 Aynara C. Wulsin
Aynara C. Wulsin James P. Herman1,2
James P. Herman1,2 Steve C. Danzer
Steve C. Danzer- 1Department of Psychiatry and Behavioral Neuroscience, College of Medicine, University of Cincinnati, Cincinnati, OH, USA
- 2Neuroscience Graduate Program, College of Medicine, University of Cincinnati, Cincinnati, OH, USA
- 3Department of Anesthesia and Pediatrics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
Status epilepticus (SE) induces rapid hyper-activation of the hypothalamo–pituitary–adrenocortical (HPA) axis. HPA axis hyperactivity results in excess exposure to high levels of circulating glucocorticoids, which are associated with neurotoxicity and depression-like behavior. These observations have led to the hypothesis that HPA axis dysfunction may exacerbate SE-induced brain injury. To test this hypothesis, we used the mouse pilocarpine model of epilepsy to determine whether use of the glucocorticoid receptor antagonist RU486 can attenuate hippocampal pathology following SE. Excess glucocorticoid secretion was evident 1 day after SE in the mice, preceding the development of spontaneous seizures (which can take weeks to develop). RU486 treatment blocked the SE-associated elevation of glucocorticoid levels in pilocarpine-treated mice. RU486 treatment also mitigated the development of hippocampal pathologies induced by SE, reducing loss of hilar mossy cells and limiting pathological cell proliferation in the dentate hilus. Mossy cell loss and accumulation of ectopic hilar cells are positively correlated with epilepsy severity, suggesting that early treatment with glucocorticoid antagonists could have anti-epileptogenic effects.
Introduction
Temporal lobe epilepsy (TLE) is commonly modeled by chemically inducing status epilepticus (SE) in rodents. SE induces widespread brain damage and neuronal restructuring, which lead to the onset of spontaneous seizures (epilepsy) a few weeks later. In particular, the loss of glutamatergic hilar mossy cells and the misplacement of newly generated dentate granule cells (DGCs) to the hilus (HIL) constitute key hippocampal pathologies that occur in synchrony with – or even precede – the development of epilepsy (1–5). Mossy cells mediate feedback inhibition of hippocampal DGCs by activating GABAergic basket cells (6), although direct connections between excitatory mossy cells and granule cells make their net contribution to dentate excitability complex (2). Ectopic granule cells, on the other hand, are hypothesized to destabilize the hippocampal network, promoting hyperexcitability (2, 7). Notably, mossy cell loss and ectopic migration of granule cells positively correlate with epilepsy severity (8), and ablating ectopic cells reduces seizure frequency (9, 10).
The hypothalamo–pituitary–adrenocortical (HPA) axis is rapidly activated by SE, manifested as glucocorticoid basal hypersecretion (11). Animal studies demonstrate that exposure to excess glucocorticoids can be detrimental in the context of epilepsy, leading to increased brain excitability (12–14) and potential damage (15). Conversely, the glucocorticoid synthesis inhibitor metyrapone reduces neuronal injury when given simultaneously with the SE-inducing convulsant kainic acid (16).
Status epilepticus increases glucocorticoid levels, and glucocorticoids can damage the brain. Collectively, these data suggest that blockade of glucocorticoid signaling may be beneficial in preventing hippocampal damage following seizures. Here, we determined whether treatment with the glucocorticoid receptor antagonist RU486, beginning 4 h after the onset of SE, can mitigate pathological changes associated with TLE.
Materials and Methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital Research Foundation and conform to NIH guidelines. Forty-eight 2-month-old C57Bl/6 male mice were obtained from Charles River. All mice received a subcutaneous (s.c.) injection of methyl scopolamine nitrate (1 mg/kg) followed by 420 mg/kg pilocarpine (n = 32) or saline (n = 16) 15 min later (8). Three hours after the onset of SE, mice received two doses of diazepam (10 mg/kg) at 15-min intervals to diminish seizure activity. Control animals also received diazepam. Pretreatment body weight was maintained using sterile Ringer’s solution s.c. to avoid potential SE-related dehydration. Mice treated with pilocarpine were monitored for behavioral seizures and the onset of SE. Only mice that experienced two or more convulsive seizures and exhibited constant behavioral seizure activity (myoclonic jerks, immobility, head bobbing, etc.) for 3 h were considered to be in SE and used in the study.
