 Pedro J. Garcia-Ruiz
Pedro J. Garcia-Ruiz Alberto J. Espay
Alberto J. Espay- 1Department of Neurology, Fundacion Jimenez Diaz, Madrid, Spain
- 2James J. and Joan A. Gardner Family Center for Parkinson’s Disease and Movement Disorders, University of Cincinnati, Cincinnati, OH, USA
There are two central premises to this evolutionary view of Parkinson disease (PD). First, PD is a specific human disease. Second, the prevalence of PD has increased over the course of human history. Several lines of evidence may explain why PD appears to be restricted to the human species. The major manifestations of PD are the consequence of degeneration in the dopamine-synthesizing neurons of the mesostriatal neuronal pathway. It is of note the enormous expansion of the human dopamine mesencephalic neurons onto the striatum compared with other mammals. Hence, an evolutionary bottle neck was reached with the expansion of the massive nigrostriatal axonal arborization. This peculiar nigral overload may partly explain the selective fragility of the human dopaminergic mesencephalic neurotransmission and the unique presence of PD in humans. On the other hand, several facts may explain the increasing prevalence of PD over the centuries. The apparently low prevalence of PD before the twentieth century may be related to the shorter life expectancy and survival compared to present times. In addition, changes in lifestyle over the course of human history might also account for the increasing burden of PD. Our hunter-gatherers ancestors invested large energy expenditure on a daily basis, a prototypical physical way of life for which our genome remains adapted. Technological advances have led to a dramatic reduction of physical exercise. Since the brain release of neurotrophic factors (including brain-derived neurotrophic factor) is partially exercise related, the marked reduction in exercise may contribute to the increasing prevalence of PD.
Parkinson disease (PD), the second most prevalent neuropsychiatric neurodegenerative disorder (1–5), increases exponentially with aging (3, 4). While PD likely represents a syndrome of several molecular subtypes, with a small proportion arising from well-defined genetic abnormalities (1, 5), the data accumulated suggest two common denominators: alpha synuclein accumulation into cytoplasmic Lewy bodies and dopamine deficiency as the casualty of neuronal loss in the nigrostriatal neurons (1, 5). The convergence of diverse biological processes, including genetic, environment, and behavioral factors in the development of PD suggests that evolutionary processes may explain in part the vulnerability to this form of neurodegeneration (6). From a phylogenetic standpoint, PD is prevalent among human primates at the top of the evolutionary chain but evidence for its presence in other species is lacking.
There are two central premises to the evolutionary view of PD. First, PD is a specific human disease. Despite the importance of animal models to the understanding of potential pathogenic mechanism in PD (5), there is no naturally occurring parkinsonism in non-human species: PD is a specific human disease. Second, the prevalence of PD has increased over the course of human history (7), and probably even over the last century (8). Several converging evolutionary theories can explain these observations.
PD is Specifically Human
Many neurological diseases can be found in non-human mammals (9–12) both acquired and hereditary (such as myelopathy, brain tumors, epilepsy, muscular dystrophy, and narcolepsy, to mention a few). However, Alzheimer disease and PD are considered specific to Homo sapiens (13–15). While there are useful animal models of PD including MPTP and alpha-synuclein-overexpressing transgenic mouse models, which may recapitulate important clinical features of the human disorders (5, 14), especially in aged monkeys (16, 17), no spontaneous akinetic-rigid syndrome is known to occur in wild mammals including non-human primates.
Several lines of evidence serve to explain why PD appears to be restricted to the human species. The major motor manifestations of PD are the consequence of degeneration in the dopamine-synthesizing neurons of the mesostriatal neuronal pathway (1, 13). In particular, the dopaminergic fields in the striatum of neurons from the substantia nigra pars compacta (SNpc) can be far more extensive than those of other neurotransmitter types (18). The elegant anatomical studies of Matsuda and colleagues illustrated that the magnitude of the SNpc-to-neostriatum relationship whereby the axon of a single tyrosine hydroxylase-positive dopaminergic neuron in the rat occupies up to 6% of the volume of the striatum (18). Conversely, the arborization of the human mesostriatal neurons occupies a much larger volume of striatum compared to other vertebrates. Vernier and colleagues suggested that the peculiar phenotype of the dopamine mesencephalic neurons, selected during vertebrate evolution, and reshaped in the human lineage, has rendered these neurons particularly prone to oxidative stress (13). Bolam and Pissadaki also stressed the enormous expansion of the human dopamine mesencephalic neurons onto the striatum compared with other mammals (19, 20). Some figures are impressive: the volume of the striatum has increased by approximately 300-fold from rats (20 mm3) to humans (6,280 mm3), but the number of dopaminergic neurons in the SNpc has increased by only 32-fold (rats, 12,000; humans, 382,000) (19). Thus, human dopamine nigral neurons must give rise to axons 10 times the size and 10 times the number of synapses compared to rats (19, 20). Pissadaki and Bolam elegantly proposed that this axonal architecture creates high-energy demands on dopamine-producing nigral neurons to maintain cell functions including the propagation of action potentials (19, 20). As these authors suggested, the nigral neurons are on the edge of an energetic catastrophe (19). Hence, an evolutionary bottle neck was reached with the expansion of the massive (and unmyelinated) nigrostriatal axonal arborization. This peculiar nigral overload may partly explain the selective fragility of the human dopaminergic mesencephalic neurotransmission and the unique presence of PD in humans (13, 19, 20).
