 Carl E. Stafstrom
Carl E. Stafstrom Verena Staedtke
Verena Staedtke Anne M. Comi
Anne M. Comi- 1Division of Pediatric Neurology, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2Department of Neurology, Kennedy Krieger Institute, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Neurocutaneous disorders are multisystem diseases affecting skin, brain, and other organs. Epilepsy is very common in the neurocutaneous disorders, affecting up to 90% of patients with tuberous sclerosis complex (TSC) and Sturge–Weber syndrome (SWS), for example. The mechanisms underlying the increased predisposition to brain hyperexcitability differ between disorders, yet some molecular pathways overlap. For instance, the mechanistic target of rapamycin (mTOR) signaling cascade plays a central role in seizures and epileptogenesis in numerous acquired and genetic disorders, including several neurocutaneous disorders. Potential routes for target-specific treatments are emerging as the genetic and molecular pathways involved in neurocutaneous disorders become increasingly understood. This review explores the clinical features and mechanisms of epilepsy in three common neurocutaneous disorders—TSC, neurofibromatosis type 1, and SWS.
What is a Neurocutaneous Disorder?
Neurocutaneous disorders are a heterogeneous group of genetic disorders characterized by abnormalities of the cutaneous and nervous systems. Tuberous sclerosis complex (TSC), neurofibromatosis type I (NF1), and Sturge–Weber syndrome (SWS) are prototypical neurocutaneous disorders in which genetic mutations in pathways regulating cell growth cause developmental dysfunction of the brain, skin, and other organs. Clinically, these neurocutaneous disorders differ significantly, but certain similarities also exist. Namely, all neurocutaneous disorders are congenital, affect several organs, are associated with learning or developmental problems, persist lifelong, are currently uncorrectable, and are optimally managed with a multidisciplinary approach in which neurologists, oncologists, educational specialists, neuropsychologists, and other therapists work together to monitor for potential complications of the respective disease and maximize abilities. In addition, there is now emerging evidence of some overlap in the cellular signaling pathways in these disorders (1–3). Epilepsy, defined as the condition of recurrent, unprovoked seizures, is a common feature of many of the neurocutaneous disorders. This chapter reviews aspects of the clinical presentation and management of epilepsy in TSC, NF1, and SWS and focuses on possible mechanisms of seizures and epilepsy in each disorder. Emphasis is placed on why epilepsy is so prevalent in these conditions.
Mechanisms of Seizures and Epilepsy
A seizure is an episode of transient neurological dysfunction due to abnormal firing of neurons. Epilepsy is the condition of recurrent, unprovoked seizures. An epilepsy syndrome involves a specific seizure type(s) plus other features such as age of onset, electroencephalographic (EEG) findings, genetics/natural history, and responsiveness to particular drugs. A seizure occurs when there is an imbalance between excitation (E) and inhibition (I) in one or more areas of the brain. An E/I imbalance can manifest at the level of subcellular signaling pathways, ion channels, synapses, or neuronal networks. Any molecular, cellular, or structural pathology that increases excitation or decreases inhibition can generate a seizure (4). For example, enhanced activity or function of excitatory glutamatergic synapses or their receptor subtypes [i.e., alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or N-methyl-d-aspartate (NMDA)] or decreased activity or function of inhibitory gamma-aminobutyric acid (GABA)-ergic neurons or receptors would favor seizure generation. Similarly, dysfunction of sodium or calcium channels (often excessive current flow through these channels) or diminished function of potassium channels may predispose to hyperexcitability and seizures.
Epilepsy or the process by which epilepsy develops (epileptogenesis) also entails E/I imbalance but other cellular pathologies can also be involved, such as structural or circuit rearrangements, inflammation, or disrupted epigenetic regulation. Therefore, the mechanisms underlying a seizure and those leading to epilepsy overlap but epileptogenesis also involves additional processes. Though admittedly oversimplified, the concept of an imbalance between excitation and inhibition is helpful when considering seizure and epilepsy mechanisms in neurocutaneous disorders, and pathophysiological changes can be considered at each of these levels of neuronal function (4, 5).
Tuberous Sclerosis Complex
Clinical Features of TSC
Tuberous sclerosis complex is a multiorgan system disorder occurring at a frequency of about 1 in every 5,000 births. The disorder is caused by a mutation in TSC1 or TSC2, which are tumor repressor genes controlling the activity of the mechanistic target of rapamycin (mTOR) signaling pathway (formerly called mammalian target of rapamycin), discussed further below. Overactivity of the mTOR pathway accelerates mRNA translation and causes excessive protein synthesis and cell growth, leading to the formation of benign tumors in several organs and giving rise to the distinctive clinical features of TSC. For this reason, TSC is considered the prototypical “mTOR-opathy.” Full diagnostic criteria for TSC have been published recently (6, 7). Classic skin findings include hypopigmented macules (ash-leaf spots) and facial angiomatoses. In addition, patients with TSC commonly have cardiac rhabdomyomas (often present at birth with subsequent involution) and renal angiomyolipomas (develop over time and frequently lead to renal insufficiency in childhood or adulthood). Here, we focus on central nervous system abnormalities in TSC that may lead to seizures.
Three major neuropathological findings characterize TSC (Figure 1A1). First, hamartomas (tubers) of widely variable size and number form in the cerebral cortex, often at the gray–white matter junction. Tubers are composed of a mixture of abnormal cells, including dysplastic, immature, cytomegalic neurons and glia, all lacking a normal lamination pattern. Importantly, tubers do not tend to expand or grow. Although tubers are considered to be static lesions, some dynamic features have been documented and tubers may appear to become more prominent over time due to interval myelination and other factors (8, 9). Tubers are extremely epileptogenic and can produce one or more seizure foci in the brain. A seizure can arise from the tuber itself or from perituberal cortex adjacent to the tuber (10, 11). Tubers differ in their epileptogenicity, even within the same patient. The precise localization of the epileptogenic focus is critical for planning tuber resection surgery. The number, volume, and possibly location of tubers correlate to some extent with the patient’s level of intellectual impairment and cognitive dysfunction (12, 13). For example, tubers located in the temporal lobe are associated with a high incidence of autism (14, 15). Second, abnormal neuronal and glial tissue, called subependymal nodules (SENs), can arise in the periventricular regions. SENs can transform into subependymal giant cell astrocytomas (SEGAs). Due to their location near the foramen of Monro, SEGAs can cause acute blockage of cerebrospinal fluid flow at that site, leading to hydrocephalus (Figure 1A2). However, owing to their cellular composition and location deep in the brain, SEGAs are not epileptogenic. Third, radially oriented heterotopias occur within white matter, consistent with disordered neuronal migration; these heterotopias have been associated with behavioral problems and may also contribute to epileptogenesis (Figure 1A1) (16). Together, these three neuropathological features account for the major age-dependent neurologic complications of TSC, namely, epilepsy (90%), autism (25–50%), and intellectual disability (40–80%). The common occurrence of neuropsychological deficits in TSC, including learning disability, attention deficits, spatial memory problems, aggressive behaviors, anxiety, and sleep disruption, has given rise to the term TAND (tuberous sclerosis-associated neuropsychiatric disorders) (17). Optimal management of patients with TSC must address TAND as well as epilepsy.
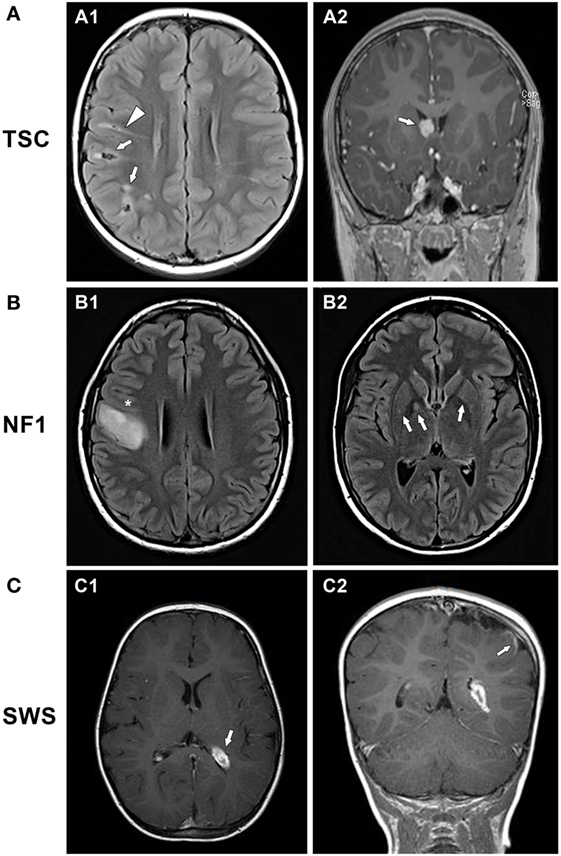
Figure 1. Examples of brain MRI scans in patients with neurocutaneous disorders. (A) 8-year-old boy with tuberous sclerosis complex. (A1) Axial fluid attenuation inversion recovery (FLAIR) and (A2) coronal contrast-enhanced T1-weighted images show multiple cortical/subcortical, partially calcified mixed FLAIR-hyper/hypointense tubers in the right front parietal lobes (arrows, A1), FLAIR-hyperintense migration lines (triangle, A1), and a homogeneously enhancing, round subependymal giant cell astrocytoma within the right foramen of Monro (arrow, A2). (B) 16-year-old girl with neurofibromatosis type 1. (B1) Axial FLAIR and T1 post-contrast images showing a large expansile lesion centered at the right precentral gyrus (*) indicating a low-grade glioma. (B2) Axial T2 FLAIR image reveals focal areas of signal intensity (arrows) in the right globus pallidus and putamen and similar but to a lesser extent on the contralateral side, representing nonspecific unidentified bright objects. (C) 21-month-old boy with Sturge–Weber syndrome. (C1) Axial T1-weighted post-contrast image showing left hemisphere atrophy with prominent choroid plexus glomus (arrow). (C2) Coronal T1-weighted post-contrast image with occipital-parietal leptomeningeal enhancement (arrow).
