 Andrzej Przekwas
Andrzej Przekwas Mahadevabharath R. Somayaji
Mahadevabharath R. Somayaji Raj K. Gupta
Raj K. Gupta- 1Computational Medicine and Biology Division, CFD Research Corporation, Huntsville, AL, USA
- 2Department of Defense Blast Injury Research Program Coordinating Office, U.S. Army Medical Research and Materiel Command, Fort Detrick, MD, USA
Blast wave-induced traumatic brain injury (TBI) is one of the most common injuries to military personnel. Brain tissue compression/tension due to blast-induced cranial deformations and shear waves due to head rotation may generate diffuse micro-damage to neuro-axonal structures and trigger a cascade of neurobiological events culminating in cognitive and neurodegenerative disorders. Although diffuse axonal injury is regarded as a signature wound of mild TBI (mTBI), blast loads may also cause synaptic injury wherein neuronal synapses are stretched and sheared. This synaptic injury may result in temporary disconnect of the neural circuitry and transient loss in neuronal communication. We hypothesize that mTBI symptoms such as loss of consciousness or dizziness, which start immediately after the insult, could be attributed to synaptic injury. Although empirical evidence is beginning to emerge; the detailed mechanisms underlying synaptic injury are still elusive. Coordinated in vitro–in vivo experiments and mathematical modeling studies can shed light into the synaptic injury mechanisms and their role in the potentiation of mTBI symptoms.
Introduction
Traumatic brain injury (TBI) has become the signature wound of military operations since service members are frequently exposed to roadside bombs and explosions. The majority of combat-related TBI cases are categorized as mild, primarily based on event history and post-injury assessment of behavioral and cognitive symptoms. Mild TBI (mTBI) typically induces a variety of heterogeneous symptoms, including concentration problems, blurred vision, irritability, headaches, sleep disorders, and depression. The symptoms may also be associated with cognitive and neurodegenerative disorders, such as post-traumatic stress disorder (PTSD) and chronic traumatic encephalopathy (CTE) (1–3). In spite of its importance and many years of research, current understanding of the primary (biomechanics) and secondary (neurobiology) brain injury mechanisms is limited. Moreover, the link between primary injury biomechanics, the neurobiology of secondary injury and repair, and the resultant neuropsychological and neurodegenerative outcomes remain elusive.
In the last few years, several hypotheses have been proposed to explain the mechanism of blast-induced primary brain injury. These include elastic and shear waves in brain, brain rotation relative to cranium, brain–skull contact, cavitation, electromagnetic pulse, axonal damage, micro hemorrhage, vascular elastic waves propagating from the blast loaded thorax to the brain, damage to the bridging veins, and others (4–9). In contrast to focal injury, diffuse axonal injury (DAI) occurs in a widespread area and is a common pathology observed in blunt and blast-induced TBI (10–13). The primary micro-damage associated with it is manifested by impairment to neurofilament units of the axonal cytoskeleton, loss of membrane integrity, and Wallerian-type axonal degeneration (14–17). Although the mechanism of DAI is thought to originate from acceleration, deceleration, and rotational forces, its pathogenesis is largely attributed to the onset of secondary injury cascade. Non-invasive brain imaging studies of humans exposed to blasts with mTBI symptoms using advanced diffusion tensor MRI (DT-MRI) have confirmed DAI signatures (12).
In the acute injury phase, diffuse structural changes to the synaptic clefts and postsynaptic densities (PSDs) cause temporary loss of neural circuit connectivity. This primary micro-damage initiates a cascade of biophysical and neurochemical events, lasting from minutes to hours, resulting in either axonal and synaptic repair or permanent damage (18, 19). Moreover, some mTBI-relevant cognitive deficits, such as loss of consciousness (LOC) or dizziness, start immediately after the insult while others, including headaches, fatigue, depression, learning/memory deficits, and neurodegeneration, take a longer time to evolve. However, it is difficult to attribute the temporal diversity of injury responses only to DAI. We posit that biomechanical micro-damage to axons as well as neuronal synapses and dendritic spines play a major role in the etiology of mTBI. Better understanding of the dynamics of diffuse synaptic injury may offer a window of opportunity in which an appropriate treatment may modify an imbalance between post-injury excitatory and inhibitory processes.
Neurotransmission and Neuroplasticity
The human brain is organized as a highly interconnected structural network of neurons, glia, and supporting cells responsible for cognitive and physiological information processing. The structural integrity of brain neurons and glia is maintained by a complex network of extracellular matrix. It is estimated that the adult human brain contains ~1011 neurons, each of them equipped with ~104 synapses (20, 21). This huge connectome of ~1015 interconnects undergoes continuous remodeling in response to a variety of stimuli, a process termed as neuroplasticity (22). These changes include structural remodeling of presynaptic terminals, PSDs, dendritic spines, adhesion molecules, and the surrounding glial cells. The magnitude and direction of these changes depend on the duration and frequency of presynaptic stimulation. Synaptic plasticity, a type of neuroplasticity, is the activity-dependent change in the synaptic strength and efficacy and forms the neurochemical basis of learning and memory. Below the level of the synapse, the physiological neuroplasticity involves complex mechanisms of gene expression, protein synthesis, receptor trafficking to and on the dendritic membrane, recruitment of new receptors, remodeling of synaptic adhesion proteins or even removal of some, and formation of new, synaptic clefts. It has been observed in magnetic resonance elastography brain imaging that brain neuronal structures are continuously modulated by physiological micromechanical loadings originating from intracranial pressure pulsations due to cardiac and respiratory rhythms as well as head movement (23). It is believed that these micro mechano-biological effects and their interaction with neurochemical pathways are essential for proper brain function and neuroprotection (24–28). On the other hand, non-physiological, high speed loadings, such as in accidental head impacts or blast waves, may cause acute injuries to neuronal microstructures, such as axons, dendritic spines, and synapses, with neuropathological implications.
