 Michele A. Calton
Michele A. Calton Jeremy R. Howard
Jeremy R. Howard Ronald M. Harper
Ronald M. Harper Dan Goldowitz
Dan Goldowitz Guy Mittleman
Guy Mittleman- 1Department of Psychology, The University of Memphis, Memphis, TN, USA
- 2Neurobiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA
- 3Centre for Molecular Medicine and Therapeutics, University of British Columbia, Vancouver, BC, Canada
- 4Department of Psychological Science, Ball State University, Muncie, IN, USA
The cerebellum assists coordination of somatomotor, respiratory, and autonomic actions. Purkinje cell alterations or loss appear in sudden infant death and sudden death in epilepsy victims, possibly contributing to the fatal event. We evaluated breathing patterns in 12 wild-type (WT) and Lurcher mutant mice with 100% developmental cerebellar Purkinje cell loss under baseline (room air), and recovery from hypercapnia, a concern in sudden death events. Six mutant and six WT mice were exposed to 4-min blocks of increasing CO2 (2, 4, 6, and 8%), separated by 4-min recovery intervals in room air. Breath-by-breath patterns, including depth of breathing and end-expiratory pause (EEP) durations during recovery, were recorded. No baseline genotypic differences emerged. However, during recovery, EEP durations significantly lengthened in mutants, compared to WT mice, following the relatively low levels of CO2 exposure. Additionally, mutant mice exhibited signs of post-sigh disordered breathing during recovery following each exposure. Developmental cerebellar Purkinje cell loss significantly affects compensatory breathing patterns following mild CO2 exposure, possibly by inhibiting recovery from elevated CO2. These data implicate cerebellar Purkinje cells in the ability to recover from hypercarbia, suggesting that neuropathologic changes or loss of these cells contribute to inadequate ventilatory recovery to increased environmental CO2. Multiple disorders, including sudden infant death syndrome (SIDS) and sudden unexpected death in epilepsy (SUDEP), appear to involve both cardiorespiratory failure and loss or injury to cerebellar Purkinje cells; the findings support the concept that such neuropathology may precede and exert a prominent role in these fatal events.
Introduction
Sudden unexpected death occurs in several syndromes, with mechanisms underlying the fatal event not yet understood. Sudden infant death syndrome (SIDS) is one such syndrome; the condition occurs in infants <1 year of age, often during sleep, and with victims succumbing suddenly with major causes excluded even after autopsy and a thorough investigation (1, 2). Sudden death in epilepsy (SUDEP), appears in adults as well as older children, often occurs during sleep, but leaves few signs of mechanisms operating to induce a fatal event. A defect in breathing, or deficient responses to a ventilatory challenge is suspected in both SIDS and SUDEP. Although an inability to respond to severe, transient hypotension is a strong possibility (3, 4), the circumstances surrounding both SIDS and SUDEP conditions suggest an unknown brain abnormality that results in an inability to appropriately compensate for, or recover from, exposure to an intrinsic or exogenous stressor (5). Here, we evaluate the role of the cerebellum in mediating the consequences of exposure to one exogenous stressor, increased levels of environmental carbon dioxide (CO2; hypercapnia).
The focus on high CO2 arises from the circumstances normally associated with SIDS or postictal cessation of breathing in epilepsy. SIDS and SUDEP victims are often found deceased in bed, in the prone position, and sometimes in soft bedding, focusing attention on exposure to elevated CO2 concentrations associated with these conditions during sleep (6, 7). Thus, SIDS likely results from an inability to respond appropriately with respiratory and cardiovascular patterns to accommodate such a breathing stressor. Similarly, postictal respiratory failure in SUDEP would elevate CO2 levels, imposing a breathing challenge that brain structures may not be able to manage.
Structures within the cerebellum play a significant role in responding to extreme respiratory or blood pressure challenge (8–11). Developmental neuropathology of the cerebellum, as well as surgical or other insult to this structure, is accompanied by various forms of disordered breathing, including apnea, obstructive apnea, and hypoventilation (12–16). Such disordered breathing induces substantial increases in markers for CO2 (17), which, if not compensated, could place the subject at risk. Cerebellar neuropathology appears in SIDS victims, with a higher incidence of delayed cerebellar cortex maturation (18), and increased Purkinje cell apoptosis than controls (19). Cerebellar injury is very common in patients with epilepsy, likely induced either by excitotoxic injury or as a consequence of long-term antiepileptic medication. Additionally, in SIDS, transcriptional nucleolar activity is significantly reduced in cerebellar Purkinje cells relative to controls, suggesting hypofunction of the Purkinje cells in these victims. The nature of mechanisms underlying these pathologies or whether they underlie failure is unknown (20).
Cerebellar Purkinje cells provide the sole output of the cerebellar cortex, and innervate the deep cerebellar nuclei which, in turn, send efferents to multiple areas of the brainstem involved in respiratory and autonomic functions (21, 22). One pair of nuclei, the fastigial nuclei (FN), is responsive to chemical respiratory challenges, with cell body lesions of these nuclei significantly attenuating compensatory respiratory responses to increased levels of environmental CO2. However, such lesions have little effect on relaxed, rhythmic breathing (10, 23). Additionally, Lurcher mutant mice (Lc/+) with global, developmental cerebellar Purkinje cell loss show reduced respiratory responses following exposure to increased environmental CO2 (24), an outcome interpreted as a reduction in sensitivity to increased blood CO2 levels (25–27).
Exposure to increased CO2 in the environment is commonly encountered in everyday life, resulting in robust physiological responses, even with small increases in CO2 (26). The phenotypic response to rising CO2 levels is to increase minute ventilation by adjusting the depth of breathing [tidal volume (TV)], the frequency of breathing, or both (25–27). Recovery from ventilatory challenges results in a gradual return to baseline minute ventilation as blood levels of CO2 decrease and blood levels of oxygen (O2) increase (26). Disordered breathing patterns during recovery from environmental stressors indicate respiratory distress and can include periods of abnormally large TVs (sighs) coupled with apneic-like periods of extended pauses between breaths [end-expiratory pause (EEP)]. Large TVs and extended EEPs limit the ability to reduce increased blood levels of CO2 by reducing the rate of gas exchange in the lungs (28). As disordered breathing patterns would reduce the ability to appropriately respond to, or recover from, exposure to exogenous stressors including hypercapnia or hypoxia, it has been suggested that such respiratory dysregulation could increase vulnerability to SIDS (29–31).