Epilepsy in TSC
Seizures are extremely common in TSC, affecting up to 90% of patients (18). In 63% of TSC patients with epilepsy, the seizures appear in the first year of life; 80% of seizures begin before 3 years of age and 70% eventually become refractory to drugs. The seizures may be focal, multifocal, infantile spasms, or a combination of these or other seizure types. Infantile spasms are a seizure type, a subtype of epileptic spasms occurring in the first year of life, and often occur as part of the electroclinical syndrome, West syndrome. Focal seizures may be isolated or occur in association with infantile spasms, and most cases of infantile spasms also include other seizure types. The incidence of infantile spasms is extremely high, affecting approximately one-third of children with TSC (18, 19). In about 5% of children with TSC, spasms continue or develop after 2 years of age, making this a very common semiology (20). With multiple tubers comprising potential seizure foci, it is not surprising that focal or multifocal seizures are present in TSC, but the frequent occurrence of infantile spasms has not been explained adequately. It is difficult to categorize infantile spasms solely as generalized or focal seizures, as both occur, even in TSC. Thus in the case of TSC, the common final output—spasms—can obviously be produced by focal or multifocal lesions. Abnormal mTOR function may be another explanation for infantile spasms, but mTOR dysfunction is seen in only a subset of the numerous etiologies of infantile spasms, so mTOR pathway dysfunction is not required for infantile spasms to occur.
Earlier seizure onset portends a less favorable outcome with regard to neuropsychiatric and developmental function (17). In TSC, the interaction between epilepsy and neuropsychiatric sequelae is complex, compounded by adverse effects of the therapies used to treat epilepsy. As opposed to classic epileptic encephalopathies in which seizures themselves cause cognitive impairment beyond the etiology, in TSC it is the molecular etiology—mTOR overactivation—that probably drives both the epilepsy and the encephalopathy/TAND (21).
The treatment of seizures in TSC involves conventional antiseizure drugs as well as non-pharmacological interventions such as the ketogenic diet, vagus nerve stimulation, or in some cases, surgical resection of the epileptogenic tubers (13, 22, 23). The percentage of patients with drug-refractory epilepsy in TSC (~70%) is much higher than the percentage of drug-refractoriness among patients with epilepsy in general (~30%) (24). Early surgical resection is recommended if a dominant tuber is identified, i.e., an area from which the majority of seizures appear to originate, but the presence of multiple tubers often makes it difficult to localize the most epileptogenic one. This identification may be aided by the use of positron emission tomography (PET) scans, which can localize brain regions with abnormal metabolism even in the absence of a structural lesion (25). The goal of surgery is palliation of seizure burden, though cognition can also improve after the targeted resection of an epileptogenic tuber (26). A meta-analysis of 229 patients in 13 studies concluded that 59% of patients were seizure free after surgery (27).
For infantile spasms in children with TSC, vigabatrin is the first choice medication, rather than adrenocorticotropic hormone or corticosteroids as used more commonly for infantile spasms due to other etiologies. The vigabatrin response rate is between 73% and 96% (28). It is unknown why vigabatrin works so well in TSC-associated infantile spasms. The therapeutic benefit could be related to vigabatrin’s main mechanism of action—inhibition of GABA transaminase, leading to increased availability of the inhibitory neurotransmitter GABA at the synapse—or to vigabatrin-induced decrease in mTOR activation (29). [However, the situation may not be so simple, as mTOR is upregulated by the excessive GABA present when the gene for succinic semialdehyde dehydrogenase is knocked out in mice (mimicking human SSADH deficiency) (30).] Since infants with TSC have such a high risk for developing infantile spasms, a novel approach is to treat infants who have epileptiform abnormalities on EEG (but no documented seizures) prophylactically with vigabatrin before spasms begin (31, 32). This approach is being tested in an ongoing European multicenter trial called EPISTOP.
Mechanistic target of rapamycin inhibition could comprise a rational, disease-modifying treatment for seizures in TSC (2, 9, 13, 23). mTOR inhibitors such as rapamycin (sirolimus) have been found to decrease the size of kidney angiomyolipomas and SEGAs (33) and are being trialed for seizures as well (23). Another mTOR inhibitor, everolimus, has shown benefit for seizures in TSC in a phase I/II clinical trial (34). In that trial, seizure frequency decreased by at least 50% in 12 of 20 participants and the median decrease in seizures was 73% in 17 of the 20 patients; in addition, there was a decrease in seizure duration and improved quality of life on several parent-reported measures. A recent phase III double-blind randomized study of 366 TSC patients with treatment-resistant focal-onset seizures (EXIST-3) compared placebo versus low- or high-dose everolimus (35). The median reduction in seizure frequency was 40% in those receiving high-dose everolimus and 28% in subjects receiving low-dose everolimus, both statistically significant compared with a 15% seizure reduction in the placebo group. Notably, children with infantile spasms were excluded from these studies. These results raise the possibility that mTOR inhibition decreases seizure frequency in TSC by targeting the molecular defect, representing a unique, disease-modifying approach. This and other therapies are urgently needed for epilepsy in TSC, emphasizing the importance of understanding the mechanisms of epileptogenesis in this disorder (36).
Molecular Basis of TSC—Overactivation of the mTOR Pathway
Tuberous sclerosis complex arises de novo in approximately 70–80% of cases; in the remainder of cases, inheritance is autosomal dominant or due to genetic mosaicism. The disorder is caused by a mutation in one of the tumor suppressor genes, TSC1 or TSC2. TSC1, located on chromosome 9, encodes the protein hamartin. TSC2, located on chromosome 16, encodes the protein tuberin. Under normal circumstances, hamartin and tuberin act together as a dimer to inhibit the mTOR signaling pathway and constrain cell growth and differentiation (37). mTOR is a serine/threonine kinase that acts as a central regulator of cell growth, differentiation, proliferation, and migration. Mutation of either TSC1 or TSC2 leads to the clinical features of TSC, which are variably expressed due to incomplete penetrance of the mutated gene. It is thought that tuber/hamartoma development requires “two hits,” whereby a germline mutation in one allele of TSC1 or TSC2 is complemented by a second somatic mutation in the other allele, leading to cell growth derangement and hamartoma formation (38). Overall, TSC2 mutations confer worse seizures and cognition than TSC1 mutations (20, 39, 40).
The mTOR signaling pathway begins at the cell membrane, where receptors respond to growth factors and nutrient/energy molecules (Figure 2). Phosphatidylinositol triphosphate 3-kinase (PI3K) activates protein kinase B (AKT) which is a serine/threonine-specific protein kinase and potent pro-survival and pro-oncogenic protein. AKT directly phosphorylates TSC2 and inhibits its function. TSC2 inactivation by AKT reduces Ras homolog enriched in brain (Rheb), a small GTPase that is a member of the Ras (rat sarcoma) superfamily. Overexpression of Rheb due to a TSC1 or TSC2 mutation leads to increased mTOR activation and excessive cell growth and proliferation. These effects are blocked by rapamycin, which inhibits the mTOR pathway by binding directly to mTOR (mTOR complex 1 or mTORC1), thereby decreasing phosphorylation of downstream mTOR effectors (41, 42). These downstream effectors include two key regulators of protein translation, ribosomal S6-kinase (S6K) and eukaryote initiation factor 4E binding protein 1 (4E-BP1). The loss of TSC1 or TSC2 function leads to selective hyperactivation of the mTOR cascade, resulting in mTOR-dependent increased phosphorylation of S6K and 4E-BP1 proteins (43). Therefore, these gene mutations provide a plausible mechanism to account for the characteristic giant cells (cytomegaly) in TSC. Inhibition of this cascade by mTOR inhibitors results in growth suppression and restricted cell size.
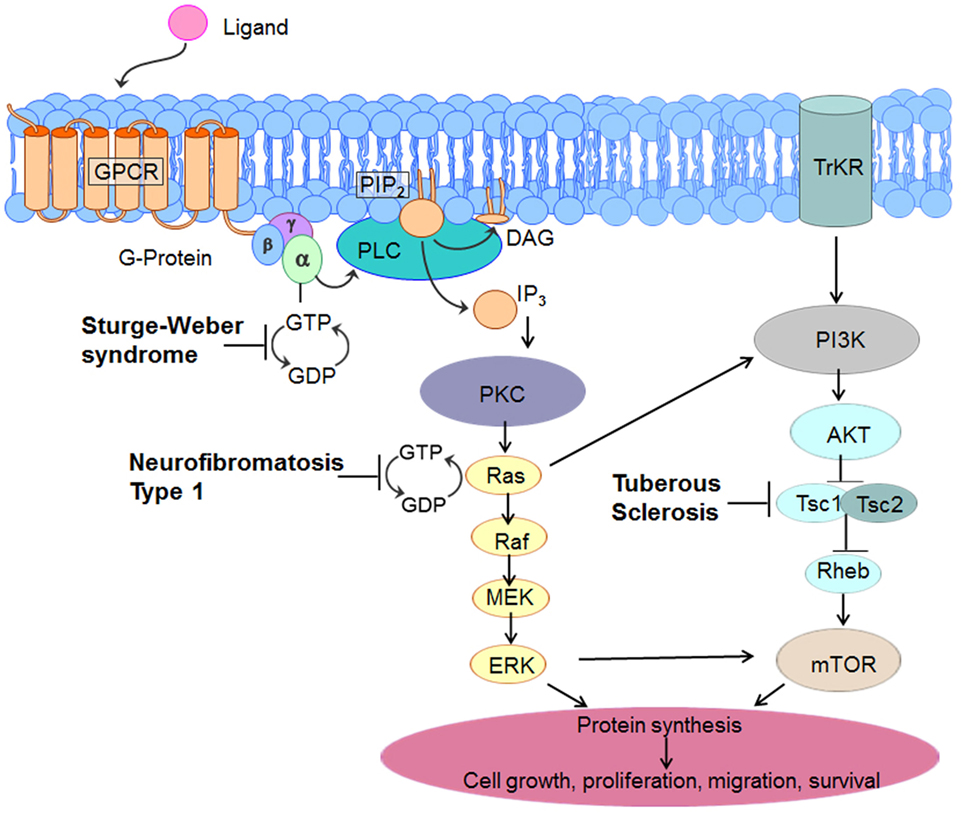
Figure 2. Molecular pathways involved in three common neurocutaneous disorders— tuberous sclerosis complex (TSC), neurofibromatosis type 1, and Sturge–Weber syndrome. As elaborated in the text, this diagram indicates unique sites of dysfunction as well as interactive and overlapping pathways that might serve as target-specific interventions.
