y, Tonchev AB, et al. Primate neurons show different vulnerability to transient ischemia and response to cathepsin inhibition. Acta Neuropathol (2002) 104:267–72.
253. Yang D, Han Y, Zhang J, Ding C, Anagli J, Seyfried DM. Improvement in recovery after experimental intracerebral hemorrhage using a selective cathepsin B and L inhibitor. J Neurosurg (2011) 114:1110–6. doi:10.3171/2010.6.JNS091856
254. Lyo V, Cattaruzza F, Kim TN, Walker AW, Paulick M, Cox D, et al. Active cathepsins B, L, and S in murine and human pancreatitis. Am J Physiol Gastrointest Liver Physiol (2012) 303:G894–903. doi:10.1152/ajpgi.00073.2012
255. Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest (2008) 118:2796–807. doi:10.1172/JCI34254
256. Hook G, Hook V, Kindy M. The cysteine protease inhibitor, E64d, reduces brain amyloid-beta and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, beta-secretase activity. J Alzheimers Dis (2011) 26:387–408. doi:10.3233/JAD-2011-110101
257. Hanada K, Tamai M, Yamagishi S, Ohmura J, Sawada I, Tanaka H. Isolation and characterization of E-64, a new thiol protease inhibitor. Agric Biol Chem (1978) 42:523–8. doi:10.1271/bbb1961.42.523
258. Parkes C, Kembhavi AA, Barrett AJ. Calpain inhibition by peptide epoxides. Biochem J (1985) 230:509–16.
259. Sasaki T, Kikuchi T, Fukui I, Murachi T. Inactivation of calpain I and calpain II by specificity-oriented tripeptidyl chloromethyl ketones. J Biochem (1986) 99:173–9.
260. Susa M, Luong-Nguyen NH, Cappellen D, Zamurovic N, Gamse R. Human primary osteoclasts: in vitro generation and applications as pharmacological and clinical assay. J Transl Med (2004) 2:6. doi:10.1186/1479-5876-2-6
261. Hashida S, Towatari T, Kominami E, Katunuma N. Inhibitions by E-64 derivatives of rat liver cathepsin B and cathepsin L in vitro and in vivo. J Biochem (1980) 88:1805–11.
262. Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K. In vitro and in vivo inhibition of cysteine proteinases by EST, a new analog of E-64. J Pharmacobiodyn (1986) 9:672–7. doi:10.1248/bpb1978.9.672
263. Tamai M, Hanada K, Adachi T, Oguma K, Kashiwagi K, Omura S, et al. Papain inhibitions by optically active E-64 analogs. J Biochem (1981) 90:255–7.
264. Murata M, Miyashita S, Yokoo C, Tamai M, Hanada K, Hatayama K, et al. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett (1991) 280:307–10. doi:10.1016/0014-5793(91)80318-W
265. Towatari T, Nikawa T, Murata M, Yokoo C, Tamai M, Hanada K, et al. Novel epoxysuccinyl peptides. A selective inhibitor of cathepsin B, in vivo. FEBS Lett (1991) 280:311–5. doi:10.1016/0014-5793(91)80319-X
266. Katunuma N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci (2011) 87:29–39. doi:10.2183/pjab.87.29
267. Montaser M, Lalmanach G, Mach L. CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol Chem (2002) 383:1305–8. doi:10.1515/BC.2002.147
268. Bogyo M, Verhelst S, Bellingard-Dubouchaud V, Toba S, Greenbaum D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem Biol (2000) 7:27–38. doi:10.1016/S1074-5521(00)00061-2
269. Buttle DJ, Murata M, Knight CG, Barrett AJ. CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch Biochem Biophys (1992) 299:377–80. doi:10.1016/0003-9861(92)90290-D
270. Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ (2003) 10:881–8. doi:10.1038/sj.cdd.4401247
271. Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett (1999) 442:117–21. doi:10.1016/S0014-5793(98)01640-8
272. Vancompernolle K, Van Herreweghe F, Pynaert G, Van De Craen M, De Vos K, Totty N, et al. Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett (1998) 438:150–8. doi:10.1016/S0014-5793(98)01275-7
273. Jacobsen W, Christians U, Benet LZ. In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab Dispos (2000) 28:1343–51.
