 Danielle E. Harlow
Danielle E. Harlow Justin M. Honce
Justin M. Honce Augusto A. Miravalle
Augusto A. Miravalle- 1Department of Cell and Developmental Biology, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
- 2Department of Radiology, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
- 3Department of Neurology, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Multiple sclerosis (MS) is an immune-mediated disorder of the central nervous system that results in destruction of the myelin sheath that surrounds axons and eventual neurodegeneration. Current treatments approved for the treatment of relapsing forms of MS target the aberrant immune response and successfully reduce the severity of attacks and frequency of relapses. Therapies are still needed that can repair damage particularly for the treatment of progressive forms of MS for which current therapies are relatively ineffective. Remyelination can restore neuronal function and prevent further neuronal loss and clinical disability. Recent advancements in our understanding of the molecular and cellular mechanisms regulating myelination, as well as the development of high-throughput screens to identify agents that enhance myelination, have lead to the identification of many potential remyelination therapies currently in preclinical and early clinical development. One problem that has plagued the development of treatments to promote remyelination is the difficulty in assessing remyelination in patients with current imaging techniques. Powerful new imaging technologies are making it easier to discern remyelination in patients, which is critical for the assessment of these new therapeutic strategies during clinical trials. This review will summarize what is currently known about remyelination failure in MS, strategies to overcome this failure, new therapeutic treatments in the pipeline for promoting remyelination in MS patients, and new imaging technologies for measuring remyelination in patients.
Introduction
The therapeutic armamentarium for multiple sclerosis (MS) has expanded significantly in the last few decades due to better understanding of the basic pathophysiological mechanisms of the disease process. However, despite the development of increasingly effective therapies, a cure for MS has not been found and MS patients continue to suffer from chronic progressive disability. Currently, approved treatments for MS work by reducing immune system activity or blocking entry of immune cells into the central nervous system (CNS). Although these treatments can reduce relapse rates and severity of attacks, they do not repair immune-mediated damage to the myelin sheaths surrounding axons. Chronic demyelination leads to degeneration of axons and eventually loss of neurons. Neuronal loss correlates highly with clinical disability, highlighting the need for treatments that promote neuronal survival in both relapsing and progressive forms of MS. Experimental models of MS (both in vitro cultures and in vivo studies) have shown that preservation of myelin and remyelination of axons can increase neuronal survival (1, 2). To protect neurons, restore function, and halt the progression of disability, additional treatments need to be developed to promote myelin repair and neuronal protection. In this article, we will review current concepts of effective remyelination in MS including proposed mechanisms of myelin regulation, emerging therapies that might contribute to repair and restore cell function in MS, and the use of magnetic resonance imaging (MRI) to measure remyelination in clinical trials.
Factors That Contribute to Remyelination Failure in MS
Understanding why endogenous remyelination often fails in MS is essential to the development of effective remyelination and repair strategies. Myelination of axons by oligodendrocytes in the CNS is a dynamic process determined by both the cytoarchitecture and microenvironment of the brain, spinal cord, and optic nerves. To ensure proper myelination of axons, oligodendrocytes regulate both their numbers and the amount of myelin each cell produces to properly match the number, diameter, and length of axons they encounter. After the completion of developmental myelination, many oligodendrocyte progenitor cells (OPCs) persist in the adult CNS. Unlike neurons, which fail to regenerate after CNS insult, adult OPCs are capable of proliferating and differentiating into mature oligodendrocytes that myelinate axons in response to injury or damage (3–5). Despite this regenerative ability, why do many axons remain demyelinated in the CNS of MS patients?
One hypothesis for remyelination failure is that the number of adult OPCs available for remyelination is depleted over time (1, 2, 4), however, post-mortem examinations of MS patients, including those in the seventh and eighth decades of life, revealed the presence of OPCs throughout the CNS, including within MS lesions (5–7). Nonetheless, many OPCs fail to mature into myelin-producing oligodendrocytes. These observations suggest that the lesion microenvironment is prohibitive to OPC differentiation and subsequent remyelination of axons.
Many changes occur in areas of demyelination that could prevent remyelination by endogenous OPCs [reviewed by Ref. (8, 9)]. Disruptions to the blood–brain barrier, basal lamina disturbances, and vasculature leakage occur (10–12). This leads to aberrant deposition of extracellular matrix (ECM) components, including fibronectin, hyaluronic acid (HA), and chondroitin sulfate proteoglycans (CSPGs), which can block the differentiation of OPCs and premyelinating oligodendrocytes (8, 9, 13–18). Demyelination can expose OPCs within lesions to other inhibitory cues, including components of damaged myelin such as the proteins MAG (myelin-associated glycoprotein), OMgp (oligodendrocyte myelin glycoprotein), and Nogo-A that signal through the Nogo receptor 1 and its co-receptors p75NTR, TROY, and LINGO-1 (leucine-rich repeat- and Ig domain-containing Nogo receptor-interacting protein 1) to inhibit both axonal regeneration as well as oligodendrocyte differentiation and remyelination (19–23). Activation of both innate CNS and peripheral immune cell populations leads to the release of soluble factors, such as pro-inflammatory cytokines, that can also negatively impact remyelination (24–27).
In particular, semaphorins, originally described as guidance molecules for axons, have been shown to play important roles in the regulation of remyelination. The soluble class III semaphorins (sema) 3A and 3F have been shown to be upregulated in active demyelinating but not chronic lesions in MS patients (28). These molecules are known to influence OPC migration, with sema 3A repelling OPCs and sema 3F attracting OPCs (29–31). Modulation of semaphorin levels within the lesion environment via viral overexpression impacts OPC migration and subsequent remyelination in animal models of demyelination suggests that relative levels of semaphorins in MS lesions may impact remyelination (31, 32). Interestingly, sema 3A and 3F are upregulated in the neuronal cell bodies of demyelinated axons far from lesions sites, raising the possibility that changes in demyelinated neurons may also influence their potential to be remyelinated (28). The transmembrane-bound semaphorin 4D (CD100) is normally expressed by mature MAG-expressing oligodendrocytes but not NG2-positive OPCs. Sema 4D is upregulated after injury and increases oligodendrocyte cell death (33, 34). Overexpression of sema 4D inhibits myelination in oligo-neuronal co-cultures while knock-down of sema 4D in a model of spinal cord injury (SCI) promotes functional recovery (35). Additionally, sema 4D expression disrupts endothelial tight junctions (35, 36). Recently, an antibody against sema 4D was used to treat animals in the experimental autoimmune encephalomyelitis (EAE) model and resulted in improved blood-brain barrier (BBB) integrity and improved OPC differentiation and axonal myelination (37). A phase I trial evaluating safety of a humanized anti-Sema4D IgG4 antibody in MS patients was recently completed (38).
As lesions develop, astrogliosis, a hallmark of MS pathology, occurs in and around lesions, resulting in both structural and biochemical changes. Astrocyte secretion of cytokines can influence lymphocyte infiltration (39, 40) while secretion of cytotoxic factors such as reactive oxygen and nitrogen species, glutamate, and ATP may directly affect oligodendrocytes and neurons (41–44). It is important to note that not all astrocyte changes are pathologic and some may even contribute to repair. For example, BDNF derived from astrocytes has been shown to play an important role in remyelination after cuprizone-induced demyelination (45). Astrocytes may also suppress inflammation and protect neurons from damaging reactive oxygen species in MS lesions via regulation of mitochondrial antioxidant enzymes (46). Clearly, the role of astrocytes in MS pathology is a complicated one and much remains to be elucidated (47, 48), but reduction of astrogliosis has beneficial effects on remyelination in several animal models of demyelination (49, 50).
Disruptions in the glial–neuronal network between astrocytes, oligodendrocytes, and neurons also lead to metabolic deficiencies across cell types that cause cellular dysfunction and death (51–56). As neurodegeneration proceeds, unhealthy axons may no longer be receptive to remyelination; therefore, strategies that improve neuronal survival may also lead to increased remyelination, and importantly, improved clinical outcomes.
Several non-disease-related factors such as age, sex, diet, and individual genetic background can also impact the efficiency of remyelination (57–61). For example, with age, remyelination occurs more slowly due to changes in the CNS environment and intrinsic epigenetic changes in oligodendrocytes, as well as age-related changes in the immune response (60, 62–66). Another interesting observation is that females are at higher risk of developing MS, yet may also remyelinate more efficiently than males, which could be due to the differential effects of sex hormones on oligodendrocyte proliferation and maturation as well as on the neuroinflammatory process (57, 58, 67, 68).
Proposed Mechanisms of CNS Repair and Remyelination in the Context of MS
There are several potential strategies to enhance the remyelination capacity of endogenous OPCs, such as manipulating intrinsic signaling pathways within oligodendrocytes to override the inhibition of remyelination or altering the lesion environment to be more permissive of OPC differentiation and remyelination. Transplantation to increase OPC numbers is probably not necessary in MS given the large numbers of OPCs present in adult brain tissue. Likewise, if endogenous OPCs sense injury and enter lesions, but cannot differentiate there, then enhancing the overall proliferation/migration of OPCs is unlikely to dramatically alter their capacity to remyelinate axons. Therefore, after dampening inflammation, it is crucial to support the survival and differentiation of endogenous adult OPCs in order to stimulate remyelination within the altered microenvironment of MS lesions, while also prolonging survival of denuded axons so that they may be effectively remyelinated.