Aside from its control of cell growth and survival, mTOR is also involved in synaptic plasticity, learning and memory (44). Long-term potentiation (LTP), a process whereby repeated activation of a neural pathway strengthens the connections between neurons, is considered to be a cellular correlate of learning and memory. mTOR activation is involved in the late phase form of LTP that is dependent on protein synthesis. The impairment of LTP by seizures is a candidate mechanism for cognitive impairment in epilepsy (45, 46). In TSC1 conditional knockout mice, TSC1+/−, the LTP deficit is partially reversed by NMDA receptor antagonists, and it has been suggested that LTP impairment is due to overactivation of the mTOR pathway or NMDA receptors (i.e., by seizures) (47, 48). Likewise, in TSC2+/− mice, rapamycin improves synaptic plasticity (LTP) and reduces behavioral deficits (49).
In addition to its pivotal role in the pathogenesis of TSC, mTOR dysfunction also plays a role in other neurologic disorders, both genetic (hemimegalencephaly, focal cortical dysplasia) and acquired (temporal lobe epilepsy, traumatic brain injury) (37, 48, 50–52). mTOR’s central action on cell growth regulation occurs in other neurocutaneous disorders as well, as discussed below for NF1 and SWS.
Epilepsy Mechanisms in TSC
In TSC, the mechanisms of E/I imbalance leading to seizure generation and epileptogenesis are complex and multifactorial, related to both the dysfunction of the mTOR signaling pathway (abnormal cellular excitation) and the neuropathological substrates (hyperexcitable circuits) (53, 54). TSC pathophysiology may be summarized as follows: abnormal molecules in abnormal cells form abnormal circuits, together leading to increased seizure propensity and epileptogenesis (55). This summary statement emphasizes the involvement of pathophysiological factors at multiple levels of brain function. In TSC, numerous features could contribute to the propensity for epilepsy by altering E/I balance, including altered cerebral cortical architecture, astrocyte proliferation, calcification, altered vascular anatomy, edema, altered neurotransmitter receptor expression, and cell proliferation and death (Figure 3) (42).
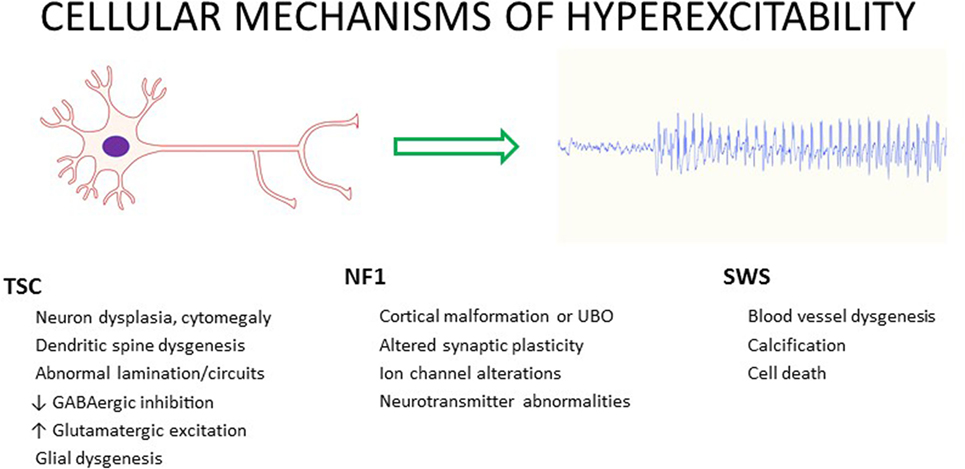
Figure 3. Possible contributors to cellular hyperexcitability and seizures in three common neurocutaneous disorders—tuberous sclerosis complex (TSC), neurofibromatosis type 1 (NF1), and Sturge–Weber syndrome (SWS).
Favoring excess excitation, glutamate receptors in dysplastic neurons and giant cells have increased expression of NMDA receptor subunits NR2B and NR2D (in newer nomenclature, GluN2B and GluN2D) and the neuronal glutamate transporter EAAC1 (56). In TSC1+/− mice, there is persistent, mTOR-dependent selective functional upregulation of the NMDA receptor subunit GluN2C, specifically in cortical layer IV neurons that are critical for thalamocortical integration of excitatory activity; this receptor subtype mediates excessive current through NMDA channels and enhances recurrent excitation and seizure propensity; furthermore, seizure generation is blocked by GluN2C antagonists (57). Tubers resected from children with TSC have a selective increase in the expression of AMPA receptor subunits GluR1 and GluR2 in dendrites of dysplastic neurons compared with normal neurons, suggesting that increased calcium permeability engendered by these receptor subunit alterations could promote epileptogenesis (58). Mice lacking TSC1 also exhibit abnormally long excitatory postsynaptic currents and epileptic discharges, like human tubers (59).
Favoring decreased inhibition, tubers have lower levels of the GABA synthetic enzyme glutamate decarboxylase 65, the vesicular GABA transporter VGAT, and GABAA receptor subunits α1 and α2 (56). In cortical specimens from patients with TSC, the number of GABAergic interneurons is decreased, consistent with diminished and aberrant local inhibition (60). Excessive depolarizing GABA responses in tuberal neurons were blocked by bumetanide, a compound that inhibits the ion transporter sodium-potassium-chloride cotransporter 1 (NKCC1) (61, 62). Microtransplantation of cortical membranes from TSC patients into Xenopus oocytes revealed a lack of expected maturation of both GABAA receptor function (delayed transition to hyperpolarizing action) and a persistently elevated ratio of NKCC1 to the potassium-chloride cotransporter 2 (KCC2, whose expression is more prominent beyond the neonatal stage and is responsible for the hyperpolarizing action of GABA receptor activation); also, AMPA receptor subunit expression (GluR1/GluR2 ratio) was increased compared to non-TSC control tissue that did undergo developmental maturation, suggesting a pattern of cerebral dysmaturity in TSC (63). Ion channel dysfunction might also cause hyperexcitability in TSC—in astrocytes of TSC1-deficient mice, there is decreased expression of potassium inward rectifier channels (Kir2.1 and Kir 6.1), limiting the passage of hyperpolarizing current (64). Collectively, these observations suggest that different tuber components possess different molecular profiles that could alter the E/I balance in favor of excitation and predispose to seizures.
In addition to molecular alterations, mTOR overactivity, via TSC1 and TSC2 mutation, leads to structural aberrations in neurons and dendrites and their growth. The soma size and dendrite size and density of hippocampal pyramidal cells are perturbed by the loss of a single copy of TSC1 (65). Additional mechanisms that may play a role in seizure predisposition in TSC include somatic and dendritic hypertrophy, aberrant basal dendritic structure, and enlargement of axon tracts. These changes lead to increased synaptogenesis and recurrent circuit formation (66). E/I imbalance may be promoted by mTOR-dependent repression of inhibitory input onto excitatory hippocampal pyramidal neurons, resulting in excitatory positive feedback (67). Attesting to the role of the mTOR pathway and its inhibition in disorders other than TSC, in a rodent model of temporal lobe epilepsy, rapamycin reduces the sprouting of mossy fiber axons that in part underlies hyperexcitability in the hippocampal dentate gyrus and may underlie epilepsy progression (68). Furthermore, rapamycin directly augments inhibition by increasing potassium channel (Kv1.1) expression (69) and decreases excitation by reducing the surface expression of AMPA receptors (70). Rapamycin also reduces the duration of epileptiform bursts induced by the GABAA receptor antagonist, bicuculline (71).
