 Yves Lecarpentier
Yves Lecarpentier Alexandre Vallée
Alexandre Vallée- 1Centre de Recherche Clinique, Hôpital de Meaux, Meaux, France
- 2CHU Amiens Picardie, Université Picardie Jules Verne, Amiens, France
- 3Experimental and Clinical Neurosciences Laboratory, INSERM U1084, University of Poitiers, Poitiers, France
The opposite interplay between peroxisome proliferator-activated receptor gamma (PPAR gamma) and Wnt/beta-catenin signaling has led to the categorization of neurodegenerative diseases (NDs) as either NDs in which PPAR gamma is downregulated while the canonical Wnt/beta-catenin pathway is upregulated [amyotrophic lateral sclerosis (ALS), Parkinson’s disease, Huntington’s disease, multiple sclerosis, Friedreich’s ataxia] or NDs in which PPAR gamma is upregulated while the canonical Wnt/beta-catenin signaling is downregulated (bipolar disorder, schizophrenia, Alzheimer’s disease). ALS, a common adult-onset debilitating ND, is characterized by a chronic and progressive degeneration of upper and lower motor neurons resulting in muscular atrophy, paralysis, and ultimately death. The intent of this review is to provide an analysis of the integration of these two opposed systems, i.e., canonical Wnt/beta-catenin and PPAR gamma, in ALS. Understanding this integration may aid in the development of novel ALS therapies. Although the canonical Wnt/beta-catenin pathway is upregulated in ALS, riluzole, an enhancer of the canonical Wnt signaling, is classically prescribed in this disease in humans. However, studies carried out on ALS transgenic mice have shown beneficial effects after treatment by PPAR gamma agonists partly due to their anti-inflammatory effects.
Introduction
Neurodegenerative diseases (NDs) are frequent and often present a bad prognosis. Both peroxisome proliferator-activated receptor (PPAR) gamma and the canonical Wnt/beta-catenin pathway play a key role in the pathophysiology of several NDs. The opposite interaction between PPAR gamma and the canonical Wnt/beta-catenin pathway has been reported in numerous studies (1–7). Certain NDs can be divided into two categories (8). On the one hand, there are NDs in which PPAR gamma is upregulated while the Wnt/beta-catenin pathway is downregulated, such as in bipolar disorder, schizophrenia, and Alzheimer’s disease. On the other hand, there are NDs in which PPAR gamma is downregulated while the Wnt/beta-catenin pathway is upregulated, such as in amyotrophic lateral sclerosis (ALS), Huntington’s disease, Parkinson’s disease, multiple sclerosis, and Friedreich’s ataxia. PPAR gamma agonists could exert protective effects in several NDs. They induce beneficial effects in ALS neurons of transgenic mice (9–12). Moreover, PPAR gamma coactivator-1alpha (PGC-1alpha) protects neurons and slows down the disease progression in ALS double transgenic mice (13, 14). PPAR gamma signaling may represent a therapeutic target in human ALS. Although the canonical Wnt/beta-catenin pathway is upregulated in ALS (15–18), riluzole, an enhancer of this system (19) represents a classically prescribed treatment of this disease in humans.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis has been first described by J. M. Charcot in 1869. ALS, a fatal neurodegenerative disorder, is characterized by a chronic and progressive degeneration of the upper and lower motor neurons. This results in muscular atrophy, paralysis, and ultimately death. ALS is a common adult-onset debilitating ND. Its prevalence is about 5 per 100,000 individuals. Today, the pathophysiology of ALS in humans is not fully elucidated and is particularly complex partly due to various interconnected pathological mechanisms (20). 82% of ALS is sporadic. The most frequent mutations in familial ALS (FALS) or inherited are found in the SOD1 gene (Cu, Zn superoxide dismutase). FALS presents glutamate toxicity, mitochondrial dysfunction, and axonal transport defects (20). Many animal studies have used the mutant SOD1 transgenic mice (21). This model is reminiscent of the human ALS phenotype by developing a progressive motor neuron degeneration (22). The human sporadic ALS differs little clinically from SOD1-related FALS. Both forms induce motor neuron degeneration, paralysis, and death within 3–5 years from the appearance of the first symptoms. No pharmacological therapy can really stop the progression of ALS. Nevertheless riluzole is approved for the treatment of ALS patients although the benefits of this drug remain marginal (23–26).
