 Sara Garofalo1,2
Sara Garofalo1,2 Azucena Justicia1
Azucena Justicia1 Gonzalo Arrondo1
Gonzalo Arrondo1 Anna O. Ermakova1Pranathi Ramachandra3
Anna O. Ermakova1Pranathi Ramachandra3 Carina Tudor-Sfetea1
Carina Tudor-Sfetea1 Trevor W. Robbins2,4
Trevor W. Robbins2,4 Roger A. Barker5
Roger A. Barker5 Paul C. Fletcher1,3,6
Paul C. Fletcher1,3,6 Graham K. Murray1,2,3,6*
Graham K. Murray1,2,3,6*
- 1Department of Psychiatry, University of Cambridge, Cambridge, UK
- 2Behavioural and Clinical Neuroscience Institute, University of Cambridge, Cambridge, UK
- 3Cambridgeshire and Peterborough NHS Foundation Trust, Cambridge, UK
- 4Department of Psychology, University of Cambridge, Cambridge, UK
- 5Department of Clinical Neuroscience, University of Cambridge, Cambridge, UK
- 6Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK
Psychotic symptoms frequently occur in Parkinson’s disease (PD), but their pathophysiology is poorly understood. According to the National Institute of Health RDoc programme, the pathophysiological basis of neuropsychiatric symptoms may be better understood in terms of dysfunction of underlying domains of neurocognition in a trans-diagnostic fashion. Abnormal cortico-striatal reward processing has been proposed as a key domain contributing to the pathogenesis of psychotic symptoms in schizophrenia. This theory has received empirical support in the study of schizophrenia spectrum disorders and preclinical models of psychosis, but has not been tested in the psychosis associated with PD. We, therefore, investigated brain responses associated with reward expectation and prediction error signaling during reinforcement learning in PD-associated psychosis. An instrumental learning task with monetary gains and losses was conducted during an fMRI study in PD patients with (n = 12), or without (n = 17), a history of psychotic symptoms, along with a sample of healthy controls (n = 24). We conducted region of interest analyses in the ventral striatum (VS), ventromedial prefrontal and posterior cingulate cortices, and whole-brain analyses. There was reduced activation in PD patients with a history of psychosis, compared to those without, in the posterior cingulate cortex and the VS during reward anticipation (p < 0.05 small volume corrected). The results suggest that cortical and striatal abnormalities in reward processing, a putative pathophysiological mechanism of psychosis in schizophrenia, may also contribute to the pathogenesis of psychotic symptoms in PD. The finding of posterior cingulate dysfunction is in keeping with prior results highlighting cortical dysfunction in the pathogenesis of PD psychosis.
Introduction
Psychotic symptoms, such as hallucinations, paranoia, and delusions, are present in about 15–40% of Parkinson’s disease (PD) patients and may have a major impact on quality of life and likelihood of nursing home placement (1–5).
Several pathophysiological mechanisms have been proposed to explain how psychotic symptoms develop in PD. It has been suggested that visual hallucinations in PD develop as a consequence of cortical degeneration in posterior cortical areas (6, 7). An alternative to a single locus account of PD psychosis (PDP) is that the psychosis emerges through interactions between subcortical and cortical pathological processes, or through the interaction of frontal and posterior cortical deficits, or through interaction between intrinsic disease-related factors and extrinsic drug factors (3, 4, 7–11).
There have been comparatively few attempts to integrate mechanistic explanations of psychotic symptoms in PD with current models of their pathophysiology in primary psychiatric illness. In spite of clear potential differences in the pathogenesis of psychiatric symptoms in PD compared to primary psychiatric illness (2, 12), there may be common pathways for the manifestation of certain neuropsychiatric symptoms across traditional diagnostic categories (6, 13). A longstanding candidate contributor cause of psychosis in PD is a dopaminergic system perturbed by a combination of intrinsic pathology and extrinsic medication, as dopaminergic drugs are strong risk factors for the manifestation of psychotic symptoms in PD: odds ratios for hallucinations are up to fivefold for dopamine agonist treatment compared to placebo in a meta-analysis (14). Furthermore, withdrawal of dopaminergic medication or dose reduction frequently brings relief from psychotic symptoms (11). There is already a large literature implicating dopamine and serotonin neurotransmission in the pathogenesis and treatment of schizophrenia spectrum psychosis and preliminary evidence linking fronto-striatal dopaminergic function to schizotypal traits in the general population (15–18). The recently identified role for serotonin in the pathology and treatment of PDP (19, 20) further weight for consideration of at least some potential commonalities in the pathophysiology of psychosis in schizophrenia spectrum conditions and PDP.
Abnormal dopaminergic (21, 22) and cortico-striatal (23, 24) function manifesting in altered processing of rewards and reward prediction, have been proposed as key factors contributing to the pathogenesis of psychotic symptoms. These accounts have received empirical support in the study of psychosis in schizophrenia spectrum disorders and preclinical models of psychosis (23, 25–31), but have not been tested in PDP. However, there is already evidence for this from a number of studies of both cortical and subcortical dysfunction during reward processing in PD (32–34). These factors merit the further investigation of cortico-striatal reward processing as a putative neurocognitive process potentially contributing to the pathogenesis of PDP.
In this study, we have focused on the neural basis of prediction of a reward and the representation of reward prediction error: processes with previous theoretical and empirical support for their involvement in the pathophysiology of psychotic symptoms in schizophrenia. A recent meta-analysis found evidence of ventral striatal dysfunction associated with both of these neurocognitive processes in schizophrenia spectrum psychosis (31), so we elected to study the ventral striatum (VS) as a region of interest (ROI) in our cohorts of PD patients. We aimed to complement this subcortical ROI with analysis within two cortical ROIs, the ventromedial prefrontal cortex and posterior cingulate cortex (PCC), where we previously identified a link between abnormal activation during reward processing and psychotic symptom expression (23).
Our hypothesis was that patients with PD psychosis should present reductions in PCC, ventromedial prefrontal cortex, and ventral striatal activations associated with reward anticipation and prediction error relative to PD patients without psychosis.
Materials and Methods
Participants
The final sample was composed of 53 participants: 17 patients with a diagnosis of PD without any psychotic symptoms (PD); 12 patients with a diagnosis of PD and a history of current or previous psychotic symptoms (lifetime CAARMS scoring equal to or greater than 3 in global and frequency scales) (PDP); 24 healthy volunteers, with no history of neurological, psychiatric, or medical disorders (controls). The initial sample was composed of 66 participants. Ten participants (8 PD, 1 PDP, 1 control) were excluded due to technical programming problems, which occurred during the task (responses not recorded, N = 9), or to the fMRI scanning (wrong scanning protocol, N = 1). Three additional subjects (2 PD, 1 control) were excluded because they did not complete the task. Patients were recruited via the PD research clinic at the John van Geest Centre for Brain Repair (VGB); all fulfilled the Queen Square Brain Bank Criteria for idiopathic PD (35) and remained on their usual medications during testing. Each patient’s dopaminergic drug regime was converted to an equivalent l-DOPA dose (36). Patients with dementia were excluded [operationalized as a Mini-Mental State Examination (37) score less than 24]. Descriptive statistics of the sample are reported in Table 1. All subjects had normal or corrected-to-normal visual acuity and were without any contraindications for MRI scanning.