Following SE, mice were housed in a 32°C incubator overnight. The next morning, mice were returned to their home cages with ad libidum food and water under a 14/10-h light/dark cycle. The final number of SE mice (n = 14) reflects 44% survival of pilocarpine-treated mice. No mortality occurred in the saline-treated group. One hour following the last administration of diazepam, mice were randomly assigned to treatment with either RU486 (20 mg/kg s.c., Sigma-Aldrich, MO, USA) or vehicle (propylene glycol). Treatment was given every morning for seven consecutive days. Groups were generated as follows: (1) SE + vehicle (n = 6), (2) SE + RU486 (n = 8), (3) no-SE + vehicle (n = 8), and (4) no-SE + RU486 (n = 8). The RU486 dose was selected based on prior studies (17). Baseline corticosterone levels were measured via radioimmunoassay (MP Biomedicals, Orangeburg, NY, USA) using blood collected by tail nick between 9:00 and 10:00 a.m. on days 1, 4, and 7 after SE. Beginning 4 days after pilocarpine or saline treatment, all mice received the first of three daily injections of 5-bromo-2′-deoxyuridine (BrdU; 50 mg/kg s.c., Sigma-Aldrich) to label dividing cells (18). Mice were perfused with 2.5% paraformaldehyde on the eighth day, and brains were prepared for histological analysis (18).
RU486
RU486 is a synthetic steroid (17 beta-hydroxy-11 beta-[4-dimethylamino phenyl] 17 alpha-[1-propynyl]estra-4,9-dien-3-one) capable of antagonizing the glucocorticoid (doses 5 mg/kg in rodents) (19) and progesterone (doses 1.5 mg/kg in rodents) (20) receptors in vitro and in vivo. It also has moderate androgen receptor antagonist properties in vitro (21); however, in vivo effects have only been noted in the testes of rats and not mice at repeated doses >25 mg/kg (22). RU486 is active as an anti-glucocorticoid at doses >5 mg/kg and readily crosses the blood–brain barrier at doses >10 mg/kg (23). RU486 binds GR in vivo 3.7× more potently than dexamethasone (24). RU486 is metabolized within 1–2 h following administration (half-life ~1 h) (25). The rate of clearance was calculated as 1.5–6 L/h/kg in rats (data not available for mice) (22). Autoradiography studies in male and female rats demonstrate selective targeting of brain regions including the hippocampus (25). For this study, a 20-mg/kg s.c. dose was selected based on prior work in mice, showing tolerability (22) and a dampening of glucocorticoid-induced effects in the hippocampus (17).
Immunohistochemistry
Free-floating brain sections (35 μm) were used for immunohistochemistry. For BrdU immunostaining, tissue was placed in 50% formamide and 1× sodium citrate buffer (0.3M NaCl, 0.03M sodium citrate) at 67°C for 2 h, followed by 30-min treatment with 2 N HCl at 37°C. Subsequently, tissue was incubated for 1 h in blocking solution (0.1% BSA, 4% NGS, 0.2% triton-X) followed by overnight incubation in rat anti-BrdU (1:800 AbD Serotec Cat# MCA2060T, RRID:AB_10015293) at 4°C. For GluR2/3 immunostaining, tissue was incubated in the same blocking solution for 1 h, followed by overnight incubation in rabbit anti-GluR2/3 (1:500 Millipore Cat# 07-598, RRID:AB_11213931). Biotinylated anti-rat (1:500 Vector Laboratories Cat# BA-9400, RRID:AB_2336202) and anti-rabbit (1:500 Vector Laboratories Cat# BA-1100, RRID:AB_2336201) secondary antibodies were used for BrdU and GluR2/3 immunostaining, respectively. This step was followed by incubation in ABC reagent (1:800, Vector Laboratories Cat# PK-7100, RRID:AB_2336827) and diaminobenzidine (Sigma-Aldrich) for peroxidase detection (26). Sections were mounted on gelatin-coated slides, co-stained with cresyl violet, dehydrated, and coverslipped.
Cell Counts
Quantification of BrdU+ and GluR2/3+ cells was done using a modification of the optical fractionator method as previously described (27). Briefly, a 1-mm thick region of dorsal hippocampus (bregma −2.80 to −3.80) was sampled by examining every sixth section (35 μm/section; four sections/mouse). Two regions of interest were examined: the dentate granule cell body layer and the HIL (Figure 2A). The dentate granule cell body layer included the subgranular zone, defined as an approximately two cell thick region between the HIL and the cell body layer. The HIL encompassed the area created between the upper and lower blades of the dentate, but excluding the subgranular zone and the portion of the CA3 pyramidal cell layer that protrudes into the HIL (28). Stereoinvestigator 5.05 (MicroBrightfield, Williston, VT, USA) set up on an Olympus BX-60 microscope equipped with a CCD video camera (HV-C20 Hitachi, San Jose, CA, USA) was used for imaging and quantification. A 5× objective was used to trace all contours, and a 60× NA 1.35 objective was used to quantify all immunopositive cells located within the region of interest. Measured tissue thickness was ~15 μm (~50% shrinkage of 35-μm sections). Optical dissector height was set at 11 μm with a 2-μm guard zone. Mossy cells were distinguished from other glutamatergic hilar neurons by their larger size (>30 μm diameter) (2). Estimated volumes were obtained using Cavalleri’s principle [sum of section areas × (15 μm × 6)] (29). Total estimated cell counts per mouse were obtained by adding the number of counted cells in each section and multiplying by six. Final values are reported as density (cells per cubic millimeter).