Figure 1 illustrates the distribution of neuronal synapses, neurotransmission, and our perspectives on synaptic injury mechanisms. Each neuron is connected to other neurons via large number of excitatory (E) and inhibitory (I) chemical synapses located on the dendritic arbor and cell body. The chemical synapse is a highly organized structure consisting of a presynaptic terminal juxtaposed across a postsynaptic button on top of a dendritic shaft and is often surrounded by astrocytic processes (astrocytes not shown in Figure 1). The action potential arriving at the presynaptic terminal causes the release of a neurotransmitter (NT) into the synaptic cleft: glutamate (Glu), norepinephrine, etc., at E-synapses and γ-aminobutyric acid (GABA) and serotonin at I-synapses. The NT diffuses through this narrow cleft and binds to ion channels and receptors on the postsynaptic neuronal membrane. The influx of ions alters the postsynaptic voltage causing either a depolarization (E signal) or hyperpolarization (I signal) in the dendritic branch of the postsynaptic neuron. The number of E and I signals that a single neuron receives dictates its excitability and function. In other words, the likelihood of firing an action potential by the receiving neuron depends on the number of E and I synaptic potentials and the somatic summation. In the case of glutamatergic synapses, the small size of the synaptic cleft (~2 attoliters) facilitates rapid rise of Glu concentration in the cleft, rapid binding to PSD receptors, and fast clearance. The small size and precise organization of the synapse facilitate high frequency neurotransmission with rapid buildup of Glu concentrations of 1–5 mM in the cleft post-stimulation, followed by a fast clearance by Glu transporters, in <1 ms (29). Mechanical extension or shear deformation of synapses post-injury may have detrimental effects on neurotransmission.
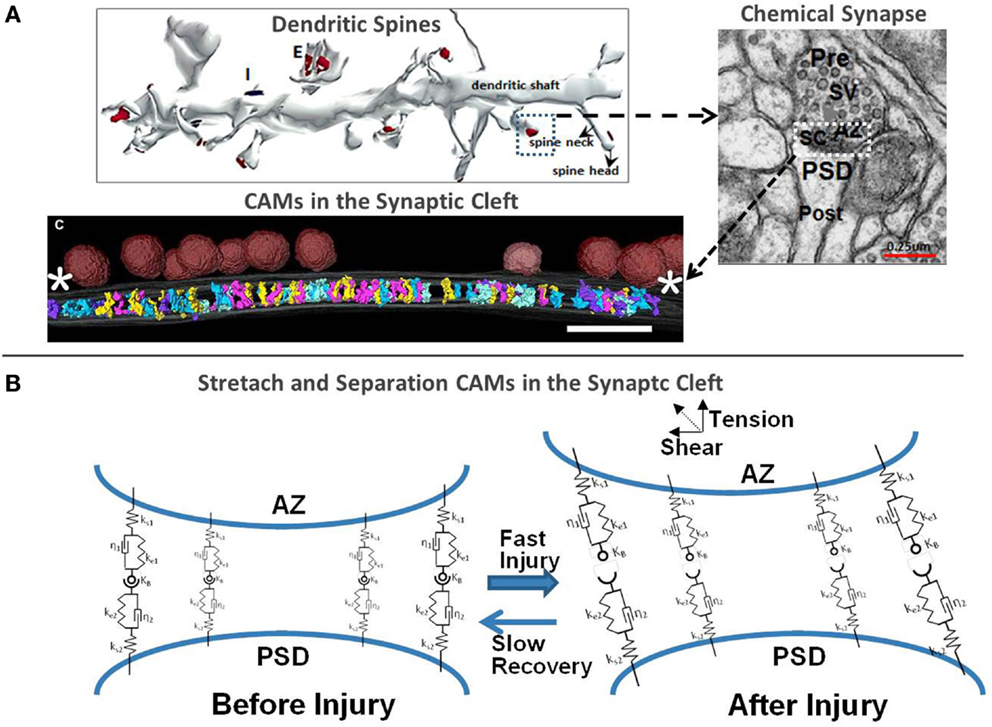
Figure 1. (A) 3D rendering of a segment of the dendritic shaft with excitatory and inhibitory chemical synapses, morphology of the synaptic cleft (30, 31) and 3D rendering of CAMs in the synaptic cleft [adapted from High et al. (32)] and (B) biomechanical representation of the cleft scaffolding proteins using spring-damper network and injury-induced perturbations.
Morphology of Dendritic Spines, E- and I-Synapses
In the central nervous system, the majority of E- and I-synapses differ in their location, composition, structure, and function (33, 34). The E-synapses are localized on dendritic spines, which are terminated with dense organelles (PSDs), hosting hundreds of scaffolding and signaling molecules involved in neurotransmission and synaptic plasticity. Most GABAergic I-synapses, on the other hand, are formed directly on dendritic shafts, as well as on the soma and proximal axonal regions. Endogenous regulatory mechanisms precisely maintain the E/I balance and any acute or chronic E/I perturbations may be responsible for various pathologies, including epilepsy, depression, and other disorders (34–37). From a mechanical perspective, the postsynaptic dendritic spine of an E synapse looks like a cantilever beam having a big heavy head with weak thin neck. The dendritic head is typically ~0.5–2 μm in diameter and is connected to the parent dendrite by a thin spine of ~0.04–1 μm in length (34). We conjecture that E-synapses located on tall dendritic spines could be more vulnerable to mechanical damage during tension and shear loadings than I-synapses positioned at dendritic roots and soma.