To determine if loss of cerebellar Purkinje cells results in distressed breathing following exposure to increased environmental CO2, we used whole body plethysmography (WBP) to investigate the hypothesis that Lurcher mutants heterozygous for the Lurcher spontaneous mutation (Grid2Lc), would exhibit disordered breathing patterns during recovery from hypercapnia, in comparison to wild-type (WT) controls. Lurcher mice were selected because we had previously observed that deficits in breath frequency appeared during recovery, following exposure to low levels of CO2 (24). In the current study, we specifically targeted two components of breath frequency, including TV and EEP periods. These dependent variables were selected because of their importance in modulating increased blood levels of CO2 by affecting the rate of gas exchange in the lungs.
Materials and Methods
Animals
Mice were bred and housed in the Animal Care Facility of the Department of Psychology at the University of Memphis. They were maintained in a temperature-controlled environment (21 ± 1°C) on a 12:12 light–dark cycle (lights on at 0700 hours) and given free access to food and water. Original Lurcher (#001046; Mouse Genome Identifier #: 1857337) breeders were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All experiments were conducted with strict adherence to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The protocol was approved by the University of Memphis Institutional Animal Care and Use Committee.
The breeding of Lurcher mice entailed filial pairing of a non-ataxic female WT (B6CBACa Aw-J/A-Grid2+) with a mutant ataxic male heterozygous for the Lurcher spontaneous mutation (B6CBACa Aw-J/A-Grid2Lc). This breeding strategy produced litters that are composed of both heterozygous Lc/+ and WT mice. The Lc/+ mouse is phenotypically distinguishable from WT littermates as early as postnatal day 12 (PND 12) with cerebellar signs, including an ataxic gait, which permits non-invasive differentiation of Lc/+ mice from their non-ataxic WT littermates (32, 33).
Animals were weaned at PND 25 ± 4 days, and sibling housed in groups of three to five in ventilated polystyrene cages. In order to avoid litter effects, six Lc/+ and six WT littermates were selected randomly, each pair from different litters and mating cages. All mice were PND 60 at testing onset with a mean weight of 20.05 g (SD = 1.79 g). The subjects consisted of 12 male mice. Male mice were chosen because the incidence of sudden death and cerebellar neuropathology is much larger in males than in females (1, 34).
Whole Body Plethysmography
Experimental data were collected during light hours using a WBP system (Emka Technologies, Falls Church, VA, USA), as previously described (24). Briefly, a transducer was mounted to the WBP device, which converted pressure differentials in the chamber caused by respiration into electrical signals that were then transmitted to, and interpreted by, the software. Data were collected for each individual breath with sampling rates set at least twice the typical eupneic breathing levels observed in mice: typical TV <30 mL, typical expired volume <30 mL, and typical breath frequency <500/bpm (35, 36), to account for changes in breathing due to the response to, and recovery from hypercapnia. Pressure differentials that met all of these requirements, as interpreted by the software, were counted as individual breaths and added to the data output. Differentials that did not meet all of these parameters were flagged as movement artifacts and not included in the data file for analysis.
Procedure
Mice were weighed prior to placement in the WBP chamber. The experimental room temperature (22 ± 3°C) and humidity (20 ± 5%) were monitored daily to ensure stability throughout the study. An in-house program coupled to EMKA software was used to assess the subjects’ respiratory responses at baseline (normal room air: 21% O2, 0% CO2, and 79% N2), under conditions of increased CO2 (2, 4, 6, and 8%), and during recovery from each CO2 challenge in normal room air. Each test day began with a 10-min habituation period, followed by a 4-min exposure to baseline (room air) period. Under all conditions, air in the WBP chamber was removed continuously (0.8–1.0 L/m), to prevent accumulation of excessive CO2 due to exhalation.
Baseline
After the 10-min habituation period, the dependent variables TV and EEP were continuously recorded, while the mice were exposed to room air for a total of 4 min (21% O2, 0% CO2, and 79% N).
Hypercapnia and Recovery
The entire hypercapnia program was 52 min in duration and consisted of an initial baseline (4 min) measured as described above, followed by four sequential challenges where CO2 content was progressively increased from 2, 4, 6, and 8% (21% O2, N2 on balance). Each of the four CO2 challenges consisted of a 2-min chamber fill period (the approximate time required for the WBP to fully achieve the desired gas percentages), followed by a 4-min exposure. To assess the recovery responses to multiple hypercapnic challenges, the program returned to room air following each challenge (again, including a 2-min chamber refill period and a 4-min recovery period). At termination of the final CO2 exposure (8%), and return to room air, the mouse was removed from the chamber.
Variables and Data Analysis
To maintain sample equality among all animals on the measure of breath frequency, the dependent variables, TVs and EEPs, were recorded for the first 510 breaths from each animal during baseline and during recovery from each of the CO2 challenge conditions. Because TV and EEP are each components of breath frequency, the breath samples from each animal were made equal at 510 breaths based on the animal with the lowest breaths per minute. Analyses were only conducted on individual breaths to specifically determine if genotypic differences in TVs and EEP durations contributed to previously observed deficits in overall breath frequency during recovery from increased levels of CO2.
Animal Weights
An independent samples t-test was performed with genotype (Lc/+ and WT) as the independent variable and weights as the dependent variable to determine whether there was a need to include weight as a covariate, since lung function is tightly correlated with body size (37).
Baseline
Baseline data were analyzed using repeated measures analysis of variance (RMANOVA), with genotype (Lc/+ and WT) as the between-subjects factor and with the first 510 breaths as the within-subjects factors. Thus, the omnibus analysis was a 2 (Genotype) × 510 (Breaths) mixed design.
Hypercapnia Recovery
The recovery conditions were analyzed using an omnibus RMANOVA with genotype (Lc/+ and WT) again serving as the between-subjects factor. Within-subjects factors included four levels of recovery from CO2 exposure (2, 4, 6, and 8% CO2), and from each recovery period, the first 510 breaths to accurately track TVs and moment-to-moment changes in EEP. Therefore, the omnibus analysis became a 2 (Genotype) × 4 (Recovery from CO2 exposure) × 510 (Breaths) mixed design. Depending on the results of the omnibus RMANOVAs, additional simple-effects tests were used to analyze interaction effects.
Results
Animals
An independent samples t-test revealed no significant difference between the body weights of the two genotypes, t(10) = −0.497, p = 0.630.