Statistical Methods
Mice were randomized to treatment groups to prevent selection bias. All immunohistochemical data were analyzed by two-way ANOVA, with condition (pilocarpine vs. saline) and treatment (RU486 vs. vehicle) as factors. Three-way repeated measures ANOVA was used to analyze baseline levels of plasma corticosterone with time (days 1, 4, and 7), treatment, and SE exposure as factors. Expected interactions were tested by Student Newman–Keuls post hoc analysis. Statistical significance was set at p ≤ 0.05. Data that violated normality assumptions were normalized by square-root transformation. Statistica software was used to perform three-way repeated measures ANOVA, while SigmaPlot (Version 13.0, Systat Software Inc.) was used for other analyses.
Results
RU486 Treatment Reduces Corticosterone Levels Following SE
Corticosterone levels were increased 1 and 4 days after pilocarpine-induced SE relative to no-SE + vehicle mice. RU486 treatment was ineffective at reducing corticosterone levels at the 1-day time point, but levels in SE + RU486 mice were similar to non-SE controls (significantly lower than SE + vehicle mice) 4 days after SE {experimental condition × treatment × time interaction [F(2,52) = 5.8019, p = 0.0053], post hoc p < 0.001} (Figure 1). In addition, RU486 treatment alone increased morning circulating corticosterone levels relative to vehicle-treated mice (post hoc p < 0.001) at day 1. This phenomenon has been previously observed in human and rodent studies and is thought to be due to the initial blockade of GR, resulting in greater amounts of circulating corticosterone secondary to an initial disruption of glucocorticoid-mediated negative feedback. At the 7-day time point, morning baseline corticosterone levels were statistically identical in all four groups.
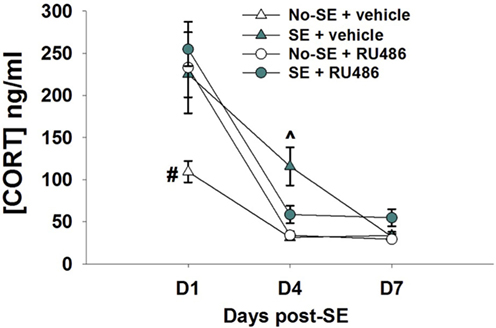
Figure 1. RU486 decreases morning baseline corticosterone in post-SE mice. Mice show increased corticosterone morning baseline secretion 24 h post-SE relative to no-SE vehicle-treated mice (#p < 0.05, no-SE vehicle different from all other groups). RU486 treatment reduced baseline corticosterone secretion in SE mice when compared to SE vehicle-treated mice (^p < 0.05, SE vehicle different from all other groups). Data presented as mean ± SEM, n = 6–8 mice per group.
RU486 Treatment Mitigates Hilar Cell Proliferation
Status epilepticus increased the density of BrdU+ cells in the dentate HIL by 17-fold in SE + vehicle mice relative to both non-SE groups (post hoc p < 0.05). RU486 treatment reduced this increase by 35% {experimental condition × treatment interaction [F(1,23) = 8.619, p = 0.007], post hoc p < 0.05}. Although the SE + RU486 group still had significantly more BrdU+ cells/HIL than the non-SE groups, counts were significantly reduced relative to SE + vehicle mice (post hoc p < 0.05) (Figures 2B,C). RU486 treatment alone (no-SE + RU486) had no effect on the density of BrdU+ hilar cells relative to the no-SE + vehicle group. Finally, SE also increased the density of BrdU+ cells within the dentate granule cell layer and subgranular zone compared to both groups of control mice. RU486 treatment had no effect on increased cell proliferation in these regions {main effect of experimental condition [F(1,23) = 24.627, p < 0.001]} (Figure 2D).
