 Gregory M. Davis
Gregory M. Davis Matilda A. Haas†
Matilda A. Haas† Roger Pocock
Roger Pocock- Development and Stem Cells Program, Monash Biomedicine Discovery Institute, Department of Anatomy and Developmental Biology, Monash University, Melbourne, VIC, Australia
MicroRNAs (miRNAs) are a class of short non-coding RNAs that operate as prominent post-transcriptional regulators of eukaryotic gene expression. miRNAs are abundantly expressed in the brain of most animals and exert diverse roles. The anatomical and functional complexity of the brain requires the precise coordination of multilayered gene regulatory networks. The flexibility, speed, and reversibility of miRNA function provide precise temporal and spatial gene regulatory capabilities that are crucial for the correct functioning of the brain. Studies have shown that the underlying molecular mechanisms controlled by miRNAs in the nervous systems of invertebrate and vertebrate models are remarkably conserved in humans. We endeavor to provide insight into the roles of miRNAs in the nervous systems of these model organisms and discuss how such information may be used to inform regarding diseases of the human brain.
Introduction
MicroRNAs (miRNAs) are non-coding RNA molecules with a length of approximately 22 nucleotides, which act as post-transcriptional regulators of gene expression (1–4). Discovered just over two decades ago, miRNAs have been found to be abundant in most organisms and critically important for post-transcriptional control of mRNAs by regulating a predicted 60% of protein-coding genes (5). Prior to the discovery of the miRNA pathway, the lin-14 gene in Caenorhabditis elegans was shown to be regulated by a 22-nucleotide partially complementary strand of RNA called lin-4 (6, 7). However, a mechanistic understanding of this process remained unclear until the let-7 gene was shown to encode a complementary sequence of lin-41 to regulate developmental timing (8). This led to a paradigm shift in how mRNA regulation was viewed, and further investigation demonstrated that the miRNA pathway was evolutionarily conserved in most eukaryotes (9). Since then miRNAs have been shown to be required for key biological processes, such as cell fate, differentiation, apoptosis, and tumor suppression (10–13).
The process of miRNA biogenesis in animals can be briefly simplified into three fundamental steps (Figure 1) [for detailed review, see Ref. (14)]. First, double-stranded primary miRNA (pri-miRNA) short hairpin structures are transcribed by RNA polymerase II. Secondly, a nuclear-localized RNA endonuclease III, Drosha, defines one end of the pri-miRNA duplex and cleaves double-stranded RNA (dsRNA) transcripts into approximately 70 nt stem loops called precursor mRNAs (pre-miRNAs) (15). These pre-miRNAs are exported to the cytoplasm by Exportin-5 (XPO5) (16) where the Dicer enzyme cleaves pre-miRNA sequences into 21–23 nt mature miRNA double-stranded duplexes (17). Such miRNA duplexes load into a pre-RISC (pre-miRNA-induced silencing complex) which is a complex of Argonaute (AGO) and other proteins (18). Within the pre-miRISC, the “passenger” strand is removed leaving just the “guide” strand in the mature miRISC. The guide strand is normally the strand with a more thermodynamically unstable 5′ end (19). The released passenger strand is either degraded or loaded into a different miRISC complex to regulate a different group of target transcripts to the guide strand. The miRISC complexes then scan the transcriptome for partially complementary mRNA sequences. The miRNA then associates with a target mRNA by imperfect base-pairing, on the most part, to its 3′UTR and mediates post-transcriptional repression (PTR) or decay of specific mRNA targets (17, 20). The partially complementary sequences of miRNAs allow them to recognize and inhibit the expression of multiple mRNA transcripts (21). mRNA recognition is primarily determined through nucleotides 2–7 of the 5′ end or “seed” region of miRNAs (3). miRNAs can also initiate mRNA degradation by recruiting the mRNA degradation machinery, or through the use of cytoplasmic RNA granules known as Processing bodies (P-bodies), which can degrade mRNAs via cap removal and 5′–3′ exonuclease activity (22).
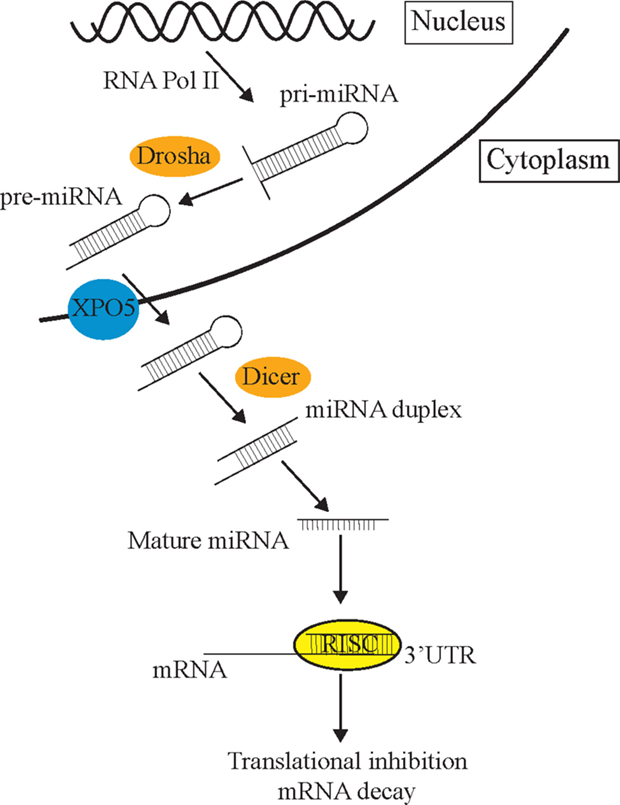
Figure 1. The miRNA pathway. Primary miRNAs (pri-miRNAs) are transcribed from the genome and form hairpin structures. Nuclear-localized Drosha endonuclease cleaves pri-miRNAs into approximately 70 nt precursor miRNAs (pre-miRNAs) which are then transported from the nucleus to the cytoplasm by Exportin-5 (XPO5) via the nuclear pore complex, where they are further cleaved by Dicer into mature 21–23 nt miRNA fragments. Once the strands separate, the guide strand is loaded into the RISC complex (AGO and different cofactors) to scan the transcriptome for partial complementary target transcripts. These sequences are either repressed by the RISC complex or degraded in P-bodies.
As mentioned above, the nature of miRNA targeting through imperfect complementarity means that single miRNAs have the potential to regulate the expression of hundreds of genes (3). In addition, certain genes have multiple miRNA-binding sites in their 3′UTRs and, therefore, multiple miRNA families potentially control their expression (3). 3′UTR length is often a determining factor as to its propensity to miRNA regulation (3). Such complex relationships between miRNAs and their targets enable exquisite control of gene regulatory networks. A better understanding of miRNA function in the control of such gene regulatory networks has been accelerated by the use of simple model organisms, such as C. elegans and Drosophila. Studies using these models are aided by their genetic amenability, short lifespans, and compact genomes. However, there are multiple mammalian-specific miRNAs for which the use of higher eukaryotes is required to study their biology.
Functions of miRNAs in the Nervous System
The human brain contains approximately 86 billion neurons and trillions of synaptic connections (23). This complex organ is an integration center where environmental information is processed and used to make an appropriate action or decision. To effect brain function as a whole, neurons are organized into circuits which communicate with each other through rapidly acting synaptic connections and slower acting neuropeptide release. An inability to regulate these molecular communication processes is causative in developmental disorders, such as autism and schizophrenia, in addition to age-related decline of brain function (24, 25). Therefore, using model organisms to dissect these mechanisms at a molecular, anatomical and functional level will provide a greater understanding of neuronal-based disease.
The ability of the nervous system to adapt to different environmental conditions and stimuli requires a well-conserved and flexible repertoire of molecular mechanisms. miRNAs offer genetic networks’ additional layers of regulatory control and are abundantly expressed in all human tissues, including the brain (26). In addition to this, many miRNAs display specific temporal and spatial patterns of expression (27). Due to the high degree of complexity of the human brain, in addition to ethical concerns, deep mechanistic understanding of how miRNAs influence neurodevelopmental and functional processes has come from model organisms. This review aims to provide examples that reveal the important roles of miRNAs in the development and function of the nervous system (Figure 2). We focus on the crucial role model organism research has played in this area to provide insight into the functions of miRNAs.