Animal models of TSC, created by deletion or knockout of TSC1 or TSC2 in certain cell types, have provided considerable insight into mechanisms by which seizures and epilepsy occur in TSC, as indicated by several experimental observations already discussed. Mice with conditional knockout of glial fibrillary acidic protein (GFAP), a marker of glial cells, have increased mTOR activity and increased SK6 leading to enhanced epileptogenicity. Importantly, brains of these knockout animals have no tubers (72). Therefore, epilepsy in these mice occurs without requiring the presence of a tuber or other macroscopic structural lesion, suggesting that alteration of the mTOR pathway itself is sufficient to generate seizures. The function of the glutamate transporters GLT and GLAST is decreased in astrocytes of these mice (73). The result is altered glutamate homeostasis with excessive extracellular and synaptic glutamate levels, leading to excitotoxicity via increased excitatory postsynaptic potentials and increased NMDA receptor activation, which increase seizure propensity. Acute seizures activate the mTOR pathway transiently (74), whereas status epilepticus leads to chronic mTOR elevation (75). There is also evidence of increased inflammatory markers such as IL-1β in GFAP knockout mice, and treatment with anti-inflammatory drugs leads to decreased seizures and increased survival (76). Timing of mTOR inhibition may be important—early treatment with rapamycin decreases mTOR activation, decreases glial proliferation, increases GLT1 expression, prevents epilepsy, and increases survival. Late rapamycin treatment, given after the mice have already developed spontaneous seizures, is also beneficial in decreasing seizure frequency and increasing survival (72). It remains a mystery why patients with TSC have such a high predilection to infantile spasms. The lack of animal models of infantile spasms hinders exploration of the underlying pathophysiology (77). In the “multiple-hit” model of infantile spasms, high-dose rapamycin delivered in daily pulses for 3 days ameliorates spasm occurrence (78); this is not a TSC model per se, rather one that mimics symptomatic spasms as a consequence of brain injury induced by three toxins. These results implicate the mTOR pathway as a possible treatment target in symptomatic infantile spasms and emphasize the possible effectiveness of pulse mTOR inhibition as opposed to continuous rapamycin administration.
Neurofibromatosis Type 1
Clinical Features of NF1
Neurofibromatosis type 1, the most common neurocutaneous disorder, occurs in approximately 1 in 3,000 births (79). Inheritance is autosomal dominant and the mutations are de novo in about half the cases. The diagnosis of NF1 is clinical, based on consensus criteria (80), and confirmation with genetic testing is available but usually not necessary (81). Disease manifestations are quite diverse, even within a family, related to variable penetrance of the mutation. Characteristic findings are hyperpigmented skin markings or café-au-lait macules (which typically develop in the first 2 years of life), axillary freckling, disease-specific Lisch nodules (hamartomas of the iris), optic pathway gliomas, and neurocognitive deficits. NF1 patients are also at risk to develop multiple benign and malignant Schwann cell neoplasms such as tumors of the peripheral nerve sheath, gliomas, pheochromocytoma, gastrointestinal stromal tumors, and other cancers including breast cancer in young women and leukemia (82). Neurofibromas, the hallmark of the disease, are benign nerve sheath tumors of two types, dermal (which can be cutaneous or subcutaneous) or plexiform. Both types may grow and cause pain and disfigurement; however, only plexiform neurofibromas can undergo malignant transformation into a malignant peripheral nerve sheath tumor, an aggressive spindle-cell sarcoma with poor prognosis (83). Optic pathway gliomas are very common, affecting up to 15% of children with NF1, the majority presenting before 7 years of age. Gliomas of the hemispheres, brainstem, or cerebellum can also occur, which are mostly pilocytic or diffuse astrocytomas (Figure 1B1). In addition to gliomas, central nervous system lesions in NF1 frequently include T2-hyperintensities on MRI scan, known as “unidentified bright objects” (UBOs) (Figure 1B2) (84). Autopsy studies have demonstrated that UBOs are vacuolar changes in myelin sheaths with dysplastic glial proliferation, often seen in the cerebellum, basal ganglia, subcortical white matter, and thalamus (85). The etiopathogenesis of UBOs is unknown; their presence may be associated with poorer cognition, but the data remains conflicted (86, 87). Intriguingly, UBOs often disappear by adulthood and their role in nervous system dysfunction is unclear (88). Also, cortical malformations, in the form of disordered cortical architecture, have been documented (85, 89).
Cognitive deficits in NF1 tend to be mild or moderate and include learning problems in more than half the patients, and these can be significant enough to affect academic performance and quality of life. Although no specific learning disability is characteristic of NF1, common deficits affect visual-spatial and visual motor abilities, executive function, verbal and nonverbal language, fine and gross motor coordination, and attention (90–92). Sleep disorders and anxiety are very common. Migraine headaches occur in up to 33%, autism in 15%, and epilepsy in 6–10% (93). Megalencephaly, manifesting as macrocephaly, is due to increased white matter volume (94). Other organ systems often involved in NF1 include skeletal (long bone dysplasia, scoliosis) and vascular (blood vessel stenosis, especially the renal artery; aneurysms; hypertension).
Epilepsy in NF1
While epilepsy is much less common in NF1 than in TSC, its incidence of 6–10% is still considerably greater than that of epilepsy in the general population (0.5%). Seizures in NF1 tend to have focal onset and commonly generalize secondarily (95–97). It is thought that seizures in NF1 arise from the numerous focal lesions that comprise the disorder, namely, tumors and malformations of cortical development (Figure 1B1). Thus, seizure occurrence requires neuroimaging, even if previous neuroimaging was normal. The relationship of UBOs to seizures is controversial, but most studies have concluded that UBOs are not associated with seizures (84, 96). Seizures in NF1 are often relatively easy to control with one or more conventional antiseizure drugs; surgical resection of offending lesions is sometimes pursued (98). Surgery has been most successful for temporal lobe gliomas (93).
Molecular Basis of NF1—Ras Overactivation
The NF1 gene on chromosome 17 encodes neurofibromin, a large cytoplasmic tumor suppressing protein. Neurofibromin, a GTPase-activating protein that suppresses the proto-oncogene Ras, is highly expressed in neurons and glia, especially oligodendrocytes. Neurofibromin ordinarily inhibits Ras activity by catalyzing the hydrolysis of GTP-bound Ras to GDP-bound Ras (converts the proto-oncogene p21-Ras from an active form to an inactive form), thereby preventing tumor formation (Figure 2) (37, 99). In NF1, the lack of neurofibromin leads to unopposed Ras (GTPase) activity, causing released control of downstream signals involved in cell growth and differentiation, such as mitogen-activated protein kinase (MAPK, also known as extracellular-signal regulated kinase or ERK) and mTOR. Activation of Ras leads to enhanced activation of Ras-PI3K, MAPK, and mTOR. Therefore, NF1 is considered a “Ras-opathy.” Neurofibromin controls mTOR via a common biochemical pathway with tuberin (100). In that sense, the molecular pathways in TSC and NF1 share some common features. mTOR overactivation has been demonstrated in NF1-associated tumors (99). However, the degree of mTOR activation in NF1 is modest compared to TSC, and it is not known how significant a role mTOR dysfunction plays in NF1 or NF1-associated seizures (3). Neurofibromin also positively regulates cyclic adenosine monophosphate (cAMP), and increased cAMP levels are associated with decreased cell growth (92).
In NF1, cognitive deficits appear to be related to synaptic dysfunction as a consequence of signaling dysfunction of Ras-ERK, cAMP, and dopamine homeostasis rather than to a macroscopic structural lesion. In mice with targeted disruption of NF1 (heterozygous null mutations, NF1+/−), Schwann cells have increased Ras activation and increased growth rate. These mice have cognitive impairments that parallel many human NF deficits in visuospatial learning, working memory, attention, and motor skills (101). Despite a lack of structural brain abnormalities, these mutant mice demonstrate several deficits in cognitive function. NF1+/− mice have impaired LTP, constituting a cellular basis for their impaired ability to perform hippocampus-based tasks involving spatial learning and memory (102). Furthermore, the cognitive deficits in NF1 are thought to arise from increased GABAergic signaling and can be reversed in animal models by pharmacologically inhibiting Ras-ERK activity with statin drugs (e.g., lovastatin, simvastatin) (103, 104). Studies in Drosophila homozygous for null NF1 gene mutations revealed a linkage between cAMP generation and neurofibromin as the underlying cause for neurobehavioral manifestations in NF1 (105), later confirmed in mammalian species (106). In these studies, decreased cAMP generation due to reduced levels of functional neurofibromin resulted in olfactory learning deficits. However, the role of cAMP in human cognitive dysfunction in NF1 patients remains less clear and further investigations are required. Lastly, reduced dopamine levels in the striatum have been described in NF1+/− mice and correction of the dopamine defect resulted in improved cognition whereas Ras inhibition was ineffective (107). Due to those multiple mechanisms underlying neurobehavioral deficits, it is not surprising that clinical trials of lovastatin in patients have had variable success (108). Some benefits in working memory and verbal memory were found in a recent 14-week randomized trial of lovastatin versus placebo (109).
Epilepsy Mechanisms in NF1
Neurofibromin plays important roles in multiple aspects of cortical development, including synaptic plasticity, learning and memory, neurotransmitter phenotype, and synapse formation (92). However, it is unclear why brains of individuals with NF1 are hyperexcitable and predisposed to seizures, and this topic is rarely discussed in the literature (110, 111). Possibilities are admittedly speculative and comprise the spectrum of pathophysiologies that disrupt the E/I balance. Of possible relevance to seizure mechanisms, it was found that GABA release and levels are enhanced in NF1+/− mice, a result of loss of neurofibromin leading to unrestrained ERK signaling and enhanced synaptic GABA release (104). While this finding was used to explain the impaired cognition, learning, and LTP of NF1+/− mice, decreased rather than increased GABA levels would be more consistent with a predisposition to epilepsy. However, augmented GABA release strategically limited to local inhibitory circuits could theoretically enhance excitability.
Ion channels are also receiving increasing attention in NF1, both in the peripheral nervous system, where enhanced pain perception is a common clinical problem, and in the central nervous system, related to abnormal LTP and cognition (112). Calcium channel opening is enhanced, and calcium currents are increased in hippocampal neurons in NF1+/− mice (113), which would increase excitability and neurotransmitter release. Dysfunction of a variety of ion channels (e.g., sodium, potassium, hyperpolarization-activated cyclic nucleotide-gated) has been reported in different brain regions and NF models (112, 114), but no consistent pattern has emerged to put forward a unified hypothesis about cortical hyperexcitability or seizures. In peripheral nociceptors, several sodium channel isoforms (NaV1.1, NaV1.7, NaV1.8) have increased expression and activity in NF1+/− mice, leading to hyperexcitability (115). These findings may have relevance to central neurons and circuits, a topic ripe for investigation in terms of epilepsy mechanisms in NF. There is no published information as to whether NF1+/− mice have altered susceptibility to seizures induced by standard experimental methods (e.g., bicuculline, kindling).