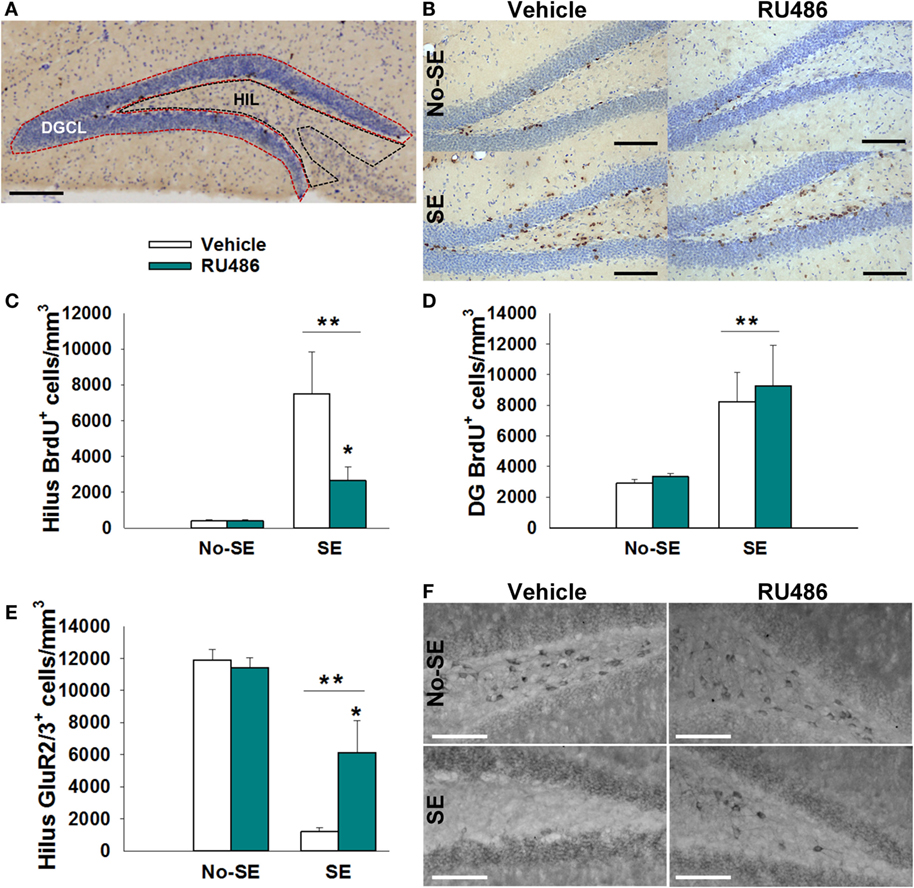
Figure 2. RU486 reduced mossy cell loss and hilar BrdU+ neurons in the hippocampus of post-SE mice. (A) Micrograph depicting the regions analyzed: dentate granule cell body layer (DGC) and the hilus (HIL). Scale bar = 200 μm. (B) Micrograph showing examples of BrdU+ staining. Scale bars = 100 μm. (C) SE leads to an increased number of BrdU+ cells in the dentate hilus (**p < 0.01 main effect of SE). RU486 treatment reduced the number of BrdU+ cells in the dentate hilus of post-SE mice relative to vehicle-treated post-SE mice (*p < 0.05, RU486 different from vehicle within SE). (D) SE leads to an increase in BrdU+ cells in the dentate gyrus relative to control mice (**p < 0.01 main effect of SE). (E) SE leads to loss of GluR2+ cells in the dentate hilus relative to control mice (**p < 0.01, main effect of SE). RU486 treatment preserves greater numbers of GluR2+ cells in the dentate hilus of post-SE mice relative to their vehicle-treated counterparts (*p < 0.05, RU486 different from vehicle within SE). (F) Micrograph depicting GluR2 immunohistochemistry in the dentate hilus. Scale bars = 100 μm. All data presented as mean ± SEM, n = 6–8 mice.
RU486 Treatment Mitigates SE-Induced Mossy Cell Loss
Status epilepticus produced an 88% reduction in the density of GluR2/3+ mossy cells in the HIL relative to control mice {main effect of pathology [F(1,21) = 70.448, p < 0.001]} (Figures 2E,F). In SE mice treated with RU486, only a 48% reduction in mossy cells/HIL was observed. While this reduction was still significant relative to non-SE groups, RU486 treatment clearly mitigated mossy cell loss relative to SE + vehicle-treated mice {pathology × treatment interaction [F(1,21) = 10.453, p = 0.004], post hoc p < 0.05}. RU486 treatment alone (no-SE + vehicle) had no effect on mossy cell numbers.
Discussion
The present study demonstrates that treatment with GR antagonist RU486 effectively blocks the SE-associated elevation of glucocorticoid levels in pilocarpine-treated mice, reduces hilar mossy cell loss, and mitigates cell proliferation in the HIL. Taken together, these findings indicate that GR blockade holds promise as a novel treatment for SE-induced brain injury.
The present findings extend prior work showing pronounced dysregulation of corticosterone levels following SE. Increased morning corticosterone secretion 24 h after SE has been observed previously (11), consistent with the current study. Here, we further demonstrate that SE-induced hypersecretion of corticosterone persists for at least 4 days but returns to baseline by 7 days. These results reveal a transient initial phase of hypersecretion. Interestingly, elevated corticosterone secretion has also been observed in rats 6 weeks after SE (30). These findings imply bimodal changes in corticosterone levels following SE, with early and late periods of hypersecretion.