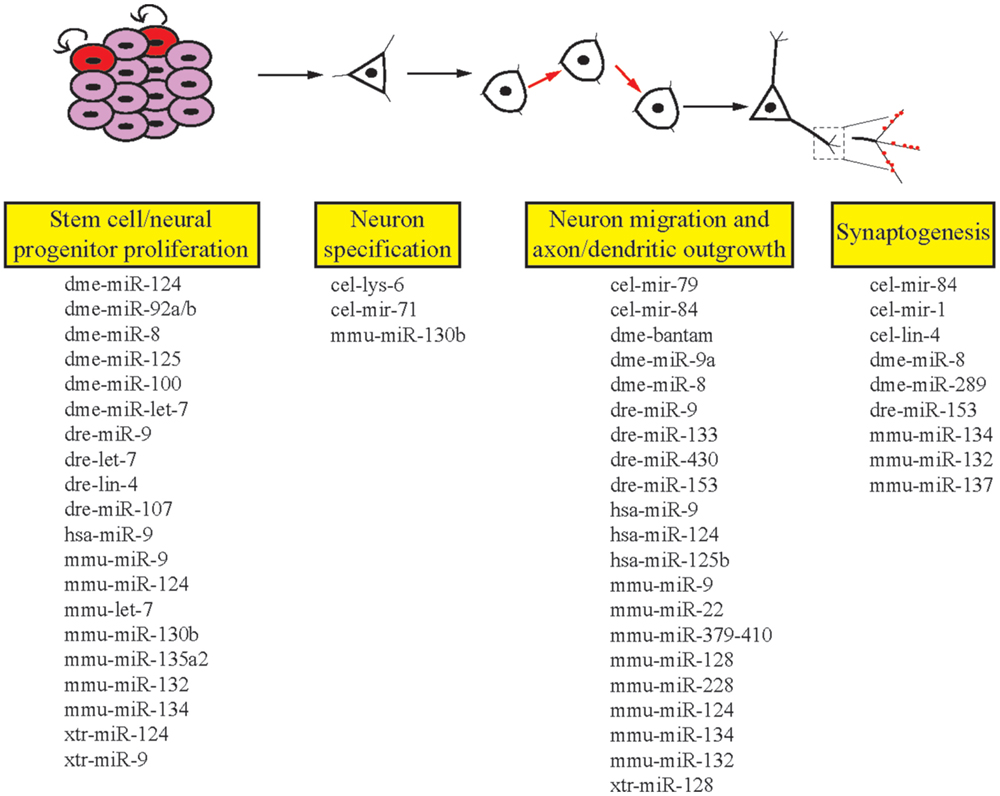
Figure 2. Roles of miRNAs in different stages of neuronal development. miRNAs are involved in the multiple stages of neuronal development in invertebrates and vertebrates. Listed here are the miRNAs we cover in this review that regulate single or multiple stages of neuronal development. cel, Caenorhabditis elegans; dme, Drosophila melanogaster; dre, Danio rerio; hsa, Homo sapiens; mmu, Mus musculus; xtr, Xenopus tropicalis.
miRNA Regulation of the Nervous System in Invertebrates
Caenorhabditis elegans
The initial discovery of miRNAs was made in the nematode C. elegans and, since that time, many fundamental studies implicating miRNAs in gene regulatory networks significant to neurodevelopment have been achieved using this model (28–30). C. elegans has proven demonstrative as many miRNAs are highly conserved throughout evolution, along with other well-known advantages of the model organism including its well-defined neuronal lineage map, neuroanatomy, and neural networks (31–35).
MicroRNAs have been shown to participate in instructing cell fate decisions made during the development of the nervous system of C. elegans. For example, the ASE neurons are a pair of morphologically similar, asymmetric gustatory sensory neurons, which have different roles: ASEL senses sodium, while ASER senses chloride (36). The neuron pair is derived from different lineages, which diverge at the four-cell stage of embryogenesis. A complex gene regulatory pathway, in which the miRNA lsy-6 plays a central role, essentially achieves the specification of this pair of neurons during development. The Nkx homeobox transcription factor COG-1, which induces ASER fate, is inhibited by lsy-6 in ASEL (37–39). It was subsequently shown that lsy-6 expression itself is controlled by a complex mechanism involving two regulatory elements, firstly where transcription factors TBX-37/38 “prime” the lsy-6 locus for expression by altering chromatin to an open state. Expression is then “boosted” by the transcription factor CHE-1, and lsy-6 induces gene expression changes associated with asymmetrical generation of the ASEL and ASER neurons (40). This “priming” and “boosting” of lsy-6 begins several cell divisions prior to the specification of ASEL neuron and is the first asymmetrically expressed gene in the ASEL and ASER neurons. This mechanism resulting in bilateral symmetry in the nervous system may provide important insights into how symmetry is established in the mammalian brain.
mir-71 is another miRNA involved in neuron specification in C. elegans. mir-71 controls cellular responses to calcium to specify asymmetry in function of the morphologically symmetrical AWCON and AWCOFF olfactory neurons (41). mir-71 controls this signaling pathway as it is expressed at a higher level in one neuron of the pair, which negatively regulates the calcium adaptor protein TIR-1/Sarm1 downstream of nsy-4/claudin and nsy-5/innexin, to specify the AWCON neuron (41). Interestingly, for this pair of neurons, cell fate is not rigid. That is, whether the left or right neuron is specified AWCON is not fixed, and if the AWCON is lost, AWCOFF can convert to AWCON (41).
Aside from neuronal specification, evidence from C. elegans indicates that miRNAs are involved in gene regulation to control neuron migration and differentiation. It was demonstrated that a regulatory pathway essential for normal neuron migration and axon guidance involves mir-79 (an ortholog of mammalian miR-9) (42). In C. elegans, loss of mir-79 from epidermal cells caused an increase in expression of SQV-5 (a chondroitin synthase) and SQV-7 (a UDP-sugar transporter), both required for the biosynthesis of glycosaminoglycan (GAG) chains that are attached to proteoglycans (43). Defective regulation of SQV-5 and SQV-7 in the epidermis caused striking defects in the migration of hermaphrodite-specific neurons (HSNs). RNAi knockdown of sqv-5 and sqv-7 in mir-79 deletion mutant animals restored the incidence of HSN defects to background levels. Additional work showed that this mechanism is required to regulate the addition of heparan sulfate chains on a glypican called LON-2. This work, therefore, defined a pathway through which mir-79 expression in epidermal cells non-cell autonomously controls HSN migration (42).
Netrin-mediated axon guidance is also influenced by miRNA expression in C. elegans to ensure the correct timing of axon termination. Cell autonomous expression of lin-4 (homologous with miR-125a/b) targets the transcription factor lin-14 at completion of extension of the anterior ventral microtubule (AVM) axon, to inhibit netrin-mediated axon attraction (44). Such temporal loss of responsiveness is due to decreased expression of the netrin receptor UNC-40 through lin-4-mediated reduction of LIN-14. The cell-autonomous regulation of lin-14 by lin-4 is also required for the timing of axon extension of the HSNs (29). In lin-4 loss of function animals, the HSNs do not extend their axons prior to the larval-adult transition, and adult axons have defective morphology. Therefore, lin-4 is required to temporally regulate extension of axons in two neuronal paradigms in C. elegans (29).
Later in neuronal development, miRNAs also control synaptogenesis and remodeling in C. elegans. For example, lin-4 targets lin-14 to remodel motor neuron synapses during the first larval stage L1, when motor neurons eliminate their synapses with ventral muscles and instead form connections with dorsal muscles. The timing of this change is regulated by the heterochronic genes lin-4–lin-14 (28) and mir-84–hbl-1 (45). mir-1 also plays important roles in synaptic function, by targeting the transcription factor MEF-2 to control neurotransmitter release at the neuromuscular junction (NMJ) (46). mir-1 also targets synaptic proteins neuroligin and neurexin (47), which in humans are two synaptic proteins that have been linked with defects in synaptic function associated with autism spectrum disorders (ASDs) (48). Elucidating these mechanisms in C. elegans may reveal conserved pathways and provide important insights into human development and mechanisms underlying neurodevelopmental disorders.
Interestingly, miRNAs have also been implicated in the developmental decline of regenerative ability of the nervous system. The miRNA let-7 is only expressed very weakly when initial axonogenesis occurs, and onset of expression with age contributes to a reduced capacity for regeneration of the AVM neurons (30). This mechanism involves a developmentally regulated loop including the TRIM protein LIN-41, among other factors. The exciting discovery that suppression of let-7 could restore regenerative capacity (30) could be conserved in vertebrates, and indeed it has since been shown that suppression of Let-7 in primary cultured rat peripheral neurons increases their regenerative response (49).
Finally, the miRNA mir-71 was found to regulate the physiology of C. elegans non-cell autonomously from the nervous system (50). The authors showed that mir-71 expression in the AB lineage (nearly all neurons) is necessary and sufficient for lifespan extension of animals lacking a germline (50). The authors showed that neuronal mir-71 regulates the localization and activity of the FOXO transcription factor DAF-16 in the intestine which acts downstream of insulin-like signaling to regulate metabolism and stress responses (50–52). Such non-cell autonomous regulation of intestinal DAF-16 by mir-71 is via an, as yet, unidentified neuronal factor.
Drosophila melanogaster
MicroRNAs in the fruit fly, Drosophila melanogaster, have been extensively investigated in various developmental processes [reviewed in Ref. (53)]. miRNAs are critical for all aspects of neuronal development, from regulating neural stem cells to regulating the events that occur at the NMJ. For example, control of neuronal progenitor proliferation is fine-tuned by the highly conserved miRNA, miR-124, which has been shown in various organisms to regulate neuronal stem cells (54–57). In Drosophila, mir-124 targets anachronism (ana), an inhibitor of neuroblast proliferation. The absence of miR-124 results in decreased proliferative activity, which is coupled with an increase in ana expression (58). In addition to this, miR-124 is required for optimal regulation of dendrite growth and targets components of the retrograde BMP signaling pathway to regulate synaptic release at the NMJ (59).