PPAR Gamma
Peroxisome proliferator-activated receptor gamma, a ligand-activated transcriptional factor, belongs to the nuclear hormone receptor superfamily. All PPARs heterodimerize with the retinoid X receptor (RXR), and they bind to peroxisome proliferator response elements, which are specific regions on the DNA of target genes. Naturally occurring agents directly activate PPAR gamma. PPAR gamma is expressed in various cell types, such as adipose tissue, immune cells, and brain cells, including astrocytes and microglia. This contributes to the anti-inflammatory response in the central nervous system (CNS) (27, 28). PPAR gamma regulates the expression of numerous genes implied in various cellular mechanisms, such as regulation of glucose homeostasis, insulin sensitivity, lipid metabolism, immune response, inflammation, and cell fate (29, 30). PPAR gamma is involved in several pathological states, such as diabetes, obesity, atherosclerosis, and cancer. PPAR gamma agonists, thiazolidinediones (TZDs), are insulin-sensitizing drugs, and certain TZDs have been used for treating type 2 diabetes. PPAR gamma diminishes inflammatory processes in many tissues. Moreover, PPAR gamma is a peripheral regulator of cardiovascular rhythms and controls circadian variations in blood pressure and heart rate through BMAL1 (31, 32). The role of PPAR gamma in neurodegeneration is well established (33). PPAR gamma agonists have been shown to be beneficial in several NDs, such as stroke, Alzheimer’s disease, ALS, Parkinson’s disease, Huntington’s disease, multiple sclerosis. PPAR gamma induces both neuroprotective and anti-inflammatory effects (27, 28). Astrocytic GLT1/EAAT2 gene, a target of PPAR gamma, induces neuroprotection by increasing glutamate uptake (34).
Canonical Wnt/Beta-Catenin Pathway
Canonical Wnt/beta-catenin signaling plays a key role in embryonic development, cell migration, cell fate, and carcinogenesis (35, 36). Activation of the canonical Wnt pathway by Wnt ligands leads to increase the cytoplasmic beta-catenin level. Beta-catenin subsequently translocates to the nucleus and activates beta-catenin-specific gene transcription (1, 37) (Figure 1). In the absence of Wnt ligand, beta-catenin is recruited into the destruction complex that contains adenomatous polyposis coli (APC), Axin, and glycogen synthase kinase-3beta (GSK-3beta), which induces phosphorylation of beta-catenin, thus targeting it for ubiquitination and proteasomal degradation (Figure 2). In the presence of Wnt ligand, the binding of Wnt to Frizzled (Fzd) activates Dishevelled (Dsh). Dsh recruits Axin, which binds to the low density lipoprotein-receptor-related proteins (LRP5–6). Activation of Dsh inhibits GSK-3beta. This reduces phosphorylation and degradation of beta-catenin. The non-phosphorylated beta-catenin translocates to the nucleus and binds to T-cell/lymphoid-enhancing binding (TCF/LEF) transcription factors. This activates numerous target genes, including c-myc, cyclin D1, Axin-2, CD44, Cox2, and PPAR beta/delta (38, 39).
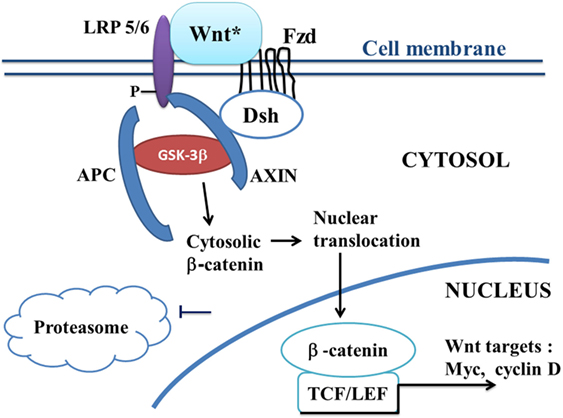
Figure 1. A schematic model of the canonical Wnt/beta-catenin pathway in ALS. In ALS, the canonical Wnt/beta-catenin pathway is upregulated (“on state”). Binding of Wnt to Fzd leads to activation of Dsh, which recruits the destruction complex to the plasma membrane. AXIN binds to the cytoplasmic tail of LRP5/6. Wnt also binds LRP5/6. This initiates LRP phosphorylation and Dsh-mediated Frizzled internalization. Activation of Dsh leads to the inhibition of GSK-3beta, which further reduces the phosphorylation and degradation of beta-catenin. The beta-catenin degradation complex AXIN/APC/GSK-3beta is inactivated with the recruitment of AXIN to the plasma membrane. Beta-catenin phosphorylation is inhibited. Then, beta-catenin accumulates into the cytosol and translocates to the nucleus to bind TCF–LEF co-transcription factors. This induces the canonical Wnt-response gene transcription (c-Myc, cyclin D, etc.). Abbreviations: APC, adenomatous polyposis coli; Dsh, Dishevelled; Fzd, Frizzled; GSK-3beta, glycogen synthase kinase-3beta; LRP5/6, low density lipoprotein receptor-related protein 5/6; TCF/LEF, T-cell factor/lymphoid enhancer factor; Wnt target genes: c-Myc, cyclin D; Wnt*, Wnt with ligands.