Finally, it is relevant to note that this phylogenetically overloaded system needed to regulate very complex motor behaviors. Motor control among mammalians became progressively more sophisticated as hominids developed such skilled motor behaviors as stone tools manufacturing (21). The basal ganglia are known to be critical for the acquisition, improvement, and sustainability of skilled motor behaviors (22, 23).
PD Prevalence has Increased Over the Course of Human Story
The “official” history of PD is quite recent. PD was named after the contribution of James Parkinson in 1817 (24). Although James Parkinson was first in bringing attention to this particular disease, aspects of the disease had been described by Galen, Sylvius, Juncker, and Cullen (24). Early artistic descriptions of PD can also be found in painting (25, 26). In any case, until the second part of the nineteenth century, PD was virtually unknown to physicians. It is relevant to ask why a motorically obvious disease such as PD was unnoticed until relatively recently. A plausible explanation is that PD was actually rare. James Parkinson described only six patients, and over the next century, even experienced neurologists such as Gowers and Wilson, and others did not report a large number of parkinsonian patients (27–30), Gowers only studied 80 cases in detail: “Of eighty cases, of which I have notes, fifty were men and thirty women” (27) and Wilson included in his textbook (1940) a table with 383 patients “… combining the collection of Erb, Peterson, Bychowski, Ruherman and Manschot” (28). Over the last few decades, the incidence and prevalence seem to have increased according to several authors (4, 8, 31), although for others, PD may have reached a nadir (31–34).
The apparently low prevalence of PD before the twentieth century may be related to the shorter life expectancy and survival compared to present times (4, 8, 31, 34). In addition, historical changes in lifestyle might also account for the increasing burden of these diseases. As O’Keefea and colleagues noted, our ancestors, surviving as hunter-gatherers, required large energy expenditures on a daily basis, and this way of life represented the prototypical physical activity regimen for which our genome adapted (35). O’Keefea et al. and Mattson suggested that technological advances (from the agricultural revolution to the industrial revolution and to the recent digital revolution) have led to progressive but dramatic reduction in physical exercise and overall activity in daily routines (35, 36). Critically, however, our inherent exercise capabilities and needs, selected after thousands of years of evolution, remain essentially unchanged as compared with those of our ancestors (35). That physical exercise is important for everyone is supported by the fact that many chronic ailments and age-related diseases including PD are associated with sedentarism (34–37). Of course, it is impossible to know exactly the collective level of daily physical exercise achieved by our stone age ancestors, but O’Keefea and colleagues suggested that the energy expenditure on physical activity of hunter-gatherers was at least four times that of the modern humans (35).
Physical activity has not only proven beneficial in preventing but also attenuating the motor and cognitive developments of PD (34, 37–40). Moderate or vigorous physical activity is associated with a >30% reduction in PD risk (40). In addition, a protective role of physical activity on PD risk is also supported by animal models (41, 42). Physical exercise seems to be one of the few practical strategies with neuroprotective potential across all neurodegenerative diseases (35, 37, 43). The putative neuroprotective benefits of physical exercise may be explained by several mechanisms including the production of neuroprotective factors such brain-derived neurotrophic factor and glial cell-derived neurotrophic factor (42–45). Vigorous exercise should, therefore, be accorded a central place in the primary prevention and secondary management of PD (35, 37–40, 43).
In summary, important evolutionary clues may explain the specificity of PD to human species, as shown in Figure 1:
1. The phenotype of the dopamine mesencephalic neurons, selected during vertebrate evolution and reshaped in the human lineage, has rendered these neurons particularly prone to oxidative stress, and thus, to the selective neurodegeneration of PD (13, 18).
2. The size and arborization of theses expanded mesencephalic neurons compared with other vertebrates have made human dopamine nigral neurons vulnerable to an energetic crisis (19, 20).
