 Prakasham Rumajogee
Prakasham Rumajogee Tatiana Bregman
Tatiana Bregman Steven P. Miller
Steven P. Miller Jerome Y. Yager3
Jerome Y. Yager3 Michael G. Fehlings
Michael G. Fehlings- 1Division of Genetics and Development, Krembil Research Institute, Toronto Western Hospital, University Health Network, Toronto, ON, Canada
- 2Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada
- 3Division of Pediatric Neurosciences, Stollery Children’s Hospital, University of Alberta, Edmonton, AB, Canada
- 4Division of Neurosurgery, Institute of Medical Science, University of Toronto, Toronto, ON, Canada
Cerebral palsy (CP) is a complex multifactorial disorder, affecting approximately 2.5–3/1000 live term births, and up to 22/1000 prematurely born babies. CP results from injury to the developing brain incurred before, during, or after birth. The most common form of this condition, spastic CP, is primarily associated with injury to the cerebral cortex and subcortical white matter as well as the deep gray matter. The major etiological factors of spastic CP are hypoxia/ischemia (HI), occurring during the last third of pregnancy and around birth age. In addition, inflammation has been found to be an important factor contributing to brain injury, especially in term infants. Other factors, including genetics, are gaining importance. The classic Rice–Vannucci HI model (in which 7-day-old rat pups undergo unilateral ligation of the common carotid artery followed by exposure to 8% oxygen hypoxic air) is a model of neonatal stroke that has greatly contributed to CP research. In this model, brain damage resembles that observed in severe CP cases. This model, and its numerous adaptations, allows one to finely tune the injury parameters to mimic, and therefore study, many of the pathophysiological processes and conditions observed in human patients. Investigators can recreate the HI and inflammation, which cause brain damage and subsequent motor and cognitive deficits. This model further enables the examination of potential approaches to achieve neural repair and regeneration. In the present review, we compare and discuss the advantages, limitations, and the translational value for CP research of HI models of perinatal brain injury.
Introduction: What is Cerebral Palsy?
Cerebral palsy (CP) is the most common pediatric neurodevelopmental physical disability, with a prevalence of 2.0–3.5/1000 births (1). This condition results in a spectrum of motor and developmental disturbances leading to movement disorders, as well as cognitive impairments, speech and language disorders, growth problems, or pain. CP is an umbrella term for several etiologic conditions. Additionally, the term CP describes a range of disturbances in motor function, with spastic CP being the most common condition accounting for 80% of cases (2).
In this review, we will provide an overview of CP, its etiology, and its main pathophysiological mechanisms. Understanding the biological mechanisms of CP has been possible through the use of various animal models, although it is beyond the scope of this review to describe all of them in detail. We will, therefore, focus on rodent hypoxic–ischemic models. Following a summary of the classic hypoxic–ischemic Rice–Vannucci rodent model, we will focus on other HI rodent models developed to mimic the pathophysiological events in humans and discuss the potential of these models in CP research.
Cerebral palsy was originally described by William Little in 1861 (3). Since that time, the definition of CP has constantly evolved alongside scientific and technical progress. In 1997, Palisano et al. described the Gross Motor Function Classification System (GMFCS), which was further revised in 2007. This reference system provides five levels of classification based on self-initiated movements, including sitting, walking, and wheeled mobility (4, 5). In 2006, “The Definition and Classification of Cerebral Palsy, April 2006,” further defined CP:
Cerebral palsy describes a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain. The motor disorders of cerebral palsy are often accompanied by disturbances of sensation, perception, cognition, communication, and behaviour, by epilepsy, and by secondary musculoskeletal problems.
The definition of CP can be approached from several angles indicating the complexity of the syndrome: (1) the anatomical site of the brain lesion; (2) the clinical symptoms and signs (e.g., spasticity, dyskinesia, or ataxia); (3) the topographical involvement of extremities (e.g., diplegia, quadriplegia, or hemiplegia); (4) the timing of presumed insult (e.g., prepartum, intrapartum, or post-neonatal); and (4) classification of the degree of muscle tone (e.g., isotonic, hypotonic, or hypertonic) (6, 7). CP patients often present with mixed symptoms. For research and knowledge transfer purposes, a standardized and more consistent classification has been proposed (1, 8). However, no simple definition of CP exists; this issue is discussed further in an excellent review by Rosenbaum and colleagues (9).
Etiology of Cerebral Palsy
The causal factors for CP are multiple and may be linked. Figure 1 summarizes the multiple-hit hypothesis of CP pathobiology. CP can follow a brain abnormality that occurs pre, peri, or postnatally or some combination thereof. Even when the injury occurs at a defined time, moderating factors may also exist (10, 11). The risk factors for CP include: preterm birth, intra-uterine growth restriction, maternal/fetal infection, inflammation, perinatal and intrapartum difficulties (e.g., hypoxic–ischemic insult), and genetic predispositions (1, 12, 13). Remarkably, intrapartum hypoxia–ischemia (HI) is thought to account for only 14.5% of CP cases (14). The importance of inflammation in CP onset has also been recently reviewed by Van Steenwinckel and colleagues [Figure 2; (15)].
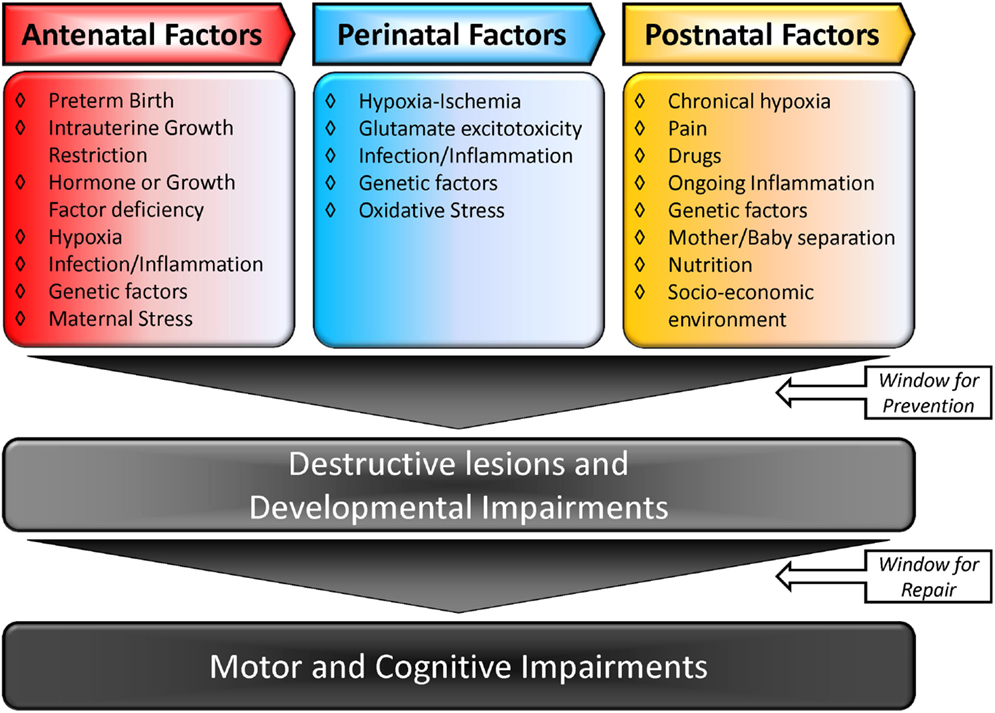
Figure 1. Multiple-hit hypothesis of cerebral palsy: a combination of two or more factors is more likely to trigger and modulate lesions in the brain, causing CP. Adapted from Ref. (10, 15).
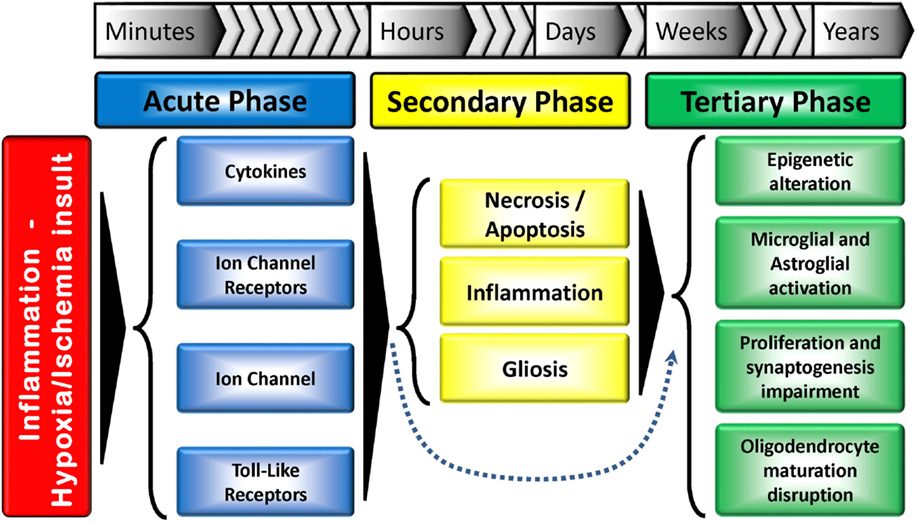
Figure 2. Tri-phasic hypothesis of the perinatal brain damage: major action elements and processes. The acute phase occurs minutes after the insult, while the secondary phase happens hours after. A tertiary phase, which can last for months/years and result in developmental disturbances has also been proposed. The secondary phase can be skipped in some situations. Adapted from Ref. (15).
A recent study has shown de novo copy number variations (CNVs) in the genome are clinically relevant to CP (16). Furthermore, genes, such as KAK1, AP4MI, or GAD1, have been shown to play a role in up to 45% of CP cases (17, 18).
It has been suggested that males and females may also respond differently to developmental brain injury. For instance, males born preterm (at 28 weeks of gestation in these studies) have been reported to be more susceptible to white matter damage (19, 20).
Developmental stage also plays a crucial role in determining the response to an insult. Generally, insults occurring during the first trimester of gestation are linked to abnormal cerebral development (e.g., schizencephaly). Insults in the late second and early third trimester typically lead to periventricular white matter injury (PVWMI), often associated with neuronal abnormalities but not necessarily neuronal loss, while insults in the late third trimester trigger cortical and deep gray matter lesions (21). Up to 24 weeks of gestation, cortical neurogenesis is defined by proliferation, migration, and formation of neural precursor cells and neurons. In the following weeks of gestation, other dynamic mechanisms take place: growth (axon/dendrites formation), differentiation (synaptogenesis, myelination), and maintenance (i.e., apoptosis, pruning) and/or specialization (development of circuitry). These mechanisms persist up to 2 years after birth (10, 22). Therefore, any injury occurring in the immature brain will trigger a cascade of developmental disturbances (Figure 2).
Pathophysiology
Characteristics of Brain Lesions: Periventricular Leukomalacia
One of the most common pathological causes of CP is periventricular leukomalacia (PVL). PVL has three forms: cystic, non-cystic, and diffuse. Cystic and non-cystic forms of PVL encompass aspects of focal necrosis and diffuse injury of the white matter. While cystic focal necrosis, which is macroscopic, turns into cysts, the microscopic non-cystic necrosis turns into glial scars (23, 24). A diffuse white matter injury is characterized by astrogliosis and microgliosis as well as hypo-myelination, due to lower amounts of pre-myelinating oligodendrocytes (OLs) (22, 24, 25). These lesions can be accompanied by hemorrhage in the germinal matrix, the periventricular zone, or into the ventricle (26–28).
There are a number of potential underlying mechanisms for diffuse PVL. The most studied mechanism is pre-oligodendrocyte (pre-OL) injury: a disruption of the OL lineage leading to serious impairment of axonal myelination and development caused by a deficit of trophic factors release (29, 30). Also, as a secondary event, axonal injury has been proposed as a mechanism leading to axonal degeneration, hypo-myelination, and decreased cortical and thalamic volumes. The subplate neurons, which are among the first neurons to appear in the cerebral cortex, play a crucial role in the development of the cerebral cortex and deep nuclei (wiring and functional maturation), and their impairment triggers secondary and maturational effects. Neonatal HI causes selective subplate neuronal death, which is responsible for abnormal cortical development (31). Subplate neuronal injury subsequently leads to plasticity impairments (32, 33). Thalamic development is also affected. In fact, thalamic neuronal injury leads to white matter axon damage and gliosis. Finally, the dorsal telencephalic subventricular zone and the late GABAergic migrating neurons, which are crucial to the structure of the upper cortical layers, are prone to primary injury [see Ref. (27)].
Davatzikos et al. report that the growth of the corpus callosum, which connects the left and right cerebral hemispheres and contains commissural myelinated fibers, is severely impaired in premature infants with PVL, though type of PVL is not specified in this work (34). This pathology preferentially occurs around the lateral ventricles, which are close to major cerebral arteries. This also affects descending corticospinal tracts (CSTs) and leads to neuromotor deficits, including CP (34–36). Subplate neurons have a fundamental role in the elaboration of connections between the thalamus and the cortex, as well as within the cortex (37). Thus, thalamocortical, corticocortical, and corticospinal synaptogenesis are impaired, leading to compromised connectivity and mapping in rodents (2, 38, 39). The most prominent anatomical feature in the onset of spastic CP is the lesion of the sensorimotor cortex and its subcortical white matter (40).
It has generally been understood that there are primary and secondary injury events in PVL. The primary event is reported as being a key contributor to the pathobiology while, in the secondary phase, developmental disturbances involving the cerebral white matter and other structures: thalamus, basal ganglia, cerebral cortex, brainstem, and cerebellum occur (27, 41). Interestingly, recent studies have shown that growth impairments are not linked to neuronal death, but rather to dendritic arbor maturation alteration, spine density dendritic network decrease (42), and disturbance in synapse formation of the cortical neurons (43).