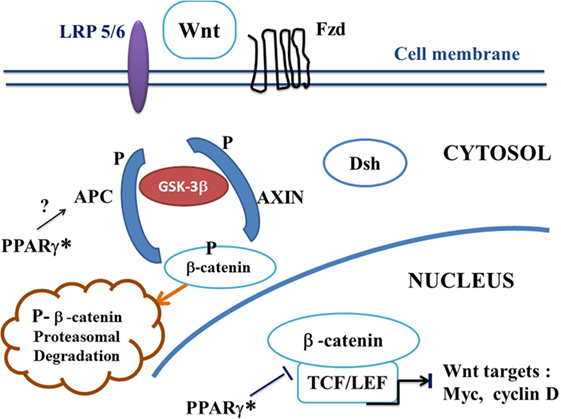
Figure 2. A potential canonical Wnt/beta-catenin pathway in ALS treated by PPAR gamma agonists. In the presence of PPAR gamma agonists, the “on state” of canonical Wnt/beta-catenin pathway may be interrupted at two potential levels. Firstly, via a hypothetical action of PPAR gamma* on APC. Inactivation of the destruction complex AXIN/APC/GSK-3beta is suppressed. Beta-catenin is phosphorylated by GSK-3beta. Thus, APC and AXIN complex with GSK-3beta and beta-catenin to enhance the destruction process of beta-catenin into the proteasome. Phosphorylated beta-catenin is recognized by an ubiquitin ligase, ubiquinated and degraded into the proteasome. The canonical Wnt pathway is in “off state.” Second, PPAR gamma can inhibit the beta-catenin/TCF/LEF complex into the nucleus, thus inhibiting the transcription of canonical Wnt target genes. Abbreviations: APC, adenomatous polyposis coli; Dsh, Dishevelled; Fzd, Frizzled; GSK-3beta, glycogen synthase kinase-3beta; LRP5/6, low density lipoprotein receptor-related protein 5/6; PPAR gamma*, PPAR gamma activated by agonists; TCF/LEF, T-cell factor/lymphoid enhancer factor; canonical Wnt target genes: c-Myc, cyclin D, etc.
Opposite Interplay Between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway
PPAR Gamma Activation Induces Repression of the Beta-Catenin Pathway
Several studies have shown the opposite interaction between beta-catenin and PPAR gamma (1, 5, 40). The TZDs troglitazone, rosiglitazone, pioglitazone, and a non-TZD PPAR gamma activator GW1929 inhibit the transcription of beta-catenin (2, 3, 5, 6). Opposite interaction between PPAR gamma and beta-catenin involves the TCF/LEF factor-binding domain of beta-catenin and a catenin-binding domain of PPAR gamma (3). The antagonism between the beta-catenin pathway and PPAR gamma has been reported in various cell types, such as adipocytes (2) and hepatocytes (6). PPAR gamma suppresses the Wnt/beta-catenin pathway during adipogenesis (5). Conversely, activation of the canonical Wnt/beta-catenin pathway inhibits PPAR gamma expression (7).
Deactivation of the Wnt/Beta-Catenin Pathway Induces Activation of PPAR Gamma
Inhibition of Wnt/beta-catenin pathway leads to increase the transcription of PPAR gamma (1). In preadipocytes, prevention of the Wnt pathway by overexpression of Axin induces differentiation into adipocytes. The canonical Wnt/beta-catenin/PPAR gamma system regulates the molecular switching of adipogenesis versus osteablastogenesis. The adipogenic pathway implies inhibition of Wnt signaling, leading to degradation of beta-catenin. This results in transcription of PPAR gamma, which initiates adipogenesis (7). Deactivation of the canonical Wnt/beta-catenin pathway and activation of PPAR gamma are also observed in arrhythmogenic right ventricular cardiomyopathy (ARVC) (1, 41). The suppression of canonical Wnt/beta-catenin signaling by plakoglobin (PG), i.e., gamma-catenin, recapitulates the phenotype of ARVC (1). The desmosomal PG has structural and functional similarities to beta-catenin (42). Gamma-catenin competes with beta-catenin through TCF/LEF transcription factors (43, 44). After competition with beta-catenin, gamma-catenin inhibits the canonical Wnt/beta-catenin-TCF/LEF pathway. This leads to enhance adipogenesis and fibrogenesis, as it is observed in human ARVC (1, 41, 45).