Another mechanically sensitive synaptic structure is the synaptic cleft, with a typical dimension of 15–25 nm height and ~300 nm diameter. It is filled with structural proteins as well as cell adhesion molecules (CAMs) that hold pre- and postsynaptic membranes together at the appropriate juxtaposition. Some of them, e.g., neurexins (NXs), neuroligins (NLs), SynCAMs, and integrins, localize at the center of the synapse, whereas others, such as Cadherins, reside at the synaptic periphery. However, transcleft elements in both E- and I-synapses typically avoid places where synaptic vesicles attach to the presynaptic membrane (32). The extracellular domains of these CAMs, protruding from the opposite sides of the synaptic terminals, are “sticky,” and are bound by hydrophobic forces to each other in either homophilic (e.g., Cadherins) or heterophilic (e.g., presynaptic NXs and postsynaptic NLs) combination and mechanically maintain the synaptic structure. Their cytoplasmic domains bind to and mechanically modulate adaptor proteins that organize the synaptic structure and function as well as the morphologies of the presynaptic active zone (AZ) and PSD. Some CAMs, such as NXs and NLs, and cadherins require extracellular calcium to maintain their elasticity and binding affinity. At physiological conditions, CAMs behave as elastic springs and are under tension force (~10–20 pN) that makes them “longer” than in crystallographic dimensions. Calcium shifts from interstitial to intracellular spaces following mechanical injury are often considered a major contributing factor to metabolic, excitatory, and apoptotic pathways in TBI (38). It is also possible that reduced synaptic calcium concentration can affect binding affinity and mechanical stiffness of Ca-dependent binding of CAMs. In the absence of depleted intra-synaptic calcium, cadherins lose their elastic strength and behave as “loose and weak rope” (39). For example, NX-1β and NL1 form a strong transsynaptic heterotetramer only in the presence of 1–3 μM free Ca2+ (40). Atomic force microscopy measurements at slow loading rates have shown that rupture forces for CAMs range between 50 and 70 pN (41–43). Although astrocytes are not directly structurally linked to the synaptic cleft, astrocytic processes are in close apposition to the synaptic structures, form tripartite synapses and mechanically interact with them through the ECM. They also act as diffusion barriers to NT spillover, facilitate NT uptake, and provide local metabolic support. To better articulate the proposed hypothesis, in the following, we will focus only on bipartite synapses.
Do Blast-Induced Forces Affect Macroscopic Brain Biomechanics and Neuronal Structures?
At the macroscopic scale, there are multiple pathways for blast-induced forces and energy content to impact the brain. Previous computational and experimental works have shown that the cranial bone is a good transmitter of elastic waves to the CSF and brain with little attenuation below 104–105 Hz (44, 45). During blast wave loading, the brain experiences two types of biomechanical events: (1) rapid elastic skull deformation causing compression/tension stress waves, which later dissipate as shear waves and (2) delayed head movement causing brain rotation relative to the skull, which then generates shear waves within the brain. Specifically, compression/tension waves propagate in the brain with the speed of sound (~1500 m/s), transform into shear waves and last only few milliseconds. Shear waves, on the other hand, are orders of magnitude slower (~10 m/s), dissipate due to viscous action and persist longer, up to hundreds of milliseconds (45). It has been documented that angular accelerations of the brain often lead to DAI, contusion, and acute subdural hematomas (14, 17, 46–53). Experimental tests on human volunteers (54) and cadavers (55, 56) have shown brain translation magnitudes of 4–5 mm and rotation of ±5° for ~300 ms at low-severity impacts in the sagittal plane. The macroscopic brain biomechanics is expected to be absorbed/dissipated at the cellular scales for potentiation of cellular injury.
At the microscopic scale, the brain tissue behaves as a heterogeneous non-linear viscoelastic material with multiple strain rates (57). All cells in the brain experience continuous mechanical forces from normal head movement and from intracranial and intracellular hydrodynamic and osmotic pressures, yet maintain their function. However, higher mechanical loads may cause inelastic structural damage to load bearing microstructures. For instance, the cantilever dendritic spines may undergo structural alterations when exposed to blast loads. Because the brain tissue is inhomogeneous, these strains concentrate at the micro-interfaces with impedance mismatch (e.g., disparate densities, morphologies) in the brain (7). In vitro and in vivo experiments show that tension and shear strains are much more damaging to the tissue than compressive strains (58). Intuitively, it can be explained that the surrounding water resists the compression and supports tissue structure while the tensile or shear force directly disrupts weaker (hydrogen, van der Waals) and stronger (covalent, ionic) bonds at the molecular level.
The rate of strain applied to viscoelastic brain tissue microstructures is also very important. At low strain rates, the tissue/cells are very ductile and can recover without damage from relatively large deformations. For example, experiments on human volunteers experiencing mild linear accelerations of ~1.5 G and angular accelerations of 120–140 rad s−2 show that significant regions of the brain exhibit 5–7% elongation strain (54, 59) and do not cause injury. However, at faster loads, the brain tissue becomes brittle and susceptible to micro-damage to brain cells/organelles, such as axons, synapses, vascular endothelium, membranes, cytoskeleton, ion channels, and other microstructures (14). This biomechanical “primary injury” lasting for a few milliseconds initiates a cascade of secondary injury (neurobiology) and recovery pathways lasting hours, days, and in some cases life times. Depending on the severity of the insult, some of the brain cells will rapidly transition to apoptosis and necrosis, while other injured cells and organelles may undergo a long lasting recovery process.