Modulation of Intrinsic Signaling Pathways
An important approach to myelin preservation and repair is the pharmacological manipulation of intrinsic signaling pathways. Small molecules that can target specific components of the signaling pathways that underlie myelination will need to be developed and tested. Fortunately, much has been learned about the many signaling pathways that govern oligodendrocyte differentiation and myelination over the last three decades (see Table 1). Several groups have recently utilized small molecules capable of modulating these pathways to enhance myelination as discussed below. Complicating matters, however, there is a tremendous amount of cross-talk among signaling pathways, and manipulation of one pathway often induces alterations in another pathway (69). The high degree of cross-talk highlights the need for continued study of the interaction of these pathways in myelinating cells to aid therapeutic intervention.
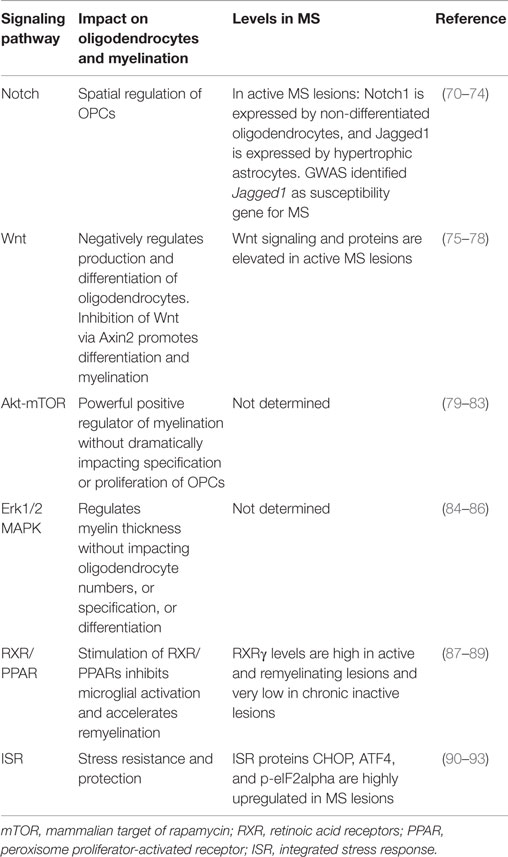
Table 1. Selected pathways and molecules that influence myelination.
Altering the Extracellular Environment
Unquestionably, the extracellular environment is altered in MS lesions, and more effective remyelination would likely be achieved if the local environment within lesions could be restored (8, 9). To this end, clearance of myelin debris and the glial scar produced by astrocytes may enhance remyelination. Attempts to modify the ECM with enzymatic digestion, particularly CSPGs and HA, in the context of SCI have generated favorable results (94–96). This is possible due to the defined local area of damage in SCI that is easily identified and accessed for the application of ECM degrading enzymes. The challenge for such an approach in MS is the disparate and currently unpredictable pattern of demyelinated regions throughout the CNS, making local delivery difficult. Therefore, strategies that directly impact cells that produce ECM components, such as astrocytes, or alter oligodendrocyte responses to aberrant ECM molecules may be more successful approaches.
Enhancement of Cell Survival
Disruptions to glial–neuronal networks and subsequent metabolic changes also impact OPCs and myelination. During acute demyelination, there is an increase in the mitochondrial content of axons early on in an attempt to keep up with the increased energy demands of denuded axons (97–99); however, over time this can lead to increased production of free radicals that can perpetuate axonal injury in the context of chronic demyelination (100). Providing metabolic support (e.g., lactate supplementation or reducing metabolic stress signaling) could preserve both neurons and oligodendroglia.
Finally, it is also important to note that acute inflammation is an important signal that activates adult OPCs to mobilize and mature, but long-term inflammation can be cytotoxic to OPCs (101, 102). Reducing the duration of astroglial and microglial activation could have several beneficial effects (e.g., reducing ECM deposition and decreasing pro-inflammatory cytokine production), which could be more conducive for OPC survival, differentiation, and remyelination. Another potential target for therapeutic intervention is to increase protection of oligodendrocytes in response to inflammation. Care must be taken, however, as the same signal can often have dual effects, either anti- or pro-inflammatory depending on the context. For example, silencing interferon-gamma (IFNγ) in astrocytes alleviates symptoms in EAE, whereas silencing of IFNγ in microglia increases disease severity (103).
Remyelination Therapies in the Pipeline for the Treatment of MS
Remyelination Therapies
Drugs that have a positive impact on remyelination and neuroprotection (Table 2) could be used as part of a combination therapy, including immunomodulatory drugs. Treatments that enhance the speed of remyelination are predicted to protect neurons from axonal degeneration. Intervention to maintain and repair myelin should occur early as chronically demyelinated axons will degenerate precluding future remyelination by existing OPCs and oligodendrocytes (104).
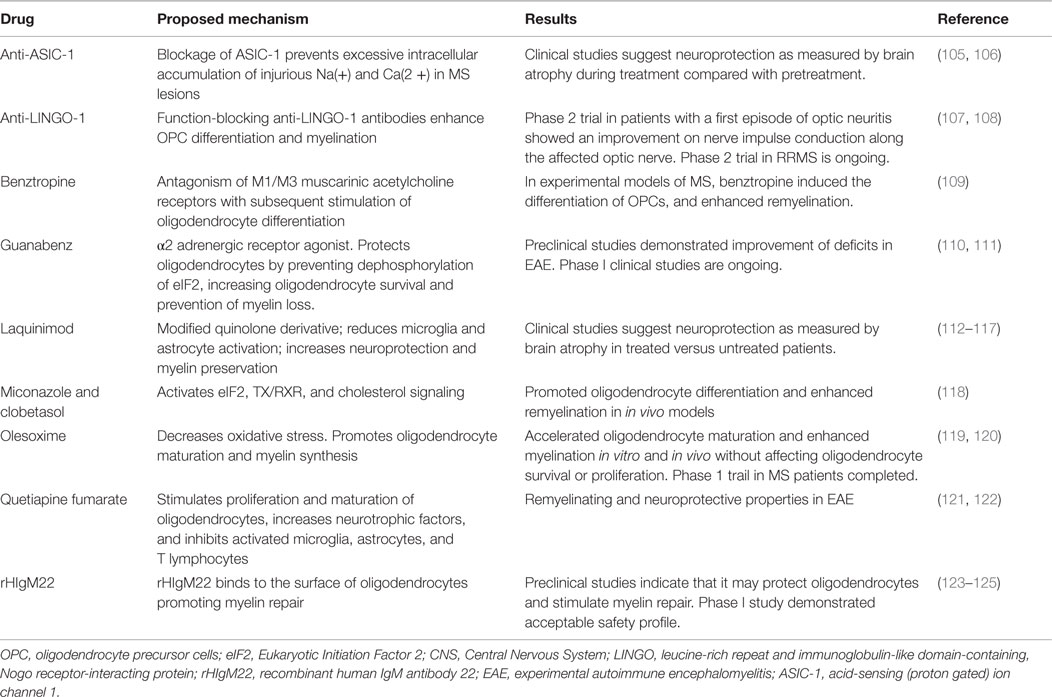
Table 2. Potential remyelinating and neuroprotective therapies in multiple sclerosis.
High-throughput screens of previously FDA-approved drugs have identified several classes of drugs that enhance OPC differentiation and myelination. Although the beneficial effects of these drugs on CNS cells are encouraging, careful study of off-target effects will need to be undertaken, given that many of these drugs were originally utilized for non-CNS targets. Anticholinergics, including the drugs benztropine (109) and clemastine (126), were identified in two separate high-throughput screens looking for agents that enhance myelination. Activation of muscarinic receptors inhibits the differentiation of oligodendrocytes (127); therefore, the pro-myelinating effects of benztropine and clemastine are likely via their antagonism of M1/M3 muscarinic acetylcholine receptors. These anti-muscarinic compounds appear to enhance remyelination through direct effects on oligodendrocytes and not immunosuppressive effects. As phase 1 safety profile testing of anticholinergics has already been done for other indications, there has been a rapid translation of the preclinical laboratory findings to phase 2 clinical trials on the efficacy of clemastine in MS (128).
A third small molecule screen identified two additional FDA-approved drugs that promote oligodendrocyte differentiation and enhance remyelination in in vivo models (118): miconazole, an antifungal agent, and clobetasol, a corticosteroid used to treat eczema and psoriasis. Both act directly on oligodendrocytes as remyelinating drugs and impact eIF2 signaling, thyroid hormone receptor/retinoic acid receptor (TX/RXR) activation, and cholesterol signaling. Interestingly, miconazole acts through MAPK signaling and has no effect on the immune system, whereas clobetasol acts through glucocorticoid receptor signaling and is also a potent immunosuppressant in addition to being a remyelinating agent. With their high safety profile already established, approval for phase 2 trials to establish efficacy in MS could occur quickly.