ALS and PPAR Gamma
Neuroinflammation is commonly reported in NDs and particularly in ALS. PPAR gamma is a regulator of neuroinflammation and possibly represents a target for new therapeutic strategies. Beneficial effects induced by PPAR gamma agonists, partly due to their anti-inflammatory effects, have been observed in NDs. PPAR gamma inhibits NF-kappa B-mediated inflammatory signaling (46, 47). In ALS transgenic mice, beneficial effects of pioglitazone have been mainly ascribed to its anti-inflammatory activity. PPAR gamma agonists reduce neuropathological damages caused by inflammation in ALS (11). The involvement of PPAR gamma in ALS progression has been demonstrated in hSOD1G93A mice using pioglitazone. Pioglitazone minimizes ALS-like symptoms (12) and increases survival in ALS transgenic mouse model (11). Pioglitazone-treated SOD1-G93A transgenic mice exhibit a delayed onset of the disease and survive significantly longer than non-treated transgenic mice. Pioglitazone induces neuroprotection in motor neurons of the spinal cord. The median fiber diameter of the quadriceps muscle is preserved. This indicates a morphological and functional protection of motor neurons due to pioglitazone (12). In neuron-specific PPAR gamma knockout mice and in response to middle cerebral artery occlusion, significant brain damages have been reported (48). Moreover, there is a reduced expression of certain genes, including SOD1 and glutathione S-transferase. Thus, in normal neurons, PPAR gamma is expressed and plays an important protective role. In addition to their anti-inflammatory effects, PPAR gamma agonists appear to be useful in neuroprotection. PPAR gamma activation induces neuroprotective effects in a Drosophila model of ALS, which recapitulates several aspects of the ALS phenotype (10). In a phase II double-blind controlled clinical trial, pioglitazone, in combination with riluzole, does not increase survival in ALS patients (49). The absence of beneficial effects in this trial might be partly due to the fact that riluzole activates the canonical Wnt/beta-catenin signaling in neuronal cells, as it will be discussed below (19). Moreover, pioglitazone is not a selective PPAR gamma agonist and belongs to the dual PPAR alpha/gamma agonists. Several studies have revealed a potential risk of bladder cancer in diabetic patients treated with pioglitazone, such as to call into question the benefit/risk ratio of this PPAR agonist (50). To highlight PPAR transcriptional activity, genetically engineered mice (PPRE-Luc mice) have been bred with the hSOD1G93AALS mouse (51). In the PPRE-Luc± hSOD1G93A± double transgenic mice, PPAR gamma transcriptional activity increases in the spinal cord (9). PPAR gamma controls natural protective mechanisms against lipid peroxidation. This effect is correlated with the upregulation of lipid detoxification enzymes, such as glutathione S-transferase and alpha-2 lipoprotein lipase. These enzymes are implied in scavenging lipid peroxidation by-products.
ALS and PPAR Gamma Coactivator-1 Alpha
Peroxisome proliferator-activated receptor gamma coactivator-1alpha, a transcriptional coactivator working together with PPAR gamma, plays an important role in numerous NDs (33). Increase in expression of PGC-1alpha in transgenic SOD1-G93A mice prevents neuronal cell death (52). SOD1-G93A mice present mitochondrial abnormalities in motor neurons of the spinal cord, and PGC-1alpha induces neuroprotection. Expression of PGC-1alpha reduces motor neuron degeneration in SOD1-G93A transgenic mice (13, 14). In PGC-1alpha transgenic mice crossed with SOD1 mutant G93A mice, there is a neuroprotection, and the progression of the disease is significantly slowed (13). Motor function and survival are improved by using a double transgenic mouse model where PGC-1alpha is overexpressed in a SOD1 transgenic mouse (14). In this double transgenic mouse model, there are decreased weight loss, improved motor performance, slowed disease progression, and reduced motor neuronal death. In a mouse model of inherited ALS, increased PGC-1alpha activity sustains mitochondrial biogenesis and muscle function without extending survival (53).