Sturge–Weber Syndrome
Clinical Features of SWS
Sturge–Weber syndrome, with an estimated incidence of 1 in 20,000 live births, is a sporadically occurring neurocutaneous disorder characterized by vascular malformations of the brain, skin, and eyes (116). The clinical hallmarks are facial angiomas (port-wine birthmarks), malformations of leptomeningeal blood vessels, and ocular angiomas causing glaucoma. Children born with a facial port-wine birth mark in the region of the forehead, temple, and upper eyelid have a greatly increased risk of also having brain or eye involvement. The cerebral vascular malformations are typically unilateral (on the same side as the port-wine birthmark) but can also occur bilaterally, which confers a worse prognosis (117). Clinical manifestations include recurrent strokes and stroke-like episodes that can lead to hemiparesis, visual field defects, cognitive deterioration, and developmental delays involving language and behavior (116). Most infants develop fairly normally for some period of time prior to the onset of seizures. Children may manifest with early handedness or a gaze preference. Seizures most commonly present in the first year of life (118). Along with the capillary-venous leptomeningeal malformations, cortical malformations such as focal cortical dysplasia or polymicrogyria can also be seen (119, 120). The cerebral atrophy, calcification, and clinical deficits can be progressive, particularly in infants and young children. Later problems can include specific learning disabilities, attention deficit disorder, and hormone deficiencies (e.g., growth hormone, thyroid hormone) (121, 122).
Epilepsy in SWS
Epilepsy occurs in 72% of SWS cases with unilateral cerebral involvement and 90% or more of those with bilateral involvement (117, 123). Seizures usually have a focal onset with secondary generalization. Although seizures often begin in the first year of life and generally by 2 years of age, about 10% can begin later in childhood, adolescence, or adulthood. The seizures commonly occur in clusters or as status epilepticus, with relatively long periods between bouts (124). The prolonged seizures in SWS are thought to worsen cognitive function (125). The combination of seizures very early in life (before 6 months of age), and extensive brain pathology portends a poor outcome. Seizures in SWS can be progressive as brain atrophy advances, leading to refractory epilepsy. Seizures sometimes respond to anticonvulsants such carbamazepine or oxcarbazepine; levetiracetam, also used commonly in SWS, can be effective at times but is associated with more frequent side effects (126). Topiramate and valproic acid are useful third-line agents. While topiramate should always be used with caution owing to the possibility that it can exacerbate glaucoma, children with SWS do not appear to be at increased risk for topiramate-associated glaucoma. In SWS, topiramate-associated glaucoma is an adverse effect seen more commonly, but not exclusively, in adults (127). Metabolic approaches such as the ketogenic or modified Atkins diet can be effective (128). Surgical options for refractory epilepsy include focal resection or hemispherectomy (129). Aspirin has been advocated to reduce stroke occurrence (130). mTOR inhibitors have not been studied in SWS. The high incidence of seizures in SWS raises the prospect that prophylactic treatment prior to the onset of seizures might improve developmental outcome, and clinical trials of this idea are needed (131).
Molecular Basis of SWS—Abnormal Blood Vessel Formation
Sturge–Weber syndrome is caused by a post-zygotic somatic mosaic mutation of GNAQ, a gene that is critical for blood vessel development (132). Interestingly, GNAQ mutations are found in individuals who have the full spectrum of SWS with skin, eye, and brain involvement as well as in patients with isolated port-wine birth marks and no brain or eye involvement. Mutations earlier in development are presumed to be associated with more severe manifestations; an animal model needs to be developed to test this hypothesis. GNAQ codes for Gαq, the alpha subunit of a heterotrimeric G protein that activates downstream pathways including the ERK and mTOR pathways (Figure 2). Most recent evidence suggests that endothelial cells in the capillary malformations are particularly enriched in GNAQ mutations (133) but the precise mechanisms by which the mutation in endothelial cells results in the vascular malformations of SWS are not known. Expression of angiogenesis factors, such as vascular endothelial growth factor, hypoxia-inducible factor α1 (134), and fibronectin (135) is altered, and these factors likely play an important role in the pathophysiology. For example, Elfn1 (extracellular-leucine-rich repeat fibronectin domain 1) is a gene enriched in hippocampal GABAergic interneurons that helps to recruit metabotropic glutamate receptors (e.g., mGluR7) to the presynaptic membrane; mutant mice with Elfn1 knocked out develop seizures (136, 137).
Epilepsy Mechanisms in SWS
The mechanism of epileptogenesis in SWS is uncertain, but abnormal blood vessel development undoubtedly figures prominently in the neuropathology and epilepsy pathogenesis (138). The clinical consequences of SWS are in part related to abnormal draining veins leading to venous stasis and congestion, which decreases regional perfusion and eventually causes hypoxic brain injury with neuronal loss and gliosis. Cortical malformations such as polymicrogyria with inherent circuit dysfunction also contribute to epilepsy pathogenesis (119). Seizures appear to originate in the cortex adjacent to the leptomeningeal angioma (139). Ictal single positron emitted computed tomography studies have demonstrated that prolonged seizures exacerbate perfusion deficits and may increase the risk of stroke (140). Diseased brain tissue shows decreased blood flow interictally and increased or decreased blood flow during a seizure. Positron emission tomography (PET) scans, which measure glucose uptake and therefore correlate with neuronal activity, have demonstrated interictal hypermetabolism in children with SWS, possibly indicating areas of incipient cortical dysfunction; these observations have implications for surgical planning (141). Fluorodeoxyglucose-PET and clinical studies show that if seizures are controlled for a prolonged period, glucose metabolism and neurodevelopmental status can improve (142). Increased vascular permeability from the leptomeningeal malformations likely leads to calcium leakage across the blood–brain barrier. While calcium itself is probably not epileptogenic, it certainly represents a marker for areas of cortical dysfunction.
An animal model would facilitate understanding of epilepsy pathogenesis in SWS, and efforts to create such a model are underway (143). However, there are several challenges to generating a model of a disorder with somatic mosaic mutations involving gain-of-function. The mutant gene must be expressed in the relevant cells and body structures only, a gene knockout approach is not applicable, and the developmental timing of the post-zygotic mutation is unknown. Nevertheless, molecular approaches to model creation using cell culture, mice, and other systems such as zebrafish should afford insights into seizure mechanisms as a consequence of endothelial cell maldevelopment (143). Continuing the theme already discussed for TSC and NF1, alterations in specific signaling molecules and pathways may engender hyperexcitable circuits in SWS, with mTOR dysfunction occupying a central location in the molecular pathways controlling numerous cell differentiation and migration (144). However, a role of mTOR in epileptogenesis in SWS has not been defined.
Conclusion
Clinical and mechanistic aspects of epilepsy in TSC, NF1, and SWS are summarized in Table 1.
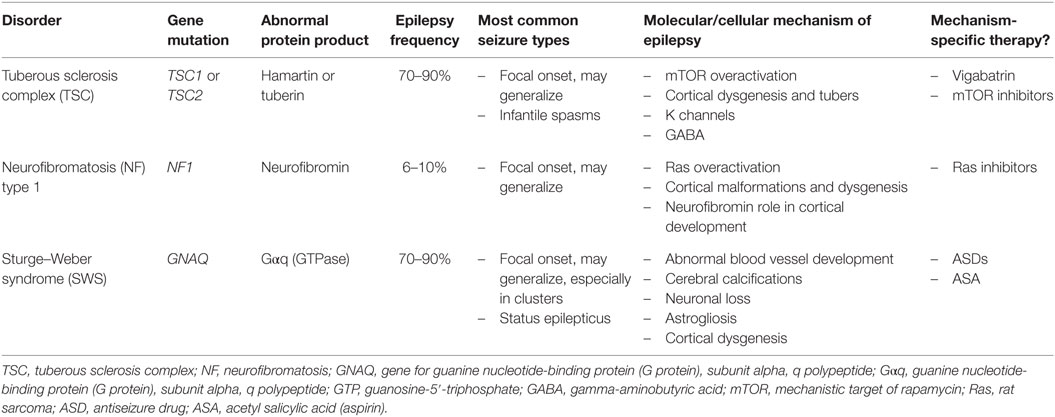
Table 1. Epilepsy in common neurocutaneous disorders.
Targeted therapies are emerging from increased molecular understanding of neurocutaneous disorders, based on both unique molecular pathways for each syndrome and commonalities among syndromes [i.e., mTOR dysfunction (2, 3, 110)]. Clues to the pathophysiology of epilepsy in TSC, NF1, and SWS may arise from the observation that in each disorder, seizures have a focal onset, focusing attention on mechanisms altering the E/I balance at discrete sites of seizure initiation and subsequent spread. Figure 3 lists some of the factors that may lead to neuronal hyperexcitability in these neurocutaneous disorders.
Other insights might be gleaned from the very rare cases in which more than one neurocutaneous syndrome exists in a single individual. For example, a child was reported with maternal inheritance of NF1 and paternal inheritance of TSC (145). This girl developed intractable epilepsy at age 5 years and felbamate was the only antiseizure drug that helped. Since felbamate’s main mechanism of action is NMDA receptor antagonism and since NMDA downregulates the mTOR pathway, it was suggested that felbamate was acting specifically to dampen the mTOR pathway and reduce neuronal excitability. Such speculations are intriguing but need to be considered cautiously. A few patients with combined SWS and TSC have been reported, but none since the discovery of the somatic mutation causing SWS (146); it would be informative to better understand these dual mutations. There is no doubt, however, that epilepsy mechanisms in neurocutaneous disorders represent a vastly understudied topic that warrants concerted clinical and laboratory investigations. Indeed, epilepsy mechanisms may reveal new insights into the relationship between cognition, behavior, seizures, and other aspects of brain function in the neurocutaneous disorders (147).