Additional miRNAs that contribute to neuronal proliferation are the fly homologs of mammalian miR-92 and miR-200 – miR-92a/b and miR-8, respectively (60, 61). miR-92a is located in the intron and miR-92b in the 3′UTR of a putative DNA-binding protein, jigr1, and they suppress this host gene to regulate neural stem cell development to prevent premature differentiation (61). Additionally, miR-8 has been implicated in regulating neuronal proliferation but is expressed in a glial cell population ensheathing the optic lobe neurepithelium (60). In the latter of these roles, miR-8 is required for the temporal and spatial control of EGFR pathway ligand, Spitz, which controls accurate neuroepithelial proliferation and neuroblast formation (60).
Drosophila has been used as a model to identify miRNAs that temporally and spatially control neuronal differentiation and specification. An example of this is with olfactory neuronal morphogenesis, which is associated with accurate miRNA function. Loss of core components of the miRNA biogenesis machinery, including Pasha or Dicer, results in abnormal olfactory neuron morphogenesis (62). The basis for these defects was defined by studies of the mushroom body neurons (MB), which mediate olfactory responses and comprise of four invariant subtypes of neurons in various insects (63–66). The generation of MB neurons requires tight post-transcriptional regulation of the BTB-zinc finger chronologically inappropriate morphogenesis (chinmo) in postmitotic neurons (66). This is achieved by a group of miRNAs that are cotranscribed from a single locus and comprise of miR-125, miR-100, and the highly conserved let-7, collectively referred to as the let-7-Complex (let-7-C) (67). The initial discovery of let-7 in C. elegans identified a heterochronic role for let-7, whereas in Drosophila let-7 expression is not enriched in early development, but upregulation of let-7-C is associated with a downregulation of chinmo (68). This suggests that the mechanism identified in C. elegans where let-7 regulates developmental timing functions in a different context in Drosophila to regulate the formation of MB neurons.
The involvement of miRNAs in dendrite growth is unclear, although a small number of studies in Drosophila have identified two miRNAs required for dendrite growth of sensory neurons. First, the miRNA, bantam, has been implicated in dendrite scaling by suppressing Akt kinase activity in nearby neurons and by regulating epithelial endoreplication (69, 70). Secondly, miR-9a (mammalian miR-9 homolog) acts from epithelial cells to fine-tune dendrite growth. This is achieved by regulating the activity of a cadherin-domain containing putative G-protein-coupled receptor, Fmi, which functions to suppress dendrite growth (71). miR-9a also acts with miR-7 to control the development of sensory organs. In Drosophila, sensory organs develop from single organ precursor cells (SOPs), which are generated from clusters of cells expressing proneural genes. This process is temporally controlled by Notch signaling and two transcription factors that regulate proneural gene expression. These consist of Senseless, a positive regulator of SOP cells, which is targeted by miR-9a, and a negative regulator, Enhancer of Split, which is targeted by miR-7 (72–74). Furthermore, miR-9a is also influenced by the RBA-binding protein, TDP-43, mutants of which display increased SOP cells coupled with decreased miR-9a expression (75). Although the exact requirement of TDP-43 in this process is unclear, genetic interaction assays suggest that SOP specification requires TDP-43 for accurate neuronal differentiation by influencing miR-9a activity.
MicroRNAs have also been shown to regulate the NMJ in Drosophila embryos and larvae. miR-8 and miR-289 are required to suppress activity-dependent synaptic growth by targeting genes involved in axon development and growth. miR-8 downregulates wingless, a presynaptic regulatory protein required for activity-dependent axon terminal growth at the NMJ (76). In this context, miR-8 regulates the timing of synaptic expansion to correlate with the growth of target muscles. Furthermore, miR-8 regulates the embryonic expression of two synaptic immunoglobulin superfamily cell adhesion molecules (IgCAMs), Fasciclin III (FasIII), and Neuroglian (Nrg) (77). Taken together, these studies lay a foundation for further study into the role(s) of miR-8 in presynaptic events, as well as the timing of synaptic assembly with neuron–muscle association.
Finally, miR-8 has also been shown to regulate apoptosis in the CNS of Drosophila (78). miR-8 regulates the expression of the transcriptional corepressor Atrophin to a particular threshold level. Loss of miR-8 results in increased atrophin levels and apoptosis; however, reduction of atrophin expression below the threshold set by miR-8 causes extra tissue being generated (78). Precise tuning of atrophin levels is, therefore, required to prevent neurodegeneration in the CNS of Drosophila.
miRNA Regulation of the Nervous System in Vertebrates
Xenopus laevis
Neuronal development and function have been extensively studied in the Xenopus laevis tadpole [reviewed in Ref. (79)]. However, in contrast to other model organisms, the influence of miRNAs during neurodevelopmental processes has received less attention. Nevertheless, certain conserved miRNAs investigated in Xenopus offer new insights to their function. For example, similar to other model organisms, miR-124 regulates early neurogenesis. However, in Xenopus, miR-124 is expressed from the beginning of eye development where it plays an important role in regulating retinal neurogenesis in the optic vesicle and forebrain (57, 80, 81). In addition to this, miR-129, miR-155, miR-214, and miR-222 contribute to developmental timing of retinal progenitor cells by regulating the activity of the transcription factors, xotx2 and xvsx1, which are both required for promoting the late-stage progenitor cells to differentiate into bipolar neurons (82).
The highly conserved miR-9 is also required for neurogenesis along the anterior–posterior axis by targeting the transcription factor, hairy1, although its function varies from the hindbrain to the forebrain. In the forebrain, regulation of hairy1 by miR-9 influences proliferation of neural progenitor cells through Fgf8 signaling, but via Wnt signaling in the hindbrain (83). This suggests positional specificity regarding miR-9 function. Defects associated with the nonsense-mediated mRNA decay (NMD) pathway result in neurological disorders in humans (84, 85). Interestingly, in Xenopus, miR-128 has been shown to repress NMD by targeting the RNA helicase, UPF1, and the exon-junction cofactor, MLN51 (86). This process allows upregulation of specific mRNAs required for differentiating neuronal cells, which are normally targeted by NMD. Moreover, this mechanism is highly conserved in mammals and represents a dual mRNA regulatory network to maintain neuron development and function (86).
Danio rerio
Danio rerio (zebrafish) is a valuable model system that has been used to uncover neurodevelopmental functions for miRNAs, with the advantage that zygotic loss of miRNAs can be examined in the absence of maternal compensation mechanisms, since the zygote develops outside of the mother (87). Embryos carrying dicer mutations display severe developmental defects, including delayed embryogenesis, perturbed neurulation, and formation of brain ventricles, as well as ill-defined anatomical boundaries, such as the midbrain-hindbrain boundary (MHB) (87). However, interestingly the same study also showed that despite the gross morphological defects, gene expression, and neuron specification were maintained within patterned regions, such as the forebrain and hindbrain rhombomeres. Later stage neuronal differentiation, such as axon extension, was also severely affected by loss of dicer (87).
In zebrafish, miRNAs are expressed in neural cells throughout the different stages of development in addition to in mature neurons. They can have ubiquitous or cell-specific expression patterns. Many conserved miRNAs are expressed at the same developmental timepoints as other vertebrates, for example, miR-9 and let-7 are expressed in both proliferating and differentiating cells (88). As in C. elegans, lin-28 and its downstream heterochronic genes let-7 and lin-4/miR-125b are expressed during development to coordinate cell proliferation (89).
In zebrafish hindbrain development, miR-107 stabilizes dicer levels, which maintains a specific level of miR-9 biogenesis to regulate optimal proliferation of neural progenitors (90). miR-9 inhibits proliferation at the MHB and hindbrain ventricular zone through targeting of proproliferation genes her5, her6/Hes1, and zic5 and then later also influences neuronal maturation by regulating elav3/HuC (91–93). Additionally, miR-9 overexpression causes a strong reduction in the MHB and cerebellum, as well as blurred somatic boundaries and altered cell fates, through downregulation of fgfr1 in the Fgf signaling pathway (91).
More recently, zebrafish hindbrain development has been used as a model system to uncover precise mechanisms of the miRNA-mediated mRNA decay pathway (94). In this study, a genetic screen implicated cnot8, which was known to have deadenylase activity in polyA tail removal in mRNA turnover. Furthermore, the role of cnot8 in the Fgf signaling pathway is responsible for hindbrain dopaminergic neuron differentiation, and application of a drug inhibiting Fgf signaling partially restored the mutant phenotype (94). Zebrafish studies also elucidated the mechanisms through which morphine influences dopaminergic neuron differentiation, since maternal influence can be removed from developmental events. Morphine downregulates miR-133, which increases pitx3 expression thereby promoting dopaminergic neuron maturation (95). This study revealed important information relating to neural networks involved in drug addiction.
In the later stages of zebrafish neurodevelopment, miR-430 controls trigeminal sensory neuron migration. These sensory neurons arise from the neural crest and placodes, and their migratory journey of up to 120 μm requires the chemokine SDF1a and its receptor Cxcr4b. The border of SDF1a expression shifts continually to make a tightly regulated chemotactic path for neurons to migrate, and miR-430 is important for clearing SDF1a from the pathway that neurons have passed through (96).
Finally, in zebrafish embryos, miRNAs are also involved in dendritic spine formation and synaptogenesis. For example, knockdown of miR-153 caused a sevenfold increase in spontaneous body movement, and the synaptic protein SNAP-25, which is involved in vesicular exocytosis, was found to be the target (97).