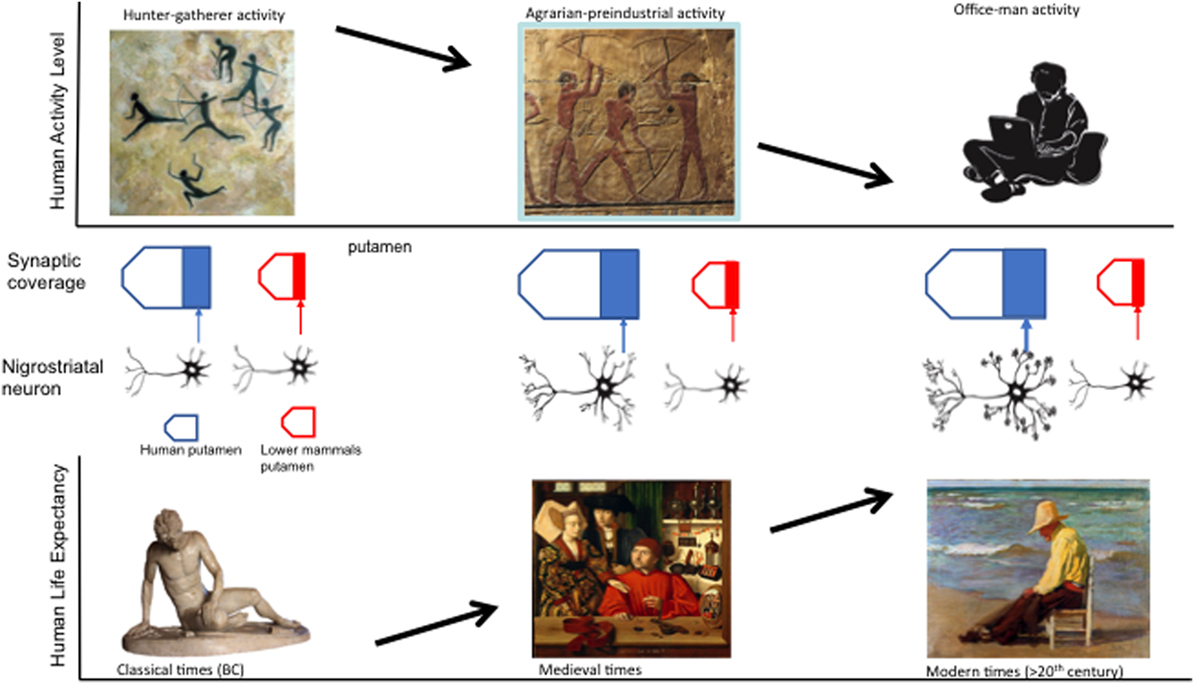
Figure 1. Evolutionary and behavioral variables that may contribute to the presence of Parkinson disease: increasing nigroestriatal arborization and synaptic putaminal coverage from lower species to humans, decreasing activity levels, and increasing human life span over the course of millennia.
Finally, other evolutionary concepts may partially explain the increasing presence of PD in our society, including the following:
1. The apparently low prevalence of PD before the twentieth century may be related to a shorter life expectancy and survival (4, 8, 31, 34).
2. Changes in lifestyle over the course of human history might also account for the increasing burden of PD. Our hunter-gatherers ancestors invested large energy expenditure on a daily basis, a prototypical physical way of life for which our genome remains adapted (35, 36).
3. Technological advances have led to a dramatic reduction of physical exercise for daily routines (35, 36).
4. Since the brain release of neurotrophic factors is partially exercise-related (44, 45), the reduction in exercise at a societal level may contribute to the increasing prevalence of PD and other neurodegenerative disorders in our era of digital revolution.
Author Contributions
PG-R: conception and design, interpretation of data, drafting the submitted material, and critical review. AE: conception and design, interpretation of data, critical revision, and supervision.
Conflict of Interest Statement
PG-R received research support from Allergan and UCB, personal compensation as a consultant/scientific advisory board from Italfarmaco, Britannia, Bial, and Zambon and speaking honoraria from Italfarmaco, UCB, Zambon, Allergan, and Abbvie. AE has received grant support from NIH, Great Lakes Neurotechnologies, and The Michael J. Fox Foundation; personal compensation as a consultant/scientific advisory board member for Abbvie, TEVA, Impax, Merz, Acadia, Cynapsus, Lundbeck, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from Abbvie, UCB, USWorldMeds, Lundbeck, Acadia, the American Academy of Neurology, and the Movement Disorders Society. He serves as Associate Editor of the Journal of Clinical Movement Disorders and on the editorial board of Parkinsonism and Related Disorders.