Baseline
The omnibus RMANOVA revealed the depth of breathing (TV) in the two groups was equivalent during normal room air [Lc/+ M = 0.158 mL, WT M = 0.170 ms, SEM 0.013 mL; Group, F(1, 10) = 0.879, p = 0.371]. Figure 1A shows the mean TVs of the two groups for the first 510 breaths. Note that breaths have been averaged into groups of 10 for clarity of presentation [Breath, F(509, 5090) = 0.970, p = 0.670; Group × Breaths, F(509, 5090) = 0.900, p = 0.940].
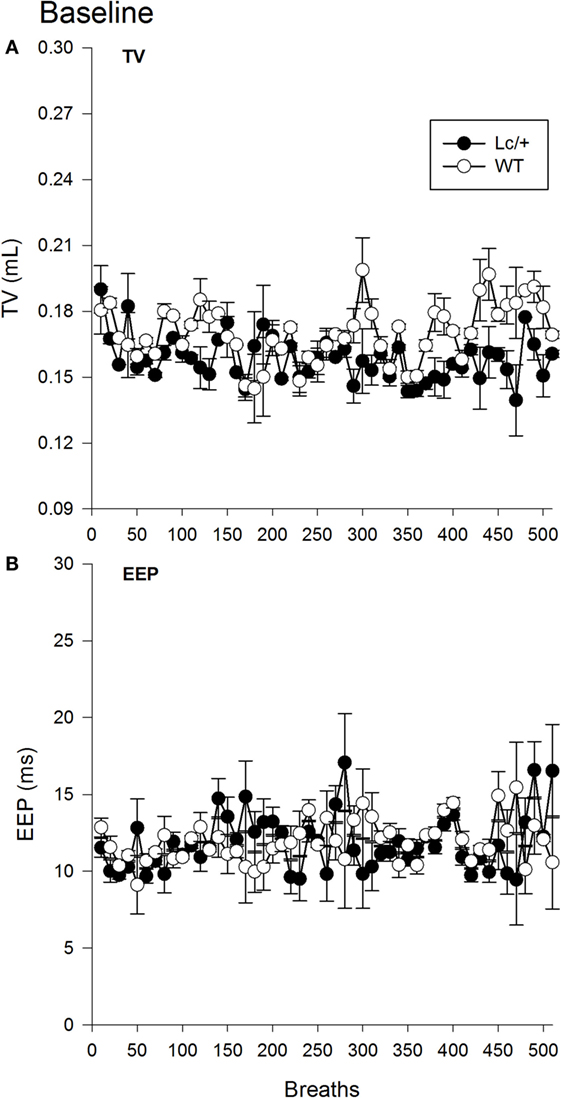
Figure 1. Baseline tidal volume (A) and end-expiratory pause (B) data for 510 breaths at normal room air in Lurcher mutant (Lc/+) and wild-type (WT) control mice. Breaths have been averaged into groups of 10 for clarity of presentation. Values represent mean ± SEM.
A second omnibus RMANOVA further revealed the two genotypes had equivalent EEP durations during exposure to room air [Lc/+ M = 11.819 ms, WT M = 11.922 ms, SEM 1.233 ms; Group, F(1, 10) = 0.007, p = 0.935]. As shown in Figure 1B, the two groups maintained equivalent EEP intervals throughout the continuum of 510 breaths and across individual breaths [Breath, F(509, 5090) = 0.891, p = 0.957; Group × Breaths, F(509, 5090) = 1.011, p = 0.427].
Hypercapnia Recovery
Figure 2A shows changes in average TV during recovery following each exposure to CO2. Changes in TV were comparable in the two genotypes. Mice in both groups showed a typical increase in TV following exposure to increasing percentages of CO2, indicating increased work to reduce the body burden of CO2 [2, 4, 6, and 8% CO2; Recovery, F(3, 30) = 3.765, p = 0.021]. Additionally, when considered across the 510 breaths within each recovery condition, both genotypes showed a gradual decline in TV, likely corresponding to gradual reductions in body levels of CO2 with the return to room air [data not shown; Breaths, F(509, 5090) = 1.420, p < 0.001].
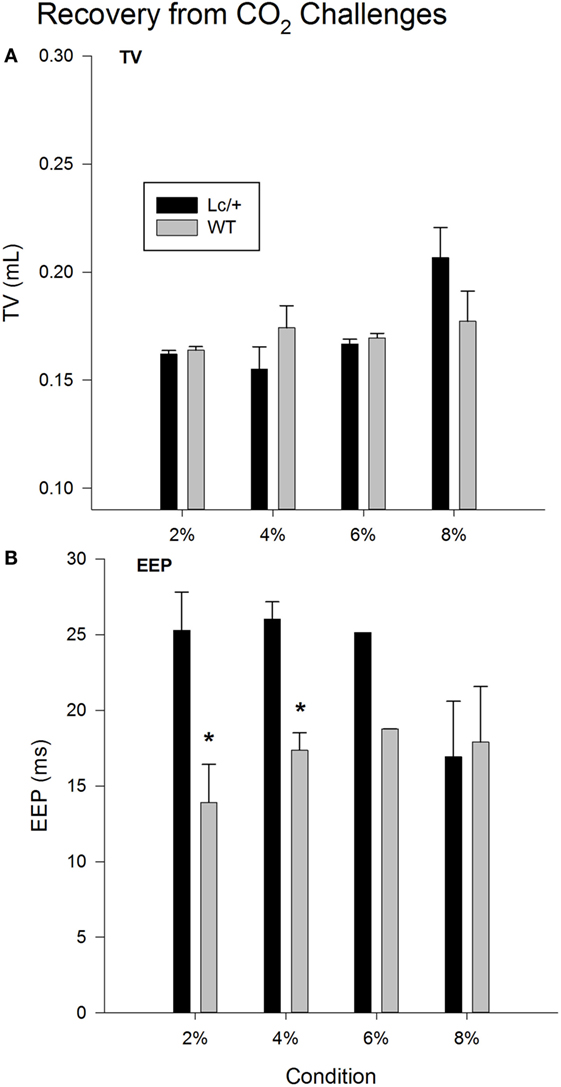
Figure 2. Recovery tidal volume (A) and end-expiratory pause (B) data, averaged across 510 breaths at room air (21% O2, 79% N2, and 0% CO2), following each CO2 exposure period in Lurcher mutant (Lc/+) and wildtype (WT) mice. Values represent mean ± SEM. Asterisks indicate significant differences found between genotypes.