Mus musculus
The earliest mammalian studies following the discovery of miRNAs in C. elegans quickly demonstrated the crucial nature of the class of non-protein-coding RNAs in mammals through the generation of mice carrying deletions for miRNA-processing pathway components including Dicer, Dgcr8, and Argonaute. Argonaute proteins are essential components of the RISC complex, facilitating translational inhibition or target mRNA cleavage, and Argonaute 2 mutants show an early and severe neurodevelopmental phenotype, with neural tube closure failing to occur (98). Deletion of another component of the miRNA-processing pathway, Dgcr8, causes microcephaly in mutant mice but is much less severe than loss of Dicer mutants (99). Under investigation is the potential importance of Dgcr8 in Di George syndrome, a multifaceted disorder where 30 genes including Dgcr8 are deleted, and has been associated with schizophrenia (100).
Loss of Dicer causes lethality in lower eukaryotes, such as C. elegans and Drosophila (101, 102). However, the neurodevelopmental consequence of Dicer loss in mice has been investigated in greater depth using conditional deletion mutants (103, 104). This led to further confirmation of both the spatial and temporal importance of miRNA-mediated pathways in multiple phases of mammalian CNS development. One study used conditional Dicer deletions generated with Emx-Cre (excitatory cerebral cortex neurons) and Nestin-Cre (all CNS neurons) mouse lines at different stages of embryonic cerebral cortex development showed that miRNAs are important for three major phases of cerebral cortex development: neuronal progenitor proliferation, neuronal migration, and differentiation (105).
Conditional mutants have been further used to confirm the importance of miRNAs in the progression of cerebral cortex development, with Cre-recombinase-driven deletion of Dicer under the control of Foxg1, Emx1, Nes, Nex, and CamkII promoters (106). These models have shown that in the early stages of cerebral cortex development, conditional deletion leads to a loss of neurons, either due to loss of the neural progenitor pool or increased apoptosis. Deletion of Dicer leads to neuron migration defects and impaired cellular differentiation, as well as cell fate changes and cortical lamination defects (106).
Conditional deletion of Dicer from the embryonic day 8 (E8) telencephalon causes a loss of radial glial progenitor markers, including nestin, Sox9, and ErbB2, which then results in an increase in basal progenitors and postmitotic neurons (107). Interestingly, increased apoptosis was also observed, and this was correlated with reduced expression of miR-9 and miR-124 (107), two miRNA families that have been widely implicated in brain development. Dicer conditionally deleted using the Nex-Cre promoter (targeting neurons of pyramidal fate) resulted in significantly smaller mouse brains, due to increased packing density of neurons, as well as abnormal neuron differentiation, but no defect in neuron production or cortical lamination (107). Conditional deletion of Dicer by α-CaMKII-Cre in the embryonic forebrain resulted in microcephaly due to increased apoptosis, rather than neuronal migration defects, since lamination appeared unaffected. Reduced dendritic branching and dendritic spine development was also observed (103), along with ataxia and reduced life span. Interestingly, Dicer deletion in neural crest cells (by Wnt1-Cre) does not affect migration and early differentiation, but miRNA pathways appear to be required for the survival of peripheral nervous system (PNS) neurons, because in Dicer deletion mutants PNS neurons are lost after completion of migration and differentiation due to apoptosis (108, 109). Thus, while Dicer mutants all point toward dysregulated nervous system development, individual studies have reported slightly different mechanisms. Whether this is due to the timing of deletion, the promoter driving Cre-recombinase or which miRNAs are being lost due to blocking the processing pathway is not clear.
Dicer deletions have been useful in determining the overall impact of loss of miRNAs on gene regulation, but more information has been gained from studying the roles of specific individual miRNAs. The let-7 family, which was one of the first described in C. elegans, was subsequently also shown in the mouse to play a significant role in maintaining the balance in neuronal progenitor proliferation and neurogenesis, since TRIM32 (110) and SOX-2 (111) influence let-7 levels to maintain cells in a proliferative state. let-7b in turn regulates neural stem cell proliferation by targeting the stem cell regulators TLX and cyclinD1 (112).
The miR-9 family is one of the most highly conserved and abundantly expressed miRNA families in the vertebrate brain and is also involved in balancing neural progenitor proliferation and controlling progenitor state (93). miR-9 regulates early progenitor proliferation in the mammalian brain through the transcription factors Hes1 (113), Foxg1, Elav2, Pax-6, as well as Gsh2 (114). Confirming its proproliferative role, loss of miR-9 suppresses neural stem cell proliferation, through stathmin (115).
The miR-124 family is also conserved from C. elegans to humans. It is expressed by differentiating neurons in the subventricular zone of the developing mouse cortex, where it controls apical/basal progenitor progression (56). It is also important for continual production of neurons in the subventricular zone of the adult mouse brain (116). In neuronal differentiation, miR-124 is involved in a mechanism with the transcriptional repressor REST, whereby REST represses miR-124a and expression of neuronal genes in non-neuronal cells and neural progenitors. But at later stages, REST ceases to repress miR-124a, allowing non-neuronal transcript degradation and thus promoting neuronal differentiation (117). REST also has miR-124-binding sites in its 3′UTR, suggesting a complex regulatory loop exists (118). Another transcriptional repressor involved in the REST complex, MeCP2, also has predicted miR-124-binding sites in its 3′UTR (118), and MeCP2 mutation or copy number variant has been implicated in multiple neurodevelopmental disorders including X-linked intellectual disability and autism (119, 120).
Therefore, the above-mentioned in vivo studies suggest that miR-9 and miR-124 are major players in the regulation of cerebral cortex development, but in vitro studies have shown that miR-9 and miR-124 can drive the neurogenic program. When Yoo and colleagues (121) added miR-9 and miR-124 precursors to cultured neonatal foreskin fibroblasts, they were able to directly convert them to neurons expressing the mature marker MAP2, albeit at a conversion rate of <5%. Neuron conversion was improved remarkably by the addition of NEUROD2, ASCL1, and MYT1L, a combination that has previously been shown to produce neurons (122). Importantly, after 4 weeks in culture, the neurons expressed synaptic markers and displayed electrophysiological properties consistent with neurons (121). This exciting study suggests that miRNAs are not only involved in controlling gene expression in neurodevelopment, but could be considered among the master regulators of neurogenesis in mammalian cells.
Many other miRNAs have been implicated in the earliest stages of mammalian brain development and regulate important pathways in development and disease. For example, miR-130b has been shown to regulate Fmr1 expression, which is lost in the disorder fragile X syndrome. Fmr1 loss causes increased progenitor proliferation and altered neuronal fate specification (123). miR-135a2 regulates Wnt signaling in midbrain dopaminergic neuron proliferation in a regulatory circuit with Lmx1b (124), and in the mouse cortex, miR-134 promotes neural progenitor cell proliferation and counteracts apoptosis and differentiation (125). miR-34, another conserved miRNA, appears to be linked with neuron proliferation, because overexpression of mir-34 in human stem cells suppressed the expression of 136 neuronal progenitor genes that possess putative miR-34 target sites. Gene ontology showed that these genes are overwhelmingly involved in cell motility and energy production (126).
In addition to being crucial for neuronal progenitor proliferation, miR-9 and miR-124 are emerging as key regulators of neuron migration. miR-9, along with miR-132, represses Foxp2 to regulate radial migration in the developing mouse cortex. Ectopic expression of Foxp2 in the developing cortex was counteracted by increased endogenous expression of miR-9 and miR-132 (127). In a different mechanism, miR-124 and miR-22 regulate cell shape changes in migrating cortical neurons by controlling expression of doublecortin, a microtubule-associated cytoskeleton protein involved in cell shape remodeling through multipolar and bipolar phases in migrating neurons, through CoREST/REST (128). Recently, the miR-379-410 cluster was shown to regulate N-cadherin expression, a crucial factor in maintaining tissue structure in the developing cortex. Overexpression of these miRNAs in radial glial cells decreased N-cadherin expression, causing increased stem cell differentiation and migration (129). miR-128 in the brain regulates Phf6, which is a mutated gene in the disorder Borjeson–Forssman–Lehmann syndrome, and ectopic expression of miR-228 in the developing brain leads to neuron migration defects, neurite outgrowth, and electrophysiological changes (130).
Specific miRNAs exhibit increased expression in the latter stages of nervous system development in the mouse and are implicated in final differentiation, neurite extension, and synapse formation. These miRNAs include miR-134 (125, 131) and miR-132 (132–134). Also, miR-124 regulates RhoG, which is a major player in the control of axon and dendrite outgrowth and complexity, in mouse hippocampal neurons (135).
Very recently, expression of a miR-137 gain-of-function construct, first in cell lines, and then in the mouse hippocampal dentate gyrus, was shown to downregulate three well-known presynaptic proteins: complexin-1 (Cplx1), N-ethylmaleimide-sensitive fusion protein (Nsf), and synaptotagmin-1 (Syt1) (136). In vivo, this was accompanied by fewer synaptic vesicles and impaired hippocampal LTP, and impaired hippocampal dependent learning in behavioral testing. Excitingly, some of these defects were rescued by codelivery of a miR-137 sponge construct, which sequestered endogenous miR-137. These findings and rescue in the mouse brain are particularly promising, given the vast data implicating miR-137 SNPs in schizophrenia (136).