Author Contributions
CS, VS, and AC each contributed to the conceptualization, design, writing, and final approval of the manuscript, and each is accountable for all aspects of the work. AC drew Figure 2.
Conflict of Interest Statement
The authors report no conflicts of interest or financial relationships relevant to this work.
Funding
VS is supported by the Francis S. Collins Scholars Program of the Neurofibromatosis Therapeutic Acceleration Program (NTAP).
References
1. Gipson TT, Johnston MV. Plasticity and mTOR: towards restoration of impaired synaptic plasticity in mTOR-related neurogenetic disorders. Neural Plast (2012) 2012:486402. doi: 10.1155/2012/486402
2. Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol (2016) 12(7):379–92. doi:10.1038/nrneurol.2016.81
3. Sato A. mTOR, a potential target to treat autism spectrum disorder. CNS Neurol Disord Drug Targets (2016) 15(5):533–43. doi:10.2174/1871527315666160413120638
4. Stafstrom CE. Recognizing seizures and epilepsy: insights from pathophysiology. In: Miller JW, Goodkin HP, editors. Epilepsy. Chichester, UK: John Wiley & Sons, Ltd (2014). p. 3–9.
5. Bonansco C, Fuenzalida M. Plasticity of hippocampal excitatory-inhibitory balance: missing the synaptic control in the epileptic brain. Neural Plast (2016) 2016:8607038. doi:10.1155/2016/8607038
6. Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol (2013) 49(4):243–54. doi:10.1016/j.pediatrneurol.2013.08.001
7. Islam MP, Roach ES. Tuberous sclerosis complex. Handb Clin Neurol (2015) 132:97–109. doi:10.1016/B978-0-444-62702-5.00006-8
8. Lee A, Maldonado M, Baybis M, Walsh CA, Scheithauer B, Yeung R, et al. Markers of cellular proliferation are expressed in cortical tubers. Ann Neurol (2003) 53(5):668–73. doi:10.1002/ana.10579
9. Curatolo P, Bombardieri R, Verdecchia M, Seri S. Intractable seizures in tuberous sclerosis complex: from molecular pathogenesis to the rationale for treatment. J Child Neurol (2005) 20:318–25. doi:10.1177/08830738050200040901
10. Mohamed AR, Bailey CA, Freeman JL, Maixner W, Jackson GD, Harvey AS. Intrinsic epileptogenicity of cortical tubers revealed by intracranial EEG monitoring. Neurology (2012) 79:2249–57. doi:10.1212/WNL.0b013e3182768923
11. Ruppe V, Dilsiz P, Reiss CS, Carlson C, Devinsky O, Zagzag D, et al. Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia (2014) 55(4):539–50. doi:10.1111/epi.12545
12. Jansen FE, Vincken KL, Algra A, Anbeek P, Braams O, Nellist M, et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology (2008) 70:916–23. doi:10.1212/01.wnl.0000280579.04974.c0
13. Jülich K, Sahin M. Mechanism-based treatment in tuberous sclerosis complex. Pediatr Neurol (2014) 50(4):290–6. doi:10.1016/j.pediatrneurol.2013.12.002
14. Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain (2002) 125:1247–55. doi:10.1093/brain/awf124
15. Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology (2011) 76(11):981–7. doi:10.1212/WNL.0b013e3182104347
16. Lewis WW, Sahin M, Scherrer B, Peters JM, Suarez RO, Vogel-Farley VK, et al. Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb Cortex (2013) 23(7):1526–32. doi:10.1093/cercor/bhs135
17. Curatolo P, Moavero R, deVries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol (2015) 14:733–45. doi:10.1016/S1474-4422(15)00069-1
18. Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia (2010) 51:1236–41. doi:10.1111/j.1528-1167.2009.02474.x
19. Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia (2010) 51(10):2168–74. doi:10.1111/j.1528-1167.2010.02695.x
20. Hsieh DT, Jennesson MM, Thiele EA. Epileptic spasms in tuberous sclerosis complex. Epilepsy Res (2013) 106(1–2):200–10. doi:10.1016/j.eplepsyres.2013.05.003
21. Curatolo P, Aronica E, Jansen A, Jansen F, Kotulska K, Lagae L, et al. Early onset epileptic encephalopathy or genetically determined encephalopathy with early onset epilepsy? Lessons learned from TSC. Eur J Paediatr Neurol (2016) 20(2):203–11. doi:10.1016/j.ejpn.2015.12.005
22. Wang S, Fallah A. Optimal management of seizures associated with tuberous sclerosis complex: current and emerging options. Neuropsychiatr Dis Treat (2014) 10:2021–30. doi:10.2147/NDT.S51789
23. Curatolo P, Bjørnvold M, Dill PE, Ferreira JC, Feucht M, Hertzberg C, et al. The role of mTOR inhibitors in the treatment of patients with tuberous sclerosis complex: evidence-based and expert opinions. Drugs (2016) 76(5):551–65. doi:10.1007/s40265-016-0552-9
24. Kwan P, Schachter SC, Brodie M. Drug-resistant epilepsy. N Engl J Med (2011) 365(10):919–26. doi:10.1056/NEJMra1004418
25. Kumar P, Chugani HT. Functional imaging: PET. Handb Clin Neurol (2013) 111:767–76. doi:10.1016/B978-0-444-52891-9.00079-8
26. Shahid A. Resecting the epileptogenic tuber: what happens in the long term? Epilepsia (2013) 54(Suppl 9):135–8. doi:10.1111/epi.12458
27. Zhang K, Hu WH, Zhang C, Meng FG, Chen N, Zhang JG. Predictors of seizure freedom after surgical management of tuberous sclerosis complex: a systematic review and meta-analysis. Epilepsy Res (2013) 105(3):377–83. doi:10.1016/j.eplepsyres.2013.02.016
28. Curatolo P, Verdecchia M, Bombardieri R. Vigabatrin for tuberous sclerosis complex. Brain Dev (2001) 23(7):649–53. doi:10.1016/S0387-7604(01)00290-X
29. Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS One (2013) 8(2):e57445. doi:10.1371/journal.pone.0057445
30. Lakhani R, Vogel KR, Till A, Liu J, Burnett SF, Gibson KM, et al. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med (2014) 6(4):551–66. doi:10.1002/emmm.201303356
31. Józwiak S, Domańska-Pakieła D, Kotulska K, Kaczorowska M. Treatment before seizures: new indications for antiepileptic therapy in children with tuberous sclerosis complex. Epilepsia (2007) 48(8):1632. doi:10.1111/j.1528-1167.2007.01178_3.x
32. Jóźwiak S, Kotulska K, Domańska-Pakieła D, Lojszczyk B, Syczewska M, Chmielewski D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol (2011) 15(5):424–31. doi:10.1016/j.ejpn.2011.03.010
33. Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol (2006) 59:490–8. doi:10.1002/ana.20784
34. Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol (2013) 74(5):679–87. doi:10.1002/ana.23960
35. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet (2016) 388(10056):2153–63. doi:10.1016/S0140-6736(16)31419-2
36. McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) in preventing epileptogenesis. Neurosci Lett (2011) 497:231–9. doi:10.1016/j.neulet.2011.02.037
37. Lipton JO, Sahin M. The neurology of mTOR. Neuron (2014) 84:275–91. doi:10.1016/j.neuron.2014.09.034
38. Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet (2000) 107(2):97–114. doi:10.1007/s004390000348
39. Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med (2007) 9(2):88–100. doi:10.1097/GIM.0b013e31803068c7
40. Jansen FE, Braams O, Vincken KL, Algra A, Anbeek P, Jennekens-Schinkel A, et al. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology (2008) 70:908–15. doi:10.1212/01.wnl.0000280578.99900.96
41. Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron (2008) 60(5):832–45. doi:10.1016/j.neuron.2008.09.037
42. Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol (2015) 52(3):281–9. doi:10.1016/j.pediatrneurol.2014.10.028
43. Inoki K, Corradetti MN, Guan K-L. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet (2005) 37:19–24. doi:10.1038/ng1494
44. Graber TE, McCamphill PK, Sossin WS. A recollection of mTOR signaling in learning and memory. Learn Mem (2013) 20(10):518–30. doi:10.1101/lm.027664
45. Meador KJ. The basic science of memory as it applies to epilepsy. Epilepsia (2007) 48(Suppl 9):23–5. doi:10.1111/j.1528-1167.2007.01396.x
46. Isaeva E, Isaev D, Holmes GL. Alteration of synaptic plasticity by neonatal seizures in rat somatosensory cortex. Epilepsy Res (2013) 106(1–2):280–3. doi:10.1016/j.eplepsyres.2013.03.011
47. Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis (2007) 28(2):184–96. doi:10.1016/j.nbd.2007.07.015
48. Takeuchi K, Gertner MJ, Zhou J, Parada LF, Bennett MV, Zukin RS. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci U S A (2013) 110(12):4738–43. doi:10.1073/pnas.1222803110
49. Ehninger D, Hans S, Shiyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/– mouse model of tuberous sclerosis. Nat Med (2008) 14:843–8. doi:10.1038/nm1788
50. Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J (2013) 36:40–50. doi:10.4103/2319-4170.110365
51. Child ND, Benarroch EE. mTOR: its role in the nervous system and involvement in neurologic disease. Neurology (2014) 83:1562–72. doi:10.1212/WNL.0000000000000922
52. Mirzaa GM, Campbell CD, Solovieff N, Goold CP, Jansen LA, Menon S, et al. Association of mTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol (2016) 73(7):836–45. doi:10.1001/jamaneurol.2016.0363