As shown in Figure 2B, when comparing recovery conditions, EEPs between the two genotypes differed significantly [Recovery × Group, F(3, 30) = 4.044, p = 0.016]. Simple-effects tests revealed that Lc/+ mice had significantly longer average EEP periods than WT mice during recovery from 2 and 4% CO2 [2% Group, F(1, 10) = 6.295, p = 0.031; 4% Group, F(1, 10) = 6.457, p = 0.029]. Although genotypic differences only approached significance following exposure to 6% CO2 [Group, F(1, 10) = 3.128, p = 0.107], there was an interval of approximately 100 breaths when Lc/+ mice had significantly longer EEPs than WT controls during recovery from 6% CO2 [data not shown; Group × Breaths, F(509, 5090) = 1.114, p = 0.046].
Figure 3, graphically illustrates the pattern of individual breaths from a representative mutant and WT mouse at the midpoint of each 4-min recovery period following successive CO2 exposures. Each panel of this figure includes the entire data form, over 10-s containing approximately 40 breaths. Visually, the two genotypes differed greatly in their breathing patterns. During the recovery periods following CO2 exposure, the Lc/+ mice exhibited a distinctly abnormal respiratory pattern, visually consistent with previous reports of post-sigh apnea in mice (38). Thus, the Lc/+ mouse had abnormally deep breaths (>100% of the average TV over the previous 10-s), which were followed by multiple breaths consisting of atypically long EEPs (>100% of the average over the previous 10-s). The WT mice, however, did not exhibit this pattern of disordered breathing, maintaining a more uniform pattern, including TVs and EEPs that did not differ in volume or length.
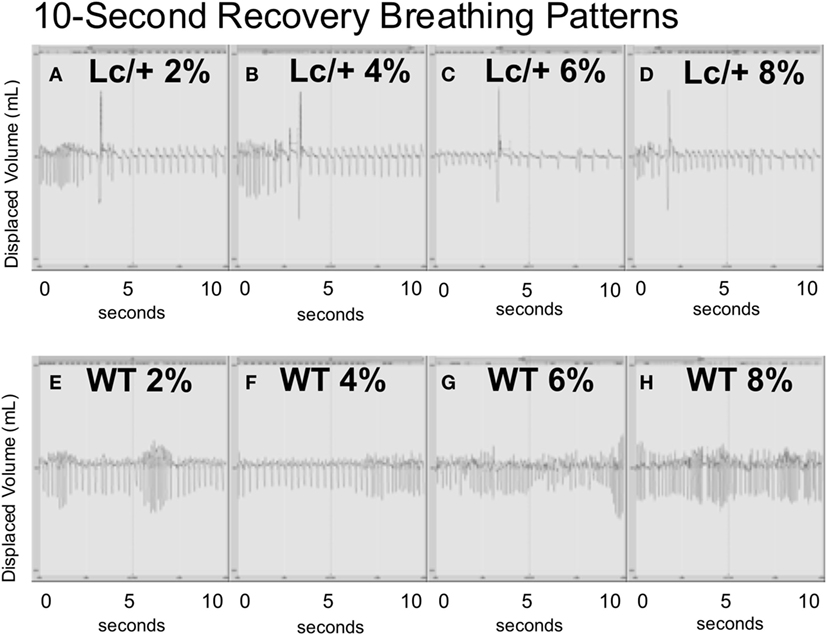
Figure 3. All data including breaths during 10-s intervals from the middle point of each recovery period (at room air) following exposure to successive CO2 challenges (2, 4, 6, and 8%) in an Lc/+ mouse (A–D), and a WT control (E–H). Data are centered on a zero line on the y-axis with inhalations falling below the zero line, and exhalations rising above the zero line. The patterns depicted with the above pair were consistent across all Lc/+ mice and wildtype controls.
Discussion
Lc/+ mice and their WT littermates showed similar patterns of respiration when breathing room air at baseline. This finding further substantiates earlier results, indicating that cerebellar Purkinje cell loss has little effect on components of eupneic breathing (11, 12, 24).
The main outcomes of this study are that developmental loss of cerebellar Purkinje cells significantly reduced the ability of mice to compensate for the increased body burden of CO2 as indicated by extended EEP periods following exposure to relatively low levels of hypercapnia. This is important because reduced compensatory responses to the increased body burden of CO2 would result in prolonged hypoxemia, which itself could facilitate the cardiorespiratory failure that likely precedes sudden death events (28, 30). A similar pattern of disordered breathing has been documented in a report of sudden onset gasping and apnea in nine infants who subsequently died from SIDS (39). Collectively this preclinical and clinical evidence indicates the importance of evaluating breathing patterns and their potential contribution to impending fatal events. That Lc/+ mutants breathing approximated WT levels at 8% further suggests that the threshold for detection for rising levels of CO2 in the mutants is much higher than that observed in control mice. This putative deficit in sensitivity to low levels of CO2 likely results in a reduced ability to compensate for rising blood levels of CO2 and the reduced ability to recover homeostatic ventilatory patterns. Since the cardiovascular system is closely integrated with breathing, disordered cardiovascular patterns, including cerebral blood flow, blood pressure, and heart rate, likely simultaneously occur (25–27). This possibility deserves further investigation.
Mechanisms potentially responsible for the patterns of disordered breathing observed in Lc/+ mice are unlikely to include peripheral muscular deficits, as Lc/+ mice show more fatigue-resistant diaphragm musculature than WT mice (40). Relatedly, a previous examination of the diaphragm in near-miss sudden death infants also revealed more fatigue-resistant diaphragm muscles compared to controls (41).
A more likely mechanism for the deficits in the recovery patterns of Lc/+ mice involves the absence of Purkinje cell input to the FN. The FN are predominately innervated by Purkinje cells of the cerebellum, especially those originating from the vermis, and serve significant roles in cardiorespiratory compensatory responses to multiple challenges, including hypercapnia and hypoxia (8, 10, 42–44). The FN are highly responsive to blood CO2 changes, and to a lesser degree, O2 changes, and modulate respiration during challenging conditions, but not during normal, rhythmic breathing. Cell body lesions of this structure attenuated responses to increased levels of environmental CO2, but had no effect on breathing patterns in room air conditions (10, 14, 23). Structurally, following the loss of 100% of cerebellar Purkinje cells, Lc/+ mice exhibit a nearly 60% reduction in FN size, making it reasonable to conclude that this loss of volume could reduce FN-mediated chemosensitivity (45, 46). Such an inability to appropriately detect and respond to increasing CO2 levels could increase the risk of a fatal outcome in chemosensitive-related extreme challenges, such as those expected in conditions such as SIDS or postictal periods in epilepsy patients.