Recently, a tertiary phase of persisting injury, lasting for months or years, linked to persistent inflammation and possibly epigenetic changes, has been identified (Figure 2). These long-term epigenetic modifications, which have been suggested as potential mechanisms involved in various pathologies, include phosphorylation, ubiquitination and acetylation of histones, and methylation of DNA and RNA. This third phase not only inhibits regeneration and/or exacerbates brain damage, but also sensitizes the brain to additional injury, such as myelin deficits. Cerebral plasticity is also reduced, further contributing to the abnormal development of the brain (20).
Disruption of Oligodendrocyte Maturation
In humans, susceptibility to PVWMI peaks between 23/24 and 32 weeks of gestation (44, 45) This vulnerability to hypoxia/ischemia/trauma has several causes, including the higher energy needs of the brain to support the rapid development of white matter. Also, as the brain grows, the distance between blood vessels increases, which makes the oxygen and blood supply more critical (46, 47). It is now apparent that OL precursor cell intrinsic hypoxia-inducible factors (HIFs) signaling is a key coupler of white matter angiogenesis and the onset of myelination postnatally (48).
Myelination impairment is linked to the disrupted OL differentiation process, characterized by a deficit of myelinating mature OL (22, 49, 50). Back et al. showed that PVL has a developmental window that starts before the beginning of myelination, when late OL progenitors are the predominant OL type. When myelination starts in the periventricular white matter, a decline of PVL incidence is also observed. This indicates that the presence of late OL progenitors (Pre-OLs) is linked to the risk of PVL and defines them as a preferential target in PVL (44, 51).
Comparative timetables between human and other species for the various developmental stages have been proposed to facilitate the understanding of PVWMI onset during development. The differences of brain structure and development between human and rodents do not allow perfect comparisons. However, Table 1 summarizes the human and rodent OL developmental stages. Briefly, embryonic day (ED) 17 to postnatal day (PND) 2 in rodents corresponds to 14–23 gestation weeks in humans. PNDs 2–3 in rodents are similar to 23–32 weeks in humans (preterm infant). PNDs 7–10 in rodents are comparable to 36–40 weeks in humans (term infant). A rodent at PNDs 20–21 is comparable to a 2–3 years old human (2, 45, 52–54). Contradictory opinions exist regarding the timing of maturation of OLs. In a review by Semple et al., it was suggested that the maturation peak of Pre-OLs occurs at PNDs 1–3 (45), by contrast, a time window of PNDs 2–5 has been suggested in a review by Clowry (2, 55). However, several reports indicate that the maturation peak occurs at PND 2 (56–58). Moreover, Dean et al. showed that immature OLs peak at PND 5 in rat, while Semple et al. suggested a predominance of immature OLs between PNDs7 and 10 (45, 58). It is now clear that OL progenitors persist into adulthood, contributing to myelination processes as well as to other mechanisms, such as neuronal activity modulation (59, 60).
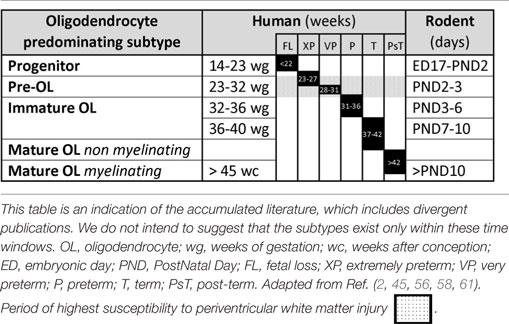
Table 1. Comparative timetable of oligodendrocyte predominating subtypes during development in human and rodent.
Cellular Mechanisms
Ischemia/oxygen glucose deprivation and inflammation are the main mechanisms that initiate PVL and they can facilitate each other (56, 62). Increase of pro-inflammatory cytokines has also been linked to PVL (63, 64). Interestingly, models of ischemic brain injury, such as the intra-uterine growth restriction model show damage that mimics CP conditions (65–67).
Other mechanisms include neurotransmitter-mediated excitotoxicity and oxidative stress (free-radical release), as well as alteration of the extracellular matrix (23, 47). Figure 3 shows the cellular and molecular mechanisms triggered by glutamate and pro-inflammatory cytokines.
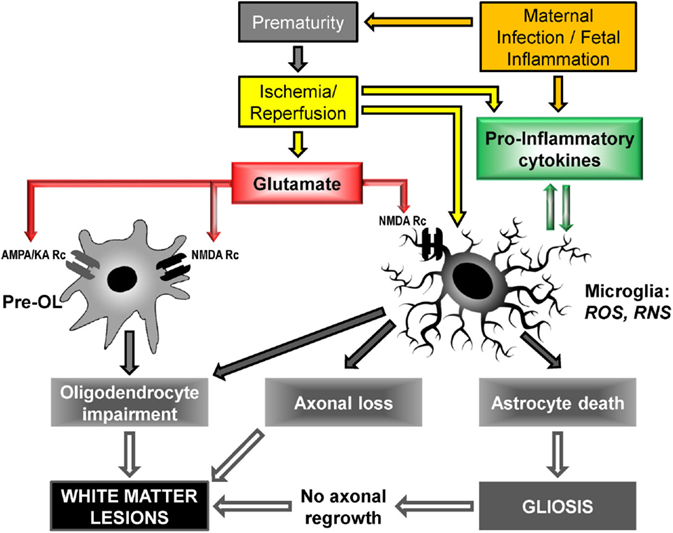
Figure 3. Cellular and molecular mechanisms to PVL onset: upstream mechanisms (cerebral hypoxia/ischemia and maternal infection/fetal inflammation) activate downstream mechanisms (glutamate excitotoxicity and free radical release) leading to cellular deficiencies, in particular oligodendrocyte maturation impairment and myelination deficit. Pre-OL, pre-oligodendrocyte; Rc, receptor; AMPA, α-3-amino-hydroxy-5-methyl-4-isoxazole propionic acid; KA, kainate; NMDA, N-methyl-D-aspartate; ROS, reactive oxygen species; RNS, reactive nitrogen species [adapted from Ref. (23, 47, 50, 68, 69)].
Pre-OLs, more so than immature OLs, are particularly vulnerable to HI insults. Early OL progenitors respond to white matter injury by a rapid and significant phase of proliferation. The pool of pre-OLs is, thus, dramatically increased. However, these cells fail to differentiate further and do not repair white matter (50, 53), resulting in development arrest that is likely linked to inhibitory factors released by reactive astrocytes, such as interleukin-1, and microglia (15, 49).
Oligodendrocytes express neurotransmitter receptors [e.g., glutamate and adenosine-triphosphate (ATP)] and respond to signals conveyed by axons. Under pathological conditions, they trigger a surge of intracellular calcium that disrupts the Ca2+ homeostasis to which OLs are highly sensitive (70).
Hypoxia–Ischemia caused by oxygen and glucose deficits leads to a decrease in ATP which in turn compromises the sodium–potassium ATP pump, crucial for membrane polarity maintenance (69). Interestingly, during ischemia, anoxic depolarization initiates extracellular ATP release from glial cells (71).
Adenosine-triphosphate-mediated toxicity, through the ionotropic P2X and the metabotropic P2Y purino-receptors (72), leads to apoptosis or necrosis and is correlated with the degree of intracellular Ca2+ surge (73).
Depolarization induces excessive glutamate release, which activates the ionotropic glutamate receptors AMPA, Kainate, and NMDA present in OLs. In addition, activation of metabotropic glutamate receptors (mGluRs), strongly expressed by pre-OLs, leads to a calcium influx (74, 75). Interestingly, the reuptake function of the glutamate transporter (GluT) is disrupted in pathological conditions after HI and the glutamate is released instead of being recaptured, which further increases its extracellular concentration (76).
Calcium is responsible for the activation of diverse enzymes, such as phospholipases, proteases, endonucleases, and neuronal nitric oxide synthase. The surge of intracellular calcium affects the mitochondria, which captures cations and subsequently produces free radicals and reactive oxygen/nitrogen species (ROS/RNS). Oxidative stress has a profound impact on enzyme function, transcription factor expression, lipid oxidation, and DNA damage (47, 69).
Various other neurotransmitter receptors, such as Dopamine D2 and D3, GABAA, and Adenosine A receptors have been shown to play a role in mature OL injury after HI (75, 77, 78). Other mechanisms include upregulation of the expression of members of the kinin protein family. OLs respond to kinins via specific receptors that elicit a calcium influx (79).
The presence of a systemic inflammatory state sensitizes the brain to injury. Increased concentrations of peripheral cytokines are a marker of cerebral inflammation since, in a neuro-inflammatory context, a sustained release of cytokines alters the blood–brain barrier. Infection can predispose the fetus to white matter lesions, leading to neurological impairments (15). Interestingly, some studies failed to show a direct causal link between chorioamnionitis or fetal infection and early brain development abnormalities in preterm infants, while neonatal infection shows higher risk of injury (80, 81). However, several other animal studies have shown a strong link between fetal infection and brain injury. For example, intraperitoneal injection of interleukin-1β (IL-1β) in newborn mice triggers systemic inflammation that modifies programs controlling brain development (82). IL-6 has been shown to be a key trophic factor for OL survival in vitro, while this effect is increased when associated with other trophic factors, such as neurotrophins (83). IL-6 and tumor necrosis factor α stimulate astrocyte proliferation and inhibit neural precursor cell proliferation in rodents. Lipopolysaccharide (LPS) has also been shown to cause susceptibility to hypoxic–ischemic insult in the newborn brain (84). Remarkably, while cytokines can be pro-inflammatory and act as mediators in the infection process, they also can have neuroprotective properties (63).
Inflammation can also trigger gliosis, which is a non-specific response to injury from glial cells, such as astrocytes and microglia. Microglia are among the first cells to react to injury and mediate neurotoxicity and they are strongly involved in the inflammatory response (85). Reactive astrocytes contribute to the glial scar and can subsequently inhibit axonal regrowth and myelination (86). Inflammation also contributes to microglia activation and augments their phagocytic capacity (20), leading, for example, to phagocytic degeneration of axons and necrotic/apoptotic cells (87). Microglia and reactive astrocytes also release interferon γ, which has a damaging effect on pre-OLs in PVL (88).
Other components, altered by the inflammation process, play an important role in glial scarring. For example, chondroitin sulfate proteoglycans (CSPGs) and immunoglobulin cell adhesion molecules (IgCAMs) inhibit regeneration, and activation of toll-like receptors (TLRs) alters the neuroimmune response and OL maturation (20, 89, 90).
A recent study, however, suggested that hyperoxia (excess of oxygen) and/or hypoxia can alter the expression of various genes, independent of any oxidative stress mechanisms (24). The level of oxygen can regulate transcription factors, such as the HIFs, which are heterodimers of an oxygen-regulated α subunit and an oxygen-insensitive β subunit. Three HIFs have been described to date. In the absence of oxygen, HIF1 and 2 are deactivated by asparaginyl and prolyl hydroxylases, two oxygen-dependent enzymes. Thus, HIFs cannot form a heterodimer with the β subunit and translocate to the nucleus to elicit specific DNA transcription (91). Therefore, postnatal myelination (starting at PND7–9 and peaking at PND15–21) is impaired if pre-OLs-encoded HIF function is altered by the absence of oxygen (48).
In summary, CP is triggered during a time of rapid brain development and maturation with many mechanisms at play, most notably an alteration of brain development. The major pathophysiological events occurring during CP onset in both rodents and humans may be seen in Table 2.
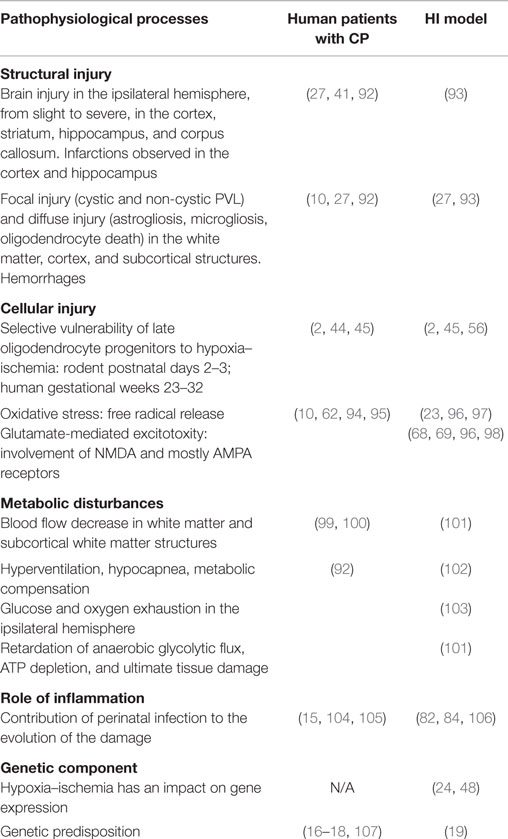
Table 2. Major pathophysiological processes in CP reproduced in the rodent HI model.
Cognitive Impairments
In addition to motor impairment, as categorized by the GMFCS, children with CP are more susceptible to several other health problems. These include orthopedic problems, epilepsy, chronic pain, as well as sensory and cognitive impairments. Indeed, children with CP often exhibit cognitive and behavioral deficits, including lower IQ, deficits in short-term memory, and hearing impairment frequently resulting in speech deficiencies and/or delayed language development (108, 109). Moreover, visual perception is often affected by CP, and is considered to be a core disorder of the CP syndrome (110).
Very preterm or very low birth weight infants are at significant risk of developing cognitive impairments (111). Neonatal brain injury in these children leads to adverse attention and processing speed outcomes at the age of 7 (112). MRI has become a tool to assess neonatal brain injury and predict potential childhood outcomes (113, 114).
Animal studies have helped to characterize these cognitive impairments. Neuro-behavioral tests in animal models of CP have shown a number of cognitive impairments, including selective and long-lasting learning and memory impairments (115), and deficits in auditory processing (116).