53. Holmes GL, Stafstrom CE, Tuberous Sclerosis Study Group. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia (2007) 48:617–30. doi:10.1111/j.1528-1167.2007.01035.x
54. Feliciano DM, Lin TV, Hartman NW, Bartley CM, Kubera C, Hsieh L, et al. A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci (2013) 31:667–78. doi:10.1016/j.ijdevneu.2013.02.008
55. Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia (2008) 49(1):8–21. doi:10.1016/j.ijdevneu.2013.02.008
56. White R, Hua Y, Scheithauer B, Lynch DR, Henske EP, Crino PB. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol (2001) 49(1):67–78. doi:10.1002/1531-8249(200101)49:1<67::AID-ANA10>3.0.CO;2-L
57. Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, et al. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat Commun (2014) 5:4563. doi:10.1038/ncomms5563
58. Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE. Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol (2008) 63:454–65. doi:10.1002/ana.21342
59. Wang Y, Greenwood JS, Calcagnotto ME, Kirsch HE, Barbaro NM, Baraban SC. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol (2007) 61(2):139–52. doi:10.1002/ana.21058
60. Valencia I, Legido A, Yelin K, Khurana D, Kothare SV, Katsetos CD. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J Child Neurol (2006) 21(12):1058–63. doi:10.1177/7010.2006.00242
61. Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, et al. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol (2012) 71(4):539–51. doi:10.1002/ana.22696
62. Cepeda C, André VM, Hauptman JS, Yamazaki I, Huynh MN, Chang JW, et al. Enhanced GABAergic network and receptor function in pediatric cortical dysplasia type IIB compared with tuberous sclerosis complex. Neurobiol Dis (2012) 45(1):310–21. doi:10.1016/j.nbd.2011.08.015
63. Ruffolo G, Iyer A, Cifelli P, Roseti C, Mühlebner A, van Scheppingen J, et al. Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol Dis (2016) 95:93–101. doi:10.1016/j.nbd.2016.07.014
64. Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia (2005) 46:1871–80. doi:10.1016/j.nbd.2016.07.014
65. Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci (2005) 8(12):1727–34. doi:10.1038/nn1566
66. Lasarge CL, Danzer SC. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci (2014) 7:18. doi:10.3389/fnmol.2014.00018
67. Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron (2013) 78:510–22. doi:10.1016/j.neuron.2013.03.017
68. Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci (2009) 29(25):8259–69. doi:10.1523/JNEUROSCI.4179-08.2009
69. Raab-Graham KF, Haddick PC, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science (2006) 314:144–8. doi:10.1126/science.1131693
70. Wang Y, Barbaro MF, Baraban SC. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett (2006) 401(1–2):35–9. doi:10.1016/j.neulet.2006.03.011
71. Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res (2007) 77(2–3):85–92. doi:10.1016/j.eplepsyres.2007.09.009
72. Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol (2008) 63:444–53. doi:10.1002/ana.21331
73. Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, et al. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol (2003) 54:251–6. doi:10.1002/ana.10648
74. Zhang B, Wong M. Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia (2012) 53(3):506–11. doi:10.1002/ana.10648
75. Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci (2009) 29:6964–72. doi:10.1523/JNEUROSCI.0066-09.2009
76. Zhang B, Zou J, Rensing NR, Yang M, Wong M. Inflammatory mechanisms contribute to the neurological manifestations of tuberous sclerosis complex. Neurobiol Dis (2015) 80:70–9. doi:10.1016/j.nbd.2015.04.016
77. Stafstrom CE. Infantile spasms: a critical review of emerging animal models. Epilepsy Curr (2009) 9(3):75–81. doi:10.1111/j.1535-7511.2009.01299.x
78. Raffo E, Coppola A, Ono T, Briggs SW, Galanopoulou AS. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis (2011) 43(2):322–9. doi:10.1016/j.nbd.2011.03.021
79. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol (2005) 141(1):71–4. doi:10.1001/archderm.141.1.71
80. Office of Medical Research Applications. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol (1988) 45:575–8. doi:10.1001/archneur.1988.00520290115023
81. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet (2007) 44:81–8. doi:10.1001/archderm.141.1.71
82. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer (2013) 108(1):193–8. doi:10.1038/bjc.2012.535
83. Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg (2009) 249:1014–22. doi:10.1097/SLA.0b013e3181a77e9a
84. Hsieh HY, Fung HC, Wang CJ, Chin SC, Wu T. Epileptic seizures in neurofibromatosis type 1 are related to intracranial tumors but not to neurofibromatosis bright objects. Seizure (2011) 20(8):606–11. doi:10.1016/j.seizure.2011.04.016
85. DiPaolo DP, Zimmerman RA, Rorke LB, Zackai EH, Bilaniuk LT, Yachnis AT. Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain. Radiology (1995) 195(3):721–4. doi:10.1148/radiology.195.3.7754001
86. Hyman SL, Gill DS, Shores EA, Steinberg A, North KN. T2 hyperintensities in children with neurofibromatosis type 1 and their relationship to cognitive functioning. J Neurol Neurosurg Psychiatry (2007) 78(10):1088–91. doi:10.1136/jnnp.2006.108134
87. Rosenbaum T, Engelbrecht V, Krölls W, van Dorsten FA, Hoehn-Berlage M, Lenard HG. MRI abnormalities in neurofibromatosis type 1 (NF1): a study of men and mice. Brain Dev (1999) 21(4):268–73. doi:10.1016/S0387-7604(99)00024-8
88. Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol (2007) 6(4):340–51. doi:10.1016/S1474-4422(07)70075-3
89. Gales J, Prayson RA. Hippocampal sclerosis and associated focal cortical dysplasia-related epilepsy in neurofibromatosis type I. J Clin Neurosci (2017) 37:15–9. doi:10.1016/j.jocn.2016.10.048
90. Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology (2005) 65(7):1037–44. doi:10.1212/01.wnl.0000179303.72345.ce
91. Hachon C, Iannuzzi S, Chaix Y. Behavioural and cognitive phenotypes in children with neurofibromatosis type 1 (NF1): the link with the neurobiological level. Brain Dev (2011) 33(1):52–61. doi:10.1016/j.braindev.2009.12.008
92. Diggs-Andrews KA, Gutmann DH. Modeling cognitive dysfunction in neurofibromatosis-1. Trends Neurosci (2013) 36(4):237–47. doi:10.1016/j.tins.2012.12.002
93. Ostendorf AP, Gutmann DH, Weisenberg JL. Epilepsy in individuals with neurofibromatosis type 1. Epilepsia (2013) 54(10):1810–4. doi:10.1111/epi.12348
94. Cutting LE, Cooper KL, Koth CW, Mostofsky SH, Kates WR, Denckla MB, et al. Megalencephaly in NF1: predominantly white matter contribution and mitigation by ADHD. Neurology (2002) 59:1388–94. doi:10.1212/01.WNL.0000032370.68306.8A
95. Korf BR, Carrazana E, Holmes GL. Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia (1993) 34(4):616–20. doi:10.1111/j.1528-1157.1993.tb00437.x
96. Vivarelli R, Grosso S, Calabrese F, Farnetani M, DiBartolo R, Morgese G, et al. Epilepsy in neurofibromatosis 1. J Child Neurol (2003) 18(5):338–42. doi:10.1177/08830738030180050501
97. Caraballo RH, Portuondo E, Fortini PS. Neurofibromatosis and epilepsy. J Pediatr Epilepsy (2016) 5(2):59–63. doi:10.1055/s-0035-1570020
98. Barba C, Jacques T, Kahane P, Polster T, Isnard J, Leijten FS, et al. Epilepsy surgery in neurofibromatosis type 1. Epilepsy Res (2013) 105(3):384–95. doi:10.1016/j.eplepsyres.2013.02.021
99. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res (2005) 65(7):2755–60. doi:10.1158/0008-5472.CAN-04-4058
100. Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A (2005) 102(24):8573–8. doi:10.1073/pnas.0503224102
101. Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, et al. A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet (1997) 15(3):281–4. doi:10.1038/ng0397-281
102. Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature (2002) 415:526–30. doi:10.1038/nature711
103. Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol (2005) 15(21):1961–7. doi:10.1016/j.cub.2005.09.043
104. Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell (2008) 135(3):549–60. doi:10.1016/j.cell.2008.09.060
105. Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature (2000) 403(6772):895–8. doi:10.1038/35002593
106. Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci (2002) 5(2):95–6. doi:10.1038/nn792
107. Brown JA, Emnett RJ, White CR, Yuede CM, Conyers SB, O’Malley KL, et al. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum Mol Genet (2010) 19(22):4515–28. doi:10.1093/hmg/ddq382