ALS, PPAR Gamma, Retinoid X Receptor, and Retinoic Acid
Peroxisome proliferator-activated receptor and RXR receptor form heterodimers to become functional. There is a convergence of the 9-cis retinoic acid and PPAR pathways through heterodimer formation of the two receptors (54). Retinoid signaling is important in the development of the CNS and contributes to regenerative processes that occur after injury. In ALS, the retinoid signaling is involved in gene expression in the spinal cord (55). Abnormalities of the anaplastic lymphoma kinase (ALK) gene are associated with sporadic ALS. Reduced ALK mRNA levels have been reported in ALS patients (56). Midkine (MK), a ligand for ALK, is decreased in patients with sporadic ALS (57). MK expression is induced by the retinoic acid signaling. Moreover, MK activates ALK. Retinoic acid gene improves survival in the ALS SOD1-G93A transgenic mice (58). In ALS patients treated with tretinoin (i.e., retinoic acid), pioglitazone combined with riluzole has demonstrated no slowing on the disease progression (59). This might be partly explained by the fact that riluzole is an enhancer of the canonical Wnt pathway, as reported below. Bexarotene (Bxt), a highly selective RXR agonist, induces neuroprotective effect in the SOD1G93A mouse model of ALS (60). Bxt significantly delays the early motor neuron degeneration in ALS transgenic mice and partially preserves the spinal motor neuron loss. Bxt ameliorates the loss of body weight and increases mouse survival up to 30% of the symptomatic period. Bxt preserves the neuromuscular function.
ALS and Canonical Wnt/Beta-Catenin Pathway
The canonical Wnt/beta-catenin signaling is involved in numerous NDs, particularly in ALS. Many studies have shown that this pathway is upregulated in motor neurons of ALS mouse model (15–18). In the spinal cord of SOD1-G93A ALS transgenic mice, mRNAs and proteins of both Wnt2 and Wnt7a are upregulated (16). Activation of the Wnt signaling inhibits the GSK-3beta activity. In the spinal cord of these mice, mRNAs and proteins of both Wnt3a and Cyclin D1 are upregulated (15). Beta-catenin translocates to the nucleus and subsequently activates transcription of the target gene Cyclin D1. Increase in Wnt5a and Fzd2 expression in the spinal cord of SOD1-G93A ALS transgenic mice, SOD1-G93A transfected NSC-34 cells, and primary cultures of astrocytes from SOD1-G93A transgenic mice have been shown to be changed (17). In the spinal cords of SOD1-G93A ALS transgenic mice, expression of Wnt1 and Fzd1 is increased (18).
ALS, Riluzole, and Lithium
No really efficient treatment exists for ALS (61, 62). However, riluzole is approved for the treatment of ALS in most countries. This is based on the results supporting a role of glutamate toxicity in ALS. Riluzole has numerous pharmacological properties, i.e., presynaptic inhibition of the glutamate release, inhibition of G-protein-dependent processes, blockade of the voltage-gated sodium channel, and modulation of N-methyl-d-aspartate ionotropic receptor (61). Two trials (23, 24) have shown a prolongation of median survival by 2–3 months by using riluzole in human ALS. Two other studies have led to similar conclusions (25, 26). Interestingly, riluzole has been shown to be an enhancer of the Wnt/β-catenin signaling in melanoma (19). Treating melanoma cells with riluzole in vitro enhances the ability of Wnt3A to regulate gene expression and decrease cell proliferation. Importantly, riluzole also enhances the Wnt/beta-catenin pathway in both HT22 neuronal cells and adult hippocampal progenitor cells (19). As the Wnt/beta-catenin pathway is upregulated, at least in genetic ALS mice (15–18), this might partly explain the poor results reported in trials testing riluzole in ALS (23–26). The GSK-3beta-inhibitor lithium, an activator of the canonical Wnt/beta-catenin signaling (63–65), has also been evaluated as a treatment for ALS (66). Surprisingly, in ALS patients treated with lithium, the disease progression appears to be markedly attenuated. In a parallel study on the genetic SOD1-G93A mouse ALS animal model, lithium induces a marked neuroprotection, delaying disease onset and duration and increasing the life span. Lithium is logically used for the treatment of bipolar disorder in which the Wnt/beta-catenin signaling is downregulated (67, 68). Thus, riluzole reduces symptoms of obsessive–compulsive disorder, unipolar and bipolar depression, and generalized anxiety disorder (69). This is expected because the Wnt/beta-catenin pathway is downregulated in bipolar syndrome and because riluzole, like lithium, are enhancers of the Wnt/β-catenin signaling (19).