Interestingly, male HI rats can learn a complex spatial memory task if exposed to gradual and repetitive situations, whereas performance is reduced if a high memory demand is requested at once (117). Furthermore, differing results have been observed according to which brain hemisphere is injured, supporting the notion that the neonatal right brain is more vulnerable to HI (118, 119). However, many human studies and animal bilateral injury models have failed to confirm the concept of brain lateralization, suggesting the need for further investigation in this area.
Potential Therapies
Therapeutic hypothermia is a standard of care procedure for term infants with moderate or severe perinatal asphyxia (120–122). This treatment is, however, not recommended in preterm infants due to potential collateral damage, such as intra-cerebral hemorrhage (122).
In an immature newborn, therapeutic strategies could interfere with the normal development of the brain making use of potential neuroprotective agents risky. Robertson et al. reviewed several compounds, including US-Food and Drug Administration (FDA) approved (i.e., Tetrahydrobiopterin, Melatonin, Erythropoietin) and not FDA approved (i.e., Xenon, Epo-mimetics), that could be used to treat the newborn infant (123).
Fleiss and colleagues (20) also reviewed the treatment options for tertiary brain damage, including targeting long-lasting inflammation, modulation of microglia or lymphocytes, and cell therapy.
The use of stem cells as a potential regenerative approach seems to be a promising strategy. Functional recovery after stem cell transplantation has been shown in many cases, including in spinal cord injury models (124, 125) and in a dysmyelinated animal model (126). However, many aspects of cell therapies, such as safety, efficacy, timing, and dose, still need to be optimized (127), while a combinatorial approach with, for example, rehabilitative strategies, use of growth factors or engineered stem cells, is likely to be the optimal strategy rather than any of these alone (128).
Clinical trials for CP encounter difficulties defining recruitment criteria and recruiting participants, mainly due to the heterogeneous patient population or inadequate group sizes. Rodent models can be of great help since they allow the examination of biological mechanisms in a safe and cost-effective manner. They are also a useful tool for testing therapeutic strategies. For example, specific microRNAs have been shown to regulate T-cell function and intracellular ATP concentrations. Furthermore, growth factors as well as drug and cell therapies are being investigated in animal models (20, 129).
Therefore, the need for rodent models of CP is great. However, each adaptation of the classic hypoxic–ischemic Rice–Vannucci model should be proven to be clinically relevant, and used appropriately once validated. The following sections discuss the advantages, limitations, and translational value of these models.
The Classic Rice–Vannucci Model
The Rice–Vannucci rat HI model (93) is the first and best studied HI neonatal model. This model allowed the study of the mechanisms underlying HI-related brain damage during development and is one of the most productive models of perinatal brain injury to have been described (130).
This model, using postnatal day 7 (PND7) neonate rats, is an adaptation of the Levine model of HI in adult rats. It is widely accepted that the 7-day-old rat has brain maturity equivalent to that of a term/near term human fetus (131).
Briefly, this model is accomplished as follows: the common carotid artery is ligated unilaterally in PND7 Sprague-Dawley rat pups. The animals rest for 4–8 h before being exposed to hypoxic air (8% oxygen) for 3.5 h at 37°C. They are subsequently returned to their dam. In this original publication, brain damage was observed after 50 h in the ipsilateral hemisphere but not in the contralateral one (93). Others have observed damage much earlier (e.g., 2 h following the insult) (132). Using these experimental conditions, hypoxia alone (without carotid occlusion) does not result in brain damage nor does carotid unilateral occlusion alone (without hypoxia) (133). After unilateral carotid occlusion alone, a regulation of the ischemic effect has been observed due to the presence of the circle of Willis that allows compensation in the blood flow between the brain hemispheres.
The injury induced in this model can be described as a spectrum, ranging from “moderate” to “severe.” Selective neuronal death or infarction can be observed, depending on the severity of the HI insult (93, 133, 134). The injuries affect the cerebral cortex, the deep gray matter (striatum of the basal ganglia and thalamus), the subcortical and periventricular white matter, and the hippocampus in the majority of the animals (90%). It should be noted that only 56% of the animals have an infarction, suggesting a variability in the technique.
A hypoxic–ischemic insult of an inadequate level to induce rapid cell death and infarction can trigger some prolonged injury mechanisms. Glutamate excitotoxicity as well as oxidative stress play crucial roles in cell death following HI insult (133).
This model has been adapted many times since it was originally described. It has been confirmed to be extremely useful in various studies of perinatal hypoxic–ischemic brain damage. Since its first description in 1981, the Rice–Vannucci model has been widely used and adapted by scientists around the world, on various species.
However, studies have revealed that the model does not mimic the human condition optimally. Specifically, the stage of white matter development in PND7 rats and the type of injury produced are not similar to that in humans. At PND7, the majority of OLs in rat brains are at the immature stage of development as defined by O4+/O1+ immunohistochemistry (57). By contrast, human injury occurs when OLs are at the earlier stage of development, which can be distinguished by O4+/O1− staining (44). O4+/O1− staining shows that the corresponding period in rodents is PND 2 (56–58). Another important consideration involves the type of injury the model aims to reproduce. Most cases of PVL resulting in CP are associated with diffuse apoptotic and relatively small necrotic areas in the infant brain, affecting mostly white matter (23, 92, 135), while the Rice–Vannucci model often results in severe injury with multiple infarctions involving both white matter and gray matter (93, 136), which accounts less than for 5% of PVL cases (137–139).
Subtypes of the Rodent HI Model
In recent years, several optimizations of the rodent HI model have been developed to match the pathophysiological processes underlying brain injury and the consequences observed in human CP patients. Major differences in brain anatomy and the speed of brain development exist between humans and rodents. Fine-tuning of the timing and the parameters of the injury insult have resulted in a more accurate reproduction of the changes observed in humans (Table 3).
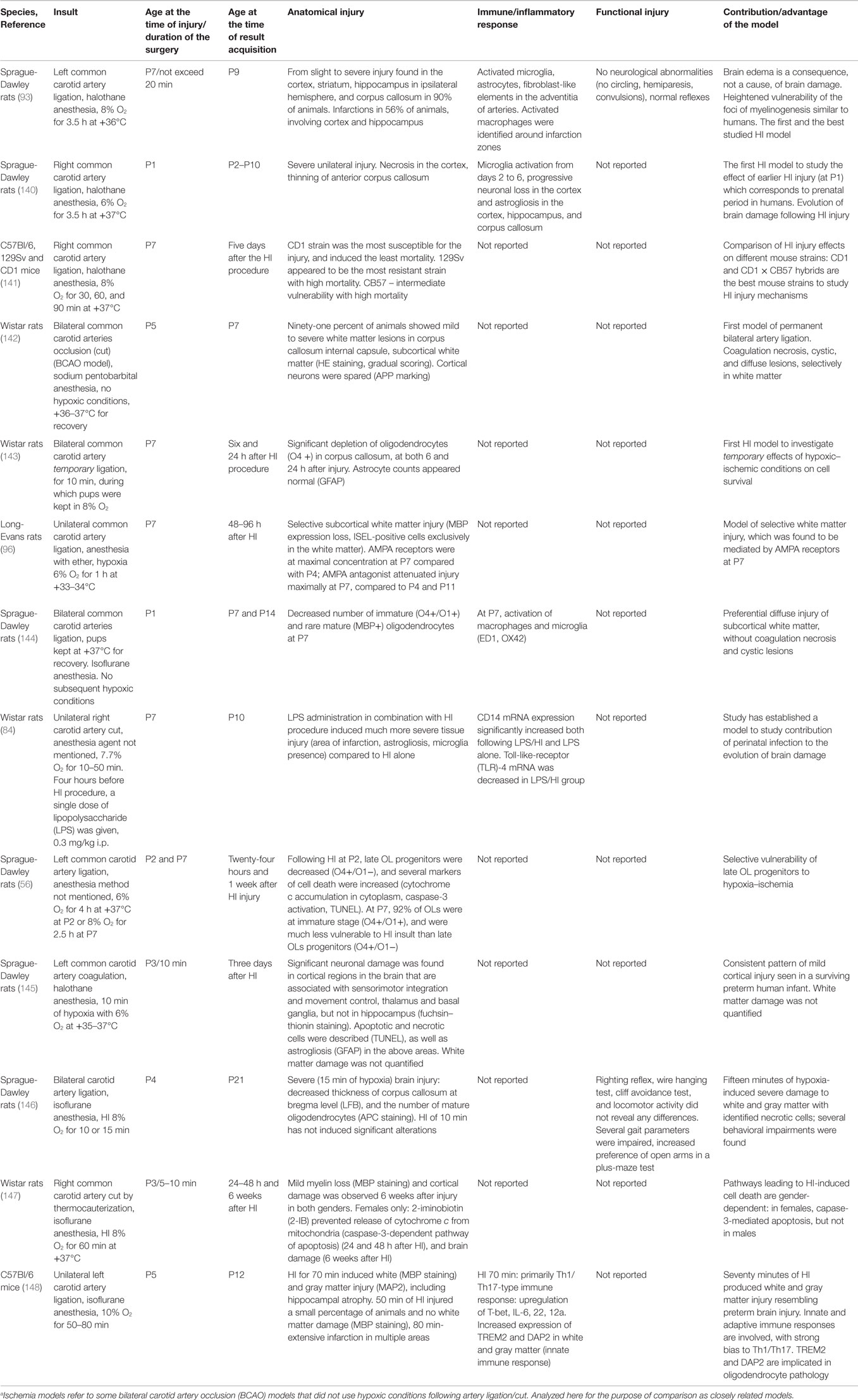
Table 3. Hypoxia–ischemia (HI) and ischemiaa rodent models developed to mimic pathophysiological processes and conditions in infants with prenatal asphyxia.
The first optimization of the HI model was done in 1996, when hypoxic–ischemic injury was induced in rat pups at PND1 (140). At PND 1, rat brain development is at a comparable stage to the human brain in the last trimester of gestation (149). Sheldon et al. showed that evolution of brain damage following HI includes progressive neuronal loss, white matter thinning, activation of microglia, and macrophages (140). Rat brains with this injury present with infarctions, necrotic areas, and cystic lesions similar to those seen in infants with severe forms of PVL (28).
As more research was undertaken using the HI model, concerns about strain differences were raised. In particular, susceptibility to the injury, mortality rates, and post-injury plasticity. Studies were commenced to obtain reproducible HI rodent models with controllable and standardized degrees of brain damage. Different strains of mice were compared in a study by Sheldon and colleagues (141), in which the CD1 strain was found to be preferable as it showed maximum brain vulnerability with little or no mortality. 129Sv and CB57 strains were found to be extremely resistant to injury and displayed very high mortality, respectively.
One year later, in 1999, a new HI model was developed which employed bilateral common carotid artery occlusion (BCAO) in Wistar rats at PND5 with no subsequent hypoxic conditions (142). The authors aimed to reproduce the ischemic conditions that occur in the human fetus, i.e., reduction of cerebrospinal fluid (CSF) flow resulting in mostly white matter lesions. In the Rice–Vannucci model, CSF flow in subcortical white matter was reduced by 60% (101), and was followed by severe white and gray matter damage. By contrast, in the BCAO model, CSF flow in subcortical white matter was reduced by 25%, which resulted in mild diffuse white matter damage in 64% of brains, or severe white matter damage with necrotic areas and cystic lesions in 36% of brains, while gray matter was spared. However, other researchers have failed to reproduce this model and the contradictory data warrant further investigation.
Also in 1999, another BCAO model was developed, which studied the effect of temporary bilateral ligation of common carotid arteries at PND7 in rats (143). Ligation was carried out for 10 min during which rat pups were kept in 8% oxygen. Progressive loss of O4 marked OLs was observed 24 h after injury. The report is very short and no other data were available, but this BCAO model is likely to produce more severe brain damage than the BCAO model by Uehara, since hypoxic conditions were added to the ischemic injury.
A study by Follett and colleagues also attempted to generate a reliable model of white matter injury (96). Parameters of the insult were changed in order to produce less severe brain damage: instead of 8% oxygen and 3.5 h of hypoxia at +37°C used in the Rice–Vannucci model, unilateral carotid artery occlusion was followed by 6% oxygen for 1 h at +33–34°C. Mild hypothermia during hypoxia has been shown to significantly reduce brain damage in rat pups when operated on at PND7 (150), and this was confirmed here. Selective mild white matter damage was seen 24–96 h after the HI procedure, as demonstrated by MBP expression loss and exclusive white matter ISEL (in situ end labeling) staining. The authors found that OL death was associated with activation of glutamate AMPA receptors, and vulnerability to AMPA activation was maximal at PND7 as compared to PND4 and PND11. At PND7, the number of AMPA receptors on OLs (most at the immature stage of development, O1+ marked) is significantly greater than that at PND4 [when pre-OLs (O4+/O1−) predominate], or at PND11 (mature cells, MBP-positive). Administration of the AMPA blocker NBQX prevented brain damage in animals at PND7, but not at PND4 and PND11. In humans, increased numbers of AMPA receptors were also found in pre-OLs and immature OLs between 24 and 32 weeks of gestation, and the mechanisms of glutamate-mediated cell death look similar (151, 152). Recent studies in rodents (153) have confirmed an important role for AMPA receptors that are located on pre-OLs and mediate their death, as opposed to immature OLs found in the study of Follett and colleagues (96). Hence, this study was important since suggested an additional possible pathophysiological mechanism for the HI-related injury and illustrated a specific vulnerability of immature OLs (O4+/O1+) compared to the younger (O4+/O1) and older OL cells (MBP+). However, this study is not without flaws and further validation of this model is required.
To study possible interactions between HI and infection/inflammation during the perinatal period, a new rodent model was introduced in 2001 (84). A single dose of LPS was given to 7-day-old rat pups 4 h before unilateral occlusion which was implemented as previously described (93). The combination of LPS with HI induced a more severe brain injury compared to HI alone. For instance, areas of infarction were enlarged, and there were increased concentrations of astrocytes and microglia around infarction zones. Mechanisms of immune system involvement in tissue damage were explored, and alterations in TLRs, and CD14 protein concentrations were found. Further research is needed to elucidate the pathways of interaction of pathophysiological processes.