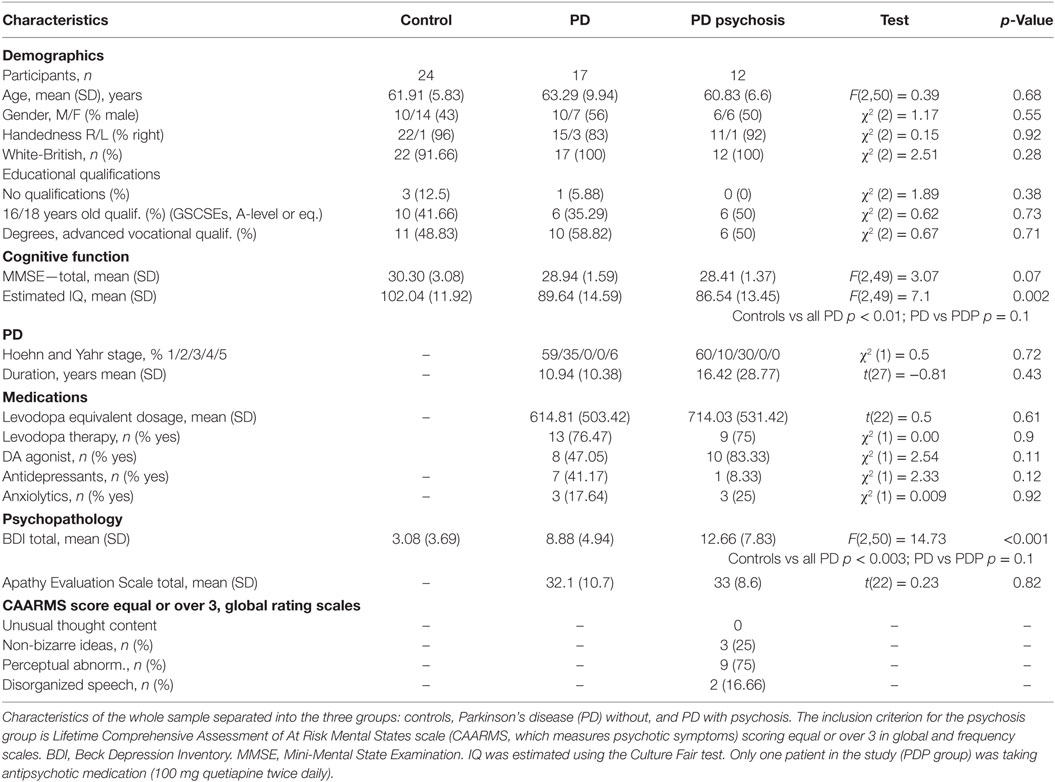
Table 1. Descriptive statistics.
Operational Definition of PDP
We defined the presence of PDP using a sensitive instrument measuring mild psychotic symptoms from the field of early detection of schizophrenia spectrum psychosis: the Comprehensive Assessment of At Risk Mental States (38, 39). This scale allows for sensitive measurement of a spectrum of psychotic experiences and includes measures on both the frequency and severity of symptoms. For example, the perceptual aberration scale scores of severity can range from 0 (no abnormality), 1 (questionable perceptual changes), 2 (heightened or dulled perceptions, illusions), through to 6 (a true hallucination that the patient believes true at the time and after the experience). The instrument takes these measures and operationalizes a categorical definition of attenuated psychosis (e.g., on the perceptual abnormalities scale, a 3 would be required for severity and frequency, indicating at least an intense illusion, “subject unsure of the nature of the experience,” with frequency of at least once per month if lasting over an hour per occasion or over 3 times per week if shorter). We reasoned that this threshold has been meaningful in clinical psychiatric practice (40) and could represent not only a sensitive but also a clinically meaningful way of classifying PD patients as having experienced psychosis or not. As we were interested in the propensity to psychosis in PD, we included patients in the PDP group with current and past (post PD onset) psychosis, even if the psychotic symptoms had resolved or improved by the time of the study.
Rating Scales
The Beck Depression Inventory (41) was used to assess depressive symptoms during the last 2 weeks. The Apathy Evaluation Scale (self-complete) assessed apathy (42). IQ was estimated using the Culture Fair test (43). The Hoehn and Yahr scale (44) was used to assess the stage of PD in patients.
fMRI Cognitive Task
During the fMRI scan, subjects performed a computerized instrumental learning task based on that used in previous studies (23, 45–48).
During the task, participants were presented with two fractal images and required to choose one of them, with a button press, in order to maximize their payoffs. Each choice was followed by a visual feedback indicating the associated outcome. There were three possible trials: during Reward Trials, one choice was associated with a £1 win in 80% of trials and with neutral feedback in 20% of trials [High-Likelihood (HL)], whereas the other choice was associated with a neutral outcome in 80% of trials and with a £1 win in 20% of trials [Low-Likelihood (LL)]; during Bivalent Trials, each choice was associated with a 50% chance of either losing or winning £1; during Neutral Trials, each choice was associated with a 80/20% chance of receiving two kinds of neutral feedback (Figure 1). Each trial type was repeated 30 times in a pseudo-random sequence, for a total of 90 trials. The order and position of the pictures presented were counterbalanced across trials of the same kind. To win money, the participants had to learn, by trial and error, to select the stimulus that was more likely to produce a reward. All participants were informed that the total amount won during the task would be paid to them at the end of the experimental session. All participants underwent a training session before entering the scan in which they received an explanation of the task, and practiced it for 5 min.
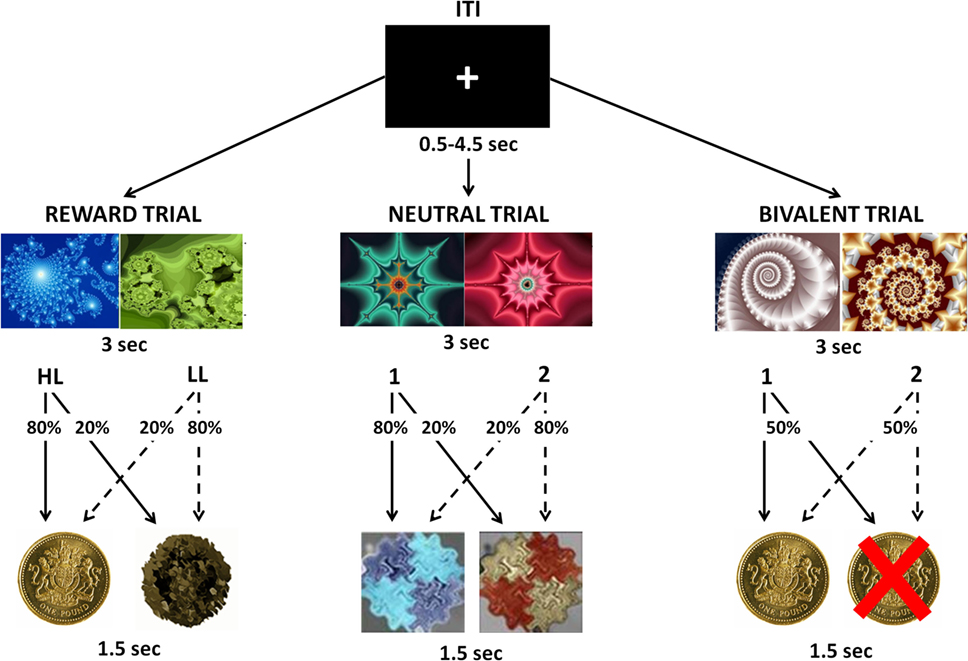
Figure 1. Visual representation of the instrumental conditioning task. Participants were presented with two fractal images and were required to choose one of them in order to maximize their payoffs. Each choice was followed by a visual feedback indicating the associated outcome. During Reward Trials, one choice was associated with an 80% chance to win £1 [High-Likelihood (HL)] and the other choice was associated with a 20% chance to win £1 [Low-Likelihood (LL)]. During Bivalent Trials, each choice was associated with a 50% chance of either losing or winning £1. During Neutral Trials, each choice was associated with an 80/20% chance of receiving two kinds of neutral feedbacks.
fMRI Data Acquisition and Analysis
A Siemens Magnetom Trio Tim operating at 3 T was used to collect imaging data. Gradient-echo, echo planar T2*-weighted images, depicting bold contrast were acquired from 35 non-contiguous oblique axial planes to minimize signal drop-out in ventral regions. TR was 1,620 ms; echo time was 30 ms; flip angle was 65°; in-plane resolution was 3.0 × 3.0, matrix size was 64 × 64; field of view was 192 mm × 192 mm, bandwidth 2,442 Hz/Px. A total of 550 volumes per subject were acquired (27 slices each of 2 mm thickness, interslice gap 1 mm). The first five volumes were discarded to allow for T1 equilibration effects. Slice-timing correction was applied.