Treatment with RU486 blocked the increase in corticosterone levels evident in untreated animals 4 days after SE. The mechanism by which chronic RU486 treatment normalizes corticosterone levels is unknown. It has been hypothesized, however, that prolonged antagonism of GR may result in upregulation of the mineralocorticoid receptor in critical hypothalamic and limbic structures. Because RU486 has no affinity for the mineralocorticoid receptor (31), glucocorticoid binding to mineralocorticoid receptors in these stress-regulatory regions may induce resetting of the HPA axis and reinstate control of baseline secretion (32–34). In addition, increased glucocorticoid binding to the mineralocorticoid receptor may oppose some of the toxic effects of GR binding (35, 36) and SE, thereby preserving key HPA axis feedback pathways.
In addition to actions on GR, RU486 also acts as a progesterone receptor antagonist (31). Although we cannot exclude the possibility that progesterone receptor antagonism is important, we favor the interpretation that reduced hippocampal pathology reflects GR antagonisms, as progesterone receptors are not widely expressed in the mouse hippocampus (37). Nonetheless, additional studies with more selective approaches will be needed to unambiguously rule out off-target effects.
RU486 treatment reduced mossy cell loss in the dentate HIL, suggesting that GR signaling during the hours and days following the insult may contribute to the development of hippocampal pathology. Findings are reminiscent of prior work showing that RU486 protects against CA1 neuronal loss following traumatic brain injury (35). GR is expressed in all hippocampal subfields and in mossy cells (38–40). Glucocorticoids, therefore, may act directly on vulnerable neurons to influence their survival. Alternatively, GR blockade on DGCs may indirectly reduce mossy cell loss. Glucocorticoids promote hypersecretion of glutamate by granule cell mossy fiber axons as early as 24 h following pilocarpine-induced SE, which can be blocked by treatment with RU486 (41). Granule cell axons directly innervate mossy cells and may contribute to excitotoxic loss of these neurons.
Status epilepticus results in large increases in cell proliferation, cell survival, and neurogenesis (42, 43). Consistent with these studies, post-SE mice had increased numbers of BrdU+ cells located in the subgranular zone and granule cell layer of the dentate gyrus. RU486 treatment did not have an effect in the number of BrdU+ cells in either of these regions. RU486 did, however, reduce the density of BrdU+ cells located in the dentate HIL. SE can induce astroglial and microglial proliferation in the HIL (44, 45), as well as the accumulation of ectopic, newly generated granule cells (46–48). Reductions in cell proliferation in the HIL imply that RU486 mitigates some or all of these proliferative changes. Indeed, glucocorticoids have been implicated in regulating the migration, maturation, and functional integration of DGCs (49, 50). Similarly, RU486 has been shown to prevent hippocampal astrogliosis in mice (51). Thus, glucocorticoid blockade following SE may prevent the aberrant migration of newborn granule cells into the dentate HIL and/or decrease astrogliosis.
Finally, it is important to note that RU486 administration to control mice did not alter any of the measures examined here and has been previously found to exert little impact on cell proliferation, survival, and inflammation in control subjects (32, 52–54). Our results are in agreement with other studies suggesting that the actions of RU486 may be specific to the pathological, high corticosterone environment (31). This specificity reflects the ability of RU486 to preferentially bind the GR over the mineralocorticoid receptor (31). Since GR’s are only fully occupied when corticosterone levels are elevated (55), the impact of blockade is most pronounced under these conditions.
Conclusion
The current study demonstrates that in the pilocarpine model of epilepsy, SE results in the rapid elevation of glucocorticoids. Treatment with the glucocorticoid antagonist RU486 following the insult blocks the SE-induced elevation of glucocorticoid baseline secretion, mitigates mossy cell loss, and reduces aberrant cell proliferation in the HIL. Our laboratory and others have implicated mossy cell loss and aberrant neuron proliferation in epileptogenesis, via mechanisms that result in hippocampal hyperexcitability (1–5). In addition, astrogliosis and brain inflammation are also associated with the pathophysiology of epilepsy (56). Together, our findings suggest that blockade of glucocorticoid signaling may be beneficial in preventing hippocampal damage following SE. To advance this line of investigation, it will be important in future studies to examine extended survival times so that the impact of RU486 on seizure frequency and animal behavior can be assessed. Time course studies to determine how long treatment can be delayed before it loses efficacy will also be important. Mossy cell loss may occur over days and weeks (57), but since this is a non-regenerative cell population, at some point loss will be irreversible. Finally, although the use of an FDA-approved drug like RU486 has obvious advantages for rapid translation, the use of more specific glucocorticoid receptor antagonists will be important to experimentally isolate glucocorticoid receptor-mediated from progesterone receptor-mediated effects.