In subsequent years, two further models of BCAO were reported, in addition to the models of Uehara (PND5) and Jelinski (PND7). In the first model (144), surgery was performed in rat pups at PND1, and no hypoxia conditions were applied. Mild diffuse injury was similar to that seen in human infants with PVL. More specifically, the injury was restricted to white matter and characterized by decreased numbers of immature and mature OLs, and activation of macrophages and microglia (144). In the second new model, surgery was undertaken in rats at PND4, and, in addition to bilateral arterial occlusion, rats were exposed to 10–15 min of 8% oxygen hypoxic conditions. Severe gray and white matter damage occurred with necrotic areas, loss of mature OLs and thinning of the corpus callosum (146).
The three models of BCAO described above produced mild and severe brain damage, with selectively affected white matter or gray matter. The only BCAO model undertaken in PND1 rats (144) produced a selective diffuse white matter injury similar to that seen in most PVL cases. The extent of the damage may be related to the younger age, different anesthesia agent used, or to the method of artery occlusion. Anesthesia duration may affect the degree of the injury (154), which is not specified in the study reports.
In 2002, Back et al. published data demonstrating that late OL progenitors (O4+/O1−) are selectively vulnerable to HI (56), similar to late OL progenitors in human infants at 24–32 weeks of gestation (44). Those OL progenitors were found to predominate at PND 2 (56–58). At PND7, immature OLs comprise 92% of all OL cells in the corpus callosum and are significantly less sensitive to HI compared to late OL progenitors (56). These findings gave rise to subsequent work using rats and mice less than 1 week old.
HI performed in rat pups at PND3 with subsequent hypoxia for 10 min produced a mild injury of cortical areas, thalamus and basal ganglia, necrotic and apoptotic cells, and astrogliosis. However, white matter damage was not quantified (145). The authors were successful in producing consistent mild brain damage due to preserving HI parameters: duration of surgery was kept within 10 min, subsequent hypoxia performed with 6–7% oxygen, +35–37°C, and 98–100% humidity.
When the surgery was done at PND3 with subsequent hypoxia using 8% oxygen for 60 min, mild white and gray matter damage was observed (147). For the first time, gender differences in HI-associated pathophysiological processes were described: post-hypoxia injections of 2-iminobiotin (2-IB) prevented brain damage in females only by suppressing release of cytochrome c from mitochondria. Results indicated that HI-induced cell death is mediated by the caspase-3 pathway in females, but not in males. This study reported a high degree of consistency in their results likely due to keeping the duration of isoflurane-maintained surgery constant (154).
A mouse model that employed HI performed at PND5 was developed recently (148). Different durations of hypoxia were investigated, and 70 min of hypoxia was found to induce a pattern of diffuse white (MBP staining) and focal gray (MAP2) matter injury similar to that in preterm infants with PVL. Innate and adaptive immune responses were recorded, and implicated in the induction of white matter damage. While MBP staining is not sufficient alone to replicate human pathology, it can indicate pathology of mature OLs and partially reveal the pathological mechanisms.
Advancements in Clinical Management of Perinatal Complications in Human Infants Based on Rodent HI Model Research
Rigorous research using HI models during the last decades have elucidated mechanisms of prenatal white and gray matter injury, and enabled the development of clinical recommendations to diminish injury consequences in premature infants. Avoidance of factors implicated in damage occurrence has led to introduction of following clinical approaches.
Maintenance of Infant Cerebral Perfusion
Maintenance of infant cerebral perfusion was found to be important based on studies done using the rodent HI model (101, 155), and significant effort has been put into the development of methods to detect infants with impaired cerebrovascular regulation. Significant decrease in cerebral blood flow during the first 12 h of life, as detected by near-infrared spectroscopy (NIRS) and color Doppler ultrasonography (cD-USG), correlates with poor neurological outcome or death (156, 157). Consequently, an array of critical care methods aimed at hypotension management has been developed (158). Prior to birth, use of magnesium sulfate, which, in addition to its vasodilation properties, has antioxidant and anticytokine effects, was found to be beneficial in preventing brain injury and is currently used in clinical practice (135, 159–162).
Post-Injury (PND7) Hypothermia
Post-injury (PND7) hypothermia (+31–34°C) in the HI model was found to reduce brain damage to the cortex (150), including hippocampus, thalamus, and basal ganglia (120), with improvement evident at PND42 (163). Similar results were found in fetal sheep (164) and newborn piglets (165), which have greater similarities to human brain maturation rates and structures than rodents (149, 166). Therapeutic hypothermia is now the standard of care for brain injury control in term infants with perinatal hypoxic–ischemic encephalopathy (167, 168); however, it is not yet recommended for preterm infants.
Prevention of Pre-OL Death
Prevention of pre-OL death (92) and maturation arrest are of greatest value. Activation of glutamate NMDA (74, 169) and AMPA (96) receptors is associated with OL death in HI However, no clinical trials investigating the use of glutamate antagonists have been commenced. Activation of another glutamate receptor type, the mGluR, which is developmentally regulated and is maximally expressed on the most vulnerable pre-OLs (170), was found to counteract oxidative stress and protect OLs (171). Others have investigated the role of the constituents of natural health products, and sulforaphane, as potential therapies to reducing the effects of oxidative stress and inflammation, in the prevention of CP (172). These promising findings are still awaiting translation to clinic.
Measures to address infection/inflammation prevention, identification, and management during pregnancy have also been considered (173). Despite the fact that the link between gestational inflammation and brain damage was not confirmed in several studies (80, 81), at present the standard of care in the US involves the administration of broad spectrum antibiotics when chorioamnionitis is diagnosed (174).
Discussion: Limitations of Rodent HI Models and Their Clinical Applicability
The focus of the present paper is on rodent HI models since rodents are the most cost-effective and the most used species in laboratory research. Furthermore, availability of antibodies and transgenic animals allows comprehensive exploration of many physiological mechanisms. However, considerable limitations of rodent models restrict their use, and the present review is intended to reveal gaps in the accumulated knowledge and to lessen redundancies in ongoing research.
Differences in Brain Organization and the Time Line of Brain Maturation in Rodents and Humans
Research undertaken in the HI rodent models during the last decades has found many similarities between the mechanisms of brain injury and damage evolution in rodents and human infants with CP. Fine-tuned HI models successfully reproduce hypoxic–ischemic conditions, the apoptotic–necrotic pattern of white and gray matter damage, and neuromotor impairments. However, the greatest limitation of the HI rodent models is the difference between rodents and humans in the overall complexity of brain organization, and the discrepancies in the rate of maturation in any time period (149).
Hypoxia–ischemia rodent models can be used to induce brain injury at a similar stage of cellular development: between 24 and 32 weeks of gestation in human, and at PND2-3 in rodents. Most of the oligodendroglial lineage cells are at the stage of pre-OLs at these time points, and these are maximally vulnerable to hypoxic–ischemic injury (Table 1). However, despite the existence of a similar cellular stage of development, the speed and duration of structural maturation of the developing brain is significantly different in rodents and humans. There is a huge difference between the size of a human and a rodent brain and, in particular, the cerebral cortex, which is associated with considerably longer neurogenesis in humans (175, 176). Accordingly, the structure and relative size of the subplate, which is the major target of hypoxic–ischemic injury, is different in humans and rodents. In humans, the subplate is a more dynamic structure, is more complex and compartmentalized, and is involved to a greater extent in the formation of the cortex pathways than in rodents (177, 178). In rodents, the subplate is a complete structure by E16–18 (179), while in humans it reaches its maximal thickness considerably later, at 24–32 weeks of gestation (37). Moreover, the increased complexity of the human subplate may contribute to differential vulnerability in response to HI in rodents and humans (180).
In an optimized HI rodent model, injury is induced at PND2–3, to match the developmental stage of the OLs to that in humans (24–32 weeks of gestation) (55–57, 96). Moreover, striking similarities between reduced cortical activity following HI and subsequent brain development both in infants and in rodents have been found (33). However, the state of subplate development is different at these time points: at PND2–3 in rodents, the subplate is already losing cells and function (181), whereas in humans at 24–32 weeks of gestation the subplate is at its maximal thickness and is actively participating in corticogenesis (37). Therefore, the consequences of subplate damage will be significantly different between humans and rodents, which indicates the limited clinical relevance of this HI rodent model (180).
Sex Differences in Response to Prenatal Injury
Another important limitation of HI models concerns sex differences relating to brain vulnerability and the ability to repair in humans and rodents. In low weight infant boys, the incidence of intraventricular hemorrhage is significantly higher (182), as is the incidence of spastic CP, compared to girls (by 20–30%) (183–186). Moreover, the recovery of brain volumes is lower in boys than in girls: low weight preterm boys still present with white matter deficits at the age of 8, while matched-weight girls do not (187).
In the rodent model, sex differences in the pathophysiological mechanisms underlying brain damage evolution and recovery were observed. In females, caspase-dependent apoptosis has been described (147, 188), while in males, cell death and subsequent brain damage is associated with a different pathway involving PARP activation (189). Hypothermia following HI injury was beneficial for females only (163); however, the finding of sex differences was not a primary aim of the study, and the results, therefore, require additional verification, as acknowledged by the authors. Despite the different sex physiological processes in response to injury, the final extent of brain damage following HI at PND7 (190, 191) or before puberty (192), was similar in males and females. However, the above studies were done in PND7 and older animals, when development of a rodent brain roughly corresponds to a term human infant. Delivery of acute HI injury at PND3 (193) or chronic HI between PND3 and PND11 (194) induced significantly greater damage of white and gray matter, greater reduction in brain volume and more cognitive deficits in males compared to females, thereby resembling the damage pattern observed in preterm infants.
In rodents and humans, sex-specific responses to hypoxic–ischemic injury are different, making the rodent model a non-ideal choice to study gender-dependent mechanisms of CP. However, sex-specific influences are complex, and the contradictory evidence (193, 194) warrants further research to elucidate sex-specific mechanisms of HI-response maturation.
Modeling Motor Impairments
Spastic CP is characterized by aberrant reflex development and the lack of voluntary control (8). Unilateral damage of the CST during the prenatal period, when motor networks are at the stage of refinement – strengthening useful and eliminating redundant connections – results in ipsilateral aberrant CST projections and abnormal reflexes that manifest in spasticity (195, 196).
Considerable data have been accumulated regarding motor impairments in the rodent HI model. Despite rodents not showing the severe spastic impairments observed in human patients, gross motor function impairments could be said to resemble those in hemi- or diplegic CP (146, 190, 197–200). Most studies were undertaken within a week of the insult, and a relatively small number of papers have addressed the correlation between specific lesions and motor performance in the long term (2). Impairments in fine skilled voluntary movement following HI are studied even more rarely (198), even though they are present in most cases of spastic CP due to corticospinal pathway damage (201–204).
There are several concerns regarding the modeling of CST injury in rodent HI models. First, the overall anatomy of the CST differs considerably from that in humans. Rodents have very few direct connections between the motor cortex and motor neurons in the ventral horn of the spinal cord (205, 206), specifically for motor neurons innervating the muscles of the fingers. Therefore, impairments of skilled voluntary movements may be less pronounced in rodents, and require sensitive behavioral tests for identification.
Another important anatomical difference is the number of axons within the CST, which decussate in the medulla to the contralateral side. In rodents 2–4% axons remain uncrossed (207), whereas in humans about 8–15% of axons stay at the ipsilateral side (195). In humans, there is an even greater amount of ipsilateral axons within the CST in the prenatal period when injury occurs. These form transient connections with spinal cord neurons and disappear during postnatal development due to the functional competition with contralateral projections. In patients with hemiplegia, those ipsilateral connections are preserved and contribute to spastic symptoms (196, 208, 209). In rodents, research suggests that the mostly contralateral pattern of innervation is determined from the onset with no transient ipsilateral projections comparable to those in humans (210, 211). However, in rodents, if brain lesions are induced in the first week after birth, when the majority of corticospinal fibers are growing into the spinal cord, considerably enlarged ipsilateral connections are formed that might resemble the aberrant ipsilateral CST projections observed in CP patients (212, 213). Indeed, mice demonstrated mirror voluntary bilateral synchronous movements following CST injury at PND0-3 that resembled mirror movements in CP patients (214).
In addition to governing voluntary fine movements of the forelimb, the CST plays an important role in the fine-tuning of spinal reflexes during spinal networks maturation, both in rodents (215, 216) and humans (211, 217, 218). However, the extent of that influence is likely different in rodents and humans, as evidenced by the effect of prenatal CST damage. More specifically, in humans this type of damage is associated with aberrant reflexes and disabling spasticity, while in rodents the effect is considerably weaker.
In summary, the anatomy of the CST is considerably different in rodents and humans, which limits clinical applicability of HI models. However, there is still a possibility of mimicking and studying specific processes involved in functional alteration and restoration, which may contribute to the development of treatment strategies in humans. For example, finding the crucial role of CST influence on fine-tuning of spinal modules during their maturation suggests that CP infants would benefit from boosting activity in the residual corticospinal innervation that might restore spinal connectivity pattern and reduce spasticity in future (211). In the rodent model, stimulating the HI-injured hemisphere will have an overall similar effect, though less pronounced in the ipsilateral side, due to less CST ipsilateral connections in rodents compared to humans.
The Rodent Model Versus Large Animal Models
The rodent HI model is the most convenient, cost-effective, and widely used animal model and has yielded a substantial body of knowledge on CP pathophysiology. However, limitations of the rodent HI model have prompted research in large animal species with greater similarity to humans. The fetal instrumented sheep model has been characterized: preterm fetal fetus (95 days of gestation, equivalent to 24–28 gestational weeks in humans) was found to display cerebral hemodynamics similar to that in the human fetus, both in normal conditions (219, 220) and in response to HI (221, 222). Moreover, the gyrencephalic structure of the sheep brain cerebrum and the major stages of neurodevelopment are similar to those in humans (223–225). In addition, the larger size of sheep allows chronic monitoring of the fetus and relevant clinical interventions have been developed based on that model (164). The major disadvantage of the instrumented fetal sheep model is the lack of opportunity to study motor deficiencies, especially dexterity movements, associated with hypoxic–ischemic insult (226).