The data were analyzed in SPM 12 software (Wellcome Department of Cognitive Neurology, London, UK). Images were realigned, spatially normalized to a standard template, and spatially smoothed with a Gaussian kernel (8 mm at full-width half-maximum). A high-pass filter was applied (128 Hz).
A within subject analysis (often referred to as “first-level” analysis) was undertaken, regressors (explanatory variables) used in the general linear model were as follows: bivalent cue presentation; neutral cue presentation; reward cue presentation; loss feedback during Bivalent Trials; win feedback during Bivalent Trials; neutral feedback 1 during Neutral Trials; neutral feedback 2 during Neutral Trials; neutral feedback during Reward Trials; and win feedback during Reward Trials. All regressors were modeled as 0 second events and convolved with the SPM canonical hemodynamic response function. Motion parameters were used as additional regressors to control for movement.
Contrasts of interest were computed at the individual subject level and then taken to a group level for statistical analysis. There were two main contrasts of interest: reward anticipation and prediction error. To measure reward anticipation, a contrast between the presentation of the reward cue minus the neutral cue was performed. To examine prediction error, a contrast between the presentation of win feedbacks during bivalent trials (where a reward is unexpected) minus a win on Reward Trials (where a win is expected) was performed.
The main contrast of interest at the group level was PDP in comparison to PD without psychosis, as this comparison controls for many of the potential confounds that would be needed in any comparison of PDP with controls. We focused on a priori-defined ROIs comprising medial orbitofrontal cortex, PCC, and bilateral VS (see Figure S1 in Supplementary Material). A single mask from these regions was created using spheres centered on the following MNI coordinates taken from the meta-analysis of reward anticipation by Liu et al. (49): 0, −30, 32 (posterior cingulate, 12 mm diameter); 0, 34, −8 (ventromedial prefrontal cortex, 8 mm diameter); and bilateral VS (12, 10, −4, 8 mm diameter, and −12, 10, −4, 8 mm diameter). Results were considered significant where peak voxel activation survived a family-wise error small volume correction across the voxels of this mask at p < 0.05. For display of the clusters that passed this small volume correction and calculation of their cluster size, the uncorrected threshold was p = 0.005.
Movement
To ensure that the three groups did not differ in the amount of movement during the fMRI scanning session, the averaged time series of translations and rotations along the three axes (x, y, z), as estimated during the realignment phase of pre-processing, were compared across groups.
Two separate mixed-effects models were used, with Group (controls/PD/PDP) as the independent variable and translations (millimeter) or rotations (degrees) as the dependent variable.
No group differences were found either in the amount of translations (p = 0.35; part. η2 = 0.04) or rotations (p = 0.32; part. η2 = 0.04).
Behavioral Analysis
To analyze participants’ performance during the task, three separate 3 × 2 mixed-effects models were performed for each trial condition (Reward/Neutral/Bivalent), using the total number of responses as the dependent variable (Figure 2A). Group (controls/PD/PDP) and Choice (HL/LL or 1/2) were used as the independent variables.
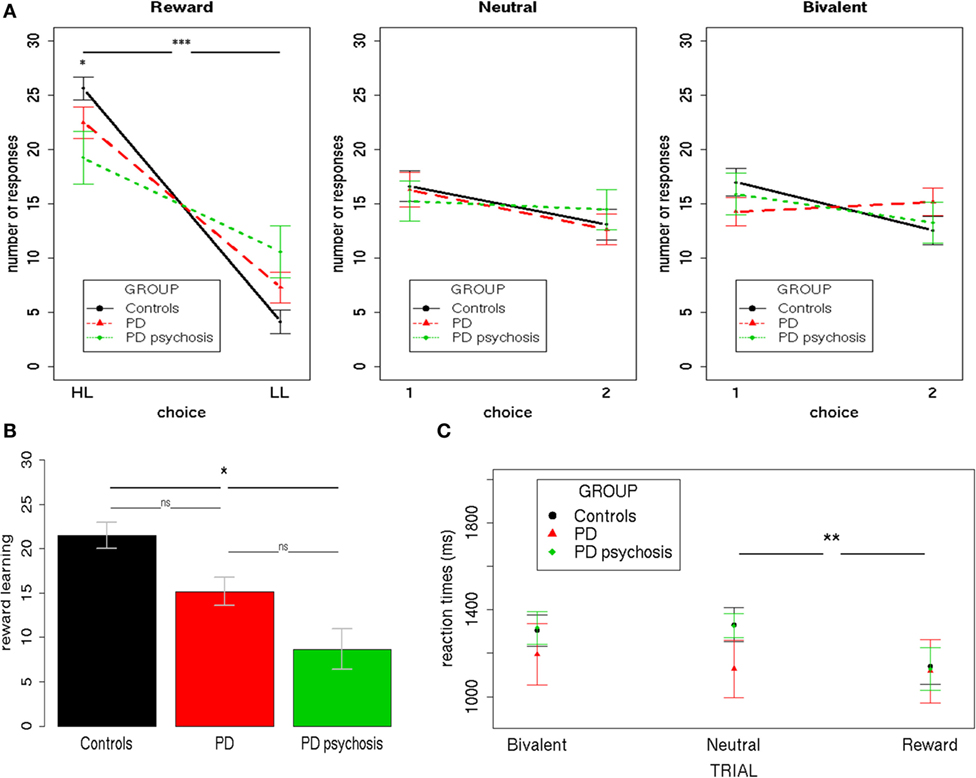
Figure 2. Behavioral performance. (A) The total number of responses during the three trial types (Reward/Neutral/Bivalent) for the three groups (controls/PD/PDP). (B) The reward learning index (HL–LL) for the three groups (controls/PD/Parkinson’s disease with psychosis). (C) Mean reaction times during the three trial types (Reward/Neutral/Bivalent) for the three groups (controls/PD/Parkinson’s disease with psychosis). HL, high-likelihood; LL, low-likelihood; 1, choice one; 2, choice two; PD, Parkinson’s disease. Bars indicate SE (***p < 0.0001; **p < 0.001; *p < 0.05).
Demographic and Clinical Analysis
Analyses of the demographic and clinical characteristics from the whole sample are reported in Table 1. χ2 was used to compare frequencies. For comparing means between groups, t-tests (comparison between PD/PDP) or ANOVAs (comparison between controls/PD/PDP) were used as appropriate. Significant differences were further investigated via Bonferroni-corrected post hoc analysis.
Results
Behavioral Results
Results from Reward Trials showed a significant main effect of Choice [F(1,50) = 83.22; two-tailed p < 0.001; part. η2 = 0.62], evidencing a higher number of HL over LL choices and a Group X Choice interaction [F(2,50) = 4,42; two-tailed p = 0.02; part. η2 = 0.15]. Bonferroni-corrected post hoc tests revealed a significant difference only between controls and PDP for both HL (p < 0.01) and LL (p < 0.01) choices. On average, HL choices were performed more by controls (mean = 25.62, SD = 5.21) than PDP (mean = 19.25, SD = 8.31); whereas LL choices were performed less by controls (mean = 4.12, SD = 5.31) than PDP (mean = 10.58, SD = 8.36).
Results from Neutral and Bivalent Trials showed no statistically significant effect (p’s > 0.1).