Deficits in serotonergic neural systems may also play a role in the current findings. Cerebral serotonin [5-hydroxytryptamine (5-HT)] has been repeatedly implicated as integral in respiratory modulation, and pathophysiological changes in the number or distribution of central 5-HT-containing neurons may play a role in disordered breathing associated with sudden unexplained death (28, 47, 48). Lc/+ mice, in comparison to controls, show cerebellar patterns of disordered 5-HT that include a redistribution of 5-HT levels, resulting in increased levels in the deep cerebellar nuclei, including the FN, as well as increased serotonin binding in these nuclei (49, 50). In normal mice, an extensive 5-HT projection to Purkinje cells, originating from medullary and pontine respiratory areas exists (51, 52). As Lc/+ mutants lack Purkinje cells, the increased 5-HT projection to the deep cerebellar nuclei possibly represents a reprograming of this serotonergic projection from the Purkinje cells to the deep cerebellar nuclei. This speculative reprograming, in turn, may provide an additional contribution to the loss of sensitivity to low levels of CO2. Thus, the differences observed in recovery patterns between Lc/+ and WT mice could result from neurochemical events that unfold relative to the developmental Purkinje cell loss in Lc/+ mice and further suggest an integral role of the cerebellum in respiratory modulation.
Other disorders also exude the characteristic traits of cerebellar neuropathology, including Purkinje cell loss, shrinkage of deep cerebellar nuclei, serotonergic disruptions, and disordered breathing patterns accompanying unexpected fatalities. Such signs are frequently found in epilepsy. People with epilepsy are roughly 24 times more likely to die unexpectedly than people without epilepsy (53), and ventilatory failure is a major suspect in the fatal event. As in SIDS, SUDEP is an exclusionary diagnosis, and SUDEP victims are typically found in the prone position in bed, which supports the possibility that death has occurred during sleep (54, 55). Postmortem examination of the brains of patients with chronic epilepsy and SUDEP victims consistently reveals cerebellar neuropathology, including cerebellar atrophy and Purkinje cell loss (56–58). Examination of the cerebellum of SUDEP victims revealed significantly higher amounts of Bergmann’s gliosis and folial atrophy compared to age- and sex-matched controls (57). Additionally, decreased cerebellar Purkinje cell densities in either the anterior or posterior lobe appear in SUDEP victims relative to controls (56). Antiepileptic drugs (AEDs), specifically phenytoin (Dilantin), exacerbate cerebellar atrophy and Purkinje cell loss found in patients with epilepsy, and cause cerebellar neuronal damage in both humans and animal models (56, 59–63). Additionally, AEDs, including phenytoin and benzodiazepines, such as clonazepam are associated with an increased incidence of sleep-disordered breathing (64).
Sleep-disordered breathing frequently presents comorbidly with epilepsy (64, 65) and even without treatment with AEDs; patients report excessive daytime sleepiness, which can be an indicator of undiagnosed sleep apnea (66–72). The relevance of these findings for the current study is that sleep-disordered breathing, even without evidence of epilepsy or AEDs, induces significant cerebellar injury (73), and such injury can further intensify disturbed respiratory patterning.
Sudden unexplained death in childhood (SUDC), another exclusionary fatal event, occurs in children >1 year of age (74). It has been speculated that the mechanisms underlying sudden death in SIDS, SUDEP, and SUDC may be closely related (75). Evidence for such a link includes the finding that SUDC victims were significantly more likely to die unexpectedly when one or more febrile seizures were experienced (76). Additionally, an inverse relationship between SIDS and SUDC cases has recently been reported, suggesting that children who die of SUDC may have escaped the window of SIDS only to fail to respond appropriately to a subsequent exogenous stressor (77). As cerebellar neuropathology is often reported in sudden death syndromes, such as SIDS and SUDEP (18, 19, 56–58), and because the cerebellum, especially the Purkinje cells, are particularly vulnerable to hypoxic insult (78), this raises the suspicion that such pathology may be present in the brains of SUDC victims. This possibility, however, has yet to be investigated as research on SUDC is still rudimentary.
Human clinical studies have yielded inconsistent support for the notion that cerebellar neuropathology is involved in sudden death events. For example, in 19 SIDS cases and 12 age-related controls, immunohistochemical staining of the cerebellar vermis revealed evidence of delayed maturation of the cerebellar cortex (18). Further, histological examination at autopsy for 35 cases of unexplained death and 20 controls revealed cerebellar cortex alterations including delayed maturation and apoptosis of the cerebellar Purkinje cells in the sudden unexplained death victims but not the controls (19). Conversely, two separate examinations of the left and right hemispheres of 12 SIDS victims and age- and sex-matched controls revealed no difference between the groups in cerebellar Purkinje cell density (79), Purkinje cell layer volume, or Purkinje cell number (80). However, as cerebellar Purkinje cells are especially vulnerable to even short periods of intermittent hypoxia (78), in the aforementioned studies, the use of controls who had died from conditions involving hypoxia (e.g., suffocation, carbon monoxide intoxication, and heart defect) leaves open the possibility that differences in Purkinje cell number between the two groups were mitigated by circumstances surrounding death. Further, these two studies did not examine the cerebellar vermis – the area of the cerebellum where the Purkinje cells that predominately innervate the FN arise from (81). As the FN is one area responsible for modulating chemosensitive responsivity in situations such as hypoxia (which are believed to occur just prior to unexpected death), it would be of value to include examination of the cerebellar vermis in future studies of sudden unexpected death victims including SIDS.
Collectively, it appears that developmental damage to the cerebellum may play an important role in the development of, or vulnerability to serious, and perhaps life-threatening breathing patterns in several patient populations. Multiple mechanisms may underlie cerebellar Purkinje cell loss, including genetic endowment, excitotoxic injury due to excessive neural activation in epilepsy, treatment with AEDs, hypoxic injury, and surgical insult. Despite various sources of damage to Purkinje cells, the present findings suggest that a potential for impaired breathing patterns exists, including a reduced ability to recover from mild hypercapnia, as demonstrated by a heightened incidence of apneic episodes (i.e., increased EEPs). Several patient populations at risk for sudden death exhibit a combination of cerebellar neuropathology and sleep-disordered breathing, a relationship that suggests that cerebellar injury may contribute to inappropriate responses following hypercapnic exposure that may lead to seriously compromised breathing. The interactions among cerebellar Purkinje cells, their output to autonomic- and respiratory-mediating FN, and integration with other neurotransmitter systems, such as serotonin, that regulate breathing patterns merits continued investigation along with the possibility of sex differences similar to those reported in the human population in sudden death events.