A non-human primate model developed in baboons at 125 days of gestation, equivalent to 26 weeks of gestation in human, has shown similarities with human preterm infants in the pattern of white matter injury (227). The primate model is associated with the necessity for artificial ventilation (228) that may resemble a specific human condition, though overall it may restrict the interpretation and applicability of the results.
A HI model in a newborn piglet has demonstrated electrophysiological and neuropathological disturbances similar to those in the asphyxiated term human infant (229). The model is able to mimic comparable changes in brain white matter to a human newborn (230). However, high mortality and discrepancies in protocols result in variability in the extent of brain injury and make it difficult to draw firm conclusions.
Intra- Versus Extra-Uterine Models
Differences in the rate of brain maturation between rodents and humans prevent concomitant times when both structural and cellular components are at the same level of development. Thus, in intra-uterine studies in rodents (E17–19), injury is induced when the brain subplate is already a complete structure (179). This period corresponds to 24–32 gestational weeks in humans when the subplate reaches its maximal thickness (37) and synaptogenesis begins both in rodents (231, 232) and in humans (233, 234). While at this stage the level of subplate development is comparable in both rodents and humans, the subsequent differentiation and function of the subplate in humans is considerably different from that in rodents (180). Moreover, maximal population of pre-OLs in rodents is found between PND2 and PND5, while in humans the peak of subplate development is between 23 and 32 weeks of gestation that coincides with the period of highest vulnerability to perinatal brain injury (32).
By contrast, extra-uterine HI models aim to induce brain injury during maximal OL vulnerability, for example, between PND2 and PND5 in rodents. Therefore, intra-uterine rodent models target different aspects of brain pathophysiology associated with HI (65). Intra-uterine growth restriction models are being developed in guinea pigs (235–237), mice (238), rabbits (239), and rats (67, 172, 240, 241).
The instrumented fetal sheep model makes it possible to induce hypoxic–ischemic injury at the stage of both the subplate and pre-OLs being at the similar stages of development in sheep and humans and, therefore, more closely mimics events accompanying hypoxic–ischemic insult (226).
Conclusion
The rodent HI model continues to contribute to CP research. It mimics many pathophysiological changes in human patients with spastic CP and allows the study of the mechanisms underlying pre- or perinatal brain injury. Unilateral injury induced at PND3-4 in combination with precisely adjusted temperature and oxygen level and surgery duration can reproduce brain damage patterns and motor impairments similar to those in hemiplegic CP patients. Moreover, the model allows investigation of new methods of brain damage prevention and repair both at the cellular (introduction of pharmacological or biotechnological agents interrupting the cascade of pathophysiological events) and the systemic level (stem cell transplantation or behavioral manipulations stimulating anatomical and functional restoration).
Research done using the HI rodent model has identified factors implicated in brain pathophysiology, and has made important contributions to the current clinical practice of treating CP patients. For instance, postnatal hypothermia, management of decreased cerebral blood flow in newborns, and management of prenatal infection are common standards of care in many countries at present. However, the model has important limitations, such as overall anatomical differences between human and rodent brains, discrepancies in the rate of maturation at any time period, and in gender-specific mechanisms. These limitations restrict the clinical applicability of the results. The rodent model is an ideal cost-effective model for proof-of-concept studies and investigation of molecular mechanisms that can be validated subsequently in large pre-clinical animal models, such as the instrumented fetal sheep (226), and the monkey model that, in addition to physiological and size similarities, allows evaluation of functional outcomes.
Method
The goal of the present study was to identify HI rodent models developed to date, analyze their contributions to the understanding of CP pathophysiological mechanisms, and to comment on the overall potential of the HI rodent model to mimic those mechanisms. A systematic literature search was conducted in May–July 2015 using PubMed, Scopus, Medline, Cochrane Library, and Web of Science databases in English. The following search strategies were used.
PubMed (All Years)
Title/Abstract (“hypoxia ischemia” OR “hypoxic ischemic” OR “BCAO”) AND Title/Abstract (“rat” OR “rats” OR “mice” OR “mouse”) AND Title/Abstract (“cerebral palsy” OR “white matter” OR “injury”) returned 1434 results.
Scopus
Title, abstract, keywords (“hypoxia ischemia” OR “hypoxic ischemic” OR BCAO) AND Title, abstract, keywords (“rat?” OR “mice” OR “mouse”) AND Title, abstract, keywords (“cerebral palsy” OR “white matter” OR “injury”) returned 1364 results.
Medline (OVID MedLine without Revisions, 1996 – July week 2, 2015)
Title/Abstract (“hypoxia ischemia” OR “hypoxic ischemic”) AND Title/Abstact (“rat” OR “rats” OR “mice” OR “mouse”) AND Title/Abstact (“cerebral palsy” OR “white matter” OR “injury”) returned 1554 results.
Cochrane Library (All Databases, All Years)
Title, abstract, keywords (“hypoxia ischemia” OR “hypoxic ischemic” OR BCAO) AND (rat OR rats OR mice OR mouse) AND (“cerebral palsy” OR “white matter” OR injury) returned 3 results.
Web of Science (without Restrictions, All Years)
Title: (“hypoxia ischemia” OR “hypoxic ischemic” OR BCAO) AND Topic: (rat OR rats OR mice OR mouse) AND Topic: (“cerebral palsy” OR “white matter” OR injury) returned 1341 results.
Titles and abstracts of found papers were screened to determine if they fell into the scope of the review. From the selected papers, only those which satisfied the following criteria were analyzed.
Criteria for Paper Inclusion into Analysis
The following papers were included: papers that were relevant to the subject, clearly described methods of research, had control groups, used appropriate outcome measures in which subjects are randomized and data scoring were blinded, and that report limitations of the research conducted. A total of 11 distinct rodent HI and two ischemic models (Table 3) were identified and analyzed in our review.
Author Contributions
PR – drafting of manuscript, revision, and editing. TB – drafting of manuscript, revision, and editing. SM – edits and revisions to manuscript. JY – edits and revisions to manuscript. MF – concept of review, editing, revision, and final approval of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Madeleine O’Higgins, Dr. Paul Bradshaw, and Dr. Nicole Forgione for editing and publication assistance.
Funding
This research received funding from NeuroDevNet. This research was conducted with the support of the Ontario Brain Institute an independent non-profit corporation funded partially by the Ontario government. The opinions results and conclusions are those of the authors and no endorsement by the Ontario Brain Institute is intended or should be inferred.
References
1. Colver A, Fairhurst C, Pharoah PO. Cerebral palsy. Lancet (2014) 383:1240–9. doi:10.1016/S0140-6736(13)61835-8
2. Clowry GJ, Basuodan R, Chan F. What are the best animal models for testing early intervention in cerebral palsy? Front Neurol (2014) 5:258. doi:10.3389/fneur.2014.00258
3. Dunn PM. Dr William Little (1810-1894) of London and cerebral palsy. Arch Dis Child Fetal Neonatal Ed (1995) 72:F209–10. doi:10.1136/fn.72.3.F209
4. Palisano R, Rosenbaum P, Walter S, Russell D, Wood E, Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol (1997) 39:214–23. doi:10.1111/j.1469-8749.1997.tb07414.x
5. Palisano RJ, Rosenbaum P, Bartlett D, Livingston MH. Content validity of the expanded and revised gross motor function classification system. Dev Med Child Neurol (2008) 50:744–50. doi:10.1111/j.1469-8749.2008.03089.x
6. Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW, Task Force on Childhood Motor, D. Classification and definition of disorders causing hypertonia in childhood. Pediatrics (2003) 111:e89–97. doi:10.1542/peds.111.1.e89
7. Rethlefsen SA, Ryan DD, Kay RM. Classification systems in cerebral palsy. Orthop Clin North Am (2010) 41:457–67. doi:10.1016/j.ocl.2010.06.005
8. Surveillance of Cerebral Palsy in Europe. Surveillance of cerebral palsy in Europe: a collaboration of cerebral palsy surveys and registers. Dev Med Child Neurol (2000) 42:816–24. doi:10.1111/j.1469-8749.2000.tb00695.x
9. Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl (2007) 109:8–14. doi:10.1111/j.1469-8749.2007.tb12610.x
10. Marret S, Vanhulle C, Laquerriere A. Pathophysiology of cerebral palsy. Handb Clin Neurol (2013) 111:169–76. doi:10.1016/B978-0-444-52891-9.00016-6
12. Stanley FJ, Blair E, Alberman ED. Cerebral Palsies: Epidemiology and Causal Pathways. London; New York: Mac Keith; Cambridge University Press (2000).
13. Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev (2002) 8:30–8. doi:10.1002/mrdd.10007
14. Graham EM, Ruis KA, Hartman AL, Northington FJ, Fox HE. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol (2008) 199:587–95. doi:10.1016/j.ajog.2008.06.094
15. Van Steenwinckel J, Schang AL, Sigaut S, Chhor V, Degos V, Hagberg H, et al. Brain damage of the preterm infant: new insights into the role of inflammation. Biochem Soc Trans (2014) 42:557–63. doi:10.1042/BST20130284
16. Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, Zarrei M, Andersen J, Wei J, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun (2015) 6:7949. doi:10.1038/ncomms8949
17. Levenson D. Genetic factors may underlie many cerebral palsy cases: new research implicates genetic variations, not lack of oxygen at birth, in disorder. Am J Med Genet A (2015) 167:vii–viii. doi:10.1002/ajmg.a.37283
18. MacLennan AH, Thompson SC, Gecz J. Cerebral palsy: causes, pathways, and the role of genetic variants. Am J Obstet Gynecol (2015) 213(6):779–88. doi:10.1016/j.ajog.2015.05.034
19. Johnston MV, Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev Med Child Neurol (2007) 49:74–8. doi:10.1017/S0012162207000199.x
20. Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol (2012) 11:556–66. doi:10.1016/S1474-4422(12)70058-3
21. Krageloh-Mann I, Cans C. Cerebral palsy update. Brain Dev (2009) 31:537–44. doi:10.1016/j.braindev.2009.03.009
22. Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, et al. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol (2006) 497:199–208. doi:10.1002/cne.20991
23. Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed (2008) 93:F153–61. doi:10.1136/adc.2006.108837
24. Wellmann S, Buhrer C, Schmitz T. Focal necrosis and disturbed myelination in the white matter of newborn infants: a tale of too much or too little oxygen. Front Pediatr (2014) 2:143. doi:10.3389/fped.2014.00143. eCollection 2014
25. Sizonenko SV, Camm EJ, Dayer A, Kiss JZ. Glial responses to neonatal hypoxic-ischemic injury in the rat cerebral cortex. Int J Dev Neurosci (2008) 26:37–45. doi:10.1016/j.ijdevneu.2007.08.014
26. Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol (2003) 62:441–50. doi:10.1093/jnen/62.5.441
27. Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol (2009) 8:110–24. doi:10.1016/S1474-4422(08)70294-1
28. Volpe JJ. The encephalopathy of prematurity – brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol (2009) 16:167–78. doi:10.1016/j.spen.2009.09.005
29. Du Y, Dreyfus CF. Oligodendrocytes as providers of growth factors. J Neurosci Res (2002) 68:647–54. doi:10.1002/jnr.10245
30. Dai X, Lercher LD, Clinton PM, Du Y, Livingston DL, Vieira C, et al. The trophic role of oligodendrocytes in the basal forebrain. J Neurosci (2003) 23:5846–53.
31. McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci (2003) 23:3308–15.
32. McQuillen PS, Ferriero DM. Perinatal subplate neuron injury: implications for cortical development and plasticity. Brain Pathol (2005) 15:250–60. doi:10.1111/j.1750-3639.2005.tb00528.x
33. Ranasinghe S, Or G, Wang EY, Ievins A, Mclean MA, Niell CM, et al. Reduced cortical activity impairs development and plasticity after neonatal hypoxia ischemia. J Neurosci (2015) 35:11946–59. doi:10.1523/JNEUROSCI.2682-14.2015
34. Davatzikos C, Barzi A, Lawrie T, Hoon AH Jr, Melhem ER. Correlation of corpus callosal morphometry with cognitive and motor function in periventricular leukomalacia. Neuropediatrics (2003) 34:247–52. doi:10.1055/s-2003-43259
35. Anderson NG, Laurent I, Cook N, Woodward L, Inder TE. Growth rate of corpus callosum in very premature infants. AJNR Am J Neuroradiol (2005) 26:2685–90.
36. Anderson NG, Laurent I, Woodward LJ, Inder TE. Detection of impaired growth of the corpus callosum in premature infants. Pediatrics (2006) 118:951–60. doi:10.1542/peds.2006-0553
37. Kostovic I, Rakic P. Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol (1990) 297:441–70. doi:10.1002/cne.902970309
38. Luhmann HJ, Kilb W, Hanganu-Opatz IL. Subplate cells: amplifiers of neuronal activity in the developing cerebral cortex. Front Neuroanat (2009) 3:19. doi:10.3389/neuro.05.019.2009
39. Kanold PO, Luhmann HJ. The subplate and early cortical circuits. Annu Rev Neurosci (2010) 33:23–48. doi:10.1146/annurev-neuro-060909-153244
40. Kostovic I, Jovanov-Milosevic N. The development of cerebral connections during the first 20-45 weeks’ gestation. Semin Fetal Neonatal Med (2006) 11:415–22. doi:10.1016/j.siny.2006.07.001
41. Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA (2006) 296:1602–8. doi:10.1001/jama.296.13.1602
42. McClendon E, Chen K, Gong X, Sharifnia E, Hagen M, Cai V, et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol (2014) 75:508–24. doi:10.1002/ana.24100
43. Dean JM, Mcclendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med (2013) 5:168ra167. doi:10.1126/scitranslmed.3004669
44. Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci (2001) 21:1302–12.
45. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol (2013) 10(6–107):1–16. doi:10.1016/j.pneurobio.2013.04.001
46. Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol (1997) 56:219–35. doi:10.1097/00005072-199703000-00001
47. Mifsud G, Zammit C, Muscat R, Di Giovanni G, Valentino M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neurosci Ther (2014) 20:603–12. doi:10.1111/cns.12263
48. Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, et al. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell (2014) 158:383–96. doi:10.1016/j.cell.2014.04.052
49. Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol (2012) 71:93–109. doi:10.1002/ana.22627
50. Back SA, Miller SP. Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Ann Neurol (2014) 75:469–86. doi:10.1002/ana.24132
51. Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci (2006) 26:3045–55. doi:10.1523/JNEUROSCI.5200-05.2006
52. Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev (2001) 81:871–927.
53. Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia (2014) 62:1790–815. doi:10.1002/glia.22658
54. Downes N, Mullins P. The development of myelin in the brain of the juvenile rat. Toxicol Pathol (2014) 42:913–22. doi:10.1177/0192623313503518
55. Gard AL, Pfeiffer SE. Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development (1989) 106:119–32.
56. Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci (2002) 22:455–63.
57. Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, et al. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol (2003) 181:231–40. doi:10.1016/S0014-4886(03)00032-3
58. Dean JM, Moravec MD, Grafe M, Abend N, Ren J, Gong X, et al. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev Neurosci (2011) 33:251–60. doi:10.1159/000327242
59. El Waly B, Macchi M, Cayre M, Durbec P. Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci (2014) 8:145. doi:10.3389/fnins.2014.00145
60. Fernandez-Castaneda A, Gaultier A. Adult oligodendrocyte progenitor cells – multifaceted regulators of the CNS in health and disease. Brain Behav Immun (2016). doi:10.1016/j.bbi.2016.01.005
61. Tucker J, McGuire W. Epidemiology of preterm birth. BMJ (2004) 329:675–8. doi:10.1136/bmj.329.7467.675
62. Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci (2000) 20:34–42.
63. Girard S, Kadhim H, Roy M, Lavoie K, Brochu ME, Larouche A, et al. Role of perinatal inflammation in cerebral palsy. Pediatr Neurol (2009) 40:168–74. doi:10.1016/j.pediatrneurol.2008.09.016
64. Burd I, Balakrishnan B, Kannan S. Models of fetal brain injury, intrauterine inflammation, and preterm birth. Am J Reprod Immunol (2012) 67:287–94. doi:10.1111/j.1600-0897.2012.01110.x
65. Basilious A, Yager J, Fehlings MG. Neurological outcomes of animal models of uterine artery ligation and relevance to human intrauterine growth restriction: a systematic review. Dev Med Child Neurol (2015) 57:420–30. doi:10.1111/dmcn.12599
66. Faulkner S, Ruff C, Foltz W, Armstrong E, Basilious A, Fan S, et al. A translational model of intrauterine growth restriction: evaluating abnormalities in neurobehavioural gait and white matter myelination. Int J Dev Neurosci (2015) 47:45. doi:10.1016/j.ijdevneu.2015.04.128
67. Ruff CA, Faulkner SD, Armstrong E, Basilious A, Fan S, Thiyagalingam S, et al. Intrauterine growth restriction-induced cerebral palsy: evaluating histological and behavioural differences in a translational rodent model. Int J Dev Neurosci (2015) 47:60–1. doi:10.1016/j.ijdevneu.2015.04.169
68. Mesples B, Plaisant F, Fontaine RH, Gressens P. Pathophysiology of neonatal brain lesions: lessons from animal models of excitotoxicity. Acta Paediatr (2005) 94:185–90. doi:10.1111/j.1651-2227.2005.tb01888.x
69. Degos V, Loron G, Mantz J, Gressens P. Neuroprotective strategies for the neonatal brain. Anesth Analg (2008) 106:1670–80. doi:10.1213/ane.0b013e3181733f6f
70. Butt AM. Neurotransmitter-mediated calcium signalling in oligodendrocyte physiology and pathology. Glia (2006) 54:666–75. doi:10.1002/glia.20424
71. Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol (2009) 7:160–79. doi:10.2174/157015909789152146
72. James G, Butt AM. P2X and P2Y purinoreceptors mediate ATP-evoked calcium signalling in optic nerve glia in situ. Cell Calcium (2001) 30:251–9. doi:10.1054/ceca.2001.0232
73. Matute C, Ransom BR. Roles of white matter in central nervous system pathophysiologies. ASN Neuro (2012) 4:e00079. doi:10.1042/AN20110060
74. Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature (2005) 438:1167–71. doi:10.1038/nature04301
75. Karadottir R, Attwell D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience (2007) 145:1426–38. doi:10.1016/j.neuroscience.2006.08.070
76. Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia (2006) 53:212–24. doi:10.1002/glia.20275
77. Rosin C, Colombo S, Calver AA, Bates TE, Skaper SD. Dopamine D2 and D3 receptor agonists limit oligodendrocyte injury caused by glutamate oxidative stress and oxygen/glucose deprivation. Glia (2005) 52:336–43. doi:10.1002/glia.20250
78. Melani A, Cipriani S, Vannucchi MG, Nosi D, Donati C, Bruni P, et al. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain (2009) 132:1480–95. doi:10.1093/brain/awp076
79. Albert-Weissenberger C, Siren AL, Kleinschnitz C. Ischemic stroke and traumatic brain injury: the role of the kallikrein-kinin system. Prog Neurobiol (2013) 10(1–102):65–82. doi:10.1016/j.pneurobio.2012.11.004
80. Reiman M, Kujari H, Maunu J, Parkkola R, Rikalainen H, Lapinleimu H, et al. Does placental inflammation relate to brain lesions and volume in preterm infants? J Pediatr (2008) 152:.e1–2. doi:10.1016/j.jpeds.2007.09.051
81. Chau V, Poskitt KJ, Mcfadden DE, Bowen-Roberts T, Synnes A, Brant R, et al. Effect of chorioamnionitis on brain development and injury in premature newborns. Ann Neurol (2009) 66:155–64. doi:10.1002/ana.21713
82. Favrais G, Van De Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol (2011) 70:550–65. doi:10.1002/ana.22489
83. Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development (1993) 118:283–95.
84. Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, et al. Bacterial endotoxin sensitizes the immature brain to hypoxic – ischaemic injury. Eur J Neurosci (2001) 13:1101–6. doi:10.1046/j.0953-816x.2001.01474.x
85. Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci (2007) 30:527–35. doi:10.1016/j.tins.2007.07.007
86. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci (2009) 32:638–47. doi:10.1016/j.tins.2009.08.002
87. Kaur C, Rathnasamy G, Ling EA. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J Neuroimmune Pharmacol (2013) 8:66–78. doi:10.1007/s11481-012-9347-2
88. Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int J Dev Neurosci (2011) 29:423–40. doi:10.1016/j.ijdevneu.2011.07.008
89. Lavdas AA, Papastefanaki F, Thomaidou D, Matsas R. Cell adhesion molecules in gene and cell therapy approaches for nervous system repair. Curr Gene Ther (2011) 11:90–100. doi:10.2174/156652311794940755
90. Maeda N, Ishii M, Nishimura K, Kamimura K. Functions of chondroitin sulfate and heparan sulfate in the developing brain. Neurochem Res (2011) 36:1228–40. doi:10.1007/s11064-010-0324-y
91. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science (2002) 295:858–61. doi:10.1126/science.1068592
92. Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res (2001) 50:553–62. doi:10.1203/00006450-200111000-00003
93. Rice JE III, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol (1981) 9:131–41. doi:10.1002/ana.410090206
94. Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci (1993) 13:1441–53.
95. Grow J, Barks JD. Pathogenesis of hypoxic-ischemic cerebral injury in the term infant: current concepts. Clin Perinatol (2002) 29:585–602,v. doi:10.1016/S0095-5108(02)00059-3
96. Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci (2000) 20:9235–41.
97. McQuillen PS, Ferriero DM. Selective vulnerability in the developing central nervous system. Pediatr Neurol (2004) 30:227–35. doi:10.1016/j.pediatrneurol.2003.10.001
98. Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, et al. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol (2001) 11:56–71. doi:10.1111/j.1750-3639.2001.tb00381.x
99. Altman DI, Powers WJ, Perlman JM, Herscovitch P, Volpe SL, Volpe JJ. Cerebral blood flow requirement for brain viability in newborn infants is lower than in adults. Ann Neurol (1988) 24:218–26. doi:10.1002/ana.410240208
100. Borch K, Greisen G. Blood flow distribution in the normal human preterm brain. Pediatr Res (1998) 43:28–33. doi:10.1203/00006450-199801000-00005
101. Vannucci RC, Lyons DT, Vasta F. Regional cerebral blood flow during hypoxia-ischemia in immature rats. Stroke (1988) 19:245–50. doi:10.1161/01.STR.19.2.245
102. Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM. Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics (1995) 95:868–74.
103. Welsh FA, Vannucci RC, Brierley JB. Columnar alterations of NADH fluorescence during hypoxia-ischemia in immature rat brain. J Cereb Blood Flow Metab (1982) 2:221–8. doi:10.1038/jcbfm.1982.22
104. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res (1997) 42:1–8. doi:10.1203/00006450-199707000-00001
105. Dammann O, Leviton A. Inflammatory brain damage in preterm newborns – dry numbers, wet lab, and causal inferences. Early Hum Dev (2004) 79:1–15. doi:10.1016/j.earlhumdev.2004.04.009
106. Rocha-Ferreira E, Hristova M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front Immunol (2015) 6:56. doi:10.3389/fimmu.2015.00056
107. Hallman M. Premature birth and diseases in premature infants: common genetic background? J Matern Fetal Neonatal Med (2012) 25(Suppl 1):21–4. doi:10.3109/14767058.2012.667600
108. Weitzdoerfer R, Pollak A, Lubec B. Perinatal asphyxia in the rat has lifelong effects on morphology, cognitive functions, and behavior. Semin Perinatol (2004) 28:249–56. doi:10.1053/j.semperi.2004.08.001
109. Eunson P. The long-term health, social, and financial burden of hypoxic-ischaemic encephalopathy. Dev Med Child Neurol (2015) 57(Suppl 3):48–50. doi:10.1111/dmcn.12727
110. Ego A, Lidzba K, Brovedani P, Belmonti V, Gonzalez-Monge S, Boudia B, et al. Visual-perceptual impairment in children with cerebral palsy: a systematic review. Dev Med Child Neurol (2015) 57(Suppl 2):46–51. doi:10.1111/dmcn.12687
111. Anderson PJ. Neuropsychological outcomes of children born very preterm. Semin Fetal Neonatal Med (2014) 19:90–6. doi:10.1016/j.siny.2013.11.012
112. Murray AL, Scratch SE, Thompson DK, Inder TE, Doyle LW, Anderson JF, et al. Neonatal brain pathology predicts adverse attention and processing speed outcomes in very preterm and/or very low birth weight children. Neuropsychology (2014) 28:552–62. doi:10.1037/neu0000071
113. Chau V, Synnes A, Grunau RE, Poskitt KJ, Brant R, Miller SP. Abnormal brain maturation in preterm neonates associated with adverse developmental outcomes. Neurology (2013) 81:2082–9. doi:10.1212/01.wnl.0000437298.43688.b9
114. Anderson PJ, Cheong JL, Thompson DK. The predictive validity of neonatal MRI for neurodevelopmental outcome in very preterm children. Semin Perinatol (2015) 39:147–58. doi:10.1053/j.semperi.2015.01.008
115. Ikeda T, Mishima K, Yoshikawa T, Iwasaki K, Fujiwara M, Xia YX, et al. Selective and long-term learning impairment following neonatal hypoxic-ischemic brain insult in rats. Behav Brain Res (2001) 118:17–25. doi:10.1016/S0166-4328(00)00287-4
116. McClure MM, Peiffer AM, Rosen GD, Fitch RH. Auditory processing deficits in rats with neonatal hypoxic-ischemic injury. Int J Dev Neurosci (2005) 23:351–62. doi:10.1016/j.ijdevneu.2004.12.008
117. Smith AL, Hill CA, Alexander M, Szalkowski CE, Chrobak JJ, Rosenkrantz TS, et al. Spatial working memory deficits in male rats following neonatal hypoxic ischemic brain injury can be attenuated by task modifications. Brain Sci (2014) 4:240–72. doi:10.3390/brainsci4020240
118. Arteni NS, Pereira LO, Rodrigues AL, Lavinsky D, Achaval ME, Netto CA. Lateralized and sex-dependent behavioral and morphological effects of unilateral neonatal cerebral hypoxia-ischemia in the rat. Behav Brain Res (2010) 210:92–8. doi:10.1016/j.bbr.2010.02.015
119. Sanches EF, Arteni N, Nicola F, Aristimunha D, Netto CA. Sexual dimorphism and brain lateralization impact behavioral and histological outcomes following hypoxia-ischemia in P3 and P7 rats. Neuroscience (2015) 290:581–93. doi:10.1016/j.neuroscience.2014.12.074
120. Thoresen M, Bagenholm R, Loberg EM, Apricena F, Kjellmer I. Posthypoxic cooling of neonatal rats provides protection against brain injury. Arch Dis Child Fetal Neonatal Ed (1996) 74:F3–9. doi:10.1136/fn.74.1.F3
121. Jacobs S, Hunt R, Tarnow-Mordi W, Inder T, Davis P. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev (2007) 4:CD003311. doi:10.1002/14651858.CD003311.pub2
122. Thoresen M. Who should we cool after perinatal asphyxia? Semin Fetal Neonatal Med (2015) 20:66–71. doi:10.1016/j.siny.2015.01.002
123. Robertson NJ, Tan S, Groenendaal F, Van Bel F, Juul SE, Bennet L, et al. Which neuroprotective agents are ready for bench to bedside translation in the newborn infant? J Pediatr (2012) 160(544–552):e544. doi:10.1016/j.jpeds.2011.12.052
124. Hawryluk GW, Spano S, Chew D, Wang S, Erwin M, Chamankhah M, et al. An examination of the mechanisms by which neural precursors augment recovery following spinal cord injury: a key role for remyelination. Cell Transplant (2014) 23:365–80. doi:10.3727/096368912X662408
125. Salewski RP, Mitchell RA, Li L, Shen C, Milekovskaia M, Nagy A, et al. Transplantation of induced pluripotent stem cell-derived neural stem cells mediate functional recovery following thoracic spinal cord injury through remyelination of axons. Stem Cells Transl Med (2015) 4:743–54. doi:10.5966/sctm.2014-0236
126. Ruff CA, Ye H, Legasto JM, Stribbell NA, Wang J, Zhang L, et al. Effects of adult neural precursor-derived myelination on axonal function in the perinatal congenitally dysmyelinated brain: optimizing time of intervention, developing accurate prediction models, and enhancing performance. J Neurosci (2013) 33:11899–915. doi:10.1523/JNEUROSCI.1131-13.2013
127. Ruff CA, Faulkner SD, Fehlings MG. The potential for stem cell therapies to have an impact on cerebral palsy: opportunities and limitations. Dev Med Child Neurol (2013) 55:689–97. doi:10.1111/dmcn.12166
128. Faulkner SD, Ruff CA, Fehlings MG. The potential for stem cells in cerebral palsy – piecing together the puzzle. Semin Pediatr Neurol (2013) 20:146–53. doi:10.1016/j.spen.2013.06.002
129. Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol (2011) 10:372–82. doi:10.1016/S1474-4422(11)70016-3
130. Northington FJ. Brief update on animal models of hypoxic-ischemic encephalopathy and neonatal stroke. ILAR J (2006) 47:32–8. doi:10.1093/ilar.47.1.32
131. Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience (2001) 105:7–17. doi:10.1016/S0306-4522(01)00171-3
132. Wang Y, Cheung PT, Shen GX, Wu EX, Cao G, Bart I, et al. Hypoxic-ischemic brain injury in the neonatal rat model: relationship between lesion size at early MR imaging and irreversible infarction. AJNR Am J Neuroradiol (2006) 27:51–4.
133. Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol (2004) 207:3149–54. doi:10.1242/jeb.01064
134. Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci (2005) 27:81–6. doi:10.1159/000085991
136. Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, et al. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res (1999) 55:158–63. doi:10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1
137. Larroque B, Marret S, Ancel PY, Arnaud C, Marpeau L, Supernant K, et al. White matter damage and intraventricular hemorrhage in very preterm infants: the EPIPAGE study. J Pediatr (2003) 143:477–83. doi:10.1067/S0022-3476(03)00417-7
138. Miller SP, Cozzio CC, Goldstein RB, Ferriero DM, Partridge JC, Vigneron DB, et al. Comparing the diagnosis of white matter injury in premature newborns with serial MR imaging and transfontanel ultrasonography findings. AJNR Am J Neuroradiol (2003) 24:1661–9.
139. Woodward LJ, Anderson PJ, Austin NC, Howard K, Inder TE. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med (2006) 355:685–94. doi:10.1056/NEJMoa053792
140. Sheldon RA, Chuai J, Ferriero DM. A rat model for hypoxic-ischemic brain damage in very premature infants. Biol Neonate (1996) 69:327–41. doi:10.1159/000244327
141. Sheldon RA, Sedik C, Ferriero DM. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res (1998) 810:114–22. doi:10.1016/S0006-8993(98)00892-0
142. Uehara H, Yoshioka H, Kawase S, Nagai H, Ohmae T, Hasegawa K, et al. A new model of white matter injury in neonatal rats with bilateral carotid artery occlusion. Brain Res (1999) 837:213–20. doi:10.1016/S0006-8993(99)01675-3
143. Jelinski SE, Yager JY, Juurlink BH. Preferential injury of oligodendroblasts by a short hypoxic-ischemic insult. Brain Res (1999) 815:150–3. doi:10.1016/S0006-8993(98)01053-1
144. Cai Z, Pang Y, Xiao F, Rhodes PG. Chronic ischemia preferentially causes white matter injury in the neonatal rat brain. Brain Res (2001) 898:126–35. doi:10.1016/S0006-8993(01)02180-1
145. Stadlin A, James A, Fiscus R, Wong YF, Rogers M, Haines C. Development of a postnatal 3-day-old rat model of mild hypoxic-ischemic brain injury. Brain Res (2003) 993:101–10. doi:10.1016/j.brainres.2003.08.058
146. Fan LW, Lin S, Pang Y, Lei M, Zhang F, Rhodes PG, et al. Hypoxia-ischemia induced neurological dysfunction and brain injury in the neonatal rat. Behav Brain Res (2005) 165:80–90. doi:10.1016/j.bbr.2005.06.033
147. Nijboer CH, Kavelaars A, Van Bel F, Heijnen CJ, Groenendaal F. Gender-dependent pathways of hypoxia-ischemia-induced cell death and neuroprotection in the immature P3 rat. Dev Neurosci (2007) 29:385–92. doi:10.1159/000105479
148. Albertsson AM, Bi D, Duan L, Zhang X, Leavenworth JW, Qiao L, et al. The immune response after hypoxia-ischemia in a mouse model of preterm brain injury. J Neuroinflammation (2014) 11:153. doi:10.1186/s12974-014-0153-z
149. Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev (1979) 3:79–83. doi:10.1016/0378-3782(79)90022-7
150. Yager J, Towfighi J, Vannucci RC. Influence of mild hypothermia on hypoxic-ischemic brain damage in the immature rat. Pediatr Res (1993) 34:525–9. doi:10.1203/00006450-199310000-00029
151. Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, et al. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci (2004) 24:4412–20. doi:10.1523/JNEUROSCI.0477-04.2004
152. Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol (2006) 497:61–77. doi:10.1002/cne.20978
153. Shen Y, Liu XB, Pleasure DE, Deng W. Axon-glia synapses are highly vulnerable to white matter injury in the developing brain. J Neurosci Res (2012) 90:105–21. doi:10.1002/jnr.22722
154. Chen H, Burris M, Fajilan A, Spagnoli F, Tang J, Zhang JH. Prolonged exposure to isoflurane ameliorates infarction severity in the rat pup model of neonatal hypoxia-ischemia. Transl Stroke Res (2011) 2:382–90. doi:10.1007/s12975-011-0081-5
155. Ohshima M, Tsuji M, Taguchi A, Kasahara Y, Ikeda T. Cerebral blood flow during reperfusion predicts later brain damage in a mouse and a rat model of neonatal hypoxic-ischemic encephalopathy. Exp Neurol (2012) 233:481–9. doi:10.1016/j.expneurol.2011.11.025
156. van Bel F, Dorrepaal CA, Benders MJ, Zeeuwe PE, Van De Bor M, Berger HM. Changes in cerebral hemodynamics and oxygenation in the first 24 hours after birth asphyxia. Pediatrics (1993) 92:365–72.
157. Kirimi E, Tuncer O, Atas B, Sakarya ME, Ceylan A. Clinical value of color Doppler ultrasonography measurements of full-term newborns with perinatal asphyxia and hypoxic ischemic encephalopathy in the first 12 hours of life and long-term prognosis. Tohoku J Exp Med (2002) 197:27–33. doi:10.1620/tjem.197.27
158. Seri I. Management of hypotension and low systemic blood flow in the very low birth weight neonate during the first postnatal week. J Perinatol (2006) 26(Suppl 1):S8–13. doi:10.1038/sj.jp.7211464
159. Magee L, Sawchuck D, Synnes A, Von Dadelszen P. SOGC clinical practice guideline. Magnesium sulphate for fetal neuroprotection. J Obstet Gynaecol Can (2011) 33:516–29. doi:10.1016/S1701-2163(16)34886-1
160. Crowther CA, Middleton PF, Bain E, Ashwood P, Bubner T, Flenady V, et al. Working to improve survival and health for babies born very preterm: the WISH project protocol. BMC Pregnancy Childbirth (2013) 13:239. doi:10.1186/1471-2393-13-239
161. Rouse DJ, Gibbins KJ. Magnesium sulfate for cerebral palsy prevention. Semin Perinatol (2013) 37:414–6. doi:10.1053/j.semperi.2013.06.025
162. Oddie S, Tuffnell DJ, Mcguire W. Antenatal magnesium sulfate: neuro-protection for preterm infants. Arch Dis Child Fetal Neonatal Ed (2015) 100(6):F553–7. doi:10.1136/archdischild-2014-307655
163. Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M. Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pediatr Res (1998) 43:738–45. doi:10.1203/00006450-199806000-00005
164. Gunn AJ, Gunn TR, De Haan HH, Williams CE, Gluckman PD. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest (1997) 99:248–56. doi:10.1172/JCI119153
165. Haaland K, Loberg EM, Steen PA, Thoresen M. Posthypoxic hypothermia in newborn piglets. Pediatr Res (1997) 41:505–12. doi:10.1203/00006450-199704000-00009
166. Dickerson JW, Dobbing J. Prenatal and postnatal growth and development of the central nervous system of the pig. Proc R Soc Lond B Biol Sci (1967) 166:384–95. doi:10.1098/rspb.1967.0002
167. Higgins RD, Raju T, Edwards AD, Azzopardi DV, Bose CL, Clark RH, et al. Hypothermia and other treatment options for neonatal encephalopathy: an executive summary of the Eunice Kennedy Shriver NICHD workshop. J Pediatr (2011) 159(851–858):e851. doi:10.1016/j.jpeds.2011.08.004
168. Gancia P, Pomero G. Therapeutic hypothermia in the prevention of hypoxic-ischaemic encephalopathy: new categories to be enrolled. J Matern Fetal Neonatal Med (2012) 25(Suppl 4):94–6. doi:10.3109/14767058.2012.715023
169. Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature (2005) 438:1162–6. doi:10.1038/nature04302
170. Jantzie LL, Talos DM, Selip DB, An L, Jackson MC, Folkerth RD, et al. Developmental regulation of group I metabotropic glutamate receptors in the premature brain and their protective role in a rodent model of periventricular leukomalacia. Neuron Glia Biol (2010) 6:277–88. doi:10.1017/S1740925X11000111
171. Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci U S A (2004) 101:7751–6. doi:10.1073/pnas.0307850101
172. Kulbatski I, Mothe AJ, Keating A, Hakamata Y, Kobayashi E, Tator CH. Oligodendrocytes and radial glia derived from adult rat spinal cord progenitors: morphological and immunocytochemical characterization. J Histochem Cytochem (2007) 55:209–22. doi:10.1369/jhc.6A7020.2006
173. Tita AT, Andrews WW. Diagnosis and management of clinical chorioamnionitis. Clin Perinatol (2010) 37:339–54. doi:10.1016/j.clp.2010.02.003
174. American College of Obstetricians and Gynecologists. ACOG practice bulletin number 47, October 2003: prophylactic antibiotics in labor and delivery. Obstet Gynecol (2003) 102:875–82. doi:10.1016/S0029-7844(03)00984-0
175. Takahashi T, Nowakowski RS, Caviness VS Jr. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci (1995) 15:6046–57.
176. Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A (1998) 95:1242–6. doi:10.1073/pnas.95.3.1242
177. Meyer G, Schaaps JP, Moreau L, Goffinet AM. Embryonic and early fetal development of the human neocortex. J Neurosci (2000) 20:1858–68.