Perspectives on Synaptic Injury in Blast-Induced mTBI
Synaptic injury mechanisms are largely unknown and have only recently begun to attract interest of neuroscientists partially because of experimental challenges at such small length and timescales (9, 25, 60–62). Recent experimental analyses have shown that the synaptic loss may be the secondary effects of DAI as a result of axonal fiber loss and synaptic terminal degeneration (63). In vitro experiments applying magnetic tweezer forces on neuronal structures have shown that mechanical damage to integrins, and potentially other CAMs, may be an important mechanism underlying the initiation of cell and sub-cellular injuries ultimately responsible for the diffuse axonal and synaptic pathology (64). Mechanical damage to neuronal micro/nano-structures, such as CAMs, cytoskeleton, membranes, and ion channels, is strain rate dependent. In sports and automobile related head injuries, the damage is caused by rotation-induced shear waves with large strains and typical strain rates of 100 s−1. Blast TBI involves very fast compression–tension wave followed by fast but slower shear waves with potentially smaller strain but much higher strain rates of the order of 1000 s−1 (65). The high strain rate of viscoelastic damage to neuronal micro/nano-structures may be more important in blast wave TBI, while slower but larger strains may be responsible for blunt and inertial TBI. It is also possible that the synaptic injury may be present in all types on mTBI. The detailed role of synaptic injury in blast and blunt loading patterns remains to be elucidated.
Experimental study of synaptic injury mechanisms is challenging. To date, it has been observed only in in vitro neuronal cultures subjected to a mechanical stretch (66, 67) and in brain slices of rodents exposed to shock waves (68, 69). Further studies of blast-induced synaptic injury mechanisms would be required using well-characterized blast waves or shock tubes for both in vitro cell/slice cultures (16, 65, 70) and in vivo animals (71, 72). Mechanical tension and shear waves may cause temporary disconnects and micro-damage of CAMs, synapses, and dendritic spines, which in turn may be manifested as temporary cognitive impairment (62, 67, 73). Moreover, reduced concentration of NTs and calcium in the deformed synapse may alter the connectivity of CAMs. As mentioned above, lower synaptic Ca2+ concentration reduces the elasticity of cadherins and diminishes the de novo hydrogen bond formation and would not allow cadherins to reassociate after injury. At the same time, it is also likely that a large number of mechanically deformed synapses in mTBI may self-restore by hydrophobic, electrokinetic, and other biophysical mechanisms due to other CAMs (39, 74).
Figure 1B schematically shows the structural response of CAMs to mechanical forces, which may cause their separation. Rapid tension or shear loads may cause separation of pre- and postsynaptic membranes, loss of contact between CAMs, plasma membrane mechanoporation and rearrangement of the cytoskeleton and scaffolding in the PSD and spinal neck. If the initial deformation is subcritical in the context of sufficient “healing” time, the CAMs may be able to reconnect and reestablish synaptic connection. This phenomenon may be one aspect of the neuro-recovery mechanisms. Transmembrane proteins, such as integrins and connexins, are responsible for structural reinforcement and alignment of both neuronal chemical synapses and electrical gap junctions. They also modulate a variety of intracellular pathways that are activated with the exertion of mechanical force on the integrin. It is likely that an inelastic damage to CAMs, integrins, and other focal adhesion molecules in brain injury affects not only the synaptic morphology but also the conductance of the ion channels and cytoskeleton remodeling. Recent experimental observations show that integrin-mediated activation of Rho may be a contributor to the DAI in mTBI (75) and suggest that a similar mechanism may be involved in synaptic injury as well (64). It is also possible that mechanical damage to N-cadherin–catenin complex, which stabilizes the cytoskeleton through Rho-family GTPases, may cause postsynaptic actomyosin contraction in dendritic spines and probable loss of excitatory synapses (76), similar to that observed in axon growth inhibition and retraction experiments (77).
The mechanical stretch or shear of the synaptic cleft will also alter the cleft volume, NT concentration, and diffusion distance and delay NT clearance. For instance, small changes in synaptic cleft height and geometry could retard Glu clearance and affect ion flux kinetics through ion channel, while higher Glu concentrations in the cleft may cause prolonged depolarization and excitotoxicity in postsynaptic neurons. Adverse neuroplasticity due to unnatural changes in synaptic morphology may affect LTP and/or LTD. Partial transient mechanoporation of dendritic and spinal membranes may cause ion leakage, affect the dynamics of de- and repolarization, induce calcium-mediated excitotoxicity, and cause increased energy demands and energetic “exhaustion” and oxidative stress. Additionally, Glu may also diffuse to a neighboring synapse and may inadvertently activate their receptors, illustrated in Figure 2.
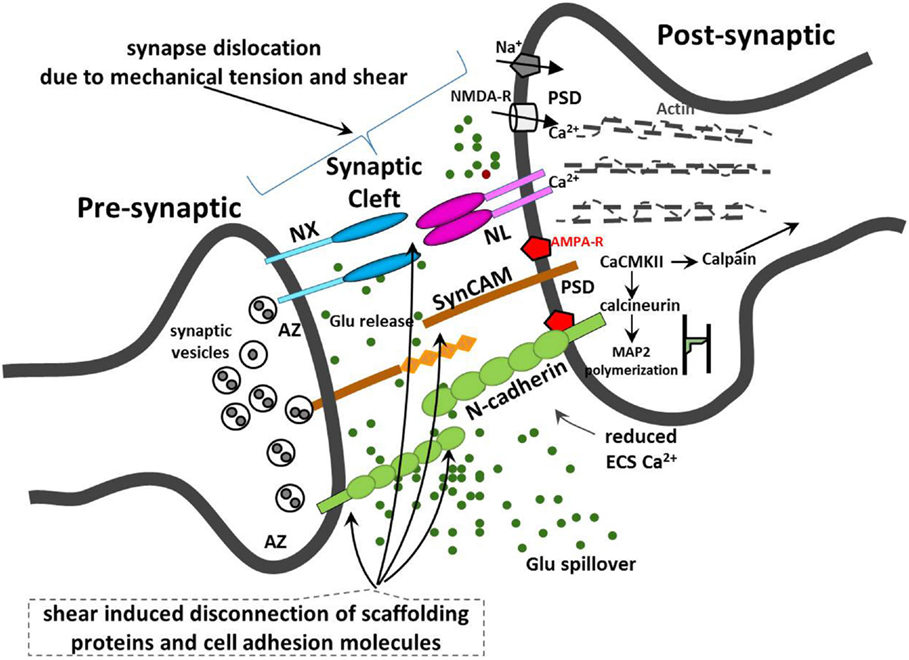
Figure 2. Illustration of synaptic injury due to mechanical tension and shear and disruption to neuronal synaptic CAMs.