Author Contributions
AW participated in experimental design, animal handling, analysis of neuroendocrine and tissue data as well as all statistical analyses for this study. In addition, AW wrote the manuscript and created all figures. JH and SD assisted with experimental design and provided editorial assistance for the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Kim Seroogy for his guidance while utilizing Stereoinvestigator software.
Funding
This work was supported by R01-NS-062806 and R01-NS-065020 to SD; T32-GM063483 and F30-NS-095578 to AW. NIH had no further role in the study design; data collection, analysis, and interpretation; and writing neither of the report nor in the decision to submit for publication.
References
1. Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia (2008) 49(Suppl 9):85–92. doi:10.1111/j.1528-1167.2008.01931.x
2. Scharfman HE, Myers CE. Hilar mossy cells of the dentate gyrus: a historical perspective. Front Neural Circuits (2012) 6:106. doi:10.3389/fncir.2012.00106
3. Myers CE, Bermudez-Hernandez K, Scharfman HE. The influence of ectopic migration of granule cells into the hilus on dentate gyrus-CA3 function. PLoS One (2013) 8:e68208. doi:10.1371/journal.pone.0068208
4. Danzer SC, He X, Loepke AW, McNamara JO. Structural plasticity of dentate granule cell mossy fibers during the development of limbic epilepsy. Hippocampus (2010) 20:113–24. doi:10.1002/hipo.20589
5. Liu J, Tang F, Liu Y. Neuron activation, degeneration and death in the hippocampus of mice after pilocarpine induced status epilepticus. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2011) 36:1071–8. doi:10.3969/j.issn.1672-7347.2011.11.007
6. Jinde S, Zsiros V, Jiang Z, Nakao K, Pickel J, Kohno K, et al. Hilar mossy cell degeneration causes transient dentate granule cell hyperexcitability and impaired pattern separation. Neuron (2012) 76:1189–200. doi:10.1016/j.neuron.2012.10.036
7. Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A (2008) 105:6179–84. doi:10.1073/pnas.0801372105
8. Hester MS, Danzer SC. Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J Neurosci (2013) 33:8926–36. doi:10.1523/JNEUROSCI.5161-12.2013
9. Cho K-O, Lybrand ZR, Ito N, Brulet R, Tafacory F, Zhang L, et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun (2015) 6:6606. doi:10.1038/ncomms7606
10. Hosford BE, Liska JP, Danzer SC. Ablation of newly generated hippocampal granule cells has disease-modifying effects in epilepsy. J Neurosci (2016) 36(43):11013–23. doi:10.1523/JNEUROSCI.1371-16.2016
11. O’Toole KK, Hooper A, Wakefield S, Maguire J. Seizure-induced disinhibition of the HPA axis increases seizure susceptibility. Epilepsy Res (2014) 108:29–43. doi:10.1016/j.eplepsyres.2013.10.013
12. Joëls M. Stress, the hippocampus, and epilepsy. Epilepsia (2009) 50:586–97. doi:10.1111/j.1528-1167.2008.01902.x
13. Kumar G, Couper A, O’Brien TJ, Salzberg MR, Jones NC, Rees SM, et al. The acceleration of amygdala kindling epileptogenesis by chronic low-dose corticosterone involves both mineralocorticoid and glucocorticoid receptors. Psychoneuroendocrinology (2007) 32:834–42. doi:10.1016/j.psyneuen.2007.05.011
14. Castro OW, Santos VR, Pun RYK, McKlveen JM, Batie M, Holland KD, et al. Impact of corticosterone treatment on spontaneous seizure frequency and epileptiform activity in mice with chronic epilepsy. PLoS One (2012) 7:e46044. doi:10.1371/journal.pone.0046044
15. Sapolsky RM, Stein-Behrens BA. Status epilepticus-induced hippocampal damage is modulated by glucose availability. Neurosci Lett (1989) 97:157–62. doi:10.1016/0304-3940(89)90156-0
16. Stein-Behrens BA, Sapolsky RM. Chemical adrenalectomy reduces hippocampal damage induced by kainic acid. Brain Res (1988) 473:175–80. doi:10.1016/0006-8993(88)90332-0
17. Llorens-Martín M, Trejo JL. Mifepristone prevents stress-induced apoptosis in newborn neurons and increases AMPA receptor expression in the dentate gyrus of C57/BL6 mice. PLoS One (2011) 6:e28376. doi:10.1371/journal.pone.0028376
18. Murphy BL, Pun RYK, Yin H, Faulkner CR, Loepke AW, Danzer SC. Heterogeneous integration of adult-generated granule cells into the epileptic brain. J Neurosci (2011) 31:105–17. doi:10.1523/JNEUROSCI.2728-10.2011
19. Nieman LK, Chrousos GP, Kellener C, Spitz IM, Nisula BC, Cutler GB, et al. Successful treatment of Cushing’s syndrome with the glucocorticoid antagonist RU 486*. J Clin Endocrinol Metab (1985) 61:536–40. doi:10.1210/jcem-61-3-536
20. Herrmann W, Wyss R, Riondel A, Philibert D, Teutsch G, Sakiz E, et al. The effects of an antiprogesterone steroid in women: interruption of the menstrual cycle and of early pregnancy. C R Seances Acad Sci III (1982) 294:933–8.
21. Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol (2004) 18:70–85. doi:10.1210/me.2003-0189
22. Brundage P. Pharmacology/toxicology NDA review and evaluation – Korlym (mifepristone). (2011). Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202107Orig1s000SumR.pdf
23. Peeters BWMM, Tonnaer JADM, Groen MB, Broekkamp CLE, van der Voort HAA, Schoonen WGFJ, et al. Short review glucocorticoid receptor antagonists: new tools to investigate disorders characterized by cortisol hypersecretion. Stress (2004) 7:233–41. doi:10.1080/10253890400019672
24. Peeters BWMM, Ruigt GSF, Craighead M, Kitchener P. Differential effects of the new glucocorticoid receptor antagonist ORG 34517 and RU486 (mifepristone) on glucocorticoid receptor nuclear translocation in the AtT20 cell line. Ann N Y Acad Sci (2008) 1148:536–41. doi:10.1196/annals.1410.072
25. Deraedt R, Bonnat C, Busigny M, Chatelet P, Cousty C, Mouren M, et al. Pharmacokinetics of RU 486. In: Baulieu E-E, Sheldon S, editors. The Antiprogestin Steroid RU 486 and Human Fertility Control. New York: Plenum Press (1985). p. 103–22.
26. Wulsin AC, Herman JP, Solomon MB. Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic-pituitary-adrenocortical axis responsiveness to stress. Psychoneuroendocrinology (2010) 35:1100–12. doi:10.1016/j.psyneuen.2010.01.011
27. Wong-Goodrich SJE, Glenn MJ, Mellott TJ, Blusztajn JK, Meck WH, Williams CL. Spatial memory and hippocampal plasticity are differentially sensitive to the availability of choline in adulthood as a function of choline supply in utero. Brain Res (2008) 1237:153–66. doi:10.1016/j.brainres.2008.08.074
28. Amaral DG. A Golgi study of cell types in the hilar region of the hippocampus in the rat. J Comp Neurol (1978) 182:851–914. doi:10.1002/cne.901820508
29. Mounton P. Principles and Practices of Unbiased Stereology. Baltimore, MD: The John Hopkins University Press (2002).
30. Mazarati AM, Shin D, Kwon YS, Bragin A, Pineda E, Tio D, et al. Elevated plasma corticosterone level and depressive behavior in experimental temporal lobe epilepsy. Neurobiol Dis (2009) 34:457–61. doi:10.1016/j.nbd.2009.02.018
31. Spitz IM, Bardin CW. Clinical pharmacology of RU486 – an antiprogestin and antiglucocorticoid. Contraception (1993) 48:403–44. doi:10.1016/0010-7824(93)90133-R
32. Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol (2001) 21:516–21. doi:10.1097/00004714-200110000-00009
33. Thomson F, Craighead M. Innovative approaches for the treatment of depression: targeting the HPA axis. Neurochem Res (2008) 33:691–707. doi:10.1007/s11064-007-9518-3
34. Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology (2006) 31:628–36. doi:10.1038/sj.npp.1300884
35. McCullers DL, Sullivan PG, Scheff SW, Herman JP. Mifepristone protects CA1 hippocampal neurons following traumatic brain injury in rat. Neuroscience (2002) 109:219–30. doi:10.1016/S0306-4522(01)00477-8
36. McCullers DL, Herman JP. Mineralocorticoid receptors regulate bcl-2 and p53 mRNA expression in hippocampus. Neuroreport (1998) 9:3085–9. doi:10.1097/00001756-199809140-00031
37. Weiland NG, Orikasa C, Hayashi S, McEwen BS. Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol (1997) 388:603–12. doi:10.1002/(SICI)1096-9861(19971201)388:4<603::AID-CNE8>3.0.CO;2-6
38. Patel A, Bulloch K. Type II glucocorticoid receptor immunoreactivity in the mossy cells of the rat and the mouse hippocampus. Hippocampus (2003) 13:59–66. doi:10.1002/hipo.10045
39. de Kloet ER, Reul JM, Sutanto W. Corticosteroids and the brain. J Steroid Biochem Mol Biol (1990) 37:387–94. doi:10.1016/0960-0760(90)90489-8
40. Garcia A, Steiner B, Kronenberg G, Bick-Sander A, Kempermann G. Age-dependent expression of glucocorticoid- and mineralocorticoid receptors on neural precursor cell populations in the adult murine hippocampus. Aging Cell (2004) 3:363–71. doi:10.1111/j.1474-9728.2004.00130.x
41. Wang C-C, Wang SJ. Modulation of presynaptic glucocorticoid receptors on glutamate release from rat hippocampal nerve terminals. Synapse (2009) 63:745–51. doi:10.1002/syn.20654
42. Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci (1997) 17:3727–38.