Conclusion
Peroxisome proliferator-activated receptor gamma agonists represent promising therapeutics for certain NDs, such as ALS. PPAR gamma agonists appear to be beneficial in several ALS transgenic animal models partly due to their anti-inflammatory properties. Moreover, PPAR gamma activation induces repression of the beta-catenin pathway (2, 3, 5, 6). This represents the rationale for the use of PPAR gamma agonists in case of downregulation of PPAR gamma and upregulation of the Wnt/beta-catenin pathway, such as in ALS. New PPAR gamma agonists, specific and devoid of adverse side effects, might represent a new therapeutic approach for the treatment of ASL. PPAR gamma agonists increase survival in ALS transgenic mice and minimize neuroinflammation. Rather than stimulation of the canonical Wnt/beta-catenin pathway by riluzole administration, inhibition of this system might also represent a therapeutic approach in ALS.
Author Contributions
The authors YL and AV contributed equally to this mini review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Michel Grivaux, Director of the Clinical Research Center, Meaux Hospital and Mr. Vincent Gobert, Administrative Manager of the Clinical Research Center, Meaux hospital, France.
References
1. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest (2006) 116:2012–21. doi:10.1172/JCI27751
2. Gerhold DL, Liu F, Jiang G, Li Z, Xu J, Lu M, et al. Gene expression profile of adipocyte differentiation and its regulation by peroxisome proliferator-activated receptor-gamma agonists. Endocrinology (2002) 143:2106–18. doi:10.1210/en.143.6.2106
3. Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol (2006) 26:5827–37. doi:10.1128/MCB.00441-06
4. Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol (2010) 636:198–202. doi:10.1016/j.ejphar.2010.03.010
5. Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, et al. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J (2003) 376:607–13. doi:10.1042/bj20030426
6. Sharma C, Pradeep A, Wong L, Rana A, Rana B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem (2004) 279:35583–94. doi:10.1074/jbc.M403143200
7. Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol (2009) 5:442–7. doi:10.1038/nrrheum.2009.137
8. Lecarpentier Y, Claes V, Duthoit G, Hebert JL. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front Physiol (2014) 5:429. doi:10.3389/fphys.2014.00429
9. Benedusi V, Martorana F, Brambilla L, Maggi A, Rossi D. The peroxisome proliferator-activated receptor gamma (PPARgamma) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis. J Biol Chem (2012) 287:35899–911. doi:10.1074/jbc.M112.366419
10. Joardar A, Menzl J, Podolsky TC, Manzo E, Estes PS, Ashford S, et al. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum Mol Genet (2015) 24:1741–54. doi:10.1093/hmg/ddu587
11. Kiaei M. Peroxisome proliferator-activated receptor-gamma in amyotrophic lateral sclerosis and huntington’s disease. PPAR Res (2008) 2008:418765. doi:10.1155/2008/418765
12. Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci (2005) 25:7805–12. doi:10.1523/JNEUROSCI.2038-05.2005
13. Liang H, Ward WF, Jang YC, Bhattacharya A, Bokov AF, Li Y, et al. PGC-1alpha protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve (2011) 44:947–56. doi:10.1002/mus.22217
14. Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener (2011) 6:51. doi:10.1186/1750-1326-6-51
15. Chen Y, Guan Y, Liu H, Wu X, Yu L, Wang S, et al. Activation of the Wnt/beta-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem Biophys Res Commun (2012) 420:397–403. doi:10.1016/j.bbrc.2012.03.006
16. Chen Y, Guan Y, Zhang Z, Liu H, Wang S, Yu L, et al. Wnt signaling pathway is involved in the pathogenesis of amyotrophic lateral sclerosis in adult transgenic mice. Neurol Res (2012) 34:390–9. doi:10.1179/1743132812Y.0000000027
17. Li X, Guan Y, Chen Y, Zhang C, Shi C, Zhou F, et al. Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int J Clin Exp Pathol (2013) 6:1245–60.