Homo sapiens
An estimated 70% of all miRNAs are expressed in the human nervous system [reviewed in Ref. (137)], although only a small number of miRNAs appear to be regulated during neuronal differentiation (138). Moreover, temporal and spatial distribution of miRNAs in human donor brains, as well as target genes associated with neurodevelopmental diseases have been identified (139). Despite this, ethical complications prevent in-depth mechanistic studies from occurring in humans. Therefore, expression of these conserved miRNAs, coupled with mechanistic studies from model organisms or cell lines, has allowed for neuronal miRNAs to be understood in greater detail [for reviews, see Ref. (140, 141)]. The majority of investigations in humans use cell lines or use screening-based approaches for miRNAs that are associated with populations of particular neurodevelopmental disease states. Cell line-based approaches, despite their in vitro limitations have characterized several miRNAs required for neuronal proliferation and differentiation. For example, overexpression of the highly conserved miR-9 promotes proliferation of neural progenitor cells in human embryonic stem cells (115). miR-9 along with miR-124 and miR-125b has also been associated with inducing human pluripotent stem cells to differentiate into neurons (142). Although described previously in Drosophila and in the mouse, miR-9 is a good example of an evolutionary conserved miRNA that contributes to various aspects of neuronal development.
Aberrant expression of miRNAs has been associated with different neurodevelopmental disorders, such as schizophrenia, autism, Down syndrome, fragile X syndrome, and Rett syndrome [reviewed in Ref. (143–145)]. Determining if the aberrant expression of all of these associated miRNAs is simply the consequence of abnormal neuronal development or the cause of the disorder itself is challenging. Nonetheless, several studies have identified numerous aberrantly expressed miRNAs associated with bipolar and schizophrenic patients (146, 147). For example, a contributing factor in schizophrenia is decreased function of the N-methyl-d-aspartate (NMDA) receptor and delivery of NMDA receptor antagonists phenocopy the conditions associated with schizophrenia (148–150). Interestingly, miR-132 is downregulated in schizophrenic patients and has also been shown to contribute to the depolarization of the NMDA receptor (151, 152), suggesting that miR-132 may be a candidate for potential therapeutics. Defects associated not only with the expression of key miRNAs, but also at a genetic level have been implicated in schizophrenia. A genome-wide study of over 40,000 schizophrenic patients identified a SNP within the putative coding region of miR-137 resulting in decreased efficiency of miR-137 function (153). This is further supported by additional studies that have shown variation of miR-137 affects brain activation and function (154–156). Unlike examples of differentially expressed miRNAs, miR-137 suggests a direct genetic-miRNA association with schizophrenia.
Neurological diseases that give rise to ASDs, such as fragile X syndrome and Rett syndrome, display elevated and depleted miRNA expression [for detailed review, see Ref. (157)]. In addition to this, mutations associated with the miRNA machinery are thought to contribute to the progression of ASDs. For example, mutations in the RNA-binding protein, fragile X mental retardation 1 protein (FMR1), are associated with fragile X syndrome patients (158). In Drosophila, FMR1 functions as a RISC cofactor to maintain miRNA function (159). Another example is associated with Rett syndrome, which is caused by mutations associated with the methyl-CpG-binding protein 2 (MECP2) (160), which prevent nuclear miRNA processing by regulating the Drosha complex (161). Despite the importance of these proteins in human disease states, most mechanistic insights regarding FMR1 and MECP2 have come from investigations employing model organisms. Furthermore, these FMR1 and MECP2 studies show how mutations in key proteins associated with miRNA targeting or processing contribute to neurological diseases that give rise to autism.
Screening-based approaches to understand ASD-associated miRNAs involve postmortem analysis of autistic patients, as well as assessing circulating miRNAs in serum and plasma. This screening approach not only has led to a greater understanding of miRNAs associated with ASDs but also has potential to use selected miRNAs as non-invasive biomarkers for ASDs. Postmortem analysis of the cerebellar cortex identified 28 dysregulated miRNAs from 13 autism patients (162). This included miRNAs that are predicted to target the synaptic scaffolding protein, SHANK3, and the presynaptic cell adhesion protein, NRXN1, which are both associated with ASDs (163, 164). More recently, two studies carried out in live patients have identified 13 miRNAs in 55 children, as well as five miRNAs in 15 patients in China that are differentially expressed in serum and plasma in children with ASDs (165, 166). These investigations have also revealed the enrichment of predicted target genes of these differentially expressed miRNAs in various neurological pathways, suggesting a potential use for diagnosis and future therapeutic approaches.
Conclusion
Since the initial discovery of miRNAs being involved in developmental timing of larval development in C. elegans (6, 8, 9), small non-coding RNAs have been implicated in a multitude of biological processes. lin-4 and let-7 were identified in unbiased forward genetic screens, as was lsy-6, the first miRNA shown to be involved in the nervous system (38). These genetic approaches enabled the identification of miRNAs with very specific roles during development. As such, these particular functions may have been overlooked when using reverse genetic techniques. However, waiting for such chance discoveries takes much time; therefore, other approaches are required to systematically drive miRNA discoveries forward. With the advent of temporal expression pattern analysis, sophisticated RNA sequencing and proteomic approaches, and miRNA prediction algorithms, the ability to move from a mutation in a specific miRNA to phenotype is a possibility using model organisms. Therefore, the meticulous scrutiny of miRNA expression patterns and screening for anatomical, functional, and behavioral phenotypes has proved fertile ground in the identification of roles for miRNAs in the brain.
In humans, the expression of a number of miRNAs has been correlated with neurodevelopmental disorders (143–145); however, the molecular mechanisms through which they act are not understood. Using model organisms as a discovery tool enables researchers to study the function of these miRNAs in depth. Due to the high degree of conservation between model organisms and humans, the elucidation of molecular mechanisms that control neuronal development using these models will help identify novel therapeutic approaches in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Agnieszka Podolska and Sandeep Gopal for constructive comments on the manuscript. RP is supported by a veski innovation fellowship (2015–2018).
References
1. Bartel DP. MicroRNAs. Genomics, biogenesis, mechanism, and function. Cell (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
2. Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet (2004) 5:396–400. doi:10.1038/nrg1328
3. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136:215–33. doi:10.1016/j.cell.2009.01.002
4. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell (2012) 149:515–24. doi:10.1016/j.cell.2012.04.005
5. Grun D, Wang YL, Langenberger D, Gunsalus KC, Rajewsky N. microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput Biol (2005) 1:e13. doi:10.1371/journal.pcbi.0010013
6. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell (1993) 75:843–54. doi:10.1016/0092-8674(93)90529-Y
7. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell (1993) 75:855–62. doi:10.1016/0092-8674(93)90530-4
8. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature (2000) 403:901–6. doi:10.1038/35002607
9. Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature (2000) 408:86–9. doi:10.1038/35040556
10. Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell-specific MicroRNAs. Dev Cell (2003) 5:351–8. doi:10.1016/S1534-5807(03)00227-2
11. Xu P, Vernooy SY, Guo M, Hay BA. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol (2003) 13:790–5. doi:10.1016/S0960-9822(03)00250-1
12. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature (2005) 435:834–8. doi:10.1038/nature03702
13. Rosa A, Spagnoli FM, Brivanlou AH. The miR-430/427/302 family controls mesendodermal fate specification via species-specific target selection. Dev Cell (2009) 16:517–27. doi:10.1016/j.devcel.2009.02.007
14. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol (2009) 11:228–34. doi:10.1038/ncb0309-228
15. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature (2003) 425:415–9. doi:10.1038/nature01957
16. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev (2003) 17:3011–6. doi:10.1101/gad.1158803
17. Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J (2002) 21:4663–70. doi:10.1093/emboj/cdf476
18. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet (2010) 11:597–610. doi:10.1038/nrg2843
19. Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell (2003) 115:209–16. doi:10.1016/S0092-8674(03)00801-8
20. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet (2012) 13:271–82. doi:10.1038/nrg3162
21. Carroll AP, Goodall GJ, Liu B. Understanding principles of miRNA target recognition and function through integrated biological and bioinformatics approaches. Wiley Interdiscip Rev RNA (2014) 5:361–79. doi:10.1002/wrna.1217
22. Jain S, Parker R. The discovery and analysis of P Bodies. Adv Exp Med Biol (2013) 768:23–43. doi:10.1007/978-1-4614-5107-5_3
23. Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol (2009) 513:532–41. doi:10.1002/cne.21974
24. Nyberg L, Lovden M, Riklund K, Lindenberger U, Backman L. Memory aging and brain maintenance. Trends Cogn Sci (2012) 16:292–305. doi:10.1016/j.tics.2012.04.005
25. Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem (2013) 126:165–82. doi:10.1111/jnc.12261
26. Narayan A, Bommakanti A, Patel AA. High-throughput RNA profiling via up-front sample parallelization. Nat Methods (2015) 12:343–6. doi:10.1038/nmeth.3311
27. Chen W, Qin C. General hallmarks of microRNAs in brain evolution and development. RNA Biol (2015) 12:701–8. doi:10.1080/15476286.2015.1048954
28. Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature (1998) 395:78–82. doi:10.1038/25757
29. Olsson-Carter K, Slack FJ. A developmental timing switch promotes axon outgrowth independent of known guidance receptors. PLoS Genet (2010) 6. doi:10.1371/journal.pgen.1001054
30. Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang CF, Chang C. Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers. Science (2013) 340:372–6. doi:10.1126/science.1231321
31. Sulston JE. Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci (1976) 275:287–97. doi:10.1098/rstb.1976.0084
32. Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol (1977) 56:110–56. doi:10.1016/0012-1606(77)90158-0
33. Sulston JE. Neuronal cell lineages in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol (1983) 48(Pt 2):443–52. doi:10.1101/SQB.1983.048.01.049
34. Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol (1983) 100:64–119. doi:10.1016/0012-1606(83)90201-4
35. Durbin R.M. Studies on the Development and Organisation of the Nervous System of Caenorhabditis Elegans. Ph.D. thesis, Cambridge: University of Cambridge (1987).