Author Contributions
MC designed the software program and oversaw data acquisition. JH was integral in data acquisition and provided revision input. RH, DG, and GM conceived this work. GM oversaw data acquisition, and performed analyses and interpretation. GM and MC cleaned data, analyzed and interpreted data, and drafted the manuscript and revisions. RH and DG provided substantial revision guidance. All authors were involved in the final approval of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was conducted with assistance from NINDS grant 1R01NS063009. RH was supported by NINDS UO1 NS090407.
References
1. Krous H, Beckwith B, Byard R, Rognum T, Bajanowski T, Corey T. Sudden infant death syndrome. Pediatrics (2004) 114(1):234–8. doi: 10.1542/peds.114.1.234
2. National Institute of Child Health and Human Development, National Institutes of Health. From Cells to Selves: Targeting Sudden Infant Death Syndrome (SIDS): A Strategic Plan. Bethesda, MD: National Institute of Child Health and Human Development (2001).
3. Harper RM, Bandler R. Finding the failure mechanism in sudden infant death syndrome. Nat Med (1998) 4(2):157–8. doi:10.1038/nm0298-157
4. Wandschneider B, Koepp M, Scott C, Micallef C, Balestrini S, Sisodiya SM, et al. Structural imaging biomarkers of sudden unexpected death in epilepsy. Brain (2015) 138(10):2907–19. doi:10.1093/brain/awv233
5. Filiano J, Kinney H. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Neonatology (1994) 65(3–4):194–7. doi:10.1159/000244052
6. Trachtenberg FL, Haas EA, Kinney HC, Stanley C, Krous HF. Risk factor changes for sudden infant death syndrome after initiation of back-to-sleep campaign. Pediatrics (2012) 129(4):630–8. doi:10.1542/peds.2011-1419
7. Zhang K, Wang X. Maternal smoking and increased risk of sudden infant death syndrome: a meta-analysis. Leg Med (2013) 15(3):115–21. doi:10.1016/j.legalmed.2012.10.007
8. Harper RM. The cerebellum and respiratory control. Cerebellum (2002) 1(1):1–2. doi:10.1080/147342202753203032
9. Harper RM, Kinney HC. Potential mechanisms of failure in the sudden infant death syndrome. Curr Pediatr Rev (2010) 6(1):39–47. doi:10.2174/157339610791317214
10. Lu L, Cao Y, Tokita K, Heck DH, Boughter JD Jr. Medial cerebellar nuclear projections and activity patterns link cerebellar output to orofacial and respiratory behavior. Front Neural Circuits (2013) 7:56. doi:10.3389/fncir.2013.00056
11. Xu F, Frazier DT. Modulation of respiratory motor output by cerebellar deep nuclei in the rat. J Appl Physiol (2000) 89(3):996–1004.
12. Chen ML, Witmans MB, Tablizo MA, Jubran RF, Turkel SB, Tavare CJ, et al. Disordered respiratory control in children with partial cerebellar resections. Pediatr Pulmonol (2005) 40(1):88–91. doi:10.1002/ppul.20225
13. Martino PF, Davis S, Opansky C, Krause K, Bonis JM, Pan LG, et al. The cerebellar fastigial nucleus contributes to CO2-H+ ventilatory sensitivity in awake goats. Respir Physiol Neurobiol (2007) 157(2–3):242–51. doi:10.1016/j.resp.2007.01.019
14. Williams JL, Robinson PJ, Lutherer LO. Inhibitory effects of cerebellar lesions on respiration in the spontaneously breathing, anesthetized cat. Brain Res (1986) 399(2):224–31. doi:10.1016/0006-8993(86)91512-X
15. Xu F, Owen J, Frazier DT. Hypoxic respiratory responses attenuated by ablation of the cerebellum or fastigial nuclei. J Appl Physiol (1995) 79(4):1181–9.
16. Xu F, Frazier DT. Role of the cerebellar deep nuclei in respiratory modulation. Cerebellum (2002) 1(1):35–40. doi:10.1080/147342202753203078
17. Wang T, Eskandari D, Zou D, Grote L, Hedner J. Increased carbonic anhydrase activity is associated with sleep apnea severity and related hypoxemia. Sleep (2015) 38(7):1067–73. doi:10.5665/sleep.4814
18. Cruz-Sanchez FF, Lucena J, Ascaso C, Tolosa E, Quintó L, Rossi ML. Cerebellar cortex delayed maturation in sudden infant death syndrome. J Neuropathol Exp Neurol (1997) 56(4):340–6. doi:10.1097/00005072-199704000-00002
19. Lavezzi AM, Ottaviani G, Mauri M, Matturri L. Alterations of biological features of the cerebellum in sudden perinatal and infant death. Curr Mol Med (2006) 6(4):429–35. doi:10.2174/156652406777435381
20. Lavezzi AM, Alfonsi G, Pusiol T, Matturri L. Decreased argyrophilic nucleolar organiser region (AgNOR) expression in Purkinje cells: first signal of neuronal damage in sudden fetal and infant death. J Clin Pathol (2016) 69(1):58–63. doi:10.1136/jclinpath-2015-202961
21. Harvey RJ, Napper RM. Quantitative studies on the mammalian cerebellum. Prog Neurobiol (1991) 36(6):437–63. doi:10.1016/0301-0082(91)90012-P
22. Napper RM, Harvey RJ. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol (1988) 274(2):168–77. doi:10.1002/cne.902740204
23. Xu F, Owen J, Frazier DT. Cerebellar modulation of ventilatory response to progressive hypercapnia. J Appl Physiol (1994) 77(3):1073–80.