References
1. Kalia LV, Lang AE. Parkinson’s disease. Lancet (2015) 386:896–912. doi: 10.1016/S0140-6736(14)61393-3
2. de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology (2004) 63:1240–4.
3. de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol (2006) 5:525–35. doi:10.1016/S1474-4422(06)70471-9
4. Savica R, Grossardt B, Bower J, Ahlskog E, Rocca W. Time trends in the incidence of Parkinson disease. JAMA Neurol (2016) 73:981–9. doi:10.1001/jamaneurol.2016.0947
5. Espay AJ, Brundin P, Lang AE. Precision medicine for disease modification in Parkinson disease. Nat Rev Neurol (2017) 13:119–26. doi:10.1038/nrneurol.2016.196
6. Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science (2004) 305:1733–6. doi:10.1126/science.1095292
8. Liu CC, Li CY, Lee PC, Sun Y. Variations in incidence and prevalence of Parkinson’s disease in Taiwan: a population-based nationwide study. Parkinsons Dis (2016) 2016:8756359. doi:10.1155/2016/8756359
9. Chen L, Brown RE, McKenna JT, McCarley RW. Animal models of narcolepsy. CNS Neurol Disord Drug Targets (2009) 8:296–308. doi:10.2174/187152709788921717
10. Schwartz M, Lamb CR, Brodbelt DC, Volk HA. Canine intracranial neoplasia: clinical risk factors for development of epileptic seizures. J Small Anim Pract (2011) 52:632–7. doi:10.1111/j.1748-5827.2011.01131.x
11. McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech (2015) 8:195–213. doi:10.1242/dmm.018424
12. Hamilton T, Glass E, Drobatz K, Agnello KA. Severity of spinal cord dysfunction and pain associated with hydrated nucleus pulposus extrusion in dogs. Vet Comp Orthop Traumatol (2014) 27:313–8. doi:10.3415/VCOT-13-06-0076
13. Vernier P, Moret F, Callier S, Snapyan M, Wersinger C, Sidhu A. The degeneration of dopamine neurons in Parkinson’s disease: insights from embryology and evolution of the mesostriatocortical system. Ann N Y Acad Sci (2004) 1035:231–49. doi:10.1196/annals.1332.015
14. Aron Badin R, Vadori M, Cozzi E, Hantraye P. Translational research for Parkinson’s disease: the value of pre-clinical primate models. Eur J Pharmacol (2015) 759:118–26. doi:10.1016/j.ejphar.2015.03.038
15. Glassa D, Arnold S. Some evolutionary perspectives on Alzheimer’s disease pathogenesis and pathology. Alzheimers Dement (2012) 8:343–51. doi:10.1016/j.jalz.2011.05.2408
16. Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, et al. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis (2007) 26:56–65. doi:10.1016/j.nbd.2006.11.013
17. Hurley PJ, Elsworth JD, Whittaker MC, Roth RH, Redmond DE Jr. Aged monkeys as a partial model for Parkinson’s disease. Pharmacol Biochem Behav (2011) 99:324–32. doi:10.1016/j.pbb.2011.05.007
18. Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci (2009) 29:444–53. doi:10.1523/JNEUROSCI.4029-08.2009
19. Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord (2012) 27:1478–83. doi:10.1002/mds.25135
20. Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci (2013) 7:13. doi:10.3389/fncom.2013.00013
21. Rein R, Bril B, Nonaka T. Coordination strategies used in stone knapping. Am J Phys Anthropol (2013) 150:539–50. doi:10.1002/ajpa.22224
22. Hikosaka O, Nakamura K, Sakai K, Nakahara H. Central mechanisms of motor skill learning. Curr Opin Neurobiol (2002) 12:217–22. doi:10.1016/S0959-4388(02)00307-0
23. Ungerleider LG, Doyon J, Karni A. Imaging brain plasticity during motor skill learning. Neurobiol Learn Mem (2002) 78:553–64. doi:10.1006/nlme.2002.4091
25. García de Yébenes J, García Ruiz P. Description of camptocormia in Parkinson disease by Francisco de Zurbarán. Neurologia (2000) 15:265–6.
26. Garcia Ruiz PJ, Ruiz Ezquerro JJ, Garcia Torres A, Fanjul S. Ancient descriptions of movement disorders: Cathedral el Burgo de Osma (Soria, Spain). J Neurol (2006) 253:731–4. doi:10.1007/s00415-006-0100-8
27. Gowers WR. Paralysis agitans. In: Gowers WR, editor. A Manual of Diseases of the Nervous System. (Vol. II), London: J &A Churchill (1886). p. 589–608.