A further 3 × 3 mixed-effects model was performed using the reaction times as the dependent variable (Figure 2C). Group (controls/PD/PDP) and Trial type (Reward/Neutral/Bivalent) were used as the independent variables. Results showed a significant main effect of Trial type [F(1,87) = 5.09; two-tailed p = 0.007; part. η2 = 0.03]. Bonferroni-corrected post hoc tests revealed a significant difference only between Reward and Neutral Trials (p = 0.006). All other effects were not significant (p’s > 0.1).
Overall, these results indicate that participants, irrespective of the group, showed contingency learning during Reward Trials, expressing both as a preference for the HL choice (i.e., more frequently associated with a reward—80%) over the LL choice (i.e., less frequently associated with a reward—20%) and at faster reaction times when choosing during such trials, as compared with Bivalent and Neutral Trials—a well-replicated finding that has been referred to as “reinforcement-related speeding” (29, 45, 50). Nevertheless, controls presented a significantly higher ability to distinguish between choices with a higher and lower chance of rewards, as compared with PD patients with psychosis. Such a difference was subsequently directly tested by creating an index of the ability to learn from rewards (reward learning), obtained by subtracting for each subject the number of LL responses from the number of HL responses (Figure 2B). A mixed-effects model was performed using this reward learning index as the dependent variable and Group (controls/PD/PDP) as the independent variable. Results showed a significant main effect of Group [F(2,50) = 4.37; two-tailed p = 0.02; part. η2 = 0.15]. This further analysis confirmed what had been observed with the previous analysis: control participants were better able to differentiate between choices with higher and lower chance of reward and to use this information to adapt their behavior. In this regard, it is important to note that PD patients with psychosis still showed a significant difference between HL and LL choices (HL > LL), so they did learn the contingencies during Reward Trials. Critically, no significant difference between PD patients with and without psychosis was found.
Neither choice preferences nor differences in reaction times were evidenced in Neutral and Bivalent Trials.
We also examined the number of actual rewards received during Reward Trials. A mixed-effects model was performed using the number of rewards received as the dependent variable and Group (controls/PD/PDP) as the independent variable. Results showed a significant main effect of Group [F(2,50) = 5.11; two-tailed p = 0.01; part. η2 = 0.17]. Bonferroni-corrected post hoc tests revealed a significant difference only between controls and PDP (p < 0.02). On average, more rewards were received by controls (mean = 21.16, SD = 0.22) relative to PDP (mean = 17.25, SD = 4.39). Critically, no significant difference between PDP (mean = 17.25, SD = 4.39) and PD (mean = 18.17, SD = 4.39) was found (p > 0.9).
Imaging Results
Reward Anticipation
PD psychosis patients, as compared with PD patients, reported a significantly reduced activation in two clusters (Figure 3): one located within the PCC sphere [peak MNI coordinates x, y, z = 2, −20, 38; peak z = 3.65; peak p = 0.031 (small volume corrected); voxels = 59] and one located in the right VS [peak MNI coordinates x, y, z = 16, 16, −2; peak z = 3.60; peak p = 0.037 (small volume corrected); voxels = 21]. A third cluster located in the left VS showed marginally significant reduced activation in PDP as compared to PD patients [peak MNI coordinates x, y, z = −16, 16, −2; peak z = 3.03; peak p = 0.077 (small volume corrected); voxels = 44].
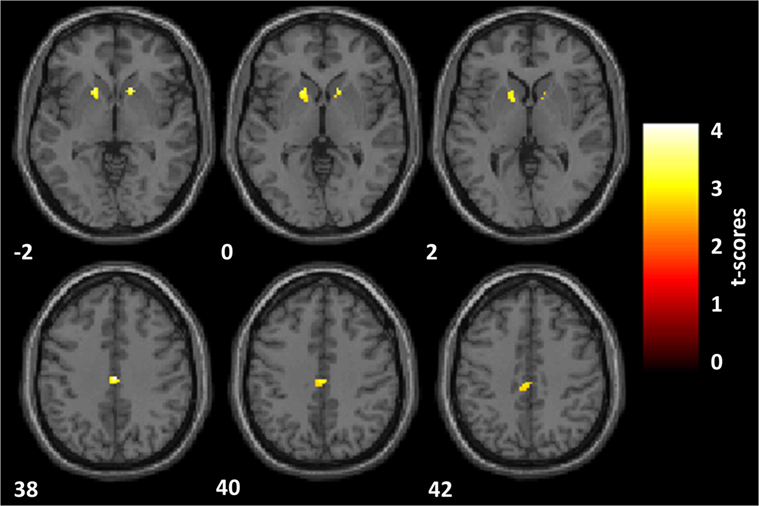
Figure 3. Reward anticipation. Statistical parametric maps of the reward anticipation calculated on the contrast of reward minus neutral cues onset. Analyses are restricted to the regions of interest and overlaid on a standard space structural image. Significant effects, thresholded at p = 0.005 and family-wise error corrected at p = 0.05, are shown in yellow. The left hemisphere is displayed on the left. MNI coordinates are reported. Parkinson’s disease patients with psychosis, as compared with Parkinson’s disease patients, showed a significantly reduced activation within the posterior cingulate cortex and the right ventral striatum (VS) and a marginally significant reduced activation in the left VS.
Parameter estimates of the two significantly different clusters (PCC and right VS) were extracted for the three groups (controls, PD, PDP) and statistically compared (Figure 4).
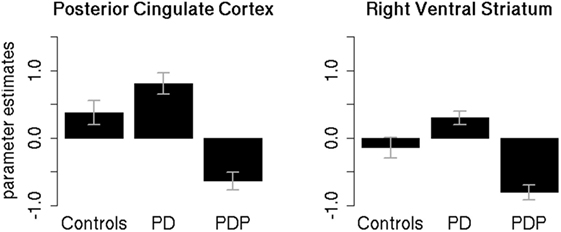
Figure 4. Mean parameter estimates in the significant clusters. Mean parameter estimates of the reward anticipation contrast, extracted for the three groups (controls, PD, PDP), from the significantly different clusters found within the posterior cingulate cortex and right ventral striatum regions of interest. PD, Parkinson’s disease patients; PDP, Parkinson’s disease patients with psychosis. Bars represent SE.
Two separate mixed-effects models, one for the PCC and one for the right VS, were performed using the extracted parameter estimates as the dependent variable. Group (controls/PD/PDP) was used as the independent variable. As the clusters are defined by differences between the PD and PDP groups, it is to be expected that these groups will show differences, but extraction of the cluster scores allows visualization of results and comparison against the healthy control group without PD.
Results in the PCC cluster showed a significant main effect of Group [F(2,50) = 4.89; two-tailed p = 0.01; part. η2 = 0.16]. Bonferroni-corrected post hoc tests revealed a significant difference between PD and PDP groups (p = 0.009). All other effects were not significant (p’s > 0.2).
Results on the right VS cluster showed a significant main effect of Group [F(2,50) = 5.54; two-tailed p = 0.007; part. η2 = 0.18]. Bonferroni-corrected post hoc tests revealed a significant difference between PD and PDP (p = 0.005) and a marginally significant difference between controls and PDP (p = 0.058). Controls and PD were not significantly different (p = 0.73).
There were no differences that survived correction for multiple comparison between PDP and PD groups on the whole-brain analysis.
Although the PD and PDP groups did not significantly differ on depressive symptoms, nevertheless, the PDP group had non-significantly more depressive symptoms than the PD group. We, therefore, compared the extracted parameter estimates between PDP and PD, controlling for depressive symptoms using ANCOVA: the difference between PDP and PD remained in the PCC cluster [F(2,26) = 10.35, p = 0.003] and in the right VS [F(2,26) = 12.92, p = 0.001].
Prediction Error
There were no differences that survived correction for multiple comparisons between PDP and PD groups on ROI or whole-brain analysis.