24. Calton M, Dickson P, Harper RM, Goldowitz D, Mittleman G. Impaired hypercarbic and hypoxic responses from developmental loss of cerebellar Purkinje neurons: implications for sudden infant death syndrome. Cerebellum (2014) 13(6):739–50. doi:10.1007/s12311-014-0592-1
25. Alheid GF, McCrimmon DR. The chemical neuroanatomy of breathing. Respir Physiol Neurobiol (2008) 164(1–2):3–11. doi:10.1016/j.resp.2008.07.014
26. Nattie E. CO2, brainstem chemoreceptors and breathing. Prog Neurobiol (1999) 59(4):299–331. doi:10.1016/S0301-0082(99)00008-8
27. Sherwood L. Human Physiology: From Cells to Systems. Belmont, CA: Wadsworth Publishing Co (1997).
28. Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci (2003) 26:239–66. doi:10.1146/annurev.neuro.26
29. Doi A, Ramirez JM. Neuromodulation and the orchestration of the respiratory rhythm. Respir Physiol Neurobiol (2008) 164(1):96–104. doi:10.1016/j.resp.2008.06.007
30. Leiter JC, Böhm I. Mechanisms of pathogenesis in the sudden infant death syndrome. Respir Physiol Neurobiol (2007) 159(2):127–38. doi:10.1016/j.resp.2007.05.014
31. Thach BT. Potential central nervous system involvement in sudden unexpected infant deaths and the sudden infant death syndrome. Compr Physiol (2015) 5(3):1061–8. doi:10.1002/cphy.c130052
32. Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos Trans R Soc Lond B Biol Sci (1979) 287(1020):167–201. doi:10.1098/rstb.1979.0055
33. Cendelín J, Vožeh F. Lurcher mouse. In: Manto M, editor. Handbook of the Cerebellum and Cerebellar Disorders. New York: Springer (2013). p. 1499–520.
34. Téllez-Zenteno JF, Ronquillo LH, Wiebe S. Sudden unexpected death in epilepsy: evidence-based analysis of incidence and risk factors. Epilepsy Res (2005) 65(1):101–15. doi:10.1016/j.eplepsyres.2005.05.004
35. Crosfill M, Widdicombe J. Physical characteristics of the chest and lungs and the work of breathing in different mammalian species. J Physiol (1961) 158(1):1–14. doi:10.1113/jphysiol.1961.sp006750
36. Lorenz JN. A practical guide to evaluating cardiovascular, renal, and pulmonary function in mice. Am J Physiol Regul Integr Comp Physiol (2002) 282(6):R1565–82. doi:10.1152/ajpregu.00759.2001
37. Zosky R. Comparative Biology of the Normal Lung. Aging of the Normal Lung. London: Elsevier (2015).
38. Nakamura A, Fukuda Y, Kuwaki T. Sleep apnea and effect of chemostimulation on breathing instability in mice. J Appl Physiol (2003) 94(2):525–32. doi:10.1152/Japplphysiol.00226.2002
39. Poets CF, Meny RG, Chobanian MR, Bonofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res (1999) 45(3):350–4. doi:10.1203/00006450-199903000-00010
40. Hartmann N, Martrette JM, Westphal A. Influence of the Lurcher mutation on myosin heavy chain expression in skeletal and cardiac muscles. J Cell Biochem (2001) 81(Suppl 36):222–31. doi:10.1002/jcb.1109
41. Scott CB, Nickerson BG, Sargent CW, Dennies PC, Platzker AC, Keens TG. Diaphragm strength in near-miss sudden infant death syndrome. Pediatrics (1982) 69(6):782–4.
42. Lutherer LO, Lutherer BC, Dormer KJ, Janssen HF, Barnes CD. Bilateral lesions of the fastigial nucleus prevent the recovery of blood pressure following hypotension induced by hemorrhage or administration of endotoxin. Brain Res (1983) 269(2):251–7. doi:10.1016/0006-8993(83)90134-8
43. Nattie E. Multiple sites for central chemoreception: their roles in response sensitivity and in sleep and wakefulness. Respir Physiol (2000) 122(2–3):223–35. doi:10.1016/S0034-5687(00)00161-4
44. Nattie E, Li AH. Central chemoreceptors: locations and functions. Compr Physiol (2012) 2(1):221–54. doi:10.1002/Cphy.C100083
45. Heckroth JA. A quantitative morphological analysis of the cerebellar nuclei in normal and Lurcher mutant mice. II. Volumetric changes in cytological components. J Comp Neurol (1994) 343(1):183–92. doi:10.1002/cne.903430114
46. Heckroth JA. Quantitative morphological analysis of the cerebellar nuclei in normal and Lurcher mutant mice. I. Morphology and cell number. J Comp Neurol (1994) 343(1):173–82. doi:10.1002/cne.903430113
47. Richerson GB, Buchanan GF. The serotonin axis: shared mechanisms in seizures, depression, and SUDEP. Epilepsia (2011) 52(Suppl 1):28–38. doi:10.1111/j.1528-1167.2010.02908.x
48. Richter DW, Manzke T, Wilken B, Ponimaskin E. Serotonin receptors: guardians of stable breathing. Trends Mol Med (2003) 9(12):542–8. doi:10.1016/j.molmed.2003.10.010
49. Le Marec N, Hebert C, Amdiss F, Botez MI, Reader TA. Regional distribution of 5-HT transporters in the brain of wild type and ‘Purkinje cell degeneration’ mutant mice: a quantitative autoradiographic study with [3H]citalopram. J Chem Neuroanat (1998) 15(3):155–71. doi:10.1016/S0891-0618(98)00041-6
50. Strazielle C, Lalonde R, Riopel L, Botez MI, Reader TA. Regional distribution of the 5-HT innervation in the brain of normal and Lurcher mice as revealed by [3H]citalopram quantitative autoradiography. J Chem Neuroanat (1996) 10(2):157–71. doi:10.1016/0891-0618(96)00115-9
51. Dieudonne S. Book review: serotonergic neuromodulation in the cerebellar cortex: cellular, synaptic, and molecular basis. Neuroscientist (2001) 7(3):207–19. doi:10.1177/107385840100700306
52. Ramos RL. Brainstem projections to molecular layer heterotopia of the cerebellar vermis: evidence from the Allen Mouse Brain Connectivity Database. PeerJ Prepr (2014):2167–9843. doi:10.7287/peerj.preprints.562v1
53. McGugan EA. Sudden unexpected deaths in epileptics – a literature review. Scot Med J (1999) 44(5):137–9.
54. Kloster R, Engelskjon T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry (1999) 67(4):439–44. doi:10.1136/jnnp.67.4.439
55. Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol (2008) 7(11):1021–31. doi:10.1016/S1474-4422(08)70202-3
56. Crooks R, Mitchell T, Thom M. Patterns of cerebellar atrophy in patients with chronic epilepsy: a quantitative neuropathological study. Epilepsy Res (2000) 41(1):63–73. doi:10.1016/S0920-1211(00)00133-9
57. Shields LB, Hunsaker DM, Hunsaker JC III, Parker JC Jr. Sudden unexpected death in epilepsy: neuropathologic findings. Am J Forensic Med Pathol (2002) 23(4):307–14. doi:10.1097/01.PAF.0000043305.50912.17
58. Thom M, Michalak Z, Wright G, Dawson T, Hilton D, Joshi A, et al. Audit of practice in sudden unexpected death in epilepsy (SUDEP) post mortems and neuropathological findings. Neuropathol Appl Neurobiol (2015). doi:10.1111/nan.12265
59. Eldridge R, Stern R, Iivanainen M, Koerber T, Wilder BJ. Baltic myoclonus epilepsy – hereditary disorder of childhood made worse by phenytoin. Lancet (1983) 2(8354):838–42. doi:10.1016/S0140-6736(83)90749-3
60. Armstrong D, Halliday W, Hawkings C, Takashima S. Pediatric Neuropathology: A Text-Atlas. New York: Springer Science & Business Media (2008). 425 p.