Another strategy for promoting remyelination in MS is altering the local environment (soluble factors released by innate CNS cells, modifying the ECM), to be more permissive to endogenous OPCs. As mentioned above, the addition of enzymes, such as chondroitinase and hyaluronidase, is not very practical in a multifocal demyelinating disease such as MS. Therefore, an alternative approach is to change cellular responses to hostile environments. One promising target is the Nogo-A co-receptor LINGO-1, which is expressed by both neurons and oligodendrocytes. LINGO-1 acts as a negative regulator of oligodendrocyte differentiation and myelination. LINGO-1 knockout mice have enhanced remyelination in several demyelination models, including EAE and toxin-induced demyelinated lesions (23, 129, 130). Function-blocking anti-LINGO-1 antibodies enhance OPC differentiation and myelination (129). Promising results were obtained in a Phase 2 trial of a human IgG1 anti-LINGO-1 monoclonal antibody (BIIB033) in patients with a first episode of optic neuritis (107). Treatment was well tolerated; and after 24 weeks, those patients given the anti-LINGO antibody had faster nerve impulse conduction along the optic nerve than before treatment compared to those on placebo, indicating myelin repair. Although visual evoked potentials were increased, neither visual acuity nor retinal neuron layer thickness were improved. Another phase 2 trial in patients with relapsing MS is ongoing with estimated completion in June 2016 (108).
Preclinical studies of a recombinant human IgM antibody (rHIgM22) indicate that it may protect oligodendrocytes and stimulate myelin repair (123, 124). rHIgM22 binds to the surface of oligodendrocytes, and prevents their apoptosis. Although the exact target is not known, rHIgM22 may bind αvß3 integrins, the vitronectin/fibronectin receptor, and aggregated fibronectin has been shown to inhibit OPC differentiation and remyelination (131, 132). rHIgM22 also seems to inhibit OPC differentiation, so how it promotes myelin repair remains unclear. rHIgM22 was found safe and tolerable in Phase 1 clinical trials after a 6-month follow-up of a single dose of patients who remained on their existing MS treatments (133). A second dose-escalating phase 1 trial is underway (125).
Oligodendrocytes have very high metabolic demands due to production of extensive amounts of lipid-rich myelin membrane. This makes oligodendrocytes particularly sensitive to disruptions in metabolic homeostasis. Hypoxia/ischemia, oxygen–glucose deprivation, viral infections, and high rates of protein synthesis in the endoplasmic reticulum (ER) can all lead to ER stress, which activates pancreatic ER kinase (PERK) phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and the integrated stress response (ISR) (90). The ISR inhibits global protein synthesis to reduce the load on the ER and also upregulates transcription factors, such as STF4, CHOP, ATF4, and p-eIF2σ that cause the cell to be in a more stress-resistant state. Upregulation of these ISR targets is seen in MS lesions, with increased PERK activity in oligodendrocytes (91). Enhancement of the ISR in oligodendrocytes could protect them and lead to increased remyelination efficiency.
Guanabenz is an α2 adrenergic receptor agonist that has been previously approved by the FDA for the treatment of hypertension (134). Guanabenz alleviates symptoms in EAE, increases oligodendrocyte survival, and reduces CD4+ T cell accumulation in the CNS (110). Guanabenz appears to work by protecting oligodendrocytes against the inflammatory CNS environment. Guanabenz enhances the protective ISR of oligodendrocytes by preventing dephosphorylation of eIF2α, increasing oligodendrocyte survival, and protecting against myelin loss (110, 135). A phase 1 safety trail of oral guanabenz in MS patients recently began recruiting patients (111).
Olesoxime, is a cholesterol-oxime compound and mitochondrial pore modulator, originally developed to treat amyotrophic lateral sclerosis that targets proteins of the outer mitochondrial membrane, preventing permeability and decreasing oxidative stress (136). Olesoxime accelerates oligodendrocyte maturation and enhanced myelination in vitro and in vivo without affecting oligodendrocyte survival or proliferation (119). A phase 1b study in MS is ongoing (120).
Stem cell-based remyelinating therapies are also a plausible alternative strategy in MS as stem cells of both neural and mesenchymal origin have the ability to facilitate endogenous reparative processes, participate directly in remyelination, and attenuate neuroinflammation. However, there are crucial questions that have to be addressed before considering clinical studies, including the determination of the optimal cellular platform, the route of cell delivery, and candidate patients for treatment (137).
Neuroprotective Strategies
In order to achieve maximal levels of remyelination, therapies that decrease axonal degeneration and increase neuronal survival in response to demyelination could be used to extend the period during which axons could be remyelinated.
Quetiapine fumarate is an atypical antipsychotic that has been shown to have both remyelinating and neuroprotective properties in EAE (121). It appears to impact various biological pathways relevant in MS with potential to stimulate proliferation and maturation of oligodendrocytes, release of neurotrophic factors, and inhibit activated microglia, astrocytes, and T lymphocytes (121). A dose-finding trial in both relapsing and progressive forms of MS is underway (122).
The acid-sensing ion channel, ASIC-1, contributes to the excessive intracellular accumulation of injurious Na(+) and Ca(2+) and is over-expressed in acute MS lesions. Blockade of ASIC1 through amiloride, a potassium-sparing diuretic that is currently licensed for hypertension and congestive cardiac failure, showed neuroprotective and myeloprotective effects in experimental models of MS (105, 106). A pilot study in primary progressive MS showed positive effects on brain volume (105) and a multi-arm randomized Phase II trial in secondary progressive MS patients is currently enrolling (138).
Promising results have been achieved with laquinimod, a small molecule that in addition to its effects on lymphocytes can also reduce glial reactivity (139, 140). Although the exact mechanism of action is not known, laquinimod, a modified quinoline derivative, appears to have direct effects on astrocytes and microglia/macrophages within the CNS (49, 117, 139, 140). This modulation of astrocyte and microglia responses appears to provide a protective effect on both neuronal function and oligodendrocytes and myelin within lesion areas (112, 141, 142). In phase 3 clinical trials of relapsing-remitting MS patients, both clinical disability and brain atrophy were reduced with laquinimod treatment indicative of a neuroprotective effect (113, 114, 143). A phase 2 trial for laquinimod in primary progressive MS is underway (115).
Assessment of Remyelination in Clinical Studies
One of the biggest difficulties in the development of remyelination therapies for MS is the demonstration of remyelination in living patients. Indirect measures, such as improvements in neurophysiological outcomes, such as electroencephalography, evoked potentials, optical coherence tomography, and transcranial magnetic stimulation may suggest, yet not confirm, remyelination (144–148). Functional improvements could indicate remyelination, but could also indicate neuronal plasticity or the spreading of sodium channels into demyelinated internodes, which could also restore conduction in unmyelinated axons (144, 149, 150). Although an extensive discussion is beyond the scope of this review, analysis of serum and cerebrospinal fluid (CSF) biomarkers could provide important and specific information regarding remyelination and repair in the future. At present, candidate biomarkers appear to relate to disease activity (interleukin-6 or its soluble receptor, nitric oxide and nitric oxide synthase, osteopontin, and fetuin-A) or neurodegeneration and blood–brain barrier dysfunction (neurofilaments, tau, 14-3-3 proteins, S-100β, GFAP), rather than repair [for review, see in Ref. (151, 152)]. Even post-mortem, histopathologic approaches to detect remyelination are inherently limited to a “snapshot” assessment of the fluctuating process of demyelination and remyelination (153).
Demyelinating lesions are readily detected by T2-weighted (T2W) and fluid attenuated inversion recovery (FLAIR) MRI (154–156). Unfortunately, these sequences are unable to differentiate remyelination from the ever-changing milieu of local inflammation, edema, and axonal and myelin damage (153, 157–159). In addition, the correlation between MRI-identified lesion load and clinical disability is weak, a phenomenon referred to as the “clinical-radiologic paradox” in MS (160). With the increasing interest in development of new therapies for MS targeting remyelination, accurate MRI measures of myelin repair are crucial for assessing the impact of these therapies (161). Advancements in MRI technology may provide an opportunity for in vivo assessment of this dynamic process in both visible lesions and the putatively “normal appearing white matter (NAWM).” Candidate imaging sequences must be sensitive and specific for changes occurring in vivo, correlate with other indicators of remyelination, and crucially for quantitative MRI, must be reproducible across imaging platforms.
Assessment of Remyelination by Advanced Imaging
Diffusion Tensor Imaging
Diffusion tensor imaging (DTI) tracks the Brownian motion of water molecules in tissue to provide quantitative data about tissue microstructure, and is particularly well-suited to highly ordered environments such as CNS white matter (162). For different aspects of white matter microstructure, DTI produces various quantitative indices, including fractional anisotropy, mean diffusivity, axial diffusivity, and radial diffusivity. Fractional anisotropy reflects the overall vector of diffusion of water molecules and, by inference, the directionality of the fiber tracts, while mean diffusivity represents the overall magnitude of diffusion, regardless of direction (162). Both mean diffusivity and fractional anisotropy correlate with the number of axons and the degree of myelination in post-mortem examination, but it is difficult, if not impossible, to distinguish remyelination from demyelination using these metrics (163, 164). In highly ordered white matter bundles, radial diffusivity is thought to represent the diffusion of water molecules perpendicular to fiber tracts, while axial diffusivity is thought to represent the diffusion of water molecules parallel to the tract, and it has been suggested that radial diffusivity might be more sensitive to myelin damage while axial diffusivity may be more sensitive to axonal injury (164–167). While this appears promising, the major limitation of the radial and axial diffusivity indices is that the models used for derivation depend on accurate calculation of the correct fiber tract orientation. Crossing fibers and disruption of the white matter microstructure by varying pathology in MS (pro-inflammatory cellular infiltration, edema, demyelination, and axonal loss) may result in inaccurate determination of the diffusion tensor and underestimation of radial diffusivity, leading to underestimation of the degree of changes in myelination (168–171). Newer techniques such as high angular resolution diffusion imaging, which is capable of resolving crossing fibers (172), and neurite orientation dispersion and density imaging, which is more specific for myelination than standard DTI indices (173), may increase the specificity of these more advanced techniques to serve as biomarkers for tracking remyelination in MS.
Magnetization Transfer Imaging
Magnetization transfer (MT) imaging exploits the exchange of magnetization that occurs between two “pools” of protons in vivo: a “free” pool of protons that are generally unbound in an aqueous environment, and a “restricted” pool of protons that are bound to high molecular weight molecules such as lipids and proteins (174, 175). While signal on conventional imaging is predominantly derived from the relaxation of protons in the “free” pool; in MT imaging, two sets of images are obtained (one with MT-weighting, and the other without) allowing imaging of both pools. An estimate of the magnetization transfer ratio (MTR) between these two pools can be calculated allowing for discrimination of subtle changes not picked up by conventional MRI.
Although edema, inflammation, and axonal density do influence MTR, the high lipid content of myelin strongly affects the MTR (170, 176–178), allowing MT imaging to be relatively sensitive in detecting changes in myelination, including remyelination (Figure 1). White matter lesions have a lower MTR than NAWM, and NAWM in MS patients has a lower MTR than NAWM in controls (179–181). There is also variation between disease subtypes: secondary progressive MS has the lowest white matter MTR measures, followed by relapsing-remitting MS, and clinically isolated syndrome (CIS) (181). Remyelinated lesions have higher MTR than unmyelinated lesions, but MTR in remyelinated lesions remains lower than NAWM (153, 182–184), suggesting either that remyelination is incomplete or that newly formed myelin in lesions has a different structure, which is consistent with reports of thinner myelin in post-mortem examination of remyelinated lesions (185). While lesions appear generally static on conventional imaging over time, MTR fluctuates within lesions, suggesting alternating waves of both demyelination and remyelination (184). Given the semi-quantitative nature of MTR measurement, recent efforts have been made to directly measure the size and relaxation characteristics of the “restricted” proton pool, to provide quantitative measurements that are even more strongly influenced by myelin in the brain (186–190). Although performing multiple MT-weighted acquisitions increases scan times, MT imaging could be advantageous in large clinical trials as quantitative metrics are more consistent across scanners.
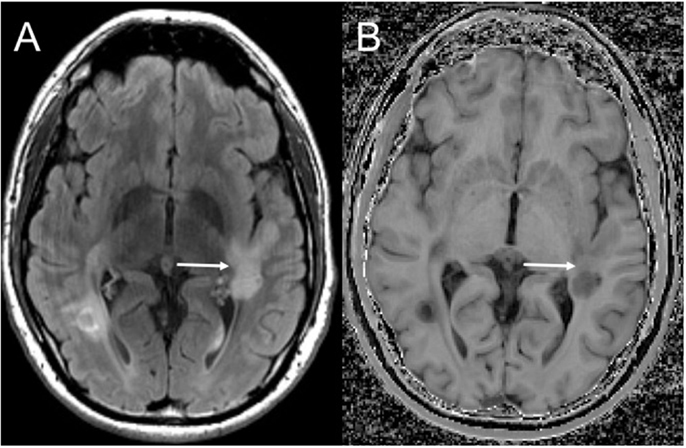
Figure 1. Magnetization transfer imaging. Axial FLAIR (A) demonstrating a large demyelinating lesion in the white matter posterior to the Sylvian fissure. Magnetization transfer image (B) demonstrates a band of normal white matter signal intensity across the mid aspect of the lesion (solid arrow) compatible with partial remyelination.
Myelin Water Fraction Imaging
Brain tissue microstructure is complex and each tissue component displays different T2 relaxation characteristics. The differences in T2 relaxation correlate with water separated into three components: a long T2 component corresponding to CSF, an intermediate T2 component corresponding to intra/extracellular water, and a short T2 relaxation time component corresponding to water trapped with layers of myelin (191). Of most interest in MS is the short T2 (myelin water) component, measured as the ratio of myelin water to the total water, i.e., the myelin water fraction (MWF). Histopathological studies have shown that the MWF correlates with myelin content, is insensitive to changes related to inflammation, and is independent of axonal loss/degeneration (186, 192).
Early techniques for measuring MWF depended on 2D multi-spin echo acquisitions, which due to time considerations permitted only incomplete coverage of the brain. To improve brain coverage and further reduce MT effects, other sequences are now available, including 3D-GRASE and most recently mcDESPOT, which allow coverage of the entire brain within reasonable acquisition times (157, 193, 194). While these techniques show much promise, work still needs to be done to confirm their tissue specificity. One recent study, for instance, indicated that in white matter mcDESPOT may not be able to precisely estimate the two-pool model with exchange (195). Despite that limitation, MWF is lower in MS patients compared to that of controls (196), correlates with disability, and decreases over time in patients with progressive MS (197). MWF imaging has the potential to follow remyelination in lesions after acute edema has resolved (198) and has been shown to increase in patients treated with alemtuzumab, suggesting it may be useful in monitoring for remyelination in treated patients (199).
Positron Emission Tomography
Positron emission tomography (PET) uses radioisotopes that directly bind to different tissue substrates to enable molecular imaging. Direct binding of imaging biomarkers specific for myelin could potentially far exceed the specificity of the other modalities discussed above and allow investigators to detect changes specific for myelin content in the brain. One such biomarker is the thioflavine-T derivative 2-(4′-methylaminophenyl)-6-hydroxybenzothiazole (PIB). 11C-PIB is currently used in the imaging of Alzheimer’s disease (200, 201) but recent studies have shown that in addition to amyloid plaques, PIB also has an affinity for CNS myelin and demonstrates differential binding to normal and demyelinated white matter (202, 203). In two RRMS patients, 11C-PIB uptake was less in enhancing lesions (203), indicating that there may be differences in the myelination of lesions at different stages. While promising, additional studies of more MS patients are required to validate these findings and not all centers will have access to such technology.
Conclusion
Repair and remyelination in MS is possible, but remyelination often fails as a consequence of failure to recruit OPCs fully into the lesions, failure of OPCs to generate mature myelinating oligodendrocytes, and failure of oligodendrocytes to remyelinate axons. Several candidates to enhance remyelination are currently under investigation with a variety of mechanisms of action. One barrier to evaluating remyelination therapies in patients is the lack of methods to accurately detect myelination, demyelination, and remyelination with standard imaging technologies. Advancements in MRI technology allow better detection of myelin-specific changes but criteria that can be readily quantified using new imaging technologies will have to be established and validated to determine the success of remyelination in clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
1. Nave K-AA. Myelination and the trophic support of long axons. Nat Rev Neurosci (2010) 11:275–83. doi:10.1038/nrn2797
2. Chari DM, Blakemore WF. New insights into remyelination failure in multiple sclerosis: implications for glial cell transplantation. Mult Scler (2002) 8:271–7. doi:10.1191/1352458502ms842oa
3. Moyon S, Dubessy AL, Aigrot M-SS, Trotter MWB, Huang JK, Dauphinot L, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci (2015) 35:4–20. doi:10.1523/JNEUROSCI.0849-14.2015
4. Franklin RJM, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol (2012) 8:624–34. doi:10.1038/nrneurol.2012.200
5. Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med (2002) 346:165–73. doi:10.1056/NEJMoa010994
6. Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci (2000) 20:6404–12.
7. Fancy SP, Zhao C, Franklin RJM. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol Cell Neurosci (2004) 27:247–54. doi:10.1016/j.mcn.2004.06.015
8. Lau LW, Cua R, Keough MB, Haylock-Jacobs S, Yong VW. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci (2013) 14:722–9. doi:10.1038/nrn3550
9. Harlow DE, Macklin WB. Inhibitors of myelination: ECM changes, CSPGs and PTPs. Exp Neurol (2014) 251:39–46. doi:10.1016/j.expneurol.2013.10.017
10. Alvarez JI, Saint-Laurent O, Godschalk A, Terouz S, Briels C, Larouche S, et al. Focal disturbances in the blood-brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol Dis (2015) 74:14–24. doi:10.1016/j.nbd.2014.09.016
11. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun (2012) 3:1227. doi:10.1038/ncomms2230
12. Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, et al. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain (2015) 138:1548–67. doi:10.1093/brain/awv077
13. Sloane JA, Batt CE, Ma Y, Harris ZM, Trapp BD, Vartanian TK. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A (2010) 107:11555–60. doi:10.1073/pnas.1006496107
14. Bugiani M, Postma N, Polder E, Dieleman N, Scheffer PG, Sim FJ, et al. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain (2013) 136:209–22. doi:10.1093/brain/aws320
15. Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med (2005) 11:966–72. doi:10.1038/nm1279
16. Haist V, Ulrich R, Kalkuhl A, Deschl U, Baumgartner W. Distinct spatio-temporal extracellular matrix accumulation within demyelinated spinal cord lesions in Theiler’s murine encephalomyelitis. Brain Pathol (2012) 22:188–204. doi:10.1111/j.1750-3639.2011.00518.x
17. van Horssen J, Bo L, Dijkstra CD, de Vries HE. Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol Dis (2006) 24:484–91. doi:10.1016/j.nbd.2006.08.005
18. Mohan H, Krumbholz M, Sharma R, Eisele S, Junker A, Sixt M, et al. Extracellular matrix in multiple sclerosis lesions: fibrillar collagens, biglycan and decorin are upregulated and associated with infiltrating immune cells. Brain Pathol (2010) 20:966–75. doi:10.1111/j.1750-3639.2010.00399.x
19. Mei F, Christin Chong SY, Chan JR. Myelin-based inhibitors of oligodendrocyte myelination: clues from axonal growth and regeneration. Neurosci Bull (2013) 29:177–88. doi:10.1007/s12264-013-1319-x
20. Kotter MR, Li WW, Zhao C, Franklin RJM. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci (2006) 26:328–32. doi:10.1523/JNEUROSCI.2615-05.2006
21. Yang Y, Liu Y, Wei P, Peng H, Winger R, Hussain RZ, et al. Silencing Nogo-A promotes functional recovery in demyelinating disease. Ann Neurol (2010) 67:498–507. doi:10.1002/ana.21935
22. Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci (2004) 7:221–8. doi:10.1038/nn1188
23. Lee X, Yang Z, Shao Z, Rosenberg SS, Levesque M, Pepinsky RB, et al. NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J Neurosci (2007) 27:220–5. doi:10.1523/JNEUROSCI.4175-06.2007
24. Rodgers JM, Miller SD. Cytokine control of inflammation and repair in the pathology of multiple sclerosis. Yale J Biol Med (2012) 85:447–68.
25. Brambilla R, Morton PD, Ashbaugh JJ, Karmally S, Lambertsen KL, Bethea JR. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia (2014) 62:452–67. doi:10.1002/glia.22616
26. Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, et al. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol (2005) 64:706–15. doi:10.1097/01.jnen.0000173893.01929.fc
27. Schmitz T, Chew LJ. Cytokines and myelination in the central nervous system. ScientificWorldJournal (2008) 8:1119–47. doi:10.1100/tsw.2008.140
28. Williams A, Piaton G, Aigrot M-SS, Belhadi A, Théaudin M, Petermann F, et al. Semaphorin 3A and 3F: key players in myelin repair in multiple sclerosis? Brain (2007) 130:2554–65. doi:10.1093/brain/awm202
29. Cohen RI, Rottkamp DM, Maric D, Barker JL, Hudson LD. A role for semaphorins and neuropilins in oligodendrocyte guidance. J Neurochem (2003) 85:1262–78. doi:10.1046/j.1471-4159.2003.01722.x
30. Spassky N, de Castro F, Le Bras B, Heydon K, Quéraud-LeSaux F, Bloch-Gallego E, et al. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J Neurosci (2002) 22:5992–6004.
31. Piaton G, Aigrot M-SS, Williams A, Moyon S, Tepavcevic V, Moutkine I, et al. Class 3 semaphorins influence oligodendrocyte precursor recruitment and remyelination in adult central nervous system. Brain (2011) 134:1156–67. doi:10.1093/brain/awr022
32. Syed YA, Hand E, Möbius W, Zhao C, Hofer M, Nave K-AA, et al. Inhibition of CNS remyelination by the presence of semaphorin 3A. J Neurosci (2011) 31:3719–28. doi:10.1523/JNEUROSCI.4930-10.2011
33. Taniguchi Y, Amazaki M, Furuyama T, Yamaguchi W, Takahara M, Saino O, et al. Sema4D deficiency results in an increase in the number of oligodendrocytes in healthy and injured mouse brains. J Neurosci Res (2009) 87:2833–41. doi:10.1002/jnr.22124
34. Yamaguchi W, Tamai R, Kageura M, Furuyama T, Inagaki S. Sema4D as an inhibitory regulator in oligodendrocyte development. Mol Cell Neurosci (2012) 49:290–9. doi:10.1016/j.mcn.2011.12.004
35. Zhang H-L, Wang J, Tang L. Sema4D knockdown in oligodendrocytes promotes functional recovery after spinal cord injury. Cell Biochem Biophys (2014) 68:489–96. doi:10.1007/s12013-013-9727-0
36. Treps L, Le Guelte A, Gavard J. Emerging roles of Semaphorins in the regulation of epithelial and endothelial junctions. Tissue Barriers (2013) 1:e23272. doi:10.4161/tisb.23272
37. Smith ES, Jonason A, Reilly C, Veeraraghavan J, Fisher T, Doherty M, et al. SEMA4D compromises blood-brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol Dis (2014) 73C:254–68. doi:10.1016/j.nbd.2014.10.008
38. Vaccinex, Inc; PRA Health Sciences. Evaluation of safety, tolerability, and PK of VX15/2503 in patients with MS. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: http://clinicaltrials.gov/ct2/show/NCT01764737
39. Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol (2009) 182:2628–40. doi:10.4049/jimmunol.0802954
40. Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity (2010) 32:414–25. doi:10.1016/j.immuni.2010.03.004
41. Hamby ME, Hewett JA, Hewett SJ. TGF-beta1 potentiates astrocytic nitric oxide production by expanding the population of astrocytes that express NOS-2. Glia (2006) 54:566–77. doi:10.1002/glia.20411
42. Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol (2001) 158:2057–66. doi:10.1016/S0002-9440(10)64677-9
43. Matute C, Sánchez-Gómez MV, Martínez-Millán L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci U S A (1997) 94:8830–5. doi:10.1073/pnas.94.16.8830
44. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med (2000) 6:67–70. doi:10.1038/71555
45. Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci (2014) 34:8186–96. doi:10.1523/JNEUROSCI.4267-13.2014
46. Nijland PG, Witte ME, van Het Hof B, van der Pol S, Bauer J, Lassmann H, et al. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: implications for multiple sclerosis. Acta Neuropathol Commun (2014) 2:170. doi:10.1186/s40478-014-0170-2
47. Williams A, Piaton G, Lubetzki C. Astrocytes – friends or foes in multiple sclerosis? Glia (2007) 55:1300–12. doi:10.1002/glia.20546
48. Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci (2008) 65:2702–20. doi:10.1007/s00018-008-8059-5
49. Brück W, Pfortner R, Pham T, Zhang J, Hayardeny L, Piryatinsky V, et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol (2012) 124:411–24. doi:10.1007/s00401-012-1009-1
50. Moore S, Khalaj AJ, Yoon J, Patel R, Hannsun G, Yoo T, et al. Therapeutic laquinimod treatment decreases inflammation, initiates axon remyelination, and improves motor deficit in a mouse model of multiple sclerosis. Brain Behav (2013) 3:664–82. doi:10.1002/brb3.174
51. Saab AS, Tzvetanova ID, Nave K-AA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol (2013) 23:1065–72. doi:10.1016/j.conb.2013.09.008
52. Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature (2012) 487:443–8. doi:10.1038/nature11314
53. Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature (2012) 485:517–21. doi:10.1038/nature11007
54. Mahad DJ, Ziabreva I, Campbell GR, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain (2009) 132:1161–74. doi:10.1093/brain/awp046
55. Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol (2006) 59:478–89. doi:10.1002/ana.20736
56. Agresti C, Meomartini ME, Amadio S, Ambrosini E, Volonté C, Aloisi F, et al. ATP regulates oligodendrocyte progenitor migration, proliferation, and differentiation: involvement of metabotropic P2 receptors. Brain Res Brain Res Rev (2005) 48:157–65. doi:10.1016/j.brainresrev.2004.12.005
57. Marin-Husstege M, Muggironi M, Raban D, Skoff RP, Casaccia-Bonnefil P. Oligodendrocyte progenitor proliferation and maturation is differentially regulated by male and female sex steroid hormones. Dev Neurosci (2004) 26:245–54. doi:10.1159/000082141
58. Kipp M, Beyer C. Impact of sex steroids on neuroinflammatory processes and experimental multiple sclerosis. Front Neuroendocrinol (2009) 30:188–200. doi:10.1016/j.yfrne.2009.04.004
59. Ligon KL, Fancy SPJ, Franklin RJM, Rowitch DH. Olig gene function in CNS development and disease. Glia (2006) 54:1–10. doi:10.1002/glia.20273
60. Sim FJ, Zhao C, Penderis J, Franklin RJM. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci (2002) 22:2451–9.
61. Adamo AM. Nutritional factors and aging in demyelinating diseases. Genes Nutr (2014) 9:360. doi:10.1007/s12263-013-0360-8
62. van Wijngaarden P, Franklin RJM. Ageing stem and progenitor cells: implications for rejuvenation of the central nervous system. Development (2013) 140:2562–75. doi:10.1242/dev.092262
63. Rist JM, Franklin RJM. Taking ageing into account in remyelination-based therapies for multiple sclerosis. J Neurol Sci (2008) 274:64–7. doi:10.1016/j.jns.2008.04.027
64. Doucette JR, Jiao R, Nazarali AJ. Age-related and cuprizone-induced changes in myelin and transcription factor gene expression and in oligodendrocyte cell densities in the rostral corpus callosum of mice. Cell Mol Neurobiol (2010) 30:607–29. doi:10.1007/s10571-009-9486-z
65. Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci (2009) 12:829–38. doi:10.1038/nn.2333
66. Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJM, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci (2008) 11:1024–34. doi:10.1038/nn.2172
67. Li WW, Penderis J, Zhao C, Schumacher M, Franklin RJM. Females remyelinate more efficiently than males following demyelination in the aged but not young adult CNS. Exp Neurol (2006) 202:250–4. doi:10.1016/j.expneurol.2006.05.012
68. Nicot A. Gender and sex hormones in multiple sclerosis pathology and therapy. Front Biosci (2009) 14:4477–515. doi:10.2741/3543
69. Dai J, Bercury KK, Macklin WB. Interaction of mTOR and Erk1/2 signaling to regulate oligodendrocyte differentiation. Glia (2014) 62:2096–109. doi:10.1002/glia.22729
70. Park HC, Appel B. Delta-Notch signaling regulates oligodendrocyte specification. Development (2003) 130:3747–55. doi:10.1242/dev.00576
71. Stidworthy MF, Genoud S, Li WW, Leone DP, Mantei N, Suter U, et al. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain (2004) 127:1928–41. doi:10.1093/brain/awh217
72. Zhang Y, Argaw AT, Gurfein BT, Zameer A, Snyder BJ, Ge C, et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci U S A (2009) 106:19162–7. doi:10.1073/pnas.0902834106
73. Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, et al. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron (1998) 21:63–75. doi:10.1016/S0896-6273(00)80515-2
74. Games Collaborative Group; Ban M, Booth D, Heard R, Stewart G, Goris A, et al. Linkage disequilibrium screening for multiple sclerosis implicates JAG1 and POU2AF1 as susceptibility genes in Europeans. J Neuroimmunol (2006) 179:108–16. doi:10.1016/j.jneuroim.2006.06.003
75. Fancy SP, Baranzini SE, Zhao C, Yuk D-I, Irvine KA, Kaing S, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev (2009) 23:1571–85. doi:10.1101/gad.1806309
76. Langseth AJ, Munji RN, Choe Y, Huynh T, Pozniak CD, Pleasure SJ. Wnts influence the timing and efficiency of oligodendrocyte precursor cell generation in the telencephalon. J Neurosci (2010) 30:13367–72. doi:10.1523/JNEUROSCI.1934-10.2010
77. Feigenson K, Reid M, See J, Crenshaw EB, Grinspan JB. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci (2009) 42:255–65. doi:10.1016/j.mcn.2009.07.010
78. Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci (2011) 14:1009–16. doi:10.1038/nn.2855
79. Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, Macklin WB. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci (2014) 34:4466–80. doi:10.1523/JNEUROSCI.4314-13.2014
80. Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, et al. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci (2009) 29:6367–78. doi:10.1523/JNEUROSCI.0234-09.2009
81. Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci (2014) 34:4453–65. doi:10.1523/JNEUROSCI.4311-13.2014
82. Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci (2009) 29:6860–70. doi:10.1523/JNEUROSCI.0232-09.2009
83. Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci (2008) 28:7174–83. doi:10.1523/JNEUROSCI.0150-08.2008
84. Fyffe-Maricich SL, Schott A, Karl M, Krasno J, Miller RH. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J Neurosci (2013) 33:18402–8. doi:10.1523/JNEUROSCI.2381-13.2013
85. Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci (2012) 32:8855–64. doi:10.1523/JNEUROSCI.0137-12.2012
86. Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci (2011) 31:843–50. doi:10.1523/JNEUROSCI.3239-10.2011
87. Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development (1994) 120:1097–108.
88. Huang JK, Jarjour AA, Nait-Oumesmar B, Kerninon C, Williams A, Krezel W, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci (2011) 14:45–53. doi:10.1038/nn.2702
89. Ahlgren SC, Wallace H, Bishop J, Neophytou C, Raff MC. Effects of thyroid hormone on embryonic oligodendrocyte precursor cell development in vivo and in vitro. Mol Cell Neurosci (1997) 9:420–32. doi:10.1006/mcne.1997.0631
90. Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, et al. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci (2013) 33:5980–91. doi:10.1523/JNEUROSCI.1636-12.2013
91. Cunnea P, Mhaille AN, McQuaid S, Farrell M, McMahon J, FitzGerald U. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult Scler J (2011) 17:808–18. doi:10.1177/1352458511399114
92. Hussien Y, Cavener DR, Popko B. Genetic inactivation of PERK signaling in mouse oligodendrocytes: normal developmental myelination with increased susceptibility to inflammatory demyelination. Glia (2014) 62:680–91. doi:10.1002/glia.22634
93. Mháille AN, McQuaid S, Windebank A, Cunnea P, McMahon J, Samali A, et al. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J Neuropathol Exp Neurol (2008) 67:200–11. doi:10.1097/NEN.0b013e318165b239
94. Starkey ML, Bartus K, Barritt AW, Bradbury EJ. Chondroitinase ABC promotes compensatory sprouting of the intact corticospinal tract and recovery of forelimb function following unilateral pyramidotomy in adult mice. Eur J Neurosci (2012) 36:3665–78. doi:10.1111/ejn.12017
95. Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature (2002) 416:636–40. doi:10.1038/416636a
96. Karimi-Abdolrezaee S, Schut D, Wang J, Fehlings MG. Chondroitinase and growth factors enhance activation and oligodendrocyte differentiation of endogenous neural precursor cells after spinal cord injury. PLoS One (2012) 7:e37589. doi:10.1371/journal.pone.0037589
97. Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci (2010) 30:6658–66. doi:10.1523/JNEUROSCI.5265-09.2010
98. Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, et al. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain (2011) 134:1901–13. doi:10.1093/brain/awr110
99. Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, et al. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A (2014) 111:9953–8. doi:10.1073/pnas.1401155111
100. Joshi DC, Zhang CL, Lin TM, Gusain A, Harris MG, Tree E, et al. Deletion of mitochondrial anchoring protects dysmyelinating shiverer: implications for progressive MS. J Neurosci (2015) 35:5293–306. doi:10.1523/JNEUROSCI.3859-14.2015
101. Setzu A, Lathia JD, Zhao C, Wells K, Rao MS, ffrench-Constant C, et al. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia (2006) 54:297–303. doi:10.1002/glia.20371
102. Blakemore WF. Regeneration and repair in multiple sclerosis: the view of experimental pathology. J Neurol Sci (2008) 265:1–4. doi:10.1016/j.jns.2007.03.006
103. Ding X, Yan Y, Li X, Li K, Ciric B, Yang J, et al. Silencing IFN-gamma binding/signaling in astrocytes versus microglia leads to opposite effects on central nervous system autoimmunity. J Immunol (2015) 194:4251–64. doi:10.4049/jimmunol.1303321
104. Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol (2001) 14:271–8. doi:10.1097/00019052-200106000-00003
105. Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI, et al. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain (2013) 136:106–15. doi:10.1093/brain/aws325
106. Yu X-W, Hu Z-L, Ni M, Fang P, Zhang P-W, Shu Q, et al. Acid-sensing ion channels promote the inflammation and migration of cultured rat microglia. Glia (2015) 63:483–96. doi:10.1002/glia.22766
107. Biogen. 215ON201 BIIB033 in acute optic neuritis (AON) (RENEW). In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT01721161
108. Biogen. Study to assess the efficacy, safety, tolerability, and pharmacokinetics of BIIB033 in participants with relapsing forms of multiple sclerosis when used concurrently with Avonex (SYNERGY). In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT01864148
109. Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, et al. A regenerative approach to the treatment of multiple sclerosis. Nature (2013) 502:327–32. doi:10.1038/nature12647
110. Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun (2015) 6:6532. doi:10.1038/ncomms7532
111. National Institute of Neurological Disorders and Stroke (NINDS); National Institutes of Health Clinical Center (CC). Oral guanabenz for multiple sclerosis. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT02423083
112. Brück W, Pförtner R, Pham T, Zhang J, Hayardeny L, Piryatinsky V, et al. Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol (2012) 124:411–24. doi:10.1007/s00401-012-1009-1
113. Vollmer TL, Sørensen PS, Selmaj K, Zipp F, Havrdova E, Cohen JA, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol (2014) 261:773–83. doi:10.1007/s00415-014-7264-4
114. Filippi M, Rocca MA, Pagani E, De Stefano N, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J Neurol Neurosurg Psychiatry (2014) 85(8):851–8. doi:10.1136/jnnp-2013-306132
115. Teva Pharmaceutical Industries. A phase 2 clinical study in subjects with primary progressive multiple sclerosis to assess the efficacy, safety and tolerability of two oral doses of laquinimod either of 0.6 mg/day or 1.5mg/day (experimental drug) as compared to placebo. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT02284568
116. Comi G, Pulizzi A, Rovaris M, Abramsky O, Arbizu T, Boiko A, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet (2008) 371:2085–92. doi:10.1016/S0140-6736(08)60918-6
117. Mishra MK, Wang J, Keough MB, Fan Y, Silva C, Sloka S, et al. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann Clin Transl Neurol (2014) 1:409–22. doi:10.1002/acn3.67
118. Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature (2015) 522:216–20. doi:10.1038/nature14335
119. Magalon K, Zimmer C, Cayre M, Khaldi J, Bourbon C, Robles I, et al. Olesoxime accelerates myelination and promotes repair in models of demyelination. Ann Neurol (2012) 71:213–26. doi:10.1002/ana.22593
120. Trophos; Hôpital de la Timone; SGS; STRAGEN Services. Safety study of olesoxime in patients with stable relapsing remitting multiple sclerosis treated with interferon beta (MSREPAIR). In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT01808885
121. Zhornitsky SWYV. Quetiapine fumarate for the treatment of multiple sclerosis: focus on myelin repair. CNS Neurosci Ther (2013) 19:737–44. doi:10.1111/cns.12154
122. University of Calgary; Multiple Sclerosis Society of Canada. Safety and tolerability of quetiapine in multiple sclerosis. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT02087631
123. Warrington AE, Bieber AJ, Ciric B, Pease LR, Van Keulen V, Rodriguez M. A recombinant human IgM promotes myelin repair after a single, very low dose. J Neurosci Res (2007) 85:967–76. doi:10.1002/jnr.21217
124. Pirko I, Ciric B, Gamez J, Bieber AJ, Warrington AE, Johnson AJ, et al. A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. FASEB J (2004) 18:1577–9. doi:10.1096/fj.04-2026fje
125. Acorda Therapeutics; PRA Health Sciences. An intravenous infusion study of rHIgM22 in patients with multiple sclerosis immediately following a relapse. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT02398461
126. Mei F, Fancy SP, Shen YA, Niu J, Zhao C, Presley B, et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med (2014) 20:954–60. doi:10.1038/nm.3618
127. De Angelis F, Bernardo A, Magnaghi V, Minghetti L, Tata AM. Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation, and differentiation. Dev Neurobiol (2012) 72:713–28. doi:10.1002/dneu.20976
128. University of California, San Francisco. Assessment of clemastine fumarate as a remyelinating agent in multiple sclerosis (ReBUILD). In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT02040298
129. Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci (2005) 8:745–51. doi:10.1038/nn1460
130. Mi S, Miller RH, Tang W, Lee X, Hu B, Wu W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol (2009) 65:304–15. doi:10.1002/ana.21581
131. Stoffels JMJ, de Jonge JC, Stancic M, Nomden A, van Strien ME, Ma D, et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain (2013) 136:116–31. doi:10.1093/brain/aws313
132. Watzlawik JO, Wootla B, Painter MM, Warrington AE, Rodriguez M. Cellular targets and mechanistic strategies of remyelination-promoting IgMs as part of the naturally occurring autoantibody repertoire. Expert Rev Neurother (2013) 13:1017–29. doi:10.1586/14737175.2013.835601
133. Acorda Therapeutics; PRA Health Sciences. An intravenous infusion study of rHIgM22 in patients with multiple sclerosis. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US) (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT01803867
134. Baum T, Shropshire AT. Studies on the centrally mediated hypotensive activity of guanabenz. Eur J Pharmacol (1976) 37:31–44. doi:10.1016/0014-2999(76)90005-4
135. Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science (2011) 332:91–4. doi:10.1126/science.1201396
136. Bordet T, Buisson B, Michaud M, Drouot C, Galea P, Delaage P, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther (2007) 322:709–20. doi:10.1124/jpet.107.123000
137. Ben-Hur T, Fainstein N, Nishri Y. Cell-based reparative therapies for multiple sclerosis. Curr Neurol Neurosci Rep (2013) 13:397. doi:10.1007/s11910-013-0397-5
138. University College, L Medical Research Council; National Institute for Health Research; United Kingdom MS Society; University of Edinburgh; Queen Mary University of London; Keele University; University of Sheffield; University of Leeds; University of Warwick London; In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US). (2000). Available from: https://clinicaltrials.gov/ct2/show/NCT01910259
139. Toubi E, Nussbaum S, Staun-Ram E, Snir A, Melamed D, Hayardeny L, et al. Laquinimod modulates B cells and their regulatory effects on T cells in multiple sclerosis. J Neuroimmunol (2012) 251:45–54. doi:10.1016/j.jneuroim.2012.07.003
140. Wegner C, Stadelmann C, Pfortner R, Raymond E, Feigelson S, Alon R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol (2010) 227:133–43. doi:10.1016/j.jneuroim.2010.07.009
141. Ruffini F, Rossi S, Bergamaschi A, Brambilla E, Finardi A, Motta C, et al. Laquinimod prevents inflammation-induced synaptic alterations occurring in experimental autoimmune encephalomyelitis. Mult Scler J (2013) 19:1084–94. doi:10.1177/1352458512469698
142. Moore CS, Cui Q-L, Warsi NM, Durafourt BA, Zorko N, Owen DR, et al. Direct and indirect effects of immune and central nervous system-resident cells on human oligodendrocyte progenitor cell differentiation. J Immunol (2015) 194:761–72. doi:10.4049/jimmunol.1401156
143. Comi G, Jeffery D, Kappos L, Montalban X, Boyko AN, Rocca MA, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med (2012) 366:1000–9. doi:10.1056/NEJMoa1104318
144. Houdayer E, Comi G, Leocani L. The neurophysiologist perspective into MS plasticity. Front Neurol (2015) 6:193. doi:10.3389/fneur.2015.00193
145. Niklas A, Sebraoui H, Hess E, Wagner A, Then Bergh F. Outcome measures for trials of remyelinating agents in multiple sclerosis: retrospective longitudinal analysis of visual evoked potential latency. Mult Scler (2009) 15:68–74. doi:10.1177/1352458508095731
146. Grecescu M. Optical Coherence Tomography versus Visual Evoked Potentials in detecting subclinical visual impairment in multiple sclerosis. J Med Life (2009) 7:538–52. doi:10.1212/WNL.0b013e3181aaea32
147. Gabelić T, Krbot Skorić M, Adamec I, Barun B, Zadro I, Habek M. The vestibular evoked myogenic potentials (VEMP) score: a promising tool for evaluation of brainstem involvement in multiple sclerosis. Eur J Neurol (2015) 22:261–9. doi:10.1111/ene.12557
148. Simpson M, Macdonell R. The use of transcranial magnetic stimulation in diagnosis, prognostication and treatment evaluation in multiple sclerosis. Mult Scler Relat Disord (2015) 4:430–6. doi:10.1016/j.msard.2015.06.014
149. Coman I, Aigrot M-SS, Seilhean D, Reynolds R, Girault JA, Zalc B, et al. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain (2006) 129:3186–95. doi:10.1093/brain/awl144
150. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A (2004) 101:8168–73. doi:10.1073/pnas.0402765101
151. Giovannoni G. Cerebrospinal fluid analysis. Handbook of Clinical Neurology (2014) 122:681–702. doi:10.1016/B978-0-444-52001-2.00029-7
152. Kroksveen AC, Opsahl JA, Guldbrandsen A. Cerebrospinal fluid proteomics in multiple sclerosis. Biochim Biophys Acta (2015) 1854:746–56. doi:10.1016/j.bbapap.2014.12.013
153. Brown RA, Narayanan S, Arnold DL. Imaging of repeated episodes of demyelination and remyelination in multiple sclerosis. Neuroimage Clin (2014) 6:20–5. doi:10.1016/j.nicl.2014.06.009
154. Simon JH, Bermel RA, Rudick RA. Simple MRI metrics contribute to optimal care of the patient with multiple sclerosis. AJNR Am J Neuroradiol (2014) 35:831–2. doi:10.3174/ajnr.A3937
155. Polak P, Magnano C, Zivadinov R, Poloni G. 3D FLAIRED: 3D fluid attenuated inversion recovery for enhanced detection of lesions in multiple sclerosis. Magn Reson Med (2012) 68:874–81. doi:10.1002/mrm.23289
156. Brex PA, Ciccarelli O, O’Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med (2002) 346:158–64. doi:10.1056/NEJMoa011341
157. Kolind S, Matthews L, Johansen-Berg H, Leite MI, Williams SC, Deoni S, et al. Myelin water imaging reflects clinical variability in multiple sclerosis. Neuroimage (2012) 60:263–70. doi:10.1016/j.neuroimage.2011.11.070
158. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain (2005) 128:2705–12. doi:10.1093/brain/awh641
159. Kipp M, Victor M, Martino G, Franklin RJM. Endogeneous remyelination: findings in human studies. CNS Neurol Disord Drug Targets (2012) 11:598–609. doi:10.2174/187152712801661257
160. Barkhof F. The clinico-radiological paradox in multiple sclerosis revisited. Curr Opin Neurol (2002) 15:239–45. doi:10.1097/00019052-200206000-00003
161. Mallik S, Samson RS, Wheeler-Kingshott CAM, Miller DH. Imaging outcomes for trials of remyelination in multiple sclerosis. J Neurol Neurosurg Psychiatr (2014) 85:1396–404. doi:10.1136/jnnp-2014-307650
162. Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, et al. Diffusion tensor imaging: concepts and applications. J Magn Reson Imaging (2001) 13:534–46. doi:10.1002/jmri.1076
163. Fox RJ, Cronin T, Lin J, Wang X, Sakaie K, Ontaneda D, et al. Measuring myelin repair and axonal loss with diffusion tensor imaging. AJNR Am J Neuroradiol (2011) 32:85–91. doi:10.3174/ajnr.A2238
164. Schmierer K, Wheeler-Kingshott CA, Boulby PA, Scaravilli F, Altmann DR, Barker GJ, et al. Diffusion tensor imaging of post mortem multiple sclerosis brain. Neuroimage (2007) 35:467–77. doi:10.1016/j.neuroimage.2006.12.010
165. Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage (2002) 17:1429–36. doi:10.1006/nimg.2002.1267
166. Song SK, Yoshino J, Le TQ, Lin SJ, Sun SW, Cross AH, et al. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage (2005) 26:132–40. doi:10.1016/j.neuroimage.2005.01.028
167. DeBoy CA, Zhang J, Dike S, Shats I, Jones M, Reich DS, et al. High resolution diffusion tensor imaging of axonal damage in focal inflammatory and demyelinating lesions in rat spinal cord. Brain (2007) 130:2199–210. doi:10.1093/brain/awm122
168. Wang Y, Wang Q, Haldar JP, Yeh FC, Xie M, Sun P, et al. Quantification of increased cellularity during inflammatory demyelination. Brain (2011) 134:3590–601. doi:10.1093/brain/awr307
169. Wheeler-Kingshott CA, Cercignani M. About “axial” and “radial” diffusivities. Magn Reson Med (2009) 61:1255–60. doi:10.1002/mrm.21965
170. Laule CEA. Magnetic resonance imaging of myelin. Neurotherapeutics (2007) 4:460–84. doi:10.1016/j.nurt.2007.05.004
171. Wheeler-Kingshott CA, Ciccarelli O, Schneider T, Alexander DC, Cercignani M. A new approach to structural integrity assessment based on axial and radial diffusivities. Funct Neurol (2012) 27:85–90.
172. Pagani E, Bammer R, Horsfield MA, Rovaris M, Gass A, Ciccarelli O, et al. Diffusion MR imaging in multiple sclerosis: technical aspects and challenges. AJNR Am J Neuroradiol (2007) 28:411–20.
173. Jespersen SN, Bjarkam CR, Nyengaard JR, Chakravarty MM, Hansen B, Vosegaard T, et al. Neurite density from magnetic resonance diffusion measurements at ultrahigh field: comparison with light microscopy and electron microscopy. Neuroimage (2010) 49:205–16. doi:10.1016/j.neuroimage.2009.08.053
174. Wolff SD, Balaban RS. Magnetization transfer imaging: practical aspects and clinical appications. Radiology (1994) 192:593–9. doi:10.1148/radiology.192.3.8058919
175. Wolff SD, Balaban RS. Magnetization transfer contrast (MTC) and tissue water proton relaxation in vivo. Magn Reson Med Sci (1989) 10:135–44. doi:10.1002/mrm.1910100113
176. van Waesberghe JH, Kamphorst W, De Groot CJ, van Walderveen MA, Castelijns JA, Ravid R, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol (1999) 46:747–54. doi:10.1002/1531-8249(199911)46:5<747::AID-ANA10>3.0.CO;2-4
177. Mottershead JP, Schmierer K, Clemence M, Thornton JS, Scaravilli F, Barker GJ, et al. High field MRI correlates of myelin content and axonal density in multiple sclerosis – a post-mortem study of the spinal cord. J Neurol (2003) 250:1293–301. doi:10.1007/s00415-003-0192-3
178. Vavasour IM, Laule C, Li DK, Traboulsee AL, MacKay AL. Is the magnetization transfer ratio a marker for myelin in multiple sclerosis? J Magn Reson Imaging (2011) 33:713–8. doi:10.1002/jmri.22441
179. Cercignani M, Bozzali M, Iannucci G, Comi G, Filippi M. Magnetisation transfer ration and mean diffusivity of normal appearing white and grey meeter from patients with multiple sclerosis. J Neurol Neurosurg Psychiatr (2001) 70:311–7. doi:10.1136/jnnp.70.3.311
180. Filippi M, Campi A, Dousset V, Baratti C, Martinelli V, Canal N, et al. A magnetization transfer imaging study of normal-appearing white matter in multiple sclerosis. Neurology (1995) 45:478–82. doi:10.1212/WNL.45.3.478
181. Filippi M, Iannucci G, Tortorella C, Minicucci L, Horsfield MA, Colombo B, et al. Comparison of MS clinical phenotypes using conventional and magnetization transfer MRI. Neurology (1999) 52:588–94. doi:10.1212/WNL.52.3.588
182. Schmierer K, Scaravilli F, Altmann DR, Barker GJ, Miller DH. Magnetization transfer ratio and myelin in postmortem multiple sclerosis brain. Ann Neurol (2004) 56:407–15. doi:10.1002/ana.20202
183. Barkhof F, Brück W, De Groot CJA, Bergers E, Hulshof S, Geurts J, et al. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol (2003) 60:1073–81. doi:10.1001/archneur.60.8.1073
184. Chen JT, Collins DL, Atkins HL, Freedman MS, Arnold DL; Canadian MS/BMT Study Group. Magnetization transfer ratio evolution with demyelination and remyelination in multiple sclerosis lesions. Ann Neurol (2008) 63:254–62. doi:10.1002/ana.21302
185. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain (2006) 129:3165–72. doi:10.1093/brain/awl217
186. Laule C, Kozlowski P, Leung E, Li DK, MacKay AL, Moore GR. Myelin water imaging of multiple sclerosis at 7 T: correlations with histopathology. Neuroimage (2008) 40:1575–80. doi:10.1016/j.neuroimage.2007.12.008
187. Levesque IR, Sled JG, Narayanan S, Giacomini PS, Ribeiro LT, Arnold DL, et al. Reproducibility of quantitative magnetization-transfer imaging parameters from repeated measurements. Magn Reson Med (2010) 64:391–400. doi:10.1002/mrm.22350
188. Schmierer K, Wheeler-Kingshott CA, Tozer DJ, Boulby PA, Parkes HG, Yousry TA, et al. Quantitative magnetic resonance of postmortem multiple sclerosis brain before and after fixation. Magn Reson Med (2008) 59:268–77. doi:10.1002/mrm.21487
189. Ou X, Sun SW, Liang HF, Song SK, Gochberg DF. Quantitative magnetization transfer measured pool-size ratio reflects optic nerve myelin content in ex vivo mice. Magn Reson Med (2009) 61:364–71. doi:10.1002/mrm.21850
190. Giacomini PS, Levesque IR, Ribeiro L, Narayanan S, Francis SJ, Pike GB, et al. Measuring demyelination and remyelination in acute multiple sclerosis lesion voxels. Arch Neurol (2009) 66:375–81. doi:10.1001/archneurol.2008.578
191. Mackay A, Whittall K, Adler J, Li D, Paty D, Graeb D. In vivo visualization of myelin water in brain by magnetic resonance. Magn Reson Med (1994) 31:673–7. doi:10.1002/mrm.1910310614
192. Gareau PJ, Rutt BK, Karlik SJ, Mitchell JR. Magnetization transfer and multicomponent T2 relaxation measurements with histopathologic correlation in an experimental model of MS. J Magn Reson Imaging (2000) 11:586–95. doi:10.1002/1522-2586(200006)11:6<586::AID-JMRI3>3.0.CO;2-V
193. Nguyen TD, Wisnieff C, Cooper MA, Kumar D, Raj A, Spincemaille P, et al. T2 prep three-dimensional spiral imaging with efficient whole brain coverage for myelin water quantification at 1.5 tesla. Magn Reson Med (2012) 67:614–21. doi:10.1002/mrm.24128
194. Prasloski T, Rauscher A, MacKay AL, Hodgson M, Vavasour IM, Laule C, et al. Rapid whole cerebrum myelin water imaging using a 3D GRASE sequence. Neuroimage (2012) 63:533–9. doi:10.1016/j.neuroimage.2012.06.064
195. Lankford CL, Does MD. On the inherent precision of mcDESPOT. Magn Reson Med (2013) 69:127–36. doi:10.1002/mrm.24241
196. Laule C, Vavasour IM, Moore GR, Oger J, Li DK, Paty DW, et al. Water content and myelin water fraction in multiple sclerosis. A T2 relaxation study. J Neurol (2004) 251:284–93. doi:10.1007/s00415-004-0306-6
197. Laule C, Vavasour IM, Zhao Y, Traboulsee AL, Oger J, Vavasour JD, et al. Two-year study of cervical cord volume and myelin water in primary progressive multiple sclerosis. Mult Scler J (2010) 16:670–7. doi:10.1177/1352458510365586
198. Vargas WS, Monohan E, Pandya S, Raj A, Vartanian T, Nguyen TD, et al. Measuring longitudinal myelin water fraction in new multiple sclerosis lesions. Neuroimage Clin (2015) 12:369–75. doi:10.1016/j.nicl.2015.09.003
199. Vavasour I, MacKay A, Li D, Laule C, Traboulsee A. Advanced imaging in lesion and normal-appearing white matter over 2 years in MS patients treated with alemtuzumab (S29.009). Neurology (2015) 69:S29.009.
200. Rabinovici GD, Furst AJ, O’Neill JP, Racine CA, Mormino EC, Baker SL, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology (2007) 68:1205–12. doi:10.1212/01.wnl.0000259035.98480.ed
201. Zhang S, editor. 11C-PIB-PET for the Early Diagnosis of Alzheimer’s Disease Dementia and Other Dementias in People with Mild Cognitive Impairment (MCI). Chichester: John Wiley & Sons, Ltd (1996).
202. Stankoff B, Wang Y, Bottlaender M, Aigrot M-SS, Dolle F, Wu C, et al. Imaging of CNS myelin by positron-emission tomography. Proc Natl Acad Sci U S A (2006) 103:9304–9. doi:10.1073/pnas.0600769103
Keywords: multiple sclerosis, remyelination, myelin repair, neuroprotection, MRI, MTI, DTI
Citation: Harlow DE, Honce JM and Miravalle AA (2015) Remyelination Therapy in Multiple Sclerosis. Front. Neurol. 6:257. doi: 10.3389/fneur.2015.00257
Received: 14 August 2015; Accepted: 23 November 2015;
Published: 10 December 2015
Edited by:
Mike P. Wattjes, VU University Medical Center, NetherlandsReviewed by:
Anders Svenningsson, Umeå University, SwedenShahrukh Mallik, University College London, UK
Copyright: © 2015 Harlow, Honce and Miravalle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danielle E. Harlow, ZGFuaWVsbGUuaGFybG93QHVjZGVudmVyLmVkdQ==