Discussion
In this study, we found abnormal brain activation in PDP associated with reward anticipation in the VS and PCC. The finding of psychosis-related abnormal striatal activity is novel for PD but in line with a significant body of literature in schizophrenia spectrum psychosis showing—across a wide range of procedures and manipulations—dysfunctional striatal processing of rewards (29, 51–56). Indeed, Radua et al. (31) recently confirmed on meta-analysis the presence of ventral striatal dysfunction associated with reward anticipation in schizophrenia spectrum psychosis.
We found a significant deficit in reward anticipation activation in the right VS in the PDP group, but only a marginally significant deficit in the left VS. This differential sensitivity might reflect important biological processes or could simply be due to chance. A recent neurofunctional meta-analysis by Radua et al. (31) directly investigated a possible VS lateralization of reward processing in psychosis. The authors analyzed 23 studies (917 patients) for reward anticipation, 9 studies (358 patients) for reward feedback, and 8 studies (314 patients) for reward prediction error, reporting no differences between left and right VS in any of the reward processes considered. Based on these results, we feel it would be premature to draw conclusions from slightly stronger group differences for the right VS found in the present study.
The functional neuroanatomical basis of psychotic symptoms in PD has received little study using fMRI. The present study is the first cognitive fMRI study, to our knowledge, to compare PD patients with and without a history of psychotic symptoms considered broadly as we have done here (although it is probable that some of the previous studies of hallucinations included patients with delusional ideation by chance). Previous studies have focused on comparing brain activation between PD patients with and without hallucinations, but only a handful have been conducted (57–61). Some studies did not make corrections for multiple comparisons, and one had a sample size of only three patients with hallucinations. Meppelink et al. (59) examined visual processing in PD patients with and without hallucinations and found robust evidence for visual processing deficits in the patients with hallucinations in the superior frontal, lingual, and fusiform gyri. There were additional areas of group difference that did not survive correction for multiple comparisons.
A number of related theories have posited that abnormal brain mechanisms of learning and predicting the environment could contribute to the pathogenesis of psychosis (21, 26, 28, 62). The striatum is key in learning and updating ones beliefs in the light of experience (63); hence striatal dysfunction is a plausible candidate process that could underpin the formation of abnormal beliefs (64). Contemporary neuroscience accounts of brain predictive processing emphasize the importance of prediction in determining perceptual experience, potentially explaining how brain predictive dysfunction could lead, not just to delusions, but also hallucinations (65). We previously showed that the degree of disruption to the representation of reward expected value in the PCC secondary to methamphetamine administration predicted the severity of psychotic symptoms induced by the drug (23); that previous study is consistent with the current results implicating posterior cingulate reward anticipation dysfunction in the pathophysiology of psychotic symptoms.
Ramírez-Ruiz et al. (60) examined activations elicited by a visual attention (face recognition) task. They found, using a cluster-correction threshold, reduced frontal activation in the superior and inferior frontal cortex in hallucinating patients compared to controls. Ramirez-Ruiz argued that their findings of frontal deficits in association with hallucination might indicate, in part, a particular difficulty in patients prone to visual hallucinations in differentiating between relevant and irrelevant visual information. A failure to differentiate between motivationally salient and irrelevant information has been argued to be a key mechanism in the pathophysiology of psychotic symptoms in schizophrenia (62). Our findings of reduced ventral striatal and PCC activation in the comparison of rewarding and neutral cues are interpretable as impairment in differential anticipation in relation to motivational salient and irrelevant information. Yao and colleagues’ (66) examination of resting-state fMRI differences between 12 PD patients with, and 12 without, visual hallucinations found resting-state differences in posterior cingulate connectivity between the groups, with no evidence of cortical thickness differences. Although we failed to find evidence of frontal reward prediction dysfunction in PDP or of prediction error-associated dysfunction in any of ROIs, this absence of evidence does not necessarily exclude these processes and regions being implicated in the pathogenesis of PDP.
Analysis of the behavioral performance in our study indicated that participants acquired a preference for the highly rewarded choice during Reward Trials (HL > LL), thus showing learning of the task contingencies. PD patients with psychosis displayed a significantly lower preference for the HL choice relative to controls. Nevertheless, in the PDP group, there was a significant preference for HL over LL choices, thus demonstrating learning. Critically, there were no differences between patients with and without psychosis, indicating that the imaging results were not confounded by learning deficits in PDP.
Although we cannot draw direct inferences about the neurochemical basis of our findings, it is possible that monoaminergic function may contribute to our findings. There is evidence that dopamine abnormalities in the striatum, and possibly also serotoninergic abnormalities, contribute to psychotic symptoms in schizophrenia (15, 17), and there are separate lines of evidence suggesting that dopaminergic and serotonergic factors may contribute to psychosis in PD (11, 19, 20). Previous studies have indicated that serotoninergic manipulations modulate fMRI activation and connectivity in the PCC (67, 68). Serotonin neurons have been shown to be important in representing reward cues and outcomes (69), and serotonin receptors, including 5HT2a, are widely expressed in the cortex, including in the posterior cingulate (70, 71). 5HT2a receptors have also been associated with more general cognitive flexibility (72–74), which could also be relevant for the generation of psychotic symptoms; one recent research study found evidence that that a failure to flexibly adapt learning to the degree of volatility in the environment could contribute to the pathogenesis of psychosis (75). Subcortical dopaminergic neurotransmission plays a key role in reward processing and motivation (76, 77), and there is also evidence for monoaminergic modulation of reward predicting activity even in posterior cortical regions (78).
Our study has a number of strength and limitations. A novel aspect of the study is the attempt to reconcile investigative methods (interview scales, fMRI paradigm) examining the physiology of psychosis in PD and schizophrenia spectrum conditions. The small number of patients with PDP is common in this research field but remains a limitation, and definitive conclusions will require a much larger sample size. Another limitation is that we made no attempt to characterize the neuropsychological performance of our participants. As the PD and PDP groups performed the fMRI task at a similar level, our results are unlikely to be driven by neuropsychological differences. Nevertheless, psychotic symptoms in PD might be associated with a specific neuropsychological profile, and characterizing this is an important goal of future research. A strength of our study is that the PD and PDP groups were well matched in terms of age and current medication: the daily l-DOPA equivalent dose did not differ across the PD groups. Levels of apathy and depression were comparable in the groups. However, a limitation of our study is that we did not record the timings of medication administration on the research day, and so we cannot eliminate the possibility that medication influence at the time of the scan might differ across the PD groups. There was no significant difference in terms of disease duration, although we note that our PDP group had a longer duration of illness than the PD group, in accordance with prior literature showing an association between psychotic symptoms and duration of illness (79–81).
Our findings suggesting a role for striatal and cingulate dysfunction in the pathogenesis of psychosis in PD should be taken in the context of an existing literature investigating PDP, mainly drawn from research modalities other than fMRI. This literature points to cortical abnormalities in PDP, such as reduced gray matter volume in the hippocampus and in frontal, parietal, and occipital cortices (6), and to the presence of Lewy bodies in frontal, cingulate, temporal, and occipital cortices on postmortem examination (9). Psychotic symptoms in PD are often comorbid with dementia and, in some cases, may presage incipient dementia (82). In the related disorder of dementia with Lewy bodies, posterior cortical–thalamic connectivity is related to hallucination severity (83). The dual syndrome hypothesis of cognitive impairments in PD (84) distinguishes between a non-dementia profile of fronto-striatal deficits (manifest in tests of executive function), and a profile of posterior cortical dysfunction with rapidly progressing cognitive decline to dementia. It is possible the psychotic symptoms in PD may emerge through either of these pathological processes or through an interaction of the two. In patients without global cognitive impairment, psychotic symptoms are often comorbid with other non-motor deficits, such as depression and sleep–wakefulness disturbance (3). A full account of psychosis in PD will need to take into account all of these complex factors in explaining why psychotic symptoms are commonly but not universally manifest in PD (12), which pathophysiological mechanisms are unique to PDP, and which are shared with mechanisms of psychotic experiences in schizophrenia or the general population (85). Our study adds to prior evidence of cortical dysfunction in PDP and suggests that there may be factors in common in the pathogenesis of schizophrenia spectrum psychosis and PD psychosis. It is possible that these abnormalities may contribute to the understanding on how monoaminergic function contributes to psychotic symptoms in PD; future pharmacological fMRI studies could specifically investigate this further by comparing, for example, the effects of 5-HT 2a inverse agonists on reward activation in PDP.