274. Boutte A, Deng-Bryant Y, Johnson D, Tortella FC, Dave J, Shear DA, et al. Serum GFAP predicts tissue GFAP break down products and therapeutic efficacy after penetrating ballistic-like brain injury. J Neurotrauma (2015). doi:10.1089/neu.2014.3672
275. Kochanek PM, Bramlett H, Dietrich WD, Dixon CE, Hayes RL, Povlishock J, et al. A novel multicenter preclinical drug screening and biomarker consortium for experimental traumatic brain injury: operation brain trauma therapy. J Trauma (2011) 71:S15–24. doi:10.1097/TA.0b013e31822117fe
276. Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol (2007) 205:154–65. doi:10.1016/j.expneurol.2007.01.023
277. Yamada KH, Kozlowski DA, Seidl SE, Lance S, Wieschhaus AJ, Sundivakkam P, et al. Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J Biol Chem (2012) 287:13182–93. doi:10.1074/jbc.M111.302612
278. Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci (2000) 20:7037–42.
279. Yanamandra N, Gumidyala KV, Waldron KG, Gujrati M, Olivero WC, Dinh DH, et al. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene (2004) 23:2224–30. doi:10.1038/sj.onc.1207338
280. Tsubokawa T, Solaroglu I, Yatsushige H, Cahill J, Yata K, Zhang JH. Cathepsin and calpain inhibitor E64d attenuates matrix metalloproteinase-9 activity after focal cerebral ischemia in rats. Stroke (2006) 37:1888–94. doi:10.1161/01.STR.0000227259.15506.24
281. Schiefer IT, Tapadar S, Litosh V, Siklos M, Scism R, Wijewickrama GT, et al. Design, synthesis, and optimization of novel epoxide incorporating peptidomimetics as selective calpain inhibitors. J Med Chem (2013) 56:6054–68. doi:10.1021/jm4006719
282. Tamai M, Omura S, Kimura M, Hanada K, Sugita H. Prolongation of life span of dystrophic hamster by cysteine proteinase inhibitor, loxistation (EST). J Pharmacobiodyn (1987) 10:678–81. doi:10.1248/bpb1978.10.678
283. Satoyoshi E. Therapeutic trials on progressive muscular dystrophy. Intern Med (1992) 31:841–6. doi:10.2169/internalmedicine.31.841
284. Watanabe T, Fukushima K, Ushiyama Y, Noda K, Suwa T. Pharmacokinetics of EST (report 5): pharmacokinetics of EST ih humans. Kiso Rinsho (1986) 20:362–6.
285. Barrett AJ, Kembhavi AA, Brown MA, Kirschke H, Knight CG, Tamai M, et al. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem J (1982) 201:189–98.
286. Suzuki K. Reaction of calcium-activated neutral protease (CANP) with an epoxysuccinyl derivative (E64c) and iodoacetic acid. J Biochem (1983) 93:1305–12.
287. Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem (2008) 77:383–414. doi:10.1146/annurev.biochem.75.101304.124125
288. Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem (2014) 83:341–77. doi:10.1146/annurev-biochem-060713-035708
289. Sanman LE, Bogyo M. Activity-based profiling of proteases. Annu Rev Biochem (2014) 83:249–73. doi:10.1146/annurev-biochem-060713-035352
290. Blum G, Von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat Chem Biol (2007) 3:668–77. doi:10.1038/nchembio.2007.26
291. Boutte AM, Friedman DB, Bogyo M, Min Y, Yang L, Lin PC. Identification of a myeloid-derived suppressor cell cystatin-like protein that inhibits metastasis. FASEB J (2011) 25:2626–37. doi:10.1096/fj.10-180604
292. Fukushima K, Yoshida H, Osabe W, Shinozaki F, Kudo K, Arai M, et al. Pharmacokinetics of EST (report 1): absorption and excretion of 14C-EST. Kiso Rinsho (1986) 20:319–27.
293. Fukushima K, Kono Y, Osabe W, Shinozaki F, Kudo K, Arai M, et al. Pharmacokinetics of EST (report 2): tissue distribution of 14C-EST. Kiso Rinsho (1986) 20:328–41.
294. Ishiura S, Hanada K, Tamai M, Kashiwagi K, Sugita H. The effect of an in vivo-injected thiol protease inhibitor, E-64-c, on the calcium-induced degeneration of myofilaments. J Biochem (1981) 90:1557–60.