62. Koller WC, Glatt SL, Perlik S, Huckman MS, Fox JH. Cerebellar atrophy demonstrated by computed tomography. Neurology (1981) 31(4):405–12. doi:10.1212/WNL.31.4_Part_2.405
63. Ohmori H, Ogura H, Yasuda M, Nakamura S, Hatta T, Kawano K, et al. Developmental neurotoxicity of phenytoin on granule cells and Purkinje cells in mouse cerebellum. J Neurochem (1999) 72(4):1497–506. doi:10.1046/j.1471-4159.1999.721497.x
64. Bazil CW. Epilepsy and sleep disturbance. Epilepsy Behav (2003) 4(Suppl 2):S39–45. doi:10.1016/j.yebeh.2003.07.005
65. Manni R, Terzaghi M. Comorbidity between epilepsy and sleep disorders. Epilepsy Res (2010) 90(3):171–7. doi:10.1016/j.eplepsyres.2010.05.006
66. Carney P, Kohrman M. Relation between epilepsy and sleep during infancy and childhood. In: Bazil CW, editor. Sleep and Epilepsy: The Clinical Spectrum. Amsterdam: Elsevier (2002). p. 359–72.
67. Declerck A, Wauquier A, Sijben-Kiggen R, Martens W. A normative study of sleep in different forms of epilepsy. In: Sterman MB, editor. Sleep and Epilepsy. Orlando, FL: Academic Press Inc (1982). p. 329–38.
68. Devinsky O, Ehrenberg B, Barthlen GM, Abramson HS, Luciano D. Epilepsy and sleep apnea syndrome. Neurology (1994) 44(11):2060–4. doi:10.1212/WNL.44.11.2060
69. Hoeppner JB, Garron DC, Cartwright RD. Self-reported sleep disorder symptoms in epilepsy. Epilepsia (1984) 25(4):434–7. doi:10.1111/j.1528-1157.1984.tb03439.x
70. Manni R, Tartara A. Evaluation of sleepiness in epilepsy. Clin Neurophysiol (2000) 111(Suppl 2):S111–4. doi:10.1016/S1388-2457(00)00410-7
71. O’Regan ME, Brown JK. Abnormalities in cardiac and respiratory function observed during seizures in childhood. Dev Med Child Neurol (2005) 47(1):4–9. doi:10.1111/j.1469-8749.2005.tb01033.x
72. Sterman MB, Shouse MN, Passouant P. Sleep and Epilepsy. Orlando, FL: Academic Press Inc (1982). 531 p.
73. Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, et al. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med (2002) 166(10):1382–7. doi:10.1164/rccm.200201-050OC
74. Krous HF, Chadwick AE, Crandall L, Nadeau-Manning JM. Sudden unexpected death in childhood: a report of 50 cases. Pediatr Dev Pathol (2005) 8(3):307–19. doi:10.1007/s10024-005-1155-8
75. Kinney HC, Rognum TO, Nattie EE, Haddad GG, Hyma B, McEntire B, et al. Sudden and unexpected death in early life: proceedings of a symposium in honor of Dr. Henry F. Krous. Forensic Sci Med Pathol (2012) 8(4):414–25. doi:10.1007/s12024-012-9376-4
76. Hesdorffer DC, Crandall LA, Friedman D, Devinsky O. Sudden unexplained death in childhood: a comparison of cases with and without a febrile seizure history. Epilepsia (2015) 56(8):1294–300. doi:10.1111/epi.13066
77. McGarvey CM, O’Regan M, Cryan J, Treacy A, Hamilton K, Devaney D, et al. Sudden unexplained death in childhood (1–4 years) in Ireland: an epidemiological profile and comparison with SIDS. Arch Dis Child (2012) 97(8):692–7. doi:10.1136/archdischild-2011-301393
78. Pae EK, Chien P, Harper RM. Intermittent hypoxia damages cerebellar cortex and deep nuclei. Neurosci Lett (2005) 375(2):123–8. doi:10.1016/j.neulet.2004.10.091
79. Oehmichen M, Wullen B, Zilles K, Saternus KS. Cytological investigations on the cerebellar cortex of sudden infant death victims. Acta Neuropathol (1989) 78(4):404–9. doi:10.1007/BF00688177
80. Kiessling MC, Bütiner A, Butti C, Müller-Starck J, Milz S, Hof PR, et al. Intact numbers of cerebellar Purkinje and granule cells in sudden infant death syndrome: a stereologic analysis and critical review of neuropathologic evidence. J Neuropathol Exp Neurol (2013) 72(9):861–70. doi:10.1097/NEN.0b013e3182a31c31
Keywords: cerebellum, Purkinje cells, disordered breathing, sudden death, SIDS, SUDC, SUDEP
Citation: Calton MA, Howard JR, Harper RM, Goldowitz D and Mittleman G (2016) The Cerebellum and SIDS: Disordered Breathing in a Mouse Model of Developmental Cerebellar Purkinje Cell Loss during Recovery from Hypercarbia. Front. Neurol. 7:78. doi: 10.3389/fneur.2016.00078
Received: 03 March 2016; Accepted: 02 May 2016;
Published: 13 May 2016
Edited by:
Anna Maria Lavezzi, Milan University, ItalyReviewed by:
Kumar Sannagowdara, Medical College of Wisconsin, USAGiuseppe Del Corno, Universita Milano-Bicocca, Italy
Copyright: © 2016 Calton, Howard, Harper, Goldowitz and Mittleman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Mittleman, Z21pdHRsZW1hbkBic3UuZWR1