Ethics Statement
The research was approved by the Cambridgeshire 2 NHS Research Ethics Committee. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
SG designed the analysis, conducted the analysis, interpreted the results, and wrote the first draft of the paper. AJ designed the data collection, recruited the participants, conducted the data collection, and revised the paper for important intellectual content. GA designed and supervised the analysis and revised the paper for important intellectual content. AE designed the data collection, conducted the data collection, designed the analysis, and revised the paper for important intellectual content. PR designed the data collection, conducted the data collection, and revised the paper for important intellectual content. CT-S designed the analysis, performed analysis, and revised the paper for important intellectual content. TR designed the study, interpreted the results, and revised the manuscript for important intellectual content. RB designed the study, recruited the participants, interpreted the results, and revised the manuscript for important intellectual content. PF designed the study, interpreted the results, and revised the manuscript for important intellectual content. GM had the idea for the study, designed the study, directed the data collection and analysis, drafted the paper, and directed the revisions of the manuscript. All authors gave final approval of the manuscript.
Conflict of Interest Statement
TR is a consultant for and receives royalties from Cambridge Cognition; is a consultant for and received a research grant from Eli Lilly; received a research grant from GlaxoSmithKline; is a consultant for and received a research grant from Lundbeck; and is a consultant for Teva, Shire Pharmaceuticals, Mundipharma, and Otsuka. PF has consulted for GlaxoSmithKline and Lundbeck and received compensation. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Naresh Subramaniam for useful discussions. The authors are grateful for the help of the John van Geest Brain Repair Centre for help with participant recruitment and Wolfson Brain Imaging Centre staff for MRI data collection support.
Funding
This study was supported by a Medical Research Council Clinician Scientist award (G0701911), and an Isaac Newton Trust award to Dr. Murray; by support to Dr. Fletcher from the Wellcome Trust and Bernard Wolfe Health Neuroscience Fund; by a Wellcome Trust strategic award to University of Cambridge (097814/Z/11), by the NIHR Cambridge Biomedical Research Centre and University of Cambridge Behavioural and Clinical Neuroscience Institute, supported by a joint award from the Medical Research Council (G1000183) and Wellcome Trust (093875/Z/10/Z).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2017.00156/full#supplementary-material.
References
1. Aarsland D. Cognitive impairment in Parkinson’s disease and dementia with Lewy bodies. Parkinsonism Relat Disord (2016) 22:S144–8. doi:10.1016/j.parkreldis.2015.09.034
2. Gallagher DA, Schrag A. Psychosis, apathy, depression and anxiety in Parkinson’s disease. Neurobiol Dis (2012) 46(3):581–9. doi:10.1016/j.nbd.2011.12.041
3. Lee AH, Weintraub D. Psychosis in Parkinson’s disease without dementia: common and comorbid with other non-motor symptoms. Mov Disord (2012) 27(7):858–63. doi:10.1002/mds.25003
4. Park A, Stacy M. Non-motor symptoms in Parkinson’s disease. J Neurol (2009) 256(Suppl 3):293–8. doi:10.1007/s00415-009-5240-1
5. Weintraub D, Hurtig HI. Presentation and management of psychosis in Parkinson’s disease and dementia with Lewy bodies. Am J Psychiatry (2007) 164(10):1491–8. doi:10.1176/appi.ajp.2007.07040715
6. Carter R, Ffytche DH. On visual hallucinations and cortical networks: a trans-diagnostic review. J Neurol (2015) 262(7):1780–90. doi:10.1007/s00415-015-7687-6
7. Ramirez-Ruiz B, Junque C, Marti M-J, Valldeoriola F, Tolosa E. Cognitive changes in Parkinson’s disease patients with visual hallucinations. Dement Geriatr Cogn Disord (2007) 23(5):281–8. doi:10.1159/000100850
8. Fénelon G. Psychosis in Parkinson’s disease: phenomenology, frequency, risk factors, and current understanding of pathophysiologic mechanisms. CNS Spectr (2008) 13(S4):18–25. doi:10.1017/S1092852900017284
9. Gallagher DA, Parkkinen L, O’Sullivan SS, Spratt A, Shah A, Davey CC, et al. Testing an aetiological model of visual hallucinations in Parkinson’s disease. Brain (2011) 134(11):3299–309. doi:10.1093/brain/awr225
10. Janzen J, van’t Ent D, Lemstra AW, Berendse HW, Barkhof F, Foncke EMJ. The pedunculopontine nucleus is related to visual hallucinations in Parkinson’s disease: preliminary results of a voxel-based morphometry study. J Neurol (2012) 259(1):147–54. doi:10.1007/s00415-011-6149-z
11. Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging (2008) 25(8):665–82. doi:10.2165/00002512-200825080-00004
12. Friedman JH, Akbar U. Psychosis in Parkinson’s disease: unexplained observations in a seemingly simple model. Expert Rev Neurother (2016) 16(6):595–6. doi:10.1586/14737175.2016.1162713
13. Insel TR. The NIMH Research Domain Criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry (2014) 171(4):395–7. doi:10.1176/appi.ajp.2014.14020138
14. Etminan M, Gill S, Samii A. Comparison of the risk of adverse events with pramipexole and ropinirole in patients with Parkinson’s disease. Drug Safety (2003) 26(6):439–44. doi:10.2165/00002018-200326060-00005
15. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull (2009) 35:549–62. doi:10.1093/schbul/sbp006
16. Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Serotonin receptors: their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(7):1159–72. doi:10.1016/j.pnpbp.2003.09.010
17. Selvaraj S, Arnone D, Cappai A, Howes O. Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci Biobehav Rev (2014) 45:233–45. doi:10.1016/j.neubiorev.2014.06.005
18. Woodward ND, Cowan RL, Park S, Ansari MS, Baldwin RM, Li R, et al. Correlation of individual differences in schizotypal personality traits with amphetamine-induced dopamine release in striatal and extrastriatal brain regions. Am J Psychiatry (2011) 168(4):418–26. doi:10.1176/appi.ajp.2010.10020165
19. Cummings J, Isaacson S, Mills R, Williams H, Chi-Burris K, Corbett A, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet (2014) 383(9916):533–40. doi:10.1016/S0140-6736(13)62106-6
20. Hacksell U, Burstein ES, McFarland K, Mills RG, Williams H. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res (2014) 39(10):2008–17. doi:10.1007/s11064-014-1293-3
21. Miller R. Schizophrenic psychology, associative learning and the role of forebrain dopamine. Med Hypotheses (1976) 2(5):203–11. doi:10.1016/0306-9877(76)90040-2
22. Robbins TW. Relationship between reward-enhancing and stereotypical effects of psychomotor stimulant drugs. Nature (1976) 264(5581):57–9. doi:10.1038/264057a0