178. Smart IH, Dehay C, Giroud P, Berland M, Kennedy H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex (2002) 12:37–53. doi:10.1093/cercor/12.1.37
179. Bayer SA, Altman J. Development of layer I and the subplate in the rat neocortex. Exp Neurol (1990) 107:48–62. doi:10.1016/0014-4886(90)90062-W
180. Wang WZ, Hoerder-Suabedissen A, Oeschger FM, Bayatti N, Ip BK, Lindsay S, et al. Subplate in the developing cortex of mouse and human. J Anat (2010) 217:368–80. doi:10.1111/j.1469-7580.2010.01274.x
181. Robertson RT, Annis CM, Baratta J, Haraldson S, Ingeman J, Kageyama GH, et al. Do subplate neurons comprise a transient population of cells in developing neocortex of rats? J Comp Neurol (2000) 426:632–50. doi:10.1002/1096-9861(20001030)426:4<632::AID-CNE10>3.0.CO;2-4
182. Tioseco JA, Aly H, Essers J, Patel K, El-Mohandes AA. Male sex and intraventricular hemorrhage. Pediatr Crit Care Med (2006) 7:40–4. doi:10.1097/01.PCC.0000192341.67078.61
183. Jarvis S, Glinianaia SV, Arnaud C, Fauconnier J, Johnson A, Mcmanus V, et al. Case gender and severity in cerebral palsy varies with intrauterine growth. Arch Dis Child (2005) 90:474–9. doi:10.1136/adc.2004.052670
184. Westbom L, Hagglund G, Nordmark E. Cerebral palsy in a total population of 4-11 year olds in southern Sweden. Prevalence and distribution according to different CP classification systems. BMC Pediatr (2007) 7:41. doi:10.1186/1471-2431-7-41
185. Chounti A, Hagglund G, Wagner P, Westbom L. Sex differences in cerebral palsy incidence and functional ability: a total population study. Acta Paediatr (2013) 102:712–7. doi:10.1111/apa.12240
186. Himmelmann K, Uvebrant P. The panorama of cerebral palsy in Sweden. XI. Changing patterns in the birth-year period 2003-2006. Acta Paediatr (2014) 103:618–24. doi:10.1111/apa.12614
187. Reiss AL, Kesler SR, Vohr B, Duncan CC, Katz KH, Pajot S, et al. Sex differences in cerebral volumes of 8-year-olds born preterm. J Pediatr (2004) 145:242–9. doi:10.1016/j.jpeds.2004.04.031
188. Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, et al. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem (2004) 279:38563–70. doi:10.1074/jbc.M405461200
189. Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem (2004) 90:1068–75. doi:10.1111/j.1471-4159.2004.02547.x
190. Jansen EM, Low WC. Long-term effects of neonatal ischemic-hypoxic brain injury on sensorimotor and locomotor tasks in rats. Behav Brain Res (1996) 78:189–94. doi:10.1016/0166-4328(95)00248-0
191. Smith AL, Alexander M, Rosenkrantz TS, Sadek ML, Fitch RH. Sex differences in behavioral outcome following neonatal hypoxia ischemia: insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Exp Neurol (2014) 254:54–67. doi:10.1016/j.expneurol.2014.01.003
192. Payan HM, Conrad JR. Carotid ligation in gerbils. Influence of age, sex, and gonads. Stroke (1977) 8:194–6. doi:10.1161/01.STR.8.2.194
193. Mayoral SR, Omar G, Penn AA. Sex differences in a hypoxia model of preterm brain damage. Pediatr Res (2009) 66:248–53. doi:10.1203/PDR.0b013e3181b1bc34
194. Lan WC, Priestley M, Mayoral SR, Tian L, Shamloo M, Penn AA. Sex-specific cognitive deficits and regional brain volume loss in mice exposed to chronic, sublethal hypoxia. Pediatr Res (2011) 70:15–20. doi:10.1038/pr.2011.240
195. Eyre JA. Corticospinal tract development and its plasticity after perinatal injury. Neurosci Biobehav Rev (2007) 31:1136–49. doi:10.1016/j.neubiorev.2007.05.011
196. Eyre JA, Smith M, Dabydeen L, Clowry GJ, Petacchi E, Battini R, et al. Is hemiplegic cerebral palsy equivalent to amblyopia of the corticospinal system? Ann Neurol (2007) 62:493–503. doi:10.1002/ana.21108
197. Bona E, Johansson BB, Hagberg H. Sensorimotor function and neuropathology five to six weeks after hypoxia-ischemia in seven-day-old rats. Pediatr Res (1997) 42:678–83. doi:10.1203/00006450-199711000-00021
198. Tomimatsu T, Fukuda H, Endoh M, Mu J, Watanabe N, Kohzuki M, et al. Effects of neonatal hypoxic-ischemic brain injury on skilled motor tasks and brainstem function in adult rats. Brain Res (2002) 926:108–17. doi:10.1016/S0006-8993(01)03311-X
199. Lubics A, Reglodi D, Tamas A, Kiss P, Szalai M, Szalontay L, et al. Neurological reflexes and early motor behavior in rats subjected to neonatal hypoxic-ischemic injury. Behav Brain Res (2005) 157:157–65. doi:10.1016/j.bbr.2004.06.019
200. Pazaiti A, Soubasi V, Spandou E, Karkavelas G, Georgiou T, Karalis P, et al. Evaluation of long-lasting sensorimotor consequences following neonatal hypoxic-ischemic brain injury in rats: the neuroprotective role of MgSO4. Neonatology (2009) 95:33–40. doi:10.1159/000151753
201. Duque J, Thonnard JL, Vandermeeren Y, Sebire G, Cosnard G, Olivier E. Correlation between impaired dexterity and corticospinal tract dysgenesis in congenital hemiplegia. Brain (2003) 126:732–47. doi:10.1093/brain/awg069
202. Arnould C, Penta M, Thonnard JL. Hand impairments and their relationship with manual ability in children with cerebral palsy. J Rehabil Med (2007) 39:708–14. doi:10.2340/16501977-0111
203. Bleyenheuft Y, Grandin CB, Cosnard G, Olivier E, Thonnard JL. Corticospinal dysgenesis and upper-limb deficits in congenital hemiplegia: a diffusion tensor imaging study. Pediatrics (2007) 120:e1502–11. doi:10.1542/peds.2007-0394
204. Son SM, Ahn YH, Sakong J, Moon HK, Ahn SH, Lee H, et al. Diffusion tensor imaging demonstrates focal lesions of the corticospinal tract in hemiparetic patients with cerebral palsy. Neurosci Lett (2007) 420:34–8. doi:10.1016/j.neulet.2007.04.054
205. Alstermark B, Ogawa J. In vivo recordings of bulbospinal excitation in adult mouse forelimb motoneurons. J Neurophysiol (2004) 92:1958–62. doi:10.1152/jn.00092.2004
206. Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med (2005) 11:1355–60. doi:10.1038/nm1331
207. Rouiller EM, Liang FY, Moret V, Wiesendanger M. Trajectory of redirected corticospinal axons after unilateral lesion of the sensorimotor cortex in neonatal rat; a phaseolus vulgaris-leucoagglutinin (PHA-L) tracing study. Exp Neurol (1991) 114:53–65. doi:10.1016/0014-4886(91)90084-P
208. Carr LJ, Harrison LM, Evans AL, Stephens JA. Patterns of central motor reorganization in hemiplegic cerebral palsy. Brain (1993) 116(Pt 5):1223–47. doi:10.1093/brain/116.5.1223
209. Eyre JA, Taylor JP, Villagra F, Smith M, Miller S. Evidence of activity-dependent withdrawal of corticospinal projections during human development. Neurology (2001) 57:1543–54. doi:10.1212/WNL.57.9.1543
210. Joosten EA, Gribnau AA, Dederen PJ. An anterograde tracer study of the developing corticospinal tract in the rat: three components. Brain Res (1987) 433:121–30. doi:10.1016/0165-3806(87)90070-8
211. Clowry GJ. The dependence of spinal cord development on corticospinal input and its significance in understanding and treating spastic cerebral palsy. Neurosci Biobehav Rev (2007) 31:1114–24. doi:10.1016/j.neubiorev.2007.04.007
212. Reinoso BS, Castro AJ. A study of corticospinal remodelling using retrograde fluorescent tracers in rats. Exp Brain Res (1989) 74:387–94. doi:10.1007/BF00248872
213. Z’Graggen WJ, Fouad K, Raineteau O, Metz GA, Schwab ME, Kartje GL. Compensatory sprouting and impulse rerouting after unilateral pyramidal tract lesion in neonatal rats. J Neurosci (2000) 20:6561–9.
214. Serradj N, Paixao S, Sobocki T, Feinberg M, Klein R, Kullander K, et al. EphA4-mediated ipsilateral corticospinal tract misprojections are necessary for bilateral voluntary movements but not bilateral stereotypic locomotion. J Neurosci (2014) 34:5211–21. doi:10.1523/JNEUROSCI.4848-13.2014
215. Gibson CL, Arnott GA, Clowry GJ. Plasticity in the rat spinal cord seen in response to lesions to the motor cortex during development but not to lesions in maturity. Exp Neurol (2000) 166:422–34. doi:10.1006/exnr.2000.7511
216. Clowry GJ, Davies BM, Upile NS, Gibson CL, Bradley PM. Spinal cord plasticity in response to unilateral inhibition of the rat motor cortex during development: changes to gene expression, muscle afferents and the ipsilateral corticospinal projection. Eur J Neurosci (2004) 20:2555–66. doi:10.1111/j.1460-9568.2004.03713.x
217. Ferrari F, Cioni G, Prechtl HF. Qualitative changes of general movements in preterm infants with brain lesions. Early Hum Dev (1990) 23:193–231. doi:10.1016/0378-3782(90)90013-9
218. O’Sullivan MC, Miller S, Ramesh V, Conway E, Gilfillan K, Mcdonough S, et al. Abnormal development of biceps brachii phasic stretch reflex and persistence of short latency heteronymous reflexes from biceps to triceps brachii in spastic cerebral palsy. Brain (1998) 121(Pt 12):2381–95. doi:10.1093/brain/121.12.2381
219. Papile LA, Rudolph AM, Heymann MA. Autoregulation of cerebral blood flow in the preterm fetal lamb. Pediatr Res (1985) 19:159–61. doi:10.1203/00006450-198502000-00001
220. Helou S, Koehler RC, Gleason CA, Jones MD Jr, Traystman RJ. Cerebrovascular autoregulation during fetal development in sheep. Am J Physiol (1994) 266:H1069–74.
221. Mallard EC, Gunn AJ, Williams CE, Johnston BM, Gluckman PD. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol (1992) 167:1423–30. doi:10.1016/S0002-9378(11)91728-1
222. Bennet L, Peebles DM, Edwards AD, Rios A, Hanson MA. The cerebral hemodynamic response to asphyxia and hypoxia in the near-term fetal sheep as measured by near infrared spectroscopy. Pediatr Res (1998) 44:951–7. doi:10.1203/00006450-199812000-00022
223. Bernhard CG, Kolmodin GM, Meyerson BA. On the prenatal development of function and structure in the somesthetic cortex of the sheep. Prog Brain Res (1967) 26:60–77. doi:10.1016/S0079-6123(08)61419-3
224. Barlow RM. The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J Comp Neurol (1969) 135:249–62. doi:10.1002/cne.901350302
225. Cook CJ, Williams C, Gluckman PD. Brainstem auditory evoked potentials in the fetal sheep, in utero. J Dev Physiol (1987) 9:429–39.
226. Back SA, Riddle A, Dean J, Hohimer AR. The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics (2012) 9:359–70. doi:10.1007/s13311-012-0108-y
227. Griffith JL, Shimony JS, Cousins SA, Rees SE, Mccurnin DC, Inder TE, et al. MR imaging correlates of white-matter pathology in a preterm baboon model. Pediatr Res (2012) 71:185–91. doi:10.1038/pr.2011.33
228. Verney C, Rees S, Biran V, Thompson M, Inder T, Gressens P. Neuronal damage in the preterm baboon: impact of the mode of ventilatory support. J Neuropathol Exp Neurol (2010) 69:473–82. doi:10.1097/NEN.0b013e3181dac07b
229. Thoresen M, Haaland K, Loberg EM, Whitelaw A, Apricena F, Hanko E, et al. A piglet survival model of posthypoxic encephalopathy. Pediatr Res (1996) 40:738–48. doi:10.1203/00006450-199611000-00014
230. Kyng KJ, Skajaa T, Kerrn-Jespersen S, Andreassen CS, Bennedsgaard K, Henriksen TB. A Piglet model of neonatal hypoxic-ischemic encephalopathy. J Vis Exp (2015) 99:e52454. doi:10.3791/52454
231. Konig N, Roch G, Marty R. The onset of synaptogenesis in rat temporal cortex. Anat Embryol (1975) 148:73–87. doi:10.1007/BF00315564
232. Higashi S, Molnar Z, Kurotani T, Toyama K. Prenatal development of neural excitation in rat thalamocortical projections studied by optical recording. Neuroscience (2002) 115:1231–46. doi:10.1016/S0306-4522(02)00418-9
233. Aboitiz F, Montiel J, Garcia RR. Ancestry of the mammalian preplate and its derivatives: evolutionary relicts or embryonic adaptations? Rev Neurosci (2005) 16:359–76. doi:10.1515/REVNEURO.2005.16.4.359
234. Bystron I, Blakemore C, Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci (2008) 9:110–22. doi:10.1038/nrn2252
235. Dieni S, Rees S. BDNF and TrkB protein expression is altered in the fetal hippocampus but not cerebellum after chronic prenatal compromise. Exp Neurol (2005) 192:265–73. doi:10.1016/j.expneurol.2004.06.003
236. Turner AJ, Trudinger BJ. A modification of the uterine artery restriction technique in the guinea pig fetus produces asymmetrical ultrasound growth. Placenta (2009) 30:236–40. doi:10.1016/j.placenta.2008.11.023
237. Tolcos M, Bateman E, O’Dowd R, Markwick R, Vrijsen K, Rehn A, et al. Intrauterine growth restriction affects the maturation of myelin. Exp Neurol (2011) 232:53–65. doi:10.1016/j.expneurol.2011.08.002
238. Catteau J, Gernet JI, Marret S, Legros H, Gressens P, Leroux P, et al. Effects of antenatal uteroplacental hypoperfusion on neonatal microvascularisation and excitotoxin sensitivity in mice. Pediatr Res (2011) 70:229–35. doi:10.1038/pr.2011.454
239. Drobyshevsky A, Derrick M, Prasad PV, Ji X, Englof I, Tan S. Fetal brain magnetic resonance imaging response acutely to hypoxia-ischemia predicts postnatal outcome. Ann Neurol (2007) 61:307–14. doi:10.1002/ana.21095
240. Wigglesworth JS. Experimental growth retardation in the foetal rat. J Pathol Bacteriol (1964) 88:1–13. doi:10.1002/path.1700880102
Keywords: spastic hemiplegic cerebral palsy, hypoxia–ischemia, HI rodent model, white matter damage, perinatal brain injury, myelination, oligodendrocyte, periventricular leukomalacia
Citation: Rumajogee P, Bregman T, Miller SP, Yager JY and Fehlings MG (2016) Rodent Hypoxia–Ischemia Models for Cerebral Palsy Research: A Systematic Review. Front. Neurol. 7:57. doi: 10.3389/fneur.2016.00057
Received: 08 September 2015; Accepted: 03 April 2016;
Published: 25 April 2016
Edited by:
Pedro M. Pimentel-Coelho, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Christopher Mario Inglese, Medical College of Wisconsin, USAShenandoah Robinson, Boston Children’s Hospital, USA
Stephen Arthur Back, Oregon Health & Science University, USA
Copyright: © 2016 Rumajogee, Bregman, Miller, Yager and Fehlings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael G. Fehlings, bWljaGFlbC5mZWhsaW5nc0B1aG4uY2E=
†Prakasham Rumajogee and Tatiana Bregman contributed equally to this work.