43. Gray WP, Sundstrom LE. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res (1998) 790:52–9. doi:10.1016/S0006-8993(98)00030-4
44. Borges K, McDermott D, Irier H, Smith Y, Dingledine R. Degeneration and proliferation of astrocytes in the mouse dentate gyrus after pilocarpine-induced status epilepticus. Exp Neurol (2006) 201:416–27. doi:10.1016/j.expneurol.2006.04.031
45. Shapiro LA, Wang L, Ribak CE. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia (2008) 49(Suppl 2):33–41. doi:10.1111/j.1528-1167.2008.01491.x
46. Cameron MC, Zhan R-Z, Nadler JV. Morphologic integration of hilar ectopic granule cells into dentate gyrus circuitry in the pilocarpine model of temporal lobe epilepsy. J Comp Neurol (2011) 519:2175–92. doi:10.1002/cne.22623
47. Shetty AK, Hattiangady B, Rao MS, Shuai B. Neurogenesis response of middle-aged hippocampus to acute seizure activity. PLoS One (2012) 7:e43286. doi:10.1371/journal.pone.0043286
48. Singh SP, LaSarge CL, An A, McAuliffe JJ, Danzer SC. Clonal analysis of newborn hippocampal dentate granule cell proliferation and development in temporal lobe epilepsy. eNeuro (2015) 2:1–13. doi:10.1523/ENEURO.0087-15.2015
49. Fitzsimons CP, van Hooijdonk LWA, Schouten M, Zalachoras I, Brinks V, Zheng T, et al. Knockdown of the glucocorticoid receptor alters functional integration of newborn neurons in the adult hippocampus and impairs fear-motivated behavior. Mol Psychiatry (2013) 18:993–1005. doi:10.1038/mp.2012.123
50. Lussier AL, Lebedeva K, Fenton EY, Guskjolen A, Caruncho HJ, Kalynchuk LE. The progressive development of depression-like behavior in corticosterone-treated rats is paralleled by slowed granule cell maturation and decreased reelin expression in the adult dentate gyrus. Neuropharmacology (2013) 71:174–83. doi:10.1016/j.neuropharm.2013.04.012
51. Revsin Y, Rekers NV, Louwe MC, Saravia FE, De Nicola AF, de Kloet ER, et al. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacology (2009) 34:747–58. doi:10.1038/npp.2008.136
52. Watson S, Gallagher P, Porter RJ, Smith MS, Herron LJ, Bulmer S, et al. A randomized trial to examine the effect of mifepristone on neuropsychological performance and mood in patients with bipolar depression. Biol Psychiatry (2012) 72:943–9. doi:10.1016/j.biopsych.2012.05.029
53. Oomen CA, Mayer JL, de Kloet ER, Joëls M, Lucassen PJ. Brief treatment with the glucocorticoid receptor antagonist mifepristone normalizes the reduction in neurogenesis after chronic stress. Eur J Neurosci (2007) 26:3395–401. doi:10.1111/j.1460-9568.2007.05972.x
54. Mayer JL, Klumpers L, Maslam S, de Kloet ER, Joëls M, Lucassen PJ. Brief treatment with the glucocorticoid receptor antagonist mifepristone normalises the corticosterone-induced reduction of adult hippocampal neurogenesis. J Neuroendocrinol (2006) 18:629–31. doi:10.1111/j.1365-2826.2006.01455.x
55. de Kloet ER. Steroids, stability and stress. Front Neuroendocrinol (1995) 16:416–25. doi:10.1006/frne.1995.1015
56. Xu D, Miller SD, Koh S. Immune mechanisms in epileptogenesis. Front Cell Neurosci (2013) 7:195. doi:10.3389/fncel.2013.00195
Keywords: RU486, mifepristone, status epilepticus, hippocampus, mossy cells
Citation: Wulsin AC, Herman JP and Danzer SC (2016) RU486 Mitigates Hippocampal Pathology Following Status Epilepticus. Front. Neurol. 7:214. doi: 10.3389/fneur.2016.00214
Received: 21 September 2016; Accepted: 14 November 2016;
Published: 28 November 2016
Edited by:
Ashok K. Shetty, Texas A&M University College of Medicine, USAReviewed by:
Mario Alonso, Instituto Nacional de Neurologia y Neurocirugia, MexicoMichele Simonato, University of Ferrara, Italy
Copyright: © 2016 Wulsin, Herman and Danzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steve C. Danzer, c3RldmUuZGFuemVyQGNjaG1jLm9yZw==