23. Bernacer J, Corlett PR, Ramachandra P, McFarlane B, Turner DC, Clark L, et al. Methamphetamine-induced disruption of frontostriatal reward learning signals: relation to psychotic symptoms. Am J Psychiatry (2013) 170(11):1326–34. doi:10.1176/appi.ajp.2013.12070978
24. Robbins TW. The case for frontostriatal dysfunction in schizophrenia. Schizophr Bull (1990) 16(3):391–402. doi:10.1093/schbul/16.3.391
25. Corlett PR, Honey GD, Aitken MRF, Dickinson A, Shanks DR, Absalom AR, et al. Frontal responses during learning predict vulnerability to the psychotogenic effects of ketamine. Arch Gen Psychiatry (2006) 63(6):611. doi:10.1001/archpsyc.63.6.611
26. Corlett PR, Murray GK, Honey GD, Aitken MRF, Shanks DR, Robbins TW, et al. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain (2007) 130(9):2387–400. doi:10.1093/brain/awm173
27. Deserno L, Boehme R, Heinz A, Schlagenhauf F. Reinforcement learning and dopamine in schizophrenia: dimensions of symptoms or specific features of a disease group? Front Psychiatry (2013) 4:172. doi:10.3389/fpsyt.2013.00172
28. Fletcher PC, Frith CD. Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci (2009) 10:48–58. doi:10.1038/nrn2536
29. Murray GK, Corlett PR, Clark L, Pessiglione M, Blackwell AD, Honey G, et al. Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Mol Psychiatry (2008) 13(3):267–76. doi:10.1038/sj.mp.4002058
30. Nielsen MØ, Rostrup E, Wulff S, Bak N, Lublin H, Kapur S, et al. Alterations of the brain reward system in antipsychotic naïve schizophrenia patients. Biol Psychiatry (2012) 71(10):898–905. doi:10.1016/j.biopsych.2012.02.007
31. Radua J, Schmidt A, Borgwardt S, Heinz A, Schlagenhauf F, McGuire P, et al. Ventral striatal activation during reward processing in psychosis. JAMA Psychiatry (2015) 72(12):1243. doi:10.1001/jamapsychiatry.2015.2196
32. Keitz M, Koerts J, Kortekaas R, Renken R, de Jong BM, Leenders KL. Prefrontal cortex and striatal activation by feedback in Parkinson’s disease. Brain Res (2008) 1236:225–33. doi:10.1016/j.brainres.2008.07.110
33. Schott BH, Niehaus L, Wittmann BC, Schütze H, Seidenbecher CI, Heinze HJ, et al. Ageing and early-stage Parkinson’s disease affect separable neural mechanisms of mesolimbic reward processing. Brain (2007) 130(9):2412–24. doi:10.1093/brain/awm147
34. Van Der Vegt JPM, Hulme OJ, Zittel S, Madsen KH, Weiss MM, Buhmann C, et al. Attenuated neural response to gamble outcomes in drug-naive patients with Parkinson’s disease. Brain (2013) 136(4):1192–203. doi:10.1093/brain/awt027
35. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry (1988) 51(6):745–52. doi:10.1136/jnnp.51.6.745
36. Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord (2010) 25(15):2649–53. doi:10.1002/mds.23429
37. Folstein MF, Folstein SE, McHugh PR. Mini-mental state. J Psychiatr Res (1975) 12(3):189–98. doi:10.1016/0022-3956(75)90026-6
38. Morrison AP, French P, Stewart SLK, Birchwood M, Fowler D, Gumley AI, et al. Early detection and intervention evaluation for people at risk of psychosis: multisite randomised controlled trial. BMJ (2012) 344:e2233. doi:10.1136/bmj.e2233
39. Yung AR, Yuen HP, McGorry PD, Phillips LJ, Kelly D, Dell’Olio M, et al. Mapping the onset of psychosis: the comprehensive assessment of at-risk mental states. Aust N Z J Psychiatry (2005) 39(11–12):964–71. doi:10.1111/j.1440-1614.2005.01714.x
40. Yung AR, Lin A. Psychotic experiences and their significance. World Psychiatry (2016) 15(2):130–1. doi:10.1002/wps.20328
41. Beck AT, Steer RA, Brown GK. Manual for the Beck Depression Inventory-II. San Antonio, TX: P. Corp (1996).
42. Marin RS, Biedrzycki RC, Firinciogullari S. Reliability and validity of the apathy evaluation scale. Psychiatry Res (1991) 38(2):143–62. doi:10.1016/0165-1781(91)90040-V
43. Cattell RB, Cattell HEP. Measuring Intelligence with the Culture Fair Tests. Champaign, IL: Institute for Personality and Ability Testing (1973).
44. Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. 1967. Neurology (1998) 50(2):318. doi:10.1212/WNL.50.2.318
45. Murray GK, Clark L, Corlett PR, Blackwell AD, Cools R, Jones PB, et al. Incentive motivation in first-episode psychosis: a behavioural study. BMC Psychiatry (2008) 8(1):34. doi:10.1186/1471-244X-8-34
46. O’Doherty JP. Reward representations and reward-related learning in the human brain: insights from neuroimaging. Curr Opin Neurobiol (2004) 14(6):769–76. doi:10.1016/j.conb.2004.10.016
47. Pessiglione M, Seymour B, Flandin G, Dolan RJ, Frith CD. Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature (2006) 442(7106):1042–5. doi:10.1038/nature05051
48. Seymour B, Daw N, Dayan P, Singer T, Dolan R. Differential encoding of losses and gains in the human striatum. J Neurosci (2007) 27(18):4826–31. doi:10.1523/JNEUROSCI.0400-07.2007
49. Liu X, Hairston J, Schrier M, Fan J. Common and distinct networks underlying reward valence and processing stages: a meta-analysis of functional neuroimaging studies. Neurosci Biobehav Rev (2011) 35(5):1219–36. doi:10.1016/j.neubiorev.2010.12.012
50. Cools R, Blackwell A, Clark L, Menzies L, Cox S, Robbins TW. Tryptophan depletion disrupts the motivational guidance of goal-directed behavior as a function of trait impulsivity. Neuropsychopharmacology (2005) 30(7):1362. doi:10.1038/sj.npp.1300704
51. de Leeuw M, Kahn RS, Vink M. Fronto-striatal dysfunction during reward processing in unaffected siblings of schizophrenia patients. Schizophr Bull (2015) 41(1):94–103. doi:10.1093/schbul/sbu153
52. Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [(18)F/(11)C]-DOPA PET studies. Schizophr Bull (2013) 39(1):33–42. doi:10.1093/schbul/sbr180
53. Juckel G, Schlagenhauf F, Koslowski M, Wüstenberg T, Villringer A, Knutson B, et al. Dysfunction of ventral striatal reward prediction in schizophrenia. Neuroimage (2006) 29(2):409–16. doi:10.1016/j.neuroimage.2005.07.051
54. Juckel G, Friedel E, Koslowski M, Witthaus H, Ozgürdal S, Gudlowski Y, et al. Ventral striatal activation during reward processing in subjects with ultra-high risk for schizophrenia. Neuropsychobiology (2012) 66(1):50–6. doi:10.1159/000337130
55. Schlagenhauf F, Sterzer P, Schmack K, Ballmaier M, Rapp M, Wrase J, et al. Reward feedback alterations in unmedicated schizophrenia patients: relevance for delusions. Biol Psychiatry (2009) 65(12):1032–9. doi:10.1016/j.biopsych.2008.12.016
56. Schlagenhauf F, Huys QJM, Deserno L, Rapp MA, Beck A, Heinze H-JJ, et al. Striatal dysfunction during reversal learning in unmedicated schizophrenia patients. Neuroimage (2014) 89:171–80. doi:10.1016/j.neuroimage.2013.11.034
57. Holroyd S, Wooten GF. Preliminary fMRI evidence of visual system dysfunction in Parkinson’s disease patients with visual hallucinations. J Neuropsychiatry Clin Neurosci (2006) 18(3):402–4. doi:10.1176/jnp.2006.18.3.402