108. Krab LC, de Goede-Bolder A, Aarsen FK, Pluijm SM, Bouman MJ, van der Geest JN, et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA (2008) 300(3):287–94. doi:10.1001/jama.300.3.287
109. Bearden CE, Hellemann GS, Rosser T, Montojo C, Jonas R, Enrique N, et al. A randomized placebo-controlled lovastatin trial for neurobehavioral function in neurofibromatosis I. Ann Clin Transl Neurol (2016) 3(4):266–79. doi:10.1002/acn3.288
110. Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia (2012) 53(7):1119–30. doi:10.1111/j.1528-1167.2012.03506.x
111. Gutmann DH, Parada LF, Silva AJ, Ratner N. Neurofibromatosis type 1: modeling CNS dysfunction. J Neurosci (2012) 32(41):14087–93. doi:10.1523/JNEUROSCI.3242-12.2012
112. Moutal A, Dustrude ET, Khanna R. Sensitization of ion channels contributes to central and peripheral dysfunction in neurofibromatosis type 1. Mol Neurobiol (2016). [Epub ahead of print]. doi:10.1007/s12035-016-9907-1
113. Wang Y, Brittain JM, Wilson SM, Hingtgen CM, Khanna R. Altered calcium currents and axonal growth in Nf1 haploinsufficient mice. Transl Neurosci (2010) 1(2):106–14. doi:10.2478/v10134-010-0025-8
114. Omrani A, van der Vaart T, Mientjes E, van Woerden GM, Hojjati MR, Li KW, et al. HCN channels are a novel therapeutic target for cognitive dysfunction in neurofibromatosis type 1. Mol Psychiatry (2015) 20(11):1311–21. doi:10.1038/mp.2015.48
115. Wang Y, Duan JH, Hingtgen CM, Nicol GD. Augmented sodium currents contribute to the enhanced excitability of small diameter capsaicin-sensitive sensory neurons isolated from Nf1+/– mice. J Neurophysiol (2010) 103(4):2085–94. doi:10.1152/jn.01010.2009
116. Comi AM. Sturge–Weber syndrome. Handb Clin Neurol (2015) 132:157–68. doi:10.1016/B978-0-444-62702-5.00011-1
117. Bebin EM, Gomez MR. Prognosis in Sturge–Weber disease: comparison of unihemispheric and bihemispheric involvement. J Child Neurol (1988) 3(3):181–4. doi:10.1177/088307388800300306
118. Sujansky E, Conradi S. Sturge–Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol (1995) 10(1):49–58. doi:10.1177/088307389501000113
119. Maton B, Krsek P, Jayakar P, Resnick T, Koehn M, Morrison G, et al. Medically intractable epilepsy in Sturge–Weber syndrome is associated with cortical malformation: implications for surgical therapy. Epilepsia (2010) 51(2):257–67. doi:10.1111/j.1528-1167.2009.02304.x
120. Murakami N, Morioka T, Suzuki SO, Hashiguchi K, Amano T, Sakata A, et al. Focal cortical dysplasia type IIa underlying epileptogenesis in patients with epilepsy associated with Sturge–Weber syndrome. Epilepsia (2012) 53(11):e184–8. doi:10.1111/j.1528-1167.2012.03628.x
121. Miller RS, Ball KL, Comi AM, Germain-Lee EL. Growth hormone deficiency in Sturge–Weber syndrome. Arch Dis Child (2006) 91(4):340–1. doi:10.1136/adc.2005.082578
122. Comi AM, Bellamkonda S, Ferenc LM, Cohen BA, Germain-Lee EL. Central hypothyroidism and Sturge–Weber syndrome. Pediatr Neurol (2008) 39(1):58–62. doi:10.1016/j.pediatrneurol.2008.03.018
123. Jagtap S, Srinivas G, Harsha KJ, Radhakrishnan N, Radhakrishnan A. Sturge–Weber syndrome: clinical spectrum, disease course, and outcome of 30 patients. J Child Neurol (2013) 28(6):725–31. doi:10.1177/0883073812451326
124. Kossoff EH, Ferenc L, Comi AM. An infantile-onset, severe, yet sporadic seizure pattern is common in Sturge–Weber syndrome. Epilepsia (2009) 50(9):2154–7. doi:10.1111/j.1528-1167.2009.02072.x
125. Comi AM. Sturge–Weber syndrome and epilepsy: an argument for aggressive seizure management in these patients. Expert Rev Neurother (2007) 7(8):951–6. doi:10.1586/14737175.7.8.951
126. Kaplan EH, Kossoff EH, Bachur CD, Gholston M, Hahn J, Widlus M, et al. Anticonvulsant efficacy in Sturge–Weber syndrome. Pediatr Neurol (2016) 58:31–6. doi:10.1016/j.pediatrneurol.2015.10.015
127. Rapoport Y, Benegas N, Kuchtey RW, Joos KM. Acute myopia and angle closure glaucoma from topiramate in a seven-year-old: a case report and review of the literature. BMC Pediatr (2014) 14:96. doi:10.1186/1471-2431-14-96
128. Kossoff EH, Borsage JL, Comi AM. A pilot study of the modified Atkins diet for Sturge–Weber syndrome. Epilepsy Res (2010) 92(2–3):240–3. doi:10.1016/j.eplepsyres.2010.09.008
129. Bourgeois M, Crimmins DW, de Oliveira RS, Arzimanoglou A, Garnett M, Roujeau T, et al. Surgical treatment of epilepsy in Sturge–Weber syndrome in children. J Neurosurg (2007) 106(1):20–8. doi:10.3171/ped.2007.106.1.20
130. Lance EI, Sreenivasan AK, Zabel TA, Kossoff EH, Comi AM. Aspirin use in Sturge–Weber syndrome: side effects and clinical outcomes. J Child Neurol (2013) 28(2):213–8. doi:10.1177/0883073812463607
131. Comi A. Current therapeutic options in Sturge–Weber syndrome. Semin Pediatr Neurol (2015) 22(4):295–301. doi:10.1016/j.spen.2015.10.005
132. Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge–Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med (2013) 368(21):1971–9. doi:10.1056/NEJMoa1213507
133. Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ mutations. Plast Reconstr Surg (2016) 137(3):1039–41. doi:10.1097/PRS.0000000000001868
134. Comati A, Beck H, Halliday W, Snipes GJ, Plate KH, Acker T. Upregulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha in leptomeningeal vascular malformations of Sturge–Weber syndrome. J Neuropathol Exp Neurol (2007) 66(1):86–97. doi:10.1097/nen.0b013e31802d9011
135. Comi AM, Weisz CJ, Highet BH, Skolasky RL, Pardo CA, Hess EJ. Sturge–Weber syndrome: altered blood vessel fibronectin expression and morphology. J Child Neurol (2005) 20(7):572–7. doi:10.1177/08830738050200070601
136. Dolan J, Mitchell KJ. Mutation of Elfn1 in mice causes seizures and hyperactivity. PLoS One (2013) 8(11):e80491. doi:10.1371/journal.pone.0080491
137. Tomioka NH, Yasuda H, Miyamoto H, Hatayama M, Morimura N, Matsumoto Y, et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat Commun (2014) 5:4501. doi:10.1038/ncomms5501
138. Pinto A, Sahin M, Pearl PL. Epileptogenesis in neurocutaneous disorders with focus in Sturge–Weber syndrome. F1000Res (2016) 5:370. doi:10.12688/f1000research.7605.1
139. Iimura Y, Sugano H, Nakajima M, Higo T, Suzuki H, Nakanishi H, et al. Analysis of epileptic discharges from implanted subdural electrodes in patients with Sturge–Weber syndrome. PLoS One (2016) 11(4):e0152992. doi:10.1371/journal.pone.0152992
140. Namer IJ, Battaglia F, Hirsch E, Constantinesco A, Marescaux C. Subtraction ictal SPECT co-registered to MRI (SISCOM) in Sturge–Weber syndrome. Clin Nucl Med (2005) 30(1):39–40. doi:10.1097/00003072-200501000-00014
141. Alkonyi B, Chugani HT, Juhász C. Transient focal cortical increase of interictal glucose metabolism in Sturge–Weber syndrome: implications for epileptogenesis. Epilepsia (2011) 52(7):1265–72. doi:10.1111/j.1528-1167.2011.03066.x
142. Juhasz C, Batista CE, Chugani DC, Muzik O, Chugani HT. Evolution of cortical metabolic abnormalities and their clinical correlates in Sturge–Weber syndrome. Eur J Paediatr Neurol (2007) 11(5):277–84. doi:10.1016/j.ejpn.2007.02.001
143. Comi AM, Sahin M, Hammill A, Kaplan EH, Juhász C, North P, et al. Leveraging a Sturge–Weber gene discovery: an agenda for future research. Pediatr Neurol (2016) 58:12–24. doi:10.1016/j.pediatrneurol.2015.11.009
144. Shirazi F, Cohen C, Fried L, Arbiser JL. Mammalian target of rapamycin (mTOR) is activated in cutaneous vascular malformations in vivo. Lymphat Res Biol (2007) 5(4):233–6. doi:10.1089/lrb.2007.1012
145. Mishal NM, Arkilo D, Tang J, Crawford JR, Wang SG. A potential role for felbamate in TSC- and NF1-related epilepsy: a case report and review of the literature. Case Rep Neurol Med (2015) 2015:960746. doi:10.1155/2015/960746
146. Curatolo P, Lo-Castro A, Pinci M, Moavero R, Bombardieri R. Neuroimaging findings of Sturge–Weber syndrome in a child with tuberous sclerosis. Brain Dev (2009) 31(5):352–5. doi:10.1016/j.braindev.2008.06.004
Keywords: seizure, epilepsy, neurocutaneous disorder, tuberous sclerosis complex, neurofibromatosis, Sturge–Weber syndrome, mechanistic target of rapamycin
Citation: Stafstrom CE, Staedtke V and Comi AM (2017) Epilepsy Mechanisms in Neurocutaneous Disorders: Tuberous Sclerosis Complex, Neurofibromatosis Type 1, and Sturge–Weber Syndrome. Front. Neurol. 8:87. doi: 10.3389/fneur.2017.00087
Received: 17 January 2017; Accepted: 24 February 2017;
Published: 17 March 2017
Edited by:
John R. Mytinger, The Research Institute at Nationwide Children’s Hospital, USAReviewed by:
Anju Aggarwal, University College of Medical Sciences, IndiaPhillip Lawrence Pearl, Boston Children’s Hospital, USA
Copyright: © 2017 Stafstrom, Staedtke and Comi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl E. Stafstrom, Y3N0YWZzdDFAamhtaS5lZHU=