Conclusion and Future Direction
It is now clear that neural synapses transmit not only biochemical and electrophysiological information but also communicate using mechanical signals. Synaptic CAMs provide direct mechanical link between presynaptic vesicular release machinery and postsynaptic cytoskeletal and molecular organization. Composition, structural and elastic properties as well as intra-synaptic distribution of CAMs may be responsible for mechanical injury and repair pathways such as those involved in CTE but may also be implicated in developmental, cognitive, and neurodegenerative diseases, including autism (78), chronic stress and depression (79, 80), Alzheimer’s disease (81), schizophrenia (82), and other diseases (83, 84). Mechanobiology of axo-glial CAMs may be also implicated in the damage of myelinated axons in response to mTBI. Mechanical damage to axo-glial CAMs in response to TBI and the subsequent de/re-myelination are yet to be explored. Emerging evidence suggests that axo-glial CAMs, including NXs, NLs, and Nectin-like molecules, located in the narrow gap between the axonal membrane and the surrounding myelin sheath, are responsible for axon myelination and the so called “white matter plasticity” (85, 86). Finally, neuronal CAMs may be a promising pharmacological target for modulating the synaptic “connectome,” impaired in brain disorders and neurotrauma (87).
Coordinated in vitro–in vivo experiments and mathematical modeling studies should be conducted to shed light into the synaptic injury mechanisms and to determine whether the diffuse synaptic injury plays a prominent etiological role in mTBI. From a modeler’s perspective, it would be beneficial to collect in vitro and in vivo experimental data of geometry, morphology, and electrophysiology of a synaptic structure at various times post-injury. State-of-the-art fixing or high-pressure freezing, tomography, and electron microscopy of ultrathin sliced cell or tissue cultures can reveal the internal structure of synapses in exceptional 3D spatial resolution (32, 88). These techniques could be used to analyze the morphology of the synaptic ultrastructure post-injury. Less precise but equally helpful experiments could use super-resolution fluorescence microscopy techniques, such as STED or RESOLFT microscopy, to obtain time resolved synaptic remodeling data (89). Experimental elasto-mechanics studies of synaptic CAMs for various strain rates could provide not only the insight into their mechanobiology but also on the elasto-dynamic constants and damage thresholds, relevant for future mathematical models of synaptic injury. Better understanding of the role of CAMs in synaptic and axo-glial injury will require “animal models” that can be “molecularly engineered.” Genetically manipulated Drosophila and mouse models have been already developed and used for TBI research (62, 90, 91). Both models should be further pursued and complemented with the corresponding computational models to expedite the development of new treatments, diagnostics, and protective measures in blast-related neurotrauma.
Author Contributions
AJP and MRS: conceived and drafted the manuscript (equal contribution). RKG: offered technical insights and reviewed the manuscript.
Disclaimer
The views expressed in this paper are those of the authors and may not necessarily be endorsed by the U.S. Army or U.S. Department of Defense.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors (AJP and MRS) would like to express their appreciation to the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) under contract W81XWH-11-2-0057 for supporting this work. The authors acknowledge Dr. Vincent Harrand, CFDRC and Prof. Beverly Rzigalinski, Virginia Tech, for reviewing the manuscript.
References
1. Belanger HG, Spiegel E, Vanderploeg RD. Neuropsychological performance following a history of multiple self-reported concussions: a meta-analysis. J Int Neuropsychol Soc (2010) 16(2):262–7. doi:10.1017/S1355617709991287
2. Bogdanova Y, Verfaellie M. Cognitive sequelae of blast-induced traumatic brain injury: recovery and rehabilitation. Neuropsychol Rev (2012) 22(1):4–20. doi:10.1007/s11065-012-9192-3
3. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med (2012) 4(134):134ra60. doi:10.1126/scitranslmed.3004862
4. Cernak I, Savic J, Ignjatovic D, Jevtic M. Blast injury from explosive munitions. J Trauma (1999) 47(1):96–103. doi:10.1097/00005373-199907000-00021
5. DePalma RG, Burris DG, Champion HR, Hodgson MJ. Blast injuries. N Engl J Med (2005) 352(13):1335–42. doi:10.1056/NEJMra042083
6. Elder GA, Cristian A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt Sinai J Med (2009) 76(2):111–8. doi:10.1002/msj.20098
7. Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, et al. Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J Neurotrauma (2011) 28(6):1101–19. doi:10.1089/neu.2010.1442
8. Risling M, Plantman S, Angeria M, Rostami E, Bellander BM, Kirkegaard M, et al. Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage (2011) 54(Sup 1):S89–97. doi:10.1016/j.neuroimage.2010.05.031
9. Gupta RK, Przekwas A. Mathematical models of blast-induced TBI: current status, challenges, and prospects. Front Neurol (2013) 4:59. doi:10.3389/fneur.2013.00059
10. Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known? J Neuropsychiatry Clin Neurosci (2006) 18(2):141–5. doi:10.1176/jnp.2006.18.2.141
11. Taber KH, Hurley RA, Haswell CS, Rowland JA, Hurt SD, Lamar CD, et al. White matter compromise in veterans exposed to primary blast forces. J Head Trauma Rehabil (2015) 30(1):E15–25. doi:10.1097/HTR.0000000000000030
12. Mac Donald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, et al. Detection of blast-related traumatic brain injury in U.S. Military personnel. N Engl J Med (2011) 364:2091–100. doi:10.1056/NEJMoa1008069
13. Jorge RE, Acion L, White T, Tordesillas-Gutierrez D, Pierson R, Crespo-Facorro B, et al. White matter abnormalities in veterans with mild traumatic brain injury. Am J Psychiatry (2012) 169(12):1284–91. doi:10.1176/appi.ajp.2012.12050600
14. Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist (2000) 6(6):483–95. doi:10.1177/107385840000600611
15. Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch Phys Med Rehabil (2001) 82(10):1461–71. doi:10.1053/apmr.2001.25137
16. Chen YC, Smith DH, Meaney DF. In-vitro approaches for studying blast-induced traumatic brain injury. J Neurotrauma (2009) 26(6):861–76. doi:10.1089/neu.2008.0645
17. Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol (2012) 233(1):364–72. doi:10.1016/j.expneurol.2011.10.030
18. Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma (2009) 26(6):925–39. doi:10.1089/neu.2008-0794
19. Morrison B III, Elkin BS, Dolle JP, Yarmush ML. In vitro models for traumatic brain injury. Annu Rev Biomed Eng (2011) 13:91–126. doi:10.1146/annurev-bioeng-071910-124706
20. Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. New York, NY: McGraw-Hill (2000).