295. Miyahara T, Shimojo S, Toyohara K, Imai T, Miyajima M, Honda H, et al. Clinical phase I trial of thiol protease inhibitor (Report 2): safety and pharmacokinetics in continuous administration. Rinsho Yakuri (1985) 16:537–46. doi:10.3999/jscpt.16.357
296. Miyahara T, Shimojo S, Toyohara K, Imai T, Miyajima M, Honda H, et al. Phase I clinical trial of thiol protease inhibitor EST (report 1): safety and pharmacokinetics with single administration. Rinsho Yakuri (1985) 16:357–65. doi:10.3999/jscpt.16.357
297. Fukushima K, Arai M, Tamai M, Yokoo C, Murata M, Suwa T, et al. Metabolic fate of loxistatin in rat. Xenobiotica (1990) 20:1043–51. doi:10.3109/00498259009046825
298. Yasui H, Goto H, Suzuki H, Sakai S, Takamura T, Nakane S. Toxicological studies on ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST)(report IX) mutagenicity study. Iyakuhin Kenkyu (1986) 17:815–25.
299. Setoyama K, Koike M, Abe S, Tsutsui Y, Tarumoto Y, Nakane S. Toxicological studies of ethyl (+)-(2S,3S)-3-[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report 1): acute toxicity studies of EST and metabolite and by-product of EST. Iyakuhin Kenkyu (1986) 17:736–43.
300. Tarumoto Y, Sakagawa T, Tsutsui Y, Kawanishi M, Kimura M, Nakane S. Toxicological studies on ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST)(report V) subacute toxicity in dogs. Iyakuhin Kenkyu (1986) 17:768–80.
301. Kimura M, Yagi K, Fujinuma S, Tsuchida T, Tarumoto Y, Noda K, et al. Toxicological studies on ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report III) subacute toxicity in rats. Iyakuhin Kenkyu (1986) 17:744–67.
302. Tarumoto Y, Sakagawa T, Tsutsui Y, Kawanishi M, Watanabe T, Nakane S. Toxicological studies on ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST)(report VII) chronic toxicity study in dogs. Iyakuhin Kenkyu (1986) 17:802–14.
303. Ohshima T, Watanabe T, Nagato C, Kimura M, Tsuchida T, Nakane S. Toxicological studies on ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST)(report VI) Chronic toxicity study in rats. Iyakuhin Kenkyu (1986) 17:781–801.
304. Fukushima K, Arai M, Kohno Y, Suwa T, Satoh T. An epoxysuccinic acid derivative(loxistatin)-induced hepatic injury in rats and hamsters. Toxicol Appl Pharmacol (1990) 105:1–12. doi:10.1016/0041-008X(90)90353-V
305. Yamada T, Nishiyama T, Ohno H, Nakane S. Reproduction studies of ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report III): study of administration to rabbits during organogenesis. Iyakuhin Kenkyu (1986) 17:632–8.
306. Yamada T, Uchida H, Inoue T, Ohba Y, Nakane S. Reproduction studies of ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report II): study of administration to rats during organogenesis. Iyakuhin Kenkyu (1986) 14:617–31.
307. Yamada T, Nishiyama T, Nakane S. Reproduction studies of ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report I): study of administration to rats prior to and in early stage of gestation. Iyakuhin Kenkyu (1986) 17:609–16.
308. Yamada T, Uchida H, Ohno H, Matsuzawa N, Nakane S. Reproduction studies of ethyl(+)-(2S,2S)-3[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoyl]-2-oxiranecarboxylate (EST) (report IV): study of administration to rats during perinatal and postnatal periods. Iyakuhin Kenkyu (1986) 17:639–51.
Keywords: traumatic brain injury, cathepsin B, protease, E64d, drug, therapeutics
Citation: Hook G, Jacobsen JS, Grabstein K, Kindy M and Hook V (2015) Cathepsin B is a New Drug Target for Traumatic Brain Injury Therapeutics: Evidence for E64d as a Promising Lead Drug Candidate. Front. Neurol. 6:178. doi: 10.3389/fneur.2015.00178
Received: 31 May 2015; Accepted: 31 July 2015;
Published: 02 September 2015
Edited by:
Angela M. Boutte, Walter Reed Army Institute of Research, USAReviewed by:
Anders Hånell, Virginia Commonwealth University, USAJohn Anagli, Henry Ford Health System, USA
Copyright: © 2015 Hook, Jacobsen, Grabstein, Kindy and Hook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivian Hook, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, 9500 Gilman Dr. MC 0719, La Jolla, San Diego, CA 92093-0719, USA,dmhvb2tAdWNzZC5lZHU=