36. Bargmann CI, Horvitz HR. Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron (1991) 7:729–42. doi:10.1016/0896-6273(91)90276-6
37. Chang S, Johnston RJ Jr, Hobert O. A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes Dev (2003) 17:2123–37. doi:10.1101/gad.1117903
38. Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature (2003) 426:845–9. doi:10.1038/nature02255
39. Sarin S, Antonio C, Tursun B, Hobert O. The C. elegans Tailless/TLX transcription factor nhr-67 controls neuronal identity and left/right asymmetric fate diversification. Development (2009) 36:2933–44. doi:10.1242/dev.040204
40. Cochella L, Hobert O. Embryonic priming of a miRNA locus predetermines postmitotic neuronal left/right asymmetry in C. elegans. Cell (2012) 151:1229–42. doi:10.1016/j.cell.2012.10.049
41. Hsieh YW, Chang C, Chuang CF. The microRNA mir-71 inhibits calcium signaling by targeting the TIR-1/Sarm1 adaptor protein to control stochastic L/R neuronal asymmetry in C. elegans. PLoS Genet (2012) 8:e1002864. doi:10.1371/journal.pgen.1002864
42. Pedersen ME, Snieckute G, Kagias K, Nehammer C, Multhaupt HA, Couchman JR, et al. An epidermal microRNA regulates neuronal migration through control of the cellular glycosylation state. Science (2013) 341:1404–8. doi:10.1126/science.1242528
43. Bulik DA, Robbins PW. The Caenorhabditis elegans sqv genes and functions of proteoglycans in development. Biochim Biophys Acta (2002) 1573:247–57. doi:10.1016/S0304-4165(02)00391-4
44. Zou Y, Chiu H, Domenger D, Chuang CF, Chang C. The lin-4 microRNA targets the LIN-14 transcription factor to inhibit netrin-mediated axon attraction. Sci Signal (2012) 5:ra43. doi:10.1126/scisignal.2002437
45. Thompson-Peer KL, Bai J, Hu Z, Kaplan JM. HBL-1 patterns synaptic remodeling in C. elegans. Neuron (2012) 73:453–65. doi:10.1016/j.neuron.2011.11.025
46. Simon DJ, Madison JM, Conery AL, Thompson-Peer KL, Soskis M, Ruvkun GB, et al. The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell (2008) 133:903–15. doi:10.1016/j.cell.2008.04.035
47. Hu Z, Hom S, Kudze T, Tong XJ, Choi S, Aramuni G, et al. Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science (2012) 337:980–4. doi:10.1126/science.1224896
48. Persico AM, Napolioni V. Autism genetics. Behav Brain Res (2013) 251:95–112. doi:10.1016/j.bbr.2013.06.012
49. Li S, Wang X, Gu Y, Chen C, Wang Y, Liu J, et al. Let-7 microRNAs regenerate peripheral nerve regeneration by targeting nerve growth factor. Mol Ther (2015) 23:423–33. doi:10.1038/mt.2014.220
50. Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab (2012) 15:439–50. doi:10.1016/j.cmet.2012.02.014
51. Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet (2001) 28:139–45. doi:10.1038/88850
52. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell (2003) 115:489–502. doi:10.1016/S0092-8674(03)00889-4
53. Chawla G, Sokol NS. MicroRNAs in Drosophila development. Int Rev Cell Mol Biol (2011) 286:1–65. doi:10.1016/B978-0-12-385859-7.00001-X
54. Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell (2007) 27:435–48. doi:10.1016/j.molcel.2007.07.015
55. Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci (2009) 12:399–408. doi:10.1038/nn.2294
56. Maiorano NA, Mallamaci A. Promotion of embryonic cortico-cerebral neuronogenesis by miR-124. Neural Dev (2009) 4:40. doi:10.1186/1749-8104-4-40
57. Liu K, Liu Y, Mo W, Qiu R, Wang X, Wu JY, et al. MiR-124 regulates early neurogenesis in the optic vesicle and forebrain, targeting NeuroD1. Nucleic Acids Res (2011) 39:2869–79. doi:10.1093/nar/gkq904
58. Weng R, Cohen SM. Drosophila miR-124 regulates neuroblast proliferation through its target anachronism. Development (2012) 139:1427–34. doi:10.1242/dev.075143
59. Sun K, Westholm JO, Tsurudome K, Hagen JW, Lu Y, Kohwi M, et al. Neurophysiological defects and neuronal gene deregulation in Drosophila mir-124 mutants. PLoS Genet (2012) 8:e1002515. doi:10.1371/journal.pgen.1002515
60. Morante J, Vallejo DM, Desplan C, Dominguez M. Conserved miR-8/miR-200 defines a glial niche that controls neuroepithelial expansion and neuroblast transition. Dev Cell (2013) 27:174–87. doi:10.1016/j.devcel.2013.09.018
61. Yuva-Aydemir Y, Xu XL, Aydemir O, Gascon E, Sayin S, Zhou W, et al. Downregulation of the host gene jigr1 by miR-92 is essential for neuroblast self-renewal in Drosophila. PLoS Genet (2015) 11:e1005264. doi:10.1371/journal.pgen.1005264
62. Berdnik D, Fan AP, Potter CJ, Luo L. MicroRNA processing pathway regulates olfactory neuron morphogenesis. Curr Biol (2008) 18:1754–9. doi:10.1016/j.cub.2008.09.045
63. Lee T, Lee A, Luo L. Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development (1999) 126:4065–76.
64. Zars T, Fischer M, Schulz R, Heisenberg M. Localization of a short-term memory in Drosophila. Science (2000) 288:672–5. doi:10.1126/science.288.5466.672
65. Zhu S, Chiang AS, Lee T. Development of the Drosophila mushroom bodies: elaboration, remodeling and spatial organization of dendrites in the calyx. Development (2003) 130:2603–10. doi:10.1242/dev.00466
66. Zhu S, Lin S, Kao CF, Awasaki T, Chiang AS, Lee T. Gradients of the Drosophila Chinmo BTB-zinc finger protein govern neuronal temporal identity. Cell (2006) 127:409–22. doi:10.1016/j.cell.2006.08.045
67. Wu YC, Chen CH, Mercer A, Sokol NS. Let-7-complex microRNAs regulate the temporal identity of Drosophila mushroom body neurons via chinmo. Dev Cell (2012) 23:202–9. doi:10.1016/j.devcel.2012.05.013
68. Chawla G, Sokol NS. Hormonal activation of let-7-C microRNAs via EcR is required for adult Drosophila melanogaster morphology and function. Development (2012) 139:1788–97. doi:10.1242/dev.077743
69. Parrish JZ, Xu P, Kim CC, Jan LY, Jan YN. The microRNA bantam functions in epithelial cells to regulate scaling growth of dendrite arbors in drosophila sensory neurons. Neuron (2009) 63:788–802. doi:10.1016/j.neuron.2009.08.006
70. Jiang N, Soba P, Parker E, Kim CC, Parrish JZ. The microRNA bantam regulates a developmental transition in epithelial cells that restricts sensory dendrite growth. Development (2014) 141:2657–68. doi:10.1242/dev.107573
71. Wang Y, Wang H, Li X, Li Y. Epithelial microRNA-9a regulates dendrite growth through Fmi-Gq signaling in Drosophila sensory neurons. Dev Neurobiol (2015). doi:10.1002/dneu.22309
72. Nolo R, Abbott LA, Bellen HJ. Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell (2000) 102:349–62. doi:10.1016/S0092-8674(00)00040-4
73. Jafar-Nejad H, Acar M, Nolo R, Lacin H, Pan H, Parkhurst SM, et al. Senseless acts as a binary switch during sensory organ precursor selection. Genes Dev (2003) 17:2966–78. doi:10.1101/gad.1122403
74. Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell (2009) 137:273–82. doi:10.1016/j.cell.2009.01.058
75. Li Z, Lu Y, Xu XL, Gao FB. The FTD/ALS-associated RNA-binding protein TDP-43 regulates the robustness of neuronal specification through microRNA-9a in Drosophila. Hum Mol Genet (2013) 22:218–25. doi:10.1093/hmg/dds420
76. Nesler KR, Sand RI, Symmes BA, Pradhan SJ, Boin NG, Laun AE, et al. The miRNA pathway controls rapid changes in activity-dependent synaptic structure at the Drosophila melanogaster neuromuscular junction. PLoS One (2013) 8:e68385. doi:10.1371/journal.pone.0068385
77. Lu CS, Zhai B, Mauss A, Landgraf M, Gygi S, Van Vactor D. MicroRNA-8 promotes robust motor axon targeting by coordinate regulation of cell adhesion molecules during synapse development. Philos Trans R Soc Lond B Biol Sci (2014) 369. doi:10.1098/rstb.2013.0517
78. Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell (2007) 131:136–45. doi:10.1016/j.cell.2007.09.020
79. Pratt KG, Khakhalin AS. Modeling human neurodevelopmental disorders in the Xenopus tadpole: from mechanisms to therapeutic targets. Dis Model Mech (2013) 6:1057–65. doi:10.1242/dmm.012138
80. Qiu R, Liu K, Liu Y, Mo W, Flynt AS, Patton JG, et al. The role of miR-124a in early development of the Xenopus eye. Mech Dev (2009) 126:804–16. doi:10.1016/j.mod.2009.08.002