18. Wang S, Guan Y, Chen Y, Li X, Zhang C, Yu L, et al. Role of Wnt1 and Fzd1 in the spinal cord pathogenesis of amyotrophic lateral sclerosis-transgenic mice. Biotechnol Lett (2013) 35:1199–207. doi:10.1007/s10529-013-1199-1
19. Biechele TL, Camp ND, Fass DM, Kulikauskas RM, Robin NC, White BD, et al. Chemical-genetic screen identifies riluzole as an enhancer of Wnt/beta-catenin signaling in melanoma. Chem Biol (2010) 17:1177–82. doi:10.1016/j.chembiol.2010.08.012
20. Redler RL, Dokholyan NV. The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog Mol Biol Transl Sci (2012) 107:215–62. doi:10.1016/B978-0-12-385883-2.00002-3
21. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 364:362. doi:10.1038/364362c0
22. Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol (2008) 85:94–134. doi:10.1016/j.pneurobio.2008.01.001
23. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/riluzole study group. N Engl J Med (1994) 330:585–91. doi:10.1056/NEJM199403033300901
24. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis/riluzole study group II. Lancet (1996) 347:1425–31. doi:10.1016/S0140-6736(96)91680-3
25. Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord (2003) 4:191–206. doi:10.1080/14660820310002601
26. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev (2012) 3:CD001447. doi:10.1002/14651858.CD001447
27. Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J Neuroinflammation (2012) 9:63. doi:10.1186/1742-2094-9-63
28. Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci (2008) 13:1813–26. doi:10.2741/2802
29. Elbrecht A, Chen Y, Cullinan CA, Hayes N, Leibowitz M, Moller DE, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun (1996) 224:431–7. doi:10.1006/bbrc.1996.1044
30. Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem (1997) 272:18779–89. doi:10.1074/jbc.272.30.18779
31. Lecarpentier Y, Claes V, Hebert JL. PPARs, cardiovascular metabolism, and function: near- or far-from-equilibrium pathways. PPAR Res (2010) 2010:783273. doi:10.1155/2010/783273
32. Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, et al. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab (2008) 8:482–91. doi:10.1016/j.cmet.2008.10.009
33. Katsouri L, Blondrath K, Sastre M. Peroxisome proliferator-activated receptor-gamma cofactors in neurodegeneration. IUBMB Life (2012) 64:958–64. doi:10.1002/iub.1097
34. Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J Cereb Blood Flow Metab (2007) 27:1327–38. doi:10.1038/sj.jcbfm.9600438
35. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell (2012) 149:1192–205. doi:10.1016/j.cell.2012.05.012
36. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet (2004) 5:691–701. doi:10.1038/nrg1427
37. Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov (2006) 5:997–1014. doi:10.1038/nrd2154
38. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science (1998) 281:1509–12. doi:10.1126/science.281.5382.1509
39. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A (1999) 96:5522–7. doi:10.1073/pnas.96.10.5522
40. Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, et al. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci U S A (2005) 102:1460–5. doi:10.1073/pnas.0405928102
41. Djouadi F, Lecarpentier Y, Hebert JL, Charron P, Bastin J, Coirault C. A potential link between peroxisome proliferator-activated receptor signalling and the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Res (2009) 84:83–90. doi:10.1093/cvr/cvp183
42. Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science (2002) 296:1644–6. doi:10.1126/science.1071549
43. Ben-Ze’ev A, Geiger B. Differential molecular interactions of beta-catenin and plakoglobin in adhesion, signaling and cancer. Curr Opin Cell Biol (1998) 10:629–39. doi:10.1016/S0955-0674(98)80039-2
44. Zhurinsky J, Shtutman M, Ben-Ze’ev A. Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol Cell Biol (2000) 20:4238–52. doi:10.1128/MCB.20.12.4238-4252.2000
45. Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol (1997) 30:1512–20. doi:10.1016/S0735-1097(97)00332-X
46. Chen YC, Wu JS, Tsai HD, Huang CY, Chen JJ, Sun GY, et al. Peroxisome proliferator-activated receptor gamma (PPAR-gamma) and neurodegenerative disorders. Mol Neurobiol (2012) 46:114–24. doi:10.1007/s12035-012-8259-8
47. Shibata N, Kawaguchi-Niida M, Yamamoto T, Toi S, Hirano A, Kobayashi M. Effects of the PPARgamma activator pioglitazone on p38 MAP kinase and IkappaBalpha in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology (2008) 28:387–98. doi:10.1111/j.1440-1789.2008.00890.x
48. Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, et al. Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci (2009) 29:6186–95. doi:10.1523/JNEUROSCI.5857-08.2009
49. Dupuis L, Dengler R, Heneka MT, Meyer T, Zierz S, Kassubek J, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS One (2012) 7:e37885. doi:10.1371/journal.pone.0037885
50. Faillie JL, Hillaire-Buys D. Examples of how the pharmaceutical industries distort the evidence of drug safety: the case of pioglitazone and the bladder cancer issue. Pharmacoepidemiol Drug Saf (2016) 25:212–4. doi:10.1002/pds.3925
51. Ciana P, Biserni A, Tatangelo L, Tiveron C, Sciarroni AF, Ottobrini L, et al. A novel peroxisome proliferator-activated receptor responsive element-luciferase reporter mouse reveals gender specificity of peroxisome proliferator-activated receptor activity in liver. Mol Endocrinol (2007) 21:388–400. doi:10.1210/me.2006-0152
52. Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiol Dis (2013) 51:72–81. doi:10.1016/j.nbd.2012.07.004
53. Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, et al. Elevated PGC-1alpha activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab (2012) 15:778–86. doi:10.1016/j.cmet.2012.03.019
54. Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature (1992) 358:771–4. doi:10.1038/358771a0
55. Malaspina A, Kaushik N, de Belleroche J. A 14-3-3 mRNA is up-regulated in amyotrophic lateral sclerosis spinal cord. J Neurochem (2000) 75:2511–20. doi:10.1046/j.1471-4159.2000.0752511.x
56. Dunckley T, Huentelman MJ, Craig DW, Pearson JV, Szelinger S, Joshipura K, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med (2007) 357:775–88. doi:10.1056/NEJMoa070174
57. Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol (2005) 57:236–51. doi:10.1002/ana.20379
58. Corcoran J, So PL, Maden M. Absence of retinoids can induce motoneuron disease in the adult rat and a retinoid defect is present in motoneuron disease patients. J Cell Sci (2002) 115:4735–41. doi:10.1242/jcs.00169
59. Levine TD, Bowser R, Hank NC, Gately S, Stephan D, Saperstein DS, et al. A pilot trial of pioglitazone HCl and tretinoin in ALS: cerebrospinal fluid biomarkers to monitor drug efficacy and predict rate of disease progression. Neurol Res Int (2012) 2012:582075. doi:10.1155/2012/582075
60. Riancho J, Ruiz-Soto M, Berciano MT, Berciano J, Lafarga M. Neuroprotective effect of bexarotene in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci (2015) 9:250. doi:10.3389/fncel.2015.00250
61. Aggarwal S, Cudkowicz M. ALS drug development: reflections from the past and a way forward. Neurotherapeutics (2008) 5:516–27. doi:10.1016/j.nurt.2008.08.002
62. Carter GT, Krivickas LS, Weydt P, Weiss MD, Miller RG. Drug therapy for amyotrophic lateral sclerosis: where are we now? IDrugs (2003) 6:147–53.
63. Galli C, Piemontese M, Lumetti S, Manfredi E, Macaluso GM, Passeri G. GSK3b-inhibitor lithium chloride enhances activation of Wnt canonical signaling and osteoblast differentiation on hydrophilic titanium surfaces. Clin Oral Implants Res (2013) 24:921–7. doi:10.1111/j.1600-0501.2012.02488.x
64. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol (1997) 185:82–91. doi:10.1006/dbio.1997.8552
65. Sinha D, Wang Z, Ruchalski KL, Levine JS, Krishnan S, Lieberthal W, et al. Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol (2005) 288:F703–13. doi:10.1152/ajprenal.00189.2004
66. Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A (2008) 105:2052–7. doi:10.1073/pnas.0708022105
67. Gould TD, Manji HK. The Wnt signaling pathway in bipolar disorder. Neuroscientist (2002) 8:497–511. doi:10.1177/107385802237176
68. Valvezan AJ, Klein PS. GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front Mol Neurosci (2012) 5:1. doi:10.3389/fnmol.2012.00001
Keywords: amyotrophic lateral sclerosis, Wnt/beta-catenin, PPAR gamma, lithium, riluzole, bexarotene, tretinoin, retinoid acid
Citation: Lecarpentier Y and Vallée A (2016) Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 7:100. doi: 10.3389/fneur.2016.00100
Received: 14 April 2016; Accepted: 15 June 2016;
Published: 28 June 2016
Edited by:
Kevin J. O’Donovan, United States Military Academy, USAReviewed by:
Roland Brandt, University of Osnabrück, GermanyRenee E. Haskew-Layton, Mercy College, USA
Copyright: © 2016 Lecarpentier and Vallée. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Lecarpentier, eXZlcy5jLmxlY2FycGVudGllckBnbWFpbC5jb20=