28. Kinnier Wilson SA. Paralysis agitans. In: Bruce N, editor. Neurology by Kinnier Wilson. (Vol. II), London: Edward Arnold & Co. (1940). p. 787–804.
29. Young AW. A clinical analysis of an extrapyramidal syndrome; paralysis agitans and postencephalitic parkinsonism. J Neurol Psychopathol (1927) 8:9–18. doi:10.1136/jnnp.s1-8.29.9
31. Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology (2007) 68:384–6. doi:10.1212/01.wnl.0000247740.47667.03
32. Rocca WA, Bower JH, McDonnell SK, Peterson BJ, Maraganore DM. Time trends in the incidence of parkinsonism in Olmsted County, Minnesota. Neurology (2001) 57:462–7. doi:10.1212/WNL.57.3.462
33. Wickremaratchi MM, Perera D, O’Loghlen C, Sastry D, Morgan E, Jones A, et al. Prevalence and age of onset of Parkinson’s disease in Cardiff: a community based cross sectional study and meta-analysis. J Neurol Neurosurg Psychiatry (2009) 80:805–7. doi:10.1136/jnnp.2008.162222
34. Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol (2016) 15:1257–72. doi:10.1016/S1474-4422(16)30230-7
35. O’Keefea JH, Vogelb R, Laviec CJ, Cordain L. Exercise like a hunter-gatherer: a prescription for organic physical fitness. Prog Cardiovasc Dis (2011) 53:471–9. doi:10.1016/j.pcad.2011.03.009
36. Mattson MP. Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res Rev (2015) 20:37–45. doi:10.1016/j.arr.2014.12.011
37. La Hue S, Comella C, Tanner CM. The best medicine? The influence of physical activity and inactivity on Parkinson’s disease. Mov Disord (2016) 31:1444–54. doi:10.1002/mds.26728
38. Corcos DM, Robichaud JA, David FJ, Leurgans SE, Vaillancourt DE, Poon C, et al. A two-year randomized controlled trial of progressive resistance exercise for Parkinson’s disease. Mov Disord (2013) 28:1230–40. doi:10.1002/mds.25380
39. David FJ, Robichaud JA, Leurgans SE, Poon C, Kohrt WM, Goldman JG, et al. Exercise improves cognition in Parkinson’s disease: the PRET-PD randomized, clinical trial. Mov Disord (2015) 30:1657–63. doi:10.1002/mds.26291
40. Yang F, Trolle Lagerros Y, Bellocco R, Adami HO, Fang F, Pedersen NL, et al. Physical activity and risk of Parkinson’s disease in the Swedish National March Cohort. Brain (2015) 138:269–75. doi:10.1093/brain/awu323
41. Klein C, Rasińska J, Empl L, Sparenberg M, Poshtiban A, Hain EG, et al. Physical exercise counteracts MPTP-induced changes in neural precursor cell proliferation in the hippocampus and restores spatial learning but not memory performance in the water maze. Behav Brain Res (2016) 307:227–38. doi:10.1016/j.bbr.2016.02.040
42. Real CC, Ferreira AF, Chaves-Kirsten GP, Torrão AS, Pires RS, Britto LR. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience (2013) 237:118–29. doi:10.1016/j.neuroscience.2013.01.060
43. Ahlskog JE. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology (2011) 77:288–94. doi:10.1212/WNL.0b013e318225ab66
44. da Silva PG, Domingues DD, de Carvalho LA, Allodi S, Correa CL. Neurotrophic factors in Parkinson’s disease are regulated by exercise: evidence-based practice. J Neurol Sci (2016) 363:5–15. doi:10.1016/j.jns.2016.02.017
Keywords: Parkinson disease, evolutionary approach, life expectancy, dopaminergic field, physical activity
Citation: Garcia-Ruiz PJ and Espay AJ (2017) Parkinson Disease: An Evolutionary Perspective. Front. Neurol. 8:157. doi: 10.3389/fneur.2017.00157
Received: 09 February 2017; Accepted: 03 April 2017;
Published: 01 May 2017
Edited by:
Oscar Arias-Carrión, Hospital General Dr. Manuel Gea González, MexicoReviewed by:
Gennaro Pagano, King’s College London, UKGraziella Madeo, National Institutes of Health (NIH), USA
Copyright: © 2017 Garcia-Ruiz and Espay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro J. Garcia-Ruiz, cGdhcmNpYUBmamQuZXM=
†These authors have equally contributed to this work.