81. Baudet ML, Zivraj KH, Abreu-Goodger C, Muldal A, Armisen J, Blenkiron C, et al. miR-124 acts through CoREST to control onset of Sema3A sensitivity in navigating retinal growth cones. Nat Neurosci (2012) 15:29–38. doi:10.1038/nn.2979
82. Decembrini S, Bressan D, Vignali R, Pitto L, Mariotti S, Rainaldi G, et al. MicroRNAs couple cell fate and developmental timing in retina. Proc Natl Acad Sci U S A (2009) 106:21179–84. doi:10.1073/pnas.0909167106
83. Bonev B, Pisco A, Papalopulu N. MicroRNA-9 reveals regional diversity of neural progenitors along the anterior-posterior axis. Dev Cell (2011) 20:19–32. doi:10.1016/j.devcel.2010.11.018
84. Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet (2007) 39:1127–33. doi:10.1038/ng2100
85. Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry (2011) 16:238–9. doi:10.1038/mp.2010.59
86. Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell (2011) 42:500–10. doi:10.1016/j.molcel.2011.04.018
87. Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson MJ, Baskerville S, et al. MicroRNAs regulate brain morphogenesis in Zebrafish. Science (2005) 308:833–8. doi:10.1126/science.1109020
88. Kapsimali M, Kloosterman WP, De Bruijn E, Rosa F, Plasterk RH, Wilson SW. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol (2007) 8:R173. doi:10.1186/gb-2007-8-8-r173
89. Ouchi Y, Yamamoto J, Iwamoto T. The heterochronic genes lin-28a and lin-28b play an essential and evolutionarily conserved role in early zebrafish development. PLoS One (2014) 9:e88086. doi:10.1371/journal.pone.0088086
90. Ristori E, Lopez-Ramirez MA, Narayanan A, Hill-Teran G, Moro A, Calvo CF, et al. A Dicer-miR-107 interaction regulates biogenesis of specific miRNAs crucial for neurogenesis. Dev Cell (2015) 32:546–60. doi:10.1016/j.devcel.2014.12.013
91. Leucht C, Stigloher C, Wizenmann A, Klafke R, Folchert A, Bally-Cuif L. MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat Neurosci (2008) 11:641–8. doi:10.1038/nn.2115
92. Coolen M, Thieffry D, Drivenes O, Becker TS, Bally-Cuif L. miR-9 controls the timing of neurogenesis through the direct inhibition of antagonistic factors. Dev Cell (2012) 22:1052–64. doi:10.1016/j.devcel.2012.03.003
93. Coolen M, Katz S, Bally-Cuif L. miR-9: a versatile regulator of neurogenesis. Front Cell Neurosci (2013) 7:220. doi:10.3389/fncel.2013.00220
94. Koch P, Lohr HB, Driever W. A mutation in cnot8, component of the Ccr4-not complex regulating transcript stability, affects expression levels of developmental regulators and reveals a role of Fgf3 in development of caudal hypothalamic dopaminergic neurons. PLoS One (2014) 9:e113829. doi:10.1371/journal.pone.0113829
95. Sanchez-Simon FM, Zhang XX, Loh HH, Law PY, Rodriguez RE. Morphine regulates dopaminergic neuron differentiation via miR-133b. Mol Pharmacol (2010) 78:935–42. doi:10.1124/mol.110.066837
96. Lewellis SW, Nagelberg D, Subedi A, Staton A, Leblanc M, Giraldez A, et al. Precise SDF1-mediated cell guidance is achieved through ligand clearance and microRNA-mediated decay. J Cell Biol (2013) 200:337–55. doi:10.1083/jcb.201207099
97. Wei C, Thatcher EJ, Olena AF, Cha DJ, Perdigoto AL, Marshall AF, et al. miR-153 regulates SNAP-25, synaptic transmission, and neuronal development. PLoS One (2013) 8:e57080. doi:10.1371/journal.pone.0057080
98. Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science (2004) 305:1437–41. doi:10.1126/science.1102513
99. Babiarz JE, Hsu R, Melton C, Thomas M, Ullian EM, Blelloch R. A role for noncanonical microRNAs in the mammalian brain revealed by phenotypic differences in Dgcr8 versus Dicer1 knockouts and small RNA sequencing. RNA (2011) 17:1489–501. doi:10.1261/rna.2442211
100. Meechan DW, Maynard TM, Tucker ES, Lamantia AS. Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: patterning, proliferation, and mitochondrial functions of 22q11 genes. Int J Dev Neurosci (2011) 29:283–94. doi:10.1016/j.ijdevneu.2010.08.005
101. Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell (2001) 106:23–34. doi:10.1016/S0092-8674(01)00431-7
102. Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell (2004) 117:69–81. doi:10.1016/S0092-8674(04)00261-2
103. Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, Mcmanus MT, et al. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci (2008) 28:4322–30. doi:10.1523/JNEUROSCI.4815-07.2008
104. Tao J, Wu H, Lin Q, Wei W, Lu XH, Cantle JP, et al. Deletion of astroglial Dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J Neurosci (2011) 31:8306–19. doi:10.1523/JNEUROSCI.0567-11.2011
105. Kawase-Koga Y, Otaegi G, Sun T. Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev Dyn (2009) 238:2800–12. doi:10.1002/dvdy.22109
106. Barca-Mayo O, De Pietri Tonelli D. Convergent microRNA actions coordinate neocortical development. Cell Mol Life Sci (2014) 71:2975–95. doi:10.1007/s00018-014-1576-5
107. Nowakowski TJ, Mysiak KS, Pratt T, Price DJ. Functional dicer is necessary for appropriate specification of radial glia during early development of mouse telencephalon. PLoS One (2011) 6:e23013. doi:10.1371/journal.pone.0023013
108. Huang T, Liu Y, Huang M, Zhao X, Cheng L. Wnt1-cre-mediated conditional loss of Dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J Mol Cell Biol (2010) 2:152–63. doi:10.1093/jmcb/mjq008
109. Zehir A, Hua LL, Maska EL, Morikawa Y, Cserjesi P. Dicer is required for survival of differentiating neural crest cells. Dev Biol (2010) 340:459–67. doi:10.1016/j.ydbio.2010.01.039
110. Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell (2009) 136:913–25. doi:10.1016/j.cell.2008.12.024
111. Cimadamore F, Amador-Arjona A, Chen C, Huang CT, Terskikh AV. SOX2-LIN28/let-7 pathway regulates proliferation and neurogenesis in neural precursors. Proc Natl Acad Sci U S A (2013) 110:E3017–26. doi:10.1073/pnas.1220176110
112. Zhao C, Sun G, Li S, Lang MF, Yang S, Li W, et al. MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc Natl Acad Sci U S A (2010) 107:1876–81. doi:10.1073/pnas.0908750107
113. Tan SL, Ohtsuka T, Gonzalez A, Kageyama R. MicroRNA9 regulates neural stem cell differentiation by controlling Hes1 expression dynamics in the developing brain. Genes Cells (2012) 17:952–61. doi:10.1111/gtc.12009
114. Shibata M, Nakao H, Kiyonari H, Abe T, Aizawa S. MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J Neurosci (2011) 31:3407–22. doi:10.1523/JNEUROSCI.5085-10.2011
115. Delaloy C, Liu L, Lee JA, Su H, Shen F, Yang GY, et al. MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell Stem Cell (2010) 6:323–35. doi:10.1016/j.stem.2010.02.015
116. Akerblom M, Sachdeva R, Barde I, Verp S, Gentner B, Trono D, et al. MicroRNA-124 is a subventricular zone neuronal fate determinant. J Neurosci (2012) 32:8879–89. doi:10.1523/JNEUROSCI.0558-12.2012
117. Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A (2006) 103:2422–7. doi:10.1073/pnas.0511041103
118. Wu J, Xie X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol (2006) 7:R85. doi:10.1186/gb-2006-7-9-r85
119. Bassani S, Zapata J, Gerosa L, Moretto E, Murru L, Passafaro M. The neurobiology of X-linked intellectual disability. Neuroscientist (2013) 19:541–52. doi:10.1177/1073858413493972
120. Rosti RO, Sadek AA, Vaux KK, Gleeson JG. The genetic landscape of autism spectrum disorders. Dev Med Child Neurol (2014) 56:12–8. doi:10.1111/dmcn.12278
121. Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature (2011) 476:228–31. doi:10.1038/nature10323
122. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature (2010) 463:1035–41. doi:10.1038/nature08797
123. Gong X, Zhang K, Wang Y, Wang J, Cui Y, Li S, et al. MicroRNA-130b targets Fmr1 and regulates embryonic neural progenitor cell proliferation and differentiation. Biochem Biophys Res Commun (2013) 439:493–500. doi:10.1016/j.bbrc.2013.08.096
124. Anderegg A, Lin HP, Chen JA, Caronia-Brown G, Cherepanova N, Yun B, et al. An Lmx1b-miR135a2 regulatory circuit modulates Wnt1/Wnt signaling and determines the size of the midbrain dopaminergic progenitor pool. PLoS Genet (2013) 9:e1003973. doi:10.1371/journal.pgen.1003973