58. Lenka A, Jhunjhunwala KR, Saini J, Pal PK. Structural and functional neuroimaging in patients with Parkinson’s disease and visual hallucinations: a critical review. Parkinsonism Relat Disord (2015) 21(7):683–91. doi:10.1016/j.parkreldis.2015.04.005
59. Meppelink AM, de Jong BM, Renken R, Leenders KL, Cornelissen FW, van Laar T. Impaired visual processing preceding image recognition in Parkinson’s disease patients with visual hallucinations. Brain (2009) 132(11):2980–93. doi:10.1093/brain/awp223
60. Ramírez-Ruiz B, Martí M-J, Tolosa E, Falcón C, Bargalló N, Valldeoriola F, et al. Brain response to complex visual stimuli in Parkinson’s patients with hallucinations: a functional magnetic resonance imaging study. Mov Disord (2008) 23(16):2335–43. doi:10.1002/mds.22258
61. Stebbins GT, Goetz CG, Carrillo MC, Bangen KJ, Turner DA, Glover GH, et al. Altered cortical visual processing in PD with hallucinations: an fMRI study. Neurology (2004) 63(8):1409–16. doi:10.1212/01.WNL.0000141853.27081.BD
62. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry (2003) 160(1):13–23. doi:10.1176/appi.ajp.160.1.13
63. O’Doherty J, Dayan P, Schultz J, Deichmann R, Friston K, Dolan RJ. Dissociable roles of ventral and dorsal striatum in instrumental conditioning. Science (2004) 304(5669):452–4. doi:10.1126/science.1094285
64. Ziauddeen H, Murray GK. The relevance of reward pathways for schizophrenia. Curr Opin Psychiatry (2010) 23(2):91–6. doi:10.1097/YCO.0b013e328336661b
65. Corlett PR, Fletcher PC. Delusions and prediction error: clarifying the roles of behavioural and brain responses. Cogn Neuropsychiatry (2015) 20(2):95–105. doi:10.1080/13546805.2014.990625
66. Yao H, Zhou B, Zhang Z, Wang P, Guo Y, Shang Y, et al. Longitudinal alteration of amygdalar functional connectivity in mild cognitive impairment subjects revealed by resting-state fMRI. Brain Connect (2014) 4(5):361–70. doi:10.1089/brain.2014.0223
67. Kraus C, Ganger S, Losak J, Hahn A, Savli M, Kranz GS, et al. Gray matter and intrinsic network changes in the posterior cingulate cortex after selective serotonin reuptake inhibitor intake. Neuroimage (2014) 84:236–44. doi:10.1016/j.neuroimage.2013.08.036
68. Matthews SC, Simmons AN, Strigo IA, Arce E, Stein MB, Paulus MP. Escitalopram attenuates posterior cingulate activity during self-evaluation in healthy volunteers. Psychiatry Res (2010) 182(2):81–7. doi:10.1016/j.pscychresns.2010.02.003
69. Luo M, Zhou J, Liu Z. Reward processing by the dorsal raphe nucleus: 5-HT and beyond. Learn Mem (2015) 22(9):452–60. doi:10.1101/lm.037317.114
70. Hall H, Farde L, Halldin C, Lundkvist C, Sedvall G. Autoradiographic localization of 5-HT(2A) receptors in the human brain using [3H]M100907 and [11C]M100907. Synapse (2000) 38(4):421–31. doi:10.1002/1098-2396(20001215)38:4<421:AID-SYN7>3.0.CO;2-X
71. Vogt BA, Plager MD, Crino PB, Bird ED. Laminar distributions of muscarinic acetylcholine, serotonin, GABA and opioid receptors in human posterior cingulate cortex. Neuroscience (1990) 36(1):165–74. doi:10.1016/0306-4522(90)90359-C
72. Macoveanu J, Hornboll B, Elliott R, Erritzoe D, Paulson OB, Siebner H, et al. Serotonin 2A receptors, citalopram and tryptophan-depletion: a multimodal imaging study of their interactions during response inhibition. Neuropsychopharmacology (2013) 38264(10):996–1005. doi:10.1038/npp.2012.264
73. Poyurovsky M, Koren D, Gonopolsky I, Schneidman M, Fuchs C, Weizman A, et al. Effect of the 5-HT2 antagonist mianserin on cognitive dysfunction in chronic schizophrenia patients: an add-on, double-blind placebo-controlled study. Eur Neuropsychopharmacol (2003) 13(2):123–8. doi:10.1016/S0924-977X(02)00155-4
74. Üçok A, Alpsan H, Çakır S, Saruhan-Direskeneli G. Association of a serotonin receptor 2A gene polymorphism with cognitive functions in patients with schizophrenia. Am J Med Genet B Neuropsychiatr Genet (2007) 144B(5):704–7. doi:10.1002/ajmg.b.30463
75. Vinckier F, Gaillard R, Palminteri S, Rigoux L, Salvador A, Fornito A, et al. Confidence and psychosis: a neuro-computational account of contingency learning disruption by NMDA blockade. Mol Psychiatry (2016) 21(7):946–55. doi:10.1038/mp.2015.73
76. Robbins TW, Everitt BJ. A role for mesencephalic dopamine in activation: commentary on Berridge (2006). Psychopharmacology (2007) 191(3):433–7. doi:10.1007/s00213-006-0528-7
77. Schultz W. Reward functions of the basal ganglia. J Neural Transm (2016) 123(7):679–93. doi:10.1007/s00702-016-1510-0
78. Apitz T, Bunzeck N. Early effects of reward anticipation are modulated by dopaminergic stimulation. PLoS One (2014) 9(10):e108886. doi:10.1371/journal.pone.0108886
79. Fénelon G, Alves G. Epidemiology of psychosis in Parkinson’s disease. J Neurol Sci (2010) 289(1–2):12–7. doi:10.1016/j.jns.2009.08.014
80. Forsaa EB, Larsen JP, Wentzel-Larsen T, Goetz CG, Stebbins GT, Aarsland D, et al. A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol (2010) 67(8):996–1001. doi:10.1001/archneurol.2010.166
81. Gibson G, Mottram PG, Burn DJ, Hindle JV, Landau S, Samuel M, et al. Frequency, prevalence, incidence and risk factors associated with visual hallucinations in a sample of patients with Parkinson’s disease: a longitudinal 4-year study. Int J Geriatr Psychiatry (2013) 28(6):626–31. doi:10.1002/gps.3869
82. Ibarretxe-Bilbao N, Ramirez-Ruiz B, Junque C, Marti MJ, Valldeoriola F, Bargallo N, et al. Differential progression of brain atrophy in Parkinson’s disease with and without visual hallucinations. J Neurol Neurosurg Psychiatry (2010) 81(6):650–7. doi:10.1136/jnnp.2009.179655
83. Delli Pizzi S, Maruotti V, Taylor J-P, Franciotti R, Caulo M, Tartaro A, et al. Relevance of subcortical visual pathways disruption to visual symptoms in dementia with Lewy bodies. Cortex (2014) 59:12–21. doi:10.1016/j.cortex.2014.07.003
84. Kehagia AA, Barker RA, Robbins TW. Cognitive impairment in Parkinson’s disease: the dual syndrome hypothesis. Neurodegener Dis (2012) 11(2):79–92. doi:10.1159/000341998
Keywords: ventral striatum, posterior cingulate cortex, prediction error, reinforcement learning, fMRI
Citation: Garofalo S, Justicia A, Arrondo G, Ermakova AO, Ramachandra P, Tudor-Sfetea C, Robbins TW, Barker RA, Fletcher PC and Murray GK (2017) Cortical and Striatal Reward Processing in Parkinson’s Disease Psychosis. Front. Neurol. 8:156. doi: 10.3389/fneur.2017.00156
Received: 15 January 2017; Accepted: 03 April 2017;
Published: 24 April 2017
Edited by:
Stefano L. Sensi, University of California Irvine, USAReviewed by:
Stefano Delli Pizzi, University of Chieti-Pescara, ItalyNicola B. Mercuri, University of Rome Tor Vergata, Italy
Copyright: © 2017 Garofalo, Justicia, Arrondo, Ermakova, Ramachandra, Tudor-Sfetea, Robbins, Barker, Fletcher and Murray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graham K. Murray, Z20yODVAY2FtLmFjLnVr