21. Herculano-Houzel S. The human brain in numbers: a linearly scaled-up primate brain. Front Hum Neurosci (2009) 3(31):1–11. doi:10.3389/neuro.09.031.2009
22. Ho VM, Lee JA, Martin KC. The cell biology of synaptic plasticity. Science (2011) 334(4):623–8. doi:10.1126/science.1209236
23. Weaver JB, Pattison AJ, McGarry MD, Perreard IM, Swienckowski JG, Eskey CJ, et al. Brain mechanical property measurement using MRE with intrinsic activation. Phys Med Biol (2012) 57(22):7275–87. doi:10.1088/0031-9155/57/22/7275
24. Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci (2002) 25(6):295–301. doi:10.1016/S0166-2236(02)02143-4
25. Tyler WJ. The mechanobiology of brain function. Nat Rev Neurosci (2012) 13:867–78. doi:10.1038/nrn3383
26. Mueller JK, Tyler WJ. A quantitative overview of biophysical forces impinging on neural function. Phys Biol (2014) 11:051001. doi:10.1088/1478-3975/11/5/051001
27. Zhu C. Mechanochemitry: a molecular biomechanics view of mechanosensing. Ann Biomed Eng (2014) 42(2):388–404. doi:10.1007/s10439-013-0904-5
28. Brockett AT, LaMarca EA, Gould E. Physical exercise enhances cognitive flexibility as well as astrocytic and synaptic markers in the medial prefrontal cortex. PLoS One (2015) 10(5):e0124859. doi:10.1371/journal.pone.0124859
29. Savtchenko LP, Rusakov DA. The optimal height of the synaptic cleft. Proc Natl Acad Sci U S A (2007) 104(6):1823–8. doi:10.1073/pnas.0606636104
30. Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev (2002) 39(1):29–54. doi:10.1016/S0165-0173(02)00158-3
31. SynapseWeb, Harris KM. (2015). Available from: http://synapses.clm.utexas.edu/
32. High B, Cole AA, Chen X, Reese TS. Electron microscopic tomography reveals discrete transcleft elements at excitatory and inhibitory synapses. Front Synaptic Neurosci (2015) 7:9. doi:10.3389/fnsyn.2015.00009
33. Levinson JN, El-Husseini A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron (2005) 48:171–4. doi:10.1016/j.neuron.2005.09.017
34. Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (2006) 21:38–47. doi:10.1152/physiol.00042.2005
35. Ramakers GJA. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci (2002) 25(4):191–9. doi:10.1016/S0166-2236(00)02118-4
36. Turrigiano G. Too many cooks? intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci (2011) 34:89–103. doi:10.1146/annurev-neuro-060909-153238
37. Froemke RC. Plasticity of cortical excitatory-inhibitory balance. Annu Rev Neurosci (2015) 38:195–219. doi:10.1146/annurev-neuro-071714-034002
38. Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol (2012) 3:60. doi:10.3389/fphar.2012.00060
39. Sotomayor M, Schulten K. The allosteric role of the Ca++ switch in adhesion and elasticity of C-cadherin. Biophys J (2008) 94(12):4621–33. doi:10.1529/biophysj.107.125591
40. Nguyen T, Sudhof TC. Binding properties of neuroligin 1 and neurexin 1β reveal function as heterophilic cell adhesion molecules. J Biol Chem (1997) 272(41):26032–9. doi:10.1074/jbc.272.41.26032
41. Maruthamuthu V, Schulten K, Leckband D. Elasticity and rupture of a multi-domain neural cell adhesion molecule complex. Biophys J. (2009) 96(8):3005–14. doi:10.1016/j.bpj.2008.12.3936
42. Leckband D. Design rules for biomolecular adhesion: lessons from force measurements. Annu Rev Chem Biomol Eng (2010) 1:365–89. doi:10.1146/annurev-chembioeng-073009-100931
43. Mikulska K, Strzelecki J, Balter A, Nowak W. Nanomechanical unfolding of α-neurexin: a major component of the synaptic junction. Chem Phys Lett (2012) 521:134–7. doi:10.1016/j.cplett.2011.11.033
44. Stenfelt S, Goode R. Transmission properties of bone conducted sound: measurements in cadaver heads. J Acoust Soc Am (2005) 118:2373–91. doi:10.1121/1.2005847
45. Tan XG, Kannan R, Przekwas AJA. Comparative study of the human body finite element model under blast loadings. IMECE2012-89072. Proceedings of the ASME 2012 International Mechanical Engineering Congress & Exposition. Houston: ASME (2012). p. 829–36.
46. Gennarelli TA, Thibault LE. Biomechanics of acute subdural hematoma. J Trauma (1982) 22(8):680–6. doi:10.1097/00005373-198208000-00005
47. King AI, Yang KH, Zhang L, Hardy W, Viano DC. Is Head Injury Caused By Linear Or Angular Acceleration? Lisbon: IRCOBI (2003). 2003 p.
48. LaPlaca MC, Prado GR. Neural mechanobiology and neural vulnerability to traumatic loading. J Biomech (2010) 43:71–8. doi:10.1016/j.jbiomech.2009.09.011
49. Kleiven S. Why most traumatic brain injuries are not caused by linear acceleration but skull fractures are. Front Bioeng Biotechnol (2013) 1:15. doi:10.3389/fbioe.2013.00015
50. Rowson S, Duma SM, Beckwith JG, Chu JJ, Greenwald RM, Crisco JJ, et al. Rotational head kinematics in football impacts: an injury risk function for concussion. Ann Biomed Eng (2012) 40:1–13. doi:10.1007/s10439-011-0392-4
51. Stemper BD, Pintar FA. Biomechanics of concussion. Prog Neurol Surg (2014) 28:14–27. doi:10.1159/000358748
52. Post A, Hoshizaki TB, Gilchrist MD, Brien S, Cusimano MD, Marshall S. The influence of dynamic response and brain deformation metrics on the occurrence of subdural hematoma in different regions of the brain. J Neurosurg (2014) 120(2):453–61. doi:10.3171/2013.10.JNS131101
53. Sullivan S, Eucker SA, Gabrieli D, Bradfield C, Coats B, Maltese MR, et al. White matter tract-oriented deformation predicts traumatic axonal brain injury and reveals rotational direction-specific vulnerabilities. Biomech Model Mechanobiol (2015) 14(4):877–96. doi:10.1007/s10237-014-0643-z
54. Feng Y, Abney TM, Okamoto RJ, Pless RB, Genin GM, Bayly PV. Relative brain displacement and deformation during constrained mild frontal head impact. J R Soc Interface (2010) 7:1677–88. doi:10.1098/rsif.2010.0210
55. Hardy WN, Foster CD, Mason MJ, Yang KH, King AI, Tashman S. Investigation of head injury mechanism using neutral density technology and high-speed biplanar X-ray. Stapp Car Crash J (2001) 45:337–68.
56. Zou H, Schmiedeler JP, Hardy WN. Separating brain motion into rigid body displacement and deformation under low-severity impacts. J Biomech (2007) 40(6):1183–91. doi:10.1016/j.jbiomech.2006.06.018
57. Rashid B, Destradeb M, Gilchrist MD. Mechanical characterization of brain tissue in tension at dynamic strain rates. J Mech Behav Biomed Mater (2014) 33:43–54. doi:10.1016/j.jmbbm.2012.07.015
58. Xi XF, Zhong P. Dynamic photoelastic study of the transient stress field in solids during shock wave lithotripsy. J Acoust Soc Am (2001) 109:1226–39. doi:10.1121/1.1349183
59. Bayly PV, Cohen TS, Leister EP, Ajo D, Leuthardt E, Genin GM. Deformation of the human brain induced by mild acceleration. J Neurotrauma (2005) 22(8):845–56. doi:10.1089/neu.2005.22.845
60. Cohen AS, Pfister BP, Schwarzbach E, Grady MS, Goforth PB, Satin LS. Injury-induced alterations in CNS electrophysiology. Prog Brain Res (2007) 161:143–69. doi:10.1016/S0079-6123(06)61010-8
61. Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma (2008) 25:513–26. doi:10.1089/neu.2007.0451
62. Park K, Biederer T. Neuronal adhesion and synapse organization in recovery after brain injury. Future Neurol (2013) 8(5):555–67. doi:10.2217/fnl.13.35
63. Merlo L, Cimino F, Angileri FF, La Torre D, Conti A, Cardali SM, et al. Alteration in synaptic junction proteins following traumatic brain injury. J Neurotrauma (2014) 31:1375–85. doi:10.1089/neu.2014.3385
64. Hemphill MA, Dauth S, Yu CJ, Dabiri BE, Parker KK. Traumatic brain injury and the neuronal microenvironment: a potential role for neuropathological mechanotransduction. Neuron (2015) 85:1177–92. doi:10.1016/j.neuron.2015.02.041
65. Panzer MB, Matthews KA, Yu AW, Morrison B III, Meaney DF, Bass CR. A multiscale approach to blast neurotrauma modeling: part I – development of novel test devices for in vivo and in vitro blast injury models. Front Neurol (2012) 3:46. doi:10.3389/fneur.2012.00046
66. Floyd CL, Lyeth BC. Astroglia: important mediators of traumatic brain injury. Prog Brain Res (2007) 161:61–79. doi:10.1016/S0079-6123(06)61005-4
67. Monnerie H, Tang-Schomer MD, Iwata A, Smith DH, Kim HA, Le Roux PD. Dendritic alterations after dynamic axonal stretch injury in vitro. Exp Neurol (2010) 224(2):415–23. doi:10.1016/j.expneurol.2010.05.001
68. Ferenc MT, Radovitzky R, Bass CR, Wood GP, Panzer M, Man HY, et al. Shock- waves Cause Synaptic Degeneration in Cultured Neurons. Boston University, MA Dep. of Biology Report for USAARO (2009). Available from: http://oai.dtic.mil/
69. Effgen GB, Vogel EW III, Lynch KA, Lobel A, Hue CD, Meaney DF, et al. Isolated primary blast alters neuronal function with minimal cell death in organotypic hippocampal slice cultures. J Neurotrauma (2014) 31(13):1202–10. doi:10.1089/neu.2013.3227
70. Zander NE, Piehler T, Boggs ME, Banton R, Benjamin R. In vitro studies of primary explosive blast loading on neurons. J Neurosci Res (2015) 93(9):1353–63. doi:10.1002/jnr.23594
71. Chavko M, Watanabe T, Adeeb S, Lankasky J, Ahlers ST, McCarron RM. Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J Neurosci Methods (2011) 195(1):61–6. doi:10.1016/j.jneumeth.2010.11.019
72. Risling M, Davidsson J. Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Front Neurol (2012) 3:30. doi:10.3389/fneur.2012.00030
73. Gao X, Deng P, Xu ZC, Chen J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One (2011) 6(9):e24566. doi:10.1371/journal.pone.0024566
74. Manibog K, Li H, Rakshit S, Sivasankar S. Resolving the molecular mechanism of cadherin catch bond formation. Nat Commun (2014) 5:3941. doi:10.1038/ncomms4941
75. Hemphill MA, Dabiri BE, Gabriele S, Kerscher L, Franck C, Goss JA, et al. A possible role for integrin signaling in diffuse axonal injury. PLoS One (2011) 6(7):e22899. doi:10.1371/journal.pone.0022899
76. Castaneda P, Munoz M, Garcia-Rojo G, Ulloa JL, Bravo JA, Marquez R, et al. Association of N-cadherin levels and downstream effectors of Rho GTPases with dendritic spine loss induced by chronic stress in rat hippocampal neurons. J Neurosci Res (2015) 93(10):1476–91. doi:10.1002/jnr.23602
77. Fujita Y, Yamashita T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front Neurosci (2014) 8:338. doi:10.3389/fnins.2014.00338
78. Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci (2015) 16:551–63. doi:10.1038/nrn3992
79. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci (2004) 5:917–30. doi:10.1038/nrn1555
80. McEwen BS, Bowles NP, Gray JD, Hill MH, Hunter RG, Karatsoreos IN, et al. Mechanisms of stress in the brain. Nat Neurosci (2015) 18(10):1353–63. doi:10.1038/nn.4086
81. Sindi IA, Tannenberg RK, Dodd PR. A role for the neurexin-neuroligin complex in Alzheimer’s disease. Neurobiol Aging (2014) 35:746–56. doi:10.1016/j.neurobiolaging.2013.09.032
82. Brennaman LH, Maness PF. NCAM in neuropsychiatric and neurodegenerative disorders. Adv Exp Med Biol (2010) 663:299–317. doi:10.1007/978-1-4419-1170-4_19
83. Berezin V, Walmod PS. Cell Adhesion Molecules, Implications in Neurological Diseases. New York, NY: Springer (2014).
84. Popoli M, Diamond D, Sanacora G. Synaptic Stress and Pathogenesis of Neuropsychiatric Disorders. New York, NY: Springer (2014).
85. Fields DR. A new mechanism of nervous system plasticity: activity-dependent myelination. Nat Rev Neurosci (2015) 16:756–67. doi:10.1038/nrn4023
86. Proctor DT, Stotz SC, Scott LOM, de la Hoz CLR, Poon KWC, Stys PK, et al. Axo-glial communication through neurexin-neuroligin signaling regulates myelination and oligodendrocyte differentiation. Glia (2015) 63:2023–39. doi:10.1002/glia.22875
87. Uhl GR, Drgonova J. Cell adhesion molecules: druggable targets for modulating the connectome and brain disorders? Neuropsychopharmacology (2014) 39:235. doi:10.1038/npp.2013.240
88. Kay KR, Smith C, Wright AK, Serrano-Pozo A, Pooler AM, Koffie R, et al. Studying synapses in human brain with array tomography and electron microscopy. Nat Protoc (2013) 8(7):1366–80. doi:10.1038/nprot.2013.078
89. Tao C, Xia C, Chen X, Zhou ZH, Bi G. Ultrastructural analysis of neuronal synapses using state-of-the-art nano-imaging techniques. Neurosci Bull (2012) 28(4):321–32. doi:10.1007/s12264-012-1249-z
90. Goldstein LE, McKee AC, Stanton PK. Considerations for animal models of blast-related traumatic brain injury and chronic traumatic encephalopathy. Alzheimers Res Ther (2014) 6:64. doi:10.1186/s13195-014-0064-3
Keywords: synapse, TBI, neuro-axonal, blast wave injury, mechanobiology, DAI
Citation: Przekwas A, Somayaji MR and Gupta RK (2016) Synaptic Mechanisms of Blast-Induced Brain Injury. Front. Neurol. 7:2. doi: 10.3389/fneur.2016.00002
Received: 20 March 2015; Accepted: 04 January 2016;
Published: 21 January 2016
Edited by:
Firas H. Kobeissy, University of Florida, USAReviewed by:
Stefania Mondello, University of Messina, ItalyKarin A. Rafaels, Army Research Laboratory, USA
Karim A. Sarhane, Johns Hopkins University, USA
Copyright: © 2016 Przekwas, Somayaji and Gupta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrzej Przekwas, YW5kcnplai5wcnpla3dhc0BjZmRyYy5jb20=