125. Gaughwin P, Ciesla M, Yang H, Lim B, Brundin P. Stage-specific modulation of cortical neuronal development by Mmu-miR-134. Cereb Cortex (2011) 21:1857–69. doi:10.1093/cercor/bhq262
126. Chang SJ, Weng SL, Hsieh JY, Wang TY, Chang MD, Wang HW. MicroRNA-34a modulates genes involved in cellular motility and oxidative phosphorylation in neural precursors derived from human umbilical cord mesenchymal stem cells. BMC Med Genomics (2011) 4:65. doi:10.1186/1755-8794-4-65
127. Clovis YM, Enard W, Marinaro F, Huttner WB, De Pietri Tonelli D. Convergent repression of Foxp2 3’UTR by miR-9 and miR-132 in embryonic mouse neocortex: implications for radial migration of neurons. Development (2012) 139:3332–42. doi:10.1242/dev.078063
128. Volvert ML, Prevot PP, Close P, Laguesse S, Pirotte S, Hemphill J, et al. MicroRNA targeting of CoREST controls polarization of migrating cortical neurons. Cell Rep (2014) 7:1168–83. doi:10.1016/j.celrep.2014.03.075
129. Rago L, Beattie R, Taylor V, Winter J. miR379-410 cluster miRNAs regulate neurogenesis and neuronal migration by fine-tuning N-cadherin. EMBO J (2014) 33:906–20. doi:10.1002/embj.201386591
130. Franzoni E, Booker SA, Parthasarathy S, Rehfeld F, Grosser S, Srivatsa S, et al. miR-128 regulates neuronal migration, outgrowth and intrinsic excitability via the intellectual disability gene Phf6. Elife (2015) 4. doi:10.7554/eLife.04263
131. Valluy J, Bicker S, Aksoy-Aksel A, Lackinger M, Sumer S, Fiore R, et al. A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat Neurosci (2015) 18:666–73. doi:10.1038/nn.3996
132. Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A (2010) 107:20382–7. doi:10.1073/pnas.1015691107
133. Pathania M, Torres-Reveron J, Yan L, Kimura T, Lin TV, Gordon V, et al. miR-132 enhances dendritic morphogenesis, spine density, synaptic integration, and survival of newborn olfactory bulb neurons. PLoS One (2012) 7:e38174. doi:10.1371/journal.pone.0038174
134. Yang D, Li T, Wang Y, Tang Y, Cui H, Tang Y, et al. miR-132 regulates the differentiation of dopamine neurons by directly targeting Nurr1 expression. J Cell Sci (2012) 125:1673–82. doi:10.1242/jcs.086421
135. Franke K, Otto W, Johannes S, Baumgart J, Nitsch R, Schumacher S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J (2012) 31:2908–21. doi:10.1038/emboj.2012.130
136. Siegert S, Seo J, Kwon EJ, Rudenko A, Cho S, Wang W, et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat Neurosci (2015) 18:1008–16. doi:10.1038/nn.4023
137. Nowak JS, Michlewski G. miRNAs in development and pathogenesis of the nervous system. Biochem Soc Trans (2013) 41:815–20. doi:10.1042/BST20130044
138. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol (2004) 5:R13. doi:10.1186/gb-2004-5-8-p13
139. Ziats MN, Rennert OM. Identification of differentially expressed microRNAs across the developing human brain. Mol Psychiatry (2014) 19:848–52. doi:10.1038/mp.2013.93
140. Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron (2009) 64:303–9. doi:10.1016/j.neuron.2009.10.020
141. Adlakha YK, Saini N. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol Cancer (2014) 13:33. doi:10.1186/1476-4598-13-33
142. Roese-Koerner B, Stappert L, Koch P, Brustle O, Borghese L. Pluripotent stem cell-derived somatic stem cells as tool to study the role of microRNAs in early human neural development. Curr Mol Med (2013) 13:707–22. doi:10.2174/1566524011313050003
143. Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci (2012) 35:325–34. doi:10.1016/j.tins.2012.01.004
144. Banerjee-Basu S, Larsen E, Muend S. Common microRNAs target established ASD genes. Front Neurol (2014) 5:205. doi:10.3389/fneur.2014.00205
145. Sun E, Shi Y. MicroRNAs: Small molecules with big roles in neurodevelopment and diseases. Exp Neurol (2015) 268:46–53. doi:10.1016/j.expneurol.2014.08.005
146. Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, et al. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol (2007) 8:R27. doi:10.1186/gb-2007-8-2-r27
147. Moreau MP, Bruse SE, David-Rus R, Buyske S, Brzustowicz LM. Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol Psychiatry (2011) 69:188–93. doi:10.1016/j.biopsych.2010.09.039
148. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51:199–214. doi:10.1001/archpsyc.1994.03950030035004
149. Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, et al. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology (1999) 20:106–18. doi:10.1016/S0893-133X(98)00067-0
150. Stone JM, Erlandsson K, Arstad E, Squassante L, Teneggi V, Bressan RA, et al. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: a [(123)I]CNS-1261 SPET study. Psychopharmacology (Berl) (2008) 197:401–8. doi:10.1007/s00213-007-1047-x
151. Cheng HY, Papp JW, Varlamova O, Dziema H, Russell B, Curfman JP, et al. microRNA modulation of circadian-clock period and entrainment. Neuron (2007) 54:813–29. doi:10.1016/j.neuron.2007.05.017
152. Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci U S A (2012) 109:3125–30. doi:10.1073/pnas.1113793109
153. Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet (2011) 43:969–76. doi:10.1038/ng.940
154. Whalley HC, Papmeyer M, Romaniuk L, Sprooten E, Johnstone EC, Hall J, et al. Impact of a microRNA MIR137 susceptibility variant on brain function in people at high genetic risk of schizophrenia or bipolar disorder. Neuropsychopharmacology (2012) 37:2720–9. doi:10.1038/npp.2012.137
155. Mothersill O, Morris DW, Kelly S, Rose EJ, Fahey C, O’brien C, et al. Effects of MIR137 on fronto-amygdala functional connectivity. Neuroimage (2014) 90:189–95. doi:10.1016/j.neuroimage.2013.12.019
156. Kuswanto CN, Sum MY, Qiu A, Sitoh YY, Liu J, Sim K. The impact of genome wide supported microRNA-137 (MIR137) risk variants on frontal and striatal white matter integrity, neurocognitive functioning, and negative symptoms in schizophrenia. Am J Med Genet B Neuropsychiatr Genet (2015) 168:317–26. doi:10.1002/ajmg.b.32314
157. Siew WH, Tan KL, Babaei MA, Cheah PS, Ling KH. MicroRNAs and intellectual disability (ID) in Down syndrome, X-linked ID, and Fragile X syndrome. Front Cell Neurosci (2013) 7:41. doi:10.3389/fncel.2013.00041
158. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell (1991) 65:905–14. doi:10.1016/0092-8674(91)90397-H
159. Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev (2002) 16:2497–508. doi:10.1101/gad.1022002
160. Amir RE, Van Den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet (1999) 23:185–8. doi:10.1038/13810
161. Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, et al. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell (2014) 28:547–60. doi:10.1016/j.devcel.2014.01.032
162. Abu-Elneel K, Liu T, Gazzaniga FS, Nishimura Y, Wall DP, Geschwind DH, et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics (2008) 9:153–61. doi:10.1007/s10048-008-0133-5
163. Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet (2007) 39:25–7. doi:10.1038/ng1933
164. Duong L, Klitten LL, Moller RS, Ingason A, Jakobsen KD, Skjodt C, et al. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am J Med Genet B Neuropsychiatr Genet (2012) 159B:354–8. doi:10.1002/ajmg.b.32036
165. Mundalil Vasu M, Anitha A, Thanseem I, Suzuki K, Yamada K, Takahashi T, et al. Serum microRNA profiles in children with autism. Mol Autism (2014) 5:40. doi:10.1186/2040-2392-5-40
Keywords: microRNA, brain, neurogenesis, axon guidance
Citation: Davis GM, Haas MA and Pocock R (2015) MicroRNAs: Not “Fine-Tuners” but Key Regulators of Neuronal Development and Function. Front. Neurol. 6:245. doi: 10.3389/fneur.2015.00245
Received: 30 July 2015; Accepted: 09 November 2015;
Published: 24 November 2015
Edited by:
Owen Murray Rennert, National Institutes of Health, USAReviewed by:
Khyobeni Mozhui, University of Tennessee Health Science Center, USATin-Lap Lee, The Chinese University of Hong Kong, Hong Kong
Copyright: © 2015 Davis, Haas and Pocock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Pocock, cm9nZXIucG9jb2NrQG1vbmFzaC5lZHU=
†Gregory Davis and Matilda Haas